
Abstract
Pyruvate carboxylase (PC), an anaplerotic enzyme, plays an essential role in various cellular metabolic pathways including gluconeogenesis, de novo fatty acid synthesis, amino acid synthesis, and glucose-induced insulin secretion. Deregulation of PC expression or activity has long been known to be associated with metabolic syndrome in several rodent models. Accumulating data in the past decade clearly showed that deregulation of PC expression is associated with type 2 diabetes in humans, while targeted inhibition of PC expression in a mouse model reduced adiposity and improved insulin sensitivity in diet-induced type 2 diabetes. More recent studies also show that PC is strongly involved in tumorigenesis in several cancers, including breast, non-small cell lung cancer, glioblastoma, renal carcinoma, and gall bladder. Systems metabolomics analysis of these cancers identified pyruvate carboxylation as an essential metabolic hub that feeds carbon skeletons of downstream metabolites of oxaloacetate into the biosynthesis of various cellular components including membrane lipids, nucleotides, amino acids, and the redox control. Inhibition or down-regulation of PC expression in several cancers markedly impairs their growth ex vivo and in vivo, drawing attention to PC as an anti-cancer target. PC has also exhibited a moonlight function by interacting with immune surveillance that can either promote or block viral infection. In certain pathogenic bacteria, PC is essential for infection, replication, and maintenance of their virulence phenotype.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Pyruvate and central metabolism
Glucose is the major fuel for most living organisms. Once transported into the cell, glucose is metabolized through glycolysis, producing pyruvate as the final product of the pathway. Under an anaerobic conditions, pyruvate is reduced in the cytoplasm by lactate dehydrogenase, producing lactate as an end product [1]. However, an accumulation of lactate can potentially lower pH of cells, perturbing cellular acid–base homeostasis. Muscle exports lactate out via the monocarboxylate transporters [2, 3] and preventing muscle cells from acidosis. In the presence of oxygen, pyruvate enters mitochondria via the mitochondrial pyruvate carrier [4, 5]. At this point, pyruvate may be further metabolized through two routes, depending on the tissues. In non-gluconeogenic tissues such as muscle, brain, etc., pyruvate is decarboxylated to form acetyl-CoA catalyzed by the pyruvate dehydrogenase complex, a multi-subunit enzyme. The acetyl carbons of acetyl-CoA subsequently enter the tricarboxylic acid (TCA) cycle, producing NADH and FADH2 that are both eventually oxidized via the respiratory chain to produce ATP. In gluconeogenic tissues where pyruvate carboxylase (PC) is highly abundant, > 80% of pyruvate entering mitochondria is carboxylated by this enzyme to form oxaloacetate [6]. Oxaloacetate formed by PC is not only used as a starting substrate for gluconeogenesis but also used for oxidation of glucose- or fatty acid-derived acetyl-CoA in the TCA cycle [7,8,9].
Pyruvate carboxylation
As mentioned, pyruvate carboxylation increases the concentration of oxaloacetate in the TCA cycle. PC is a member of the biotin-dependent enzyme family that contains biotin as a prosthetic group [8]. Native mammalian PC is a tetramer composed of four identical subunits each of 1178 amino acids (129 kDa). Each subunit contains four functional domains, the biotin carboxylase, carboxyltransferase, biotin carboxyl carrier, and allosteric or tetramerization domains [10, 11]. PC catalyzes the conversion of pyruvate to oxaloacetate via a two-step reaction. The first step occurs in the biotin carboxylase domain where biotin is carboxylated via a carboxyphosphate intermediate formed in a reaction between ATP and bicarbonate [8]. The carboxybiotin formed in the first step then swings to the carboxyltransferase domain where –CO2− is transferred to pyruvate, forming oxaloacetate [8]. The reaction involves intermolecular catalysis where the biotin of one subunit is carboxylated in that subunit’s biotin carboxylase domain, but transfers its –CO2− to pyruvate in a partner subunit’s carboxyltransferase domain [10]. In brain, PC supports the biosynthesis of neurotransmitter substances including glutamate and γ-amino-butyric acid, while in adipose tissue, PC supports de novo fatty acid synthesis and glyceroneogenesis [7].
In many but not all organisms, PC activity is modulated by allosteric activation by acetyl CoA which may accumulate through excessive fatty acid oxidation [12] or through diminution of TCA cycle intermediates owing to removal for biosynthetic purpose [13]. Mammalian PCs are extremely sensitive to acetyl CoA and most show only very low levels of activation in its absence [14]. Similarly, many bacterial PCs are highly dependent on acetyl CoA for activity, e.g., Rhizobium etli PC, however, some bacterial PCs are not regulated by acetyl CoA at all, e.g., Pseudomonas aeruginosa [14].
On the other hand, accumulation of the TCA cycle intermediate, α-ketoglutarate or l-glutamate derived by reductive amination of α-ketoglutarate are allosteric inhibitors of mammalian PCs [15]. l-Aspartate that is formed directly from oxaloacetate by transamination acts as an allosteric inhibitor of many bacterial PCs [15, 16].
PC deficiency and deregulation
Given the importance of oxaloacetate in various biochemical pathways, perturbation of oxaloacetate production by PC can produce serious diseases such as type 2 diabetes, neurological disorder, or cancers. In certain pathogenic bacteria, PC is a vital gene, enabling them to survive in mammalian hosts, e.g., Listeria monocytogenes [17] as discussed below.
Diabetes
The control of systemic glucose homeostasis is pivotal to the body because it enables various tissues or organs to maintain their normal function during the feeding and starvation cycle. In response to low plasma glucose, glucagon is released and acts metabolically by stimulating hepatic glycogenolysis and gluconeogenesis, increasing plasma glucose [18, 19]. This glucagon-induced hepatic glucose production protects brain and red blood cells from starvation-induced organ failure. An opposite situation occurs during the feeding period when insulin is released in response to elevated plasma glucose and acts by increasing the rate of glucose uptake into peripheral tissues while suppressing hepatic gluconeogenesis [20, 21]. Therefore, insulin resistance results in the failure of peripheral tissues to uptake glucose and de-repression of gluconeogenesis during the postprandial period, contributing to hyperglycemia [22, 23] (Fig. 1). Studies performed in rodent models showed that insulin strongly inhibits transcription of genes encoding cytosolic phosphoenolpyruvate carboxykinase (PEPCK-C) and glucose-6-phosphatase 1 (G6Pase1), but this applies only weakly to PC [24]. During the feeding period, insulin blocks lipolysis in adipose tissue by inhibiting adipocyte triglyceride lipase and hormone-sensitive lipase activities, lowering delivery of free fatty acids to liver. However, under insulin resistance, in which the rate of lipolysis from adipose tissue becomes high, more free fatty acids are delivered to the liver for oxidation, thereby producing large amounts of acetyl-CoA (Fig. 2). High levels of hepatic acetyl-CoA in turn allosterically activate PC activity, increasing the rate of hepatic glucose production [12, 22]. This lipolysis-driven hepatic gluconeogenesis also operates in a rodent model of type 1 diabetes where the leptin level is low [12]. Low circulating leptin under type 1 diabetic conditions, in turn, induces the activity of the hypothalamic–pituitary–adrenal axis, causing the secretion of more corticosterone. This hormone enhances whole body lipolysis and fatty acid oxidation, producing a large amount of acetyl-CoA, which in turn activates PC activity and increases hepatic gluconeogenesis [25]. In addition to allosteric regulation by acetyl-CoA, the acetylation status of PC protein is also an important posttranslational mechanism during the development of diabetes. Kendrick et al. [26] have shown that high fat diet-induced insulin-resistant mice display increased acetylation of PC which might contribute to enhanced PC activity during the development of diabetes.

Lipolysis-induced hepatic PC overexpression. Insulin resistance of white adipose tissue results in excessive lipolysis contributing to elevated levels of plasma free fatty acids (FFA) [14]. Oxidation of FFA in liver produces acetyl-CoA which acts as an allosteric activator of PC. Increased PC activity increases gluconeogenesis, contributing to hyperglycemia. Elevated circulating plasma glucose and triglycerides cause glucotoxicity and lipotoxicity that impairs glucose-induced insulin secretion from pancreatic beta cells and exacerbate diabetes [23]

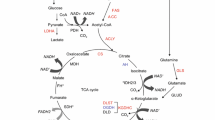
Anaplerotic role of PC in supporting growth of various cancer models. Glycolysis and glutaminolysis are two major biochemical reactions that provide carbon skeletons for cellular biosynthesis. Pyruvate carboxylation (green arrow route) by PC feeds glycolysis-derived pyruvate to oxaloacetate into TCA cycle. Oxaloacetate can be converted to aspartate via direct transamination. In contrast, reductive carboxylation (blue arrow route) feeds carbon skeletons from glutamine to isocitrate via IDH1 or IDH2. Isocitrate is further converted to citrate before decarboxylated to acetyl-CoA, a precursor of long chain fatty acid synthesis. a In NSCLC and breast cancer, pyruvate carboxylation is a major anaplerotic reaction that supports cancer growth while glutaminolysis is a minor pathway. b In glioma, glutaminolysis is the major anaplerotic reaction that supports cancer growth while pyruvate carboxylase is essential during glutamine-deprivation condition. c In low-grade glioma carrying loss-of-function mutations of IDH1 or IDH2, reductive carboxylation of glutamine-derived α-ketoglutarate to citrate is inhibited, forcing cancer cells to rely on pyruvate carboxylation to support biosynthesis. d In cancers carrying loss-of-function mutations of SDH or FH gene, cells rely on pyruvate carboxylation and reductive carboxylation from glutamine to support their growth
Consistent with the importance of PC in controlling hepatic glucose production, Lee et al. [27] have shown that pyruvate carboxylation flux in liver of high fat diet-induced insulin-resistant mice is increased, accompanied by increased hepatic glucose production. In agreement with this study, Burgess et al. [28] have shown that PEPCK-C does not solely regulate overall rate of hepatic gluconeogenesis because a 90% reduction of PEPCK-C expression in liver of mice only mildly affects hepatic glucose output. A more conclusive report was obtained from the study performed in humans which demonstrated that PC but not PEPCK-C or G6Pase1 is associated with the glycemic index of type 2 diabetic patients [29, 30]. A later study by the same group of investigators showed that targeted inhibition of hepatic PC expression can lower plasma glucose levels, adiposity, and improved hepatic insulin sensitivity in high fat diet-induced diabetic rat, underscoring the importance of PC during the development of type 2 diabetes [30].
Not only does PC control hepatic gluconeogenesis, it also serves a non-gluconeogenic role in pancreatic islets. In this cell type, oxaloacetate is not used as the starting gluconeogenic substrate because this tissue does not contain all the gluconeogenic enzymes [31, 32]. Instead, rapid formation of oxaloacetate in pancreatic beta cells drives the export of citrate and malate out of the mitochondria into the cytosol where both metabolites are eventually converted back to pyruvate by cytosolic malic enzyme, concomitant with the production of NADPH. Pyruvate then re-enters the mitochondria in a cycle, producing a large amount of NADPH that acts as a coupling factor to drive insulin exocytosis during glucose stimulation [33, 34]. As the rate of pyruvate cycling and PC activity are directly proportional to the rate of glucose entry into beta cells, this indicates that PC is part of the glucose sensing system that enables them to secrete insulin when extracellular concentrations of glucose are above the physiological range [35, 36]. Suppression of PC expression in insulinoma cells impairs glucose-induced insulin release [37, 38]. Lowered levels of PC expression in pancreatic beta cells are also observed in genetically type 2 diabetic rodents and type 2 diabetic patients, suggesting that impaired insulin release in both rodents and humans may be secondary to the deregulation of PC expression [39,40,41]. Regulation of PC expression in pancreatic beta cells is also positively controlled by several transcription factors such as carbohydrate-responsive binding protein, PPAR-α, FoxA2, MAFA [42,43,44,45,46]. Tumor suppressor protein p53 has recently been reported to negatively regulate PC expression in pancreatic beta cells [47].
Cancers
In response to rapid proliferation, most cancers rewire their cellular metabolism in order to meet the demand for the structural components of the cells. One of such changes is the oxidation of glucose via lactic acid production, regardless of the presence of oxygen, known as aerobic glycolysis or Warburg’s effect [48]. Consequently, cancers slow down the use of mitochondria to oxidize pyruvate as a result of redirecting pyruvate toward aerobic glycolysis. Inhibiting pyruvate oxidation in mitochondria provides several benefits for cancer cells. Firstly, rapid entry of glucose into the glycolytic pathway increases the abundance of glycolytic intermediates, enabling them to flow into pentose phosphate pathway that provides the ribose sugar for nucleotide synthesis and NADPH required in redox maintenance and in de novo fatty acid synthesis [49, 50]. Secondly, inhibiting pyruvate entry into mitochondria allows TCA cycle intermediates to be available for biosynthetic purposes because several TCA cycle intermediates are also used for biosynthesis. For example, citrate is used as an acetyl-group donor for de novo fatty acid or cholesterol synthesis. Oxaloacetate is also used as a substrate of aspartate production while α-ketoglutarate can be directly converted to glutamate. Therefore, pyruvate carboxylation ensures that the availability of these substrates is not limited during cancer proliferation. For this reason, cancer cells increase the replenishment of TCA cycle intermediates by increasing anaplerosis that comprises two reactions. The first one is glutaminolysis that feeds 5-carbon skeletons from glutamine via glutamate. Glutaminolysis involves a two-step reaction, one of which involves the conversion of glutamine to glutamate by glutaminase followed by the second reaction involving oxidative deamination of glutamate to α-ketoglutarate by glutamate dehydrogenase (Fig. 2a). This anaplerotic reaction is crucial for many cancers because carbon skeletons in glutamine are used as biosynthetic precursors via the TCA cycle. Inhibition of glutaminolysis markedly inhibits cancer growth and survival. While glutaminolysis has long been known to be crucial [51], accumulating evidence has now revealed that pyruvate carboxylation is also crucial in many cancers, depending on genetic backgrounds and metabolic conditions.
As shown in Fig. 2a, the non-small cell lung cancer (NSCLC) uses pyruvate carboxylation as the main anaplerotic reaction to support its growth while glutaminolysis only plays a supportive role [52, 53]. A later study from the same group showed that silencing PC expression reduced tumor growth ex vivo and in a xenograft mouse model. This tumor growth restriction is accompanied by impaired mitochondrial anaplerosis and lowered levels of nucleotides, aspartate, and glutathione [53]. The genetic background and tumor tissue origin can also strongly influence metabolic reprogramming in many cases. For example, K-ras oncogene mutations can strongly influence metabolic phenotypes of many cancers [54]. Davidson et al. [55] showed that K-ras-driven NSCLC metabolizes nutrients differently when they are grown ex vivo and in lung. When of lung origin, NSCLC relies on pyruvate carboxylation to support its growth while it relies on glutaminolysis during ex vivo growth. Suppression of PC expression also blocks the ability of NSCLC to induce tumor formation in lung tissue of mice [56]. Similar to NSCLC, the role of PC has been well studied in breast cancer. Phannasil et al. (2015) [57] showed that the expression of PC is highly correlated with the tumor’s mass and advanced stage, independent of the status of their estrogen, progesterone, or epidermal growth factor receptors. Suppression of PC expression in highly invasive cells impaired mitochondrial anaplerosis, perturbing several biosynthetic pathways including those of non-essential amino acids, nucleotides, and fatty acids [58]. Tumor microenvironment also contributes to metabolic phenotype to some extent. Christen et al. [56] showed that breast cancer metastasized to lung tissue possesses increased levels of mitochondrial pyruvate accompanied by increased levels of PC mRNA and enzyme activity. Up-regulation of PC expression enables breast cancer cells to become more resistant to glutamine deprivation. This study indicates that pyruvate carboxylation becomes a major anaplerotic route when glutamine is restricted. Accumulation of mitochondrial pyruvate might also serve as a metabolic signal that dictates aggressive phenotype as observed in certain cancers [59, 60].
The use of pyruvate carboxylation to support growth was also well studied in cancers bearing loss of function mutations of TCA cycle enzymes. Mutations of isocitrate dehydrogenase (IDH) are found to make a big impact on cellular energetics [61]. IDH comprises three isoenzymes produced from different genes. IDH3 is a mitochondrial enzyme that catalyzes the non-reversible decarboxylation of isocitrate to α-ketoglutarate, concomitant with NADH production. IDH2, which is also a mitochondrial enzyme, catalyzes the reversible decarboxylation of isocitrate to α-ketoglutarate, concomitant with production of NADPH. In contrast, IDH1 is a cytosolic isoenzyme which can also catalyze the reversible conversion of isocitrate to α-ketoglutarate. As shown in Fig. 2a, the presence of IDH2 allows glutamine to directly convert to citrate in the reverse direction of TCA cycle without going through succinate dehydrogenase (SDH) which is normally inhibited during hypoxic conditions [62]. The ability of cancer cells to convert glutamine to citrate via IDH1 or IDH2 is known as the reductive carboxylation [63]. It is interesting that non-defective IDH1/2-glioblastoma uses glutaminolysis during glutamine-dependent growth condition while using pyruvate carboxylation to support its growth during glutamine-deprivation condition [64] (Fig. 2b). However, low-grade glioblastomas, which often carry loss-of-function mutations of the IDH1/IDH2 gene, adapt to this metabolic defect by up-regulating expression of PC to bypass the reductive carboxylation from glutamine. This metabolic flexibility allows IDH1/2-defective gliomas to produce citrate from oxaloacetate provided by PC [65]. In this manner, most glucose-derived pyruvate enters the pyruvate carboxylation flux because of inactivation of PDH flux by the pyruvate dehydrogenase kinase (Fig. 2c). The use of PC as the major anaplerotic reaction was also reported in renal carcinoma bearing mutations of SDH which functions in converting succinate to fumarate. A loss-of-function mutation of the SDH gene would also truncate the TCA cycle, inhibiting conversion of glutamine-derived α-ketoglutarate to citrate via succinate due to the absence of SDH, as shown in Fig. 2d. However, SDH-deficient parenchymal or renal cancers up-regulate the expression of PC, enabling them to bypass the defect in the conversion of glutamine to citrate because oxaloacetate formed by PC can be combined with two carbons from acetyl-CoA to form citrate, hence replacing production of citrate from glutamine [66, 67]. Remarkably, suppression of PC expression in this SDH-deficient renal carcinoma markedly inhibits growth although this tumor contains wild-type IDH2 and IDH3 that should ideally allow glutamine to support tumor growth via reductive carboxylation. Significantly, a growth defect of SDH-defective renal carcinoma deprived of PC expression can be fully rescued by exogenous aspartate, indicating that aspartate production from oxaloacetate is absolutely crucial for cellular proliferation, whereas the reductive carboxylation from glutamine cannot fulfill aspartate production in SDH-defective tumors when PC is absent. The crucial role of aspartate in supporting growth of SDH-defective renal carcinoma is also consistent with the studies in breast cancer and NSCLC that knocking down expression of PC markedly lowers cellular aspartate levels which in turn affects biosynthesis of nucleotides and restricts their growth. As noted in Fig. 2d, SDH-deficient renal cancer redirects glucose-derived pyruvate into mitochondria in order to produce enough pyruvate as the starting substrate for citrate production [66].
Not only in cancer cells, PC also appears to support growth of the surrounding fibroblast known as the cancer-associated fibroblasts (CAFs). Linares et al. [68] have recently reported that p62-deficient prostate cancer stroma, grown under glutamine deprivation conditions, up-regulates expression of PC and asparagine synthase (ASN) to support itself and prostate cancer. In this manner, PC provides oxaloacetate which is further converted to aspartate and subsequently converted to asparagine by ASN. These two amino acids are crucial nitrogen sources that support proliferation of both CAFs and cancer cells. Silencing expression of PC in CAFs markedly inhibits their growth under this nitrogen-limited condition [68]. In gall bladder cancer, the levels of PC protein and mRNA expression were regulated via the interaction with the long non-coding RNA, GSASPC [69]. This long non-coding RNA binds to PC, thus sequestering it from being a tumor-promoting enzyme. In patients with highly invasive gall bladder cancer, GSASPC was down-regulated and accompanied by increased PC expression. Although this mechanism is thought to be a key step to regulate the abundance of PC in supporting anaplerosis in gall bladder cancer, the detailed mechanism is not well understood. Although PC plays an essential role in supporting growth in many cancers, it does not appear to be important in melanoma which prefers glutaminolysis rather than pyruvate carboxylation as the main anaplerotic route to support fatty acid synthesis [70].
The use of pyruvate carboxylation as an escape route of survival was also reported in some cancers that were treated with the monocarboxylic acid transporters (MCTs). MCTs function as bidirectional lactate pumps, allowing lactate/pyruvate to transport in and out of cells [3]. Acute exposure of human lymphoma and colon carcinoma with AZD3965, an MCT1 inhibitor, causes the accumulation of intracellular lactate, lowering pH of the cells and also blocks regeneration of NAD+, thereby disrupting glycolysis. However, chronic exposure of these cancers to this agent triggers the re-oxidation of lactate to pyruvate accompanied by increased expression of PC and PDH, which allows pyruvate to enter the mitochondria via pyruvate carboxylation and pyruvate dehydrogenation, respectively, to overcome energy deprivation caused by inhibition of glycolysis by MCT blockage. This evidence demonstrates that PC at least in part is necessary to maintain cell survival under drug stress [71]. Reed et al. [72] also reported that the anti-proliferative action of anti-cancer drugs, bezafibrate and medroxyprogesterone, on primary acute myeloid leukemia is facilitated by pyruvate carboxylation. Exposure of this cancer to these drugs triggers the production of malonate, a reactive oxygen species (ROS) that is primarily produced from oxaloacetate through pyruvate carboxylation. This drug-induced cell death is not only associated with production of ROS but is also associated with the massive draining of oxaloacetate from the TCA cycle toward malonate production [72]. Accumulation of malonate can potentially inhibit SDH activity [73], further impairing cellular energetics, leading to cell death. Wilmanski et al. [74, 75] also reported that PC is part of the anti-oxidant system in the k-ras-transformed MCF-10A breast cancer cell line by working in concert with malic enzyme in providing NADPH, which is used to detoxify ROS-induced during vitamin D (1α,25-dihydroxyvitamin D) treatment. Chronic treatment of cells with vitamin D down-regulates PC expression, rendering cells susceptible to oxidative damage.
At present, it is not known what cellular signaling drives PC expression during tumorigenesis in many cancers. Lee et al. (2013) [76] show that the Wnt signaling pathway during epithelial–mesenchymal transition contributes to up-regulation of PC because treatment of the MCF-7 breast cancer cell line with Wnt ligand markedly increased expression of PC. In a human prostate cancer model, Linares et al. [68] showed that endoplasmic reticulum (ER) stress-responsive transcription factor ATF4 partly contributes to up-regulation of PC in p62-deficient prostate cancer-associated fibroblasts. The renal primary epithelial cells expressing constitutively active forms of HIF1α or HIF2, mimicking clear cell renal carcinoma, show that PC is highly inducible by these two isoforms of HIF [77].
Microbial infection
Although PC is well known to support intermediary metabolism during various physiological conditions, recent studies have shown that host PC is required to support viral infection and replication. Vastag et al. (2011) [78] showed that herpes simplex viruses type I (HSV1) reprograms metabolic pathways in host cells to support their replication. One of these changes is an increased TCA cycle activity via up-regulation of PC expression. Oxaloacetate produced by PC would replenish the removal of downstream TCA cycle intermediates for biosynthesis of viral structural components including nucleic acids and membrane proteins (Fig. 3b). Suppression of PC expression in host cells markedly inhibits replication of HSV1, clearly demonstrating the crucial role of PC in supporting viral replication [78]. Similarly, Grady et al. (2013) [79] performed siRNA screening of metabolic genes that are essential for HSV1 replication and found that this virus induces a robust activation of PC and glutamic-oxaloacetic transaminase 2. The latter enzyme works in concert with PC by direct transamination of amino groups from glutamate to oxaloacetate, producing aspartate which is essential for nucleotide synthesis (Fig. 3b).

Role of PC during viral infection and replication. During influenza A virus (IAV), human enterovirus 71 (EV71), and vesicular stomatitis virus (VSV) infection, viral protein induces PC mRNA of infected cells through unknown signal or molecule. Uncoated viral RNA also triggers RIG-I-like receptor-mediated antiviral innate immune response that involves activation of mitochondrial antiviral signaling protein (MAVS)-mediated NF-κB activation. During infection by these viruses, PC was translocated to interact with MAVS at the outer membrane of mitochondria. This interaction is essential to trigger NF-κB-mediated activation of pro-inflammatory cytokine production (a). During HSV1, HCV, or H5N1 infection, host PC protein or activity is up-regulated to increase the supply of oxaloacetate which is further converted to aspartate, an important precursor for nucleotide synthesis of viral genome (b)
Similarly, in influenza A virus H1N1-infected MDCK kidney cells, infection by this virus increases PC activity while decreasing PDH activity, indicating that pyruvate is specifically channeled to anaplerosis rather than oxidation via PDH. Interestingly, glutaminase activity is also increased in parallel with increased PC activity, demonstrating the importance of both pyruvate carboxylation and glutaminolysis during viral infection. An increase in anaplerosis is accompanied by increased activity of ATP-citrate lyase that generates acetyl groups for de novo fatty acid synthesis together with increased activity of malic enzyme and glucose-6-phosphate dehydrogenase, both of which produce NADPH that supports de novo fatty acid synthesis [80]. Yim et al. [80] also reported the supporting role of host PC during hepatitis C virus (HCV) replication. During infection, the non-structural protein NS5A enters the mitochondria and interacts with PC. Silencing of PC expression impairs viral maturation and release from the host cells though it did not affect viral DNA or protein synthesis. The authors point out that host PC may be required to support membrane lipid synthesis during viral maturation from host cells [81]. Although the above evidence demonstrates that host PC is essential to support viral replication, this seems to be opposite in the RNA viruses, including influenza A virus, human enterovirus 71, and vesicular stomatitis virus [81]. Infection of cells with these RNA viruses triggers the interaction between PC and mitochondrial antiviral signaling protein (MAVS), one component of the RIG-I-like receptor-mediated antiviral innate immune response [82]. During infection by these RNA viruses, PC is translocated to the outer membrane and interacts with MAVS together with its associated protein TRAF6, resulting in activation of NF-κΒ. Activated NF-κΒ in turn binds to the promoter of its target genes including interferon-γ, interleukins, and TNF-α to suppress viral infection (Fig. 3a). Suppression of PC or MAVS expression eliminates the ability of host cells to eliminate infection by these viruses, demonstrating that interaction of both proteins is required to maintain this innate immune response against these RNA viruses [83].
In pathogenic bacteria such as Staphylococcus aureus, disruption of the PC gene attenuates its ability to cause systemic and abscess infection [84]. Similarly, in Listeria monocytogenes, which is a pathogenic bacterium that can enter many mammalian cells, inactivation of PC gene results in marked reduction of the pathogen’s growth rate and diminishes its ability to invade and replicate inside host cells [17]. The phenotypic defects of these two pathogenic bacteria are associated with the lowered synthesis rate of small molecules that support bacterial growth, including aspartate and glutamate, which are precursors of nucleotide synthesis. A recent study in L. monocytogenes also demonstrates that PC activity of this bacterium is inhibited by the cyclic di-adenosine monophosphate (c-di-AMP), an important physiological regulator of many bacteria. Inactivation of L. monocytogenes PC via binding of this allosteric ligand is proposed to prevent the overproduction of aspartate, leading to metabolic imbalance during intracellular growth [85].
In conclusion, PC is an important anaplerotic enzyme that maintains proper levels of oxaloacetate and its downstream metabolites. Deregulation of hepatic PC expression caused by peripheral insulin resistance enhances hepatic gluconeogenesis, contributing to type 2 diabetes in humans. Furthermore, up-regulation of PC expression is one of the key mechanisms that many tumors use to increase the cellular oxaloacetate pool that supports various biosynthetic pathways during rapid cellular proliferation. The role of PC in supporting growth also extends to certain DNA viruses as this enzyme is hijacked to support their replication, infection, or maturation, while PC in pathogenic bacteria appears to be essential in maintaining their virulence. Down-regulation of PC expression may provide a therapeutic means for the treatment of type 2 diabetes and cancers, or of some microbial infections if selective inhibitors can be devised. Furthermore, the availability of the recent crystal structures of PC in humans and pathogens [10, 11, 86] and identification of its allosteric binding site [87,88,89] will provide information necessary to design small molecules that can interfere with substrate or allosteric binding sites and modulate PC activity.
References
Adeva-Andany M, López-Ojén M, Funcasta-Calderón R, Ameneiros-Rodríguez E, Donapetry-García C, Vila-Altesor M, Rodríguez-Seijas J (2014) Comprehensive review on lactate metabolism in human health. Mitochondrion 17:76–100
Halestrap AP, Price NT (1999) The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem J 34:281–299
Halestrap AP (2012) The monocarboxylate transporter family—structure and functional characterization. IUBMB Life 64(1):1–9
McCommis KS, Finck BN (2015) Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J 466(3):443–454
Vanderperre B, Bender T, Kunji ER, Martinou JC (2015) Mitochondrial pyruvate import and its effects on homeostasis. Curr Opin Cell Biol 33:35–41
Merritt ME, Harrison C, Sherry AD, Malloy CR, Burgess SC (2011) Flux through hepatic pyruvate carboxylase and phosphoenolpyruvate carboxykinase detected by hyperpolarized 13C magnetic resonance. Proc Natl Acad Sci U S A 108(47):19084–19089
Jitrapakdee S, Vidal-Puig A, Wallace JC (2006) Anaplerotic roles of pyruvate carboxylase in mammalian tissues. Cell Mol Life Sci 63(7-8):843–854
Jitrapakdee S, St Maurice M, Rayment I, Cleland WW, Wallace JC, Attwood PV (2008) Structure, mechanism and regulation of pyruvate carboxylase. Biochem J 413(3):369–387
Owen OE, Kalhan SC, Hanson RW (2002) The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 277(34):30409–30412
St Maurice M, Reinhardt L, Surinya KH, Attwood PV, Wallace JC, Cleland WW, Rayment I (2007) Domain architecture of pyruvate carboxylase, a biotin-dependent multifunctional enzyme. Science 317(5841):1076–1079
Xiang S, Tong L (2008) Crystal structures of human and Staphylococcus aureus pyruvate carboxylase and molecular insights into the carboxyltransfer reaction. Nat Struct Mol Biol 15(3):295–302
Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM et al (2015) Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 160(4):745–758
Adina-Zada A, Zeczycki TN, Attwood PV (2012) Regulation of the structure and activity of pyruvate carboxylase by acetyl CoA. Arch Biochem Biophys 519(2):118–130
Wallace JC (1985) Distribution and biological functions of pyruvate carboxylase in nature. In: Keech D, Wallace J (eds) Pyruvate carboxylase. CRC Press, Boca Raton, pp 5–64
Zeczycki TN, St. Maurice M, Attwood PV (2010) Inhibitors of pyruvate carboxylase. Open Enzyme Inhib J 3(1):8–26
Sirithanakorn C, Adina-Zada A, Wallace JC, Jitrapakdee S, Attwood PV (2014) Mechanisms of inhibition of Rhizobium etli pyruvate carboxylase by L-aspartate. Biochemistry 53(45):7100–7106
Schär J, Stoll R, Schauer K, Loeffler DI, Eylert E, Joseph B, Eisenreich W, Fuchs TM, Goebel W (2010) Pyruvate carboxylase plays a crucial role in carbon metabolism of extra- and intracellularly replicating Listeria monocytogenes. J Bacteriol 192(7):1774–1784
Nordlie RC, Foster JD, Lange AJ (1999) Regulation of glucose production by the liver. Annu Rev Nutr 19(1):379–406
Barthel A, Schmoll D (2003) Novel concepts in insulin regulation of hepatic gluconeogenesis. Am J Physiol Endocrinol Metab 285(4):E685–E692
Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414(6865):799–806
Jitrapakdee S (2012) Transcription factors and coactivators controlling nutrient and hormonal regulation of hepatic gluconeogenesis. Int J Biochem Cell Biol 44(1):33–45
Samuel VT, Shulman GI (2016) The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest 126(1):12–22
Hatting M, Tavares CDJ, Sharabi K, Rines AK, Puigserver P (2017) Insulin regulation of gluconeogenesis. Ann N Y Acad Sci. https://doi.org/10.1111/nyas.13435
Thonpho A, Sereeruk C, Rojvirat P, Jitrapakdee S (2010) Identification of the cyclic AMP responsive element (CRE) that mediates transcriptional regulation of the pyruvate carboxylase gene in HepG2 cells. Biochem Biophys Res Commun 393(4):714–719
Perry RJ, Zhang XM, Zhang D, Kumashiro N, Camporez JP, Cline GW, Rothman DL, Shulman GI (2014) Leptin reverses diabetes by suppression of the hypothalamic–pituitary–adrenal axis. Nat Med 20(7):759–763
Kendrick AA, Choudhury M, Rahman SM, McCurdy CE, Friederich M, Van Hove JL, Watson PA, Birdsey N, Bao J, Gius D et al (2011) Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J 433(3):505–514
Lee P, Leong W, Tan T, Lim M, Han W, Radda GK (2013) In vivo hyperpolarized carbon-13 magnetic resonance spectroscopy reveals increased pyruvate carboxylase flux in an insulin-resistant mouse model. Hepatology 57(2):515–524
Burgess SC, He T, Yan Z, Lindner J, Sherry AD, Malloy CR, Browning JD, Magnuson MA (2007) Cytosolic phosphoenolpyruvate carboxykinase does not solely control the rate of hepatic gluconeogenesis in the intact mouse liver. Cell Metab 5(4):313–320
Samuel VT, Beddow SA, Iwasaki T, Zhang XM, Chu X, Still CD, Gerhard GS, Shulman GI (2009) Fasting hyperglycemia is not associated with increased expression of PEPCK or G6Pc in patients with type 2 diabetes. Proc Natl Acad Sci U S A 106(29):12121–12126
Kumashiro N, Beddow SA, Vatner DF, Majumdar SK, Cantley JL, Guebre-Egziabher F, Fat I, Guigni B, Jurczak MJ, Birkenfeld AL et al (2013) Targeting pyruvate carboxylase reduces gluconeogenesis and adiposity and improves insulin resistance. Diabetes 62(7):2183–2194
MacDonald MJ, Kaysen JH, Moran SM, Pomije CE (1991) Pyruvate dehydrogenase and pyruvate carboxylase. Sites of pre-translational regulation by glucose of glucose-induced insulin release in pancreatic islets. J Biol Chem 266(33):22392–22397
MacDonald MJ, McKenzie DI, Walker TM, Kaysen JH (1992) Lack of glyconeogenesis in pancreatic islets: expression of gluconeogenic enzyme genes in islets. Horm Metab Res 24(04):158–160
MacDonald MJ (1995) Feasibility of a mitochondrial pyruvate malate shuttle in pancreatic islets. Further implication of cytosolic NADPH in insulin secretion. J Biol Chem 270(34):20051–20058
Farfari S, Schulz V, Corkey B, Prentki M (2000) Glucose-regulated anaplerosis and cataplerosis in pancreatic beta-cells: possible implication of a pyruvate/citrate shuttle in insulin secretion. Diabetes 49(5):718–726
MacDonald MJ, Fahien LA, Brown LJ, Hasan NM, Buss JD, Kendrick MA (2005) Perspective: emerging evidence for signaling roles of mitochondrial anaplerotic products in insulin secretion. Am J Physiol Endocrinol Metab 288(1):E1–15
Jitrapakdee S, Wutthisathapornchai A, Wallace JC, MacDonald MJ (2010) Regulation of insulin secretion: role of mitochondrial signalling. Diabetologia 53(6):1019–1032
Hasan NM, Longacre MJ, Stoker SW, Boonsaen T, Jitrapakdee S, Kendrick MA, Wallace JC, MacDonald MJ (2008) Impaired anaplerosis and insulin secretion in insulinoma cells caused by small interfering RNA-mediated suppression of pyruvate carboxylase. J Biol Chem 283(42):28048–28059
Xu J, Han J, Long YS, Epstein PN, Liu YQ (2008) The role of pyruvate carboxylase in insulin secretion and proliferation in rat pancreatic beta cells. Diabetologia 51(11):2022–2030
MacDonald MJ, Tang J, Polonsky KS (1996) Low mitochondrial glycerol phosphate dehydrogenase and pyruvate carboxylase in pancreatic islets of Zucker diabetic fatty rats. Diabetes 45(11):1626–1630
MacDonald MJ, Efendić S, Ostenson CG (1996) Normalization by insulin treatment of low mitochondrial glycerol phosphate dehydrogenase and pyruvate carboxylase in pancreatic islets of the GK rat. Diabetes 45(7):886–890
MacDonald MJ, Longacre MJ, Langberg EC, Tibell A, Kendrick MA, Fukao T, Ostenson CG (2009) Decreased levels of metabolic enzymes in pancreatic islets of patients with type 2 diabetes. Diabetologia 52(6):1087–1091
Boonsaen T, Rojvirat P, Surinya KH, Wallace JC, Jitrapakdee S (2007) Transcriptional regulation of the distal promoter of the rat pyruvate carboxylase gene by hepatocyte nuclear factor 3beta/Foxa2 and upstream stimulatory factors in insulinoma cells. Biochem J 405(2):359–367
Wang H, Brun T, Kataoka K, Sharma AJ, Wollheim CB (2007) MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia 50(2):348–358
Pedersen KB, Buckley RS, Scioneaux R (2010) Glucose induces expression of rat pyruvate carboxylase through a carbohydrate response element in the distal gene promoter. Biochem J 426(2):159–170
Gupta D, Leahy AA, Monga N, Peshavaria M, Jetton TL, Leahy JL (2013) Peroxisome proliferator-activated receptor γ (PPARγ) and its target genes are downstream effectors of FoxO1 protein in islet β-cells: mechanism of β-cell compensation and failure. J Biol Chem 288(35):25440–25449
Wutthisathapornchai A, Vongpipatana T, Muangsawat S, Boonsaen T, MacDonald MJ, Jitrapakdee S (2014) Multiple E-boxes in the distal promoter of the rat pyruvate carboxylase gene function as a glucose-responsive element. PLoS One 9(7):e102730
Li X, Cheng KK, Liu Z, Yang JK, Wang B, Jiang X, Zhou Y, Hallenborg P, Hoo RL, Lam KS et al (2016) The MDM2-p53-pyruvate carboxylase signalling axis couples mitochondrial metabolism to glucose-stimulated insulin secretion in pancreatic β-cells. Nat Commun 7:11740
Warburg O (1956) On respiratory impairment in cancer cells. Science 124(3215):269–270
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324(5930):1029–1033
Ward PS, Thompson CB (2012) Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21(3):297–308
Altman BJ, Stine ZE, Dang CV (2016) From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 16(10):619–634
Fan TW, Lane AN, Higashi RM, Farag MA, Gao H, Bousamra M, Miller DM (2009) Altered regulation of metabolic pathways in human lung cancer discerned by (13)C stable isotope-resolved metabolomics (SIRM). Mol Cancer 8(1):41
Sellers K, Fox MP, Bousamra M 2nd, Slone SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R et al (2015) Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest 125(2):687–698
Vizan P, Boros LG, Figueras A, Capella G, Mangues R, Bassilian S, Lim S, Lee WN, Cascante M (2005) K-ras codon-specific mutations produce distinctive metabolic phenotypes in NIH3T3 mice fibroblasts. Cancer Res 65(13):5512–5515
Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA et al (2016) Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab 23(3):517–528
Christen S, Lorendeau D, Schmieder R, Broekaert D, Metzger K, Veys K, Elia I, Buescher JM, Orth MF, Davidson SM et al (2016) Breast cancer-derived lung metastases show increased pyruvate carboxylase-dependent Anaplerosis. Cell Rep 17(3):837–848
Phannasil P, Thuwajit C, Warnnissorn M, Wallace JC, MacDonald MJ, Jitrapakdee S (2015) Pyruvate carboxylase is up-regulated in breast cancer and essential to support growth and invasion of MDA-MB-231 cells. PLoS One 10(6):e0129848
Phannasil P, Ansari IH, El Azzouny M, Longacre MJ, Rattanapornsompong K, Burant CF, MacDonald MJ, Jitrapakdee S (2017) Mass spectrometry analysis shows the biosynthetic pathways supported by pyruvate carboxylase in highly invasive breast cancer cells. Biochim Biophys Acta 1863(2):537–551
Caneba CA, Bellance N, Yang L, Pabst L, Nagrath D (2012) Pyruvate uptake is increased in highly invasive ovarian cancer cells under anoikis conditions for anaplerosis, mitochondrial function, and migration. Am J Physiol Endocrinol Metab 303(8):E1036–E1052
Kamarajugadda S, Stemboroski L, Cai Q, Simpson NE, Nayak S, Tan M, Lu J (2012) Glucose oxidation modulates anoikis and tumor metastasis. Mol Cell Biol 32(10):1893–1907
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE et al (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17(3):225–234
King A, Selak MA, Gottlieb B (2006) Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 25(34):4675–4682
Yang M, Soga T, Pollard PJ (2013) Oncometabolites: linking altered metabolism with cancer. J Clin Invest 123(9):3652–3658
Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Matés JM, DeBerardinis RJ (2011) Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A 108(21):8674–8679
Izquierdo-Garcia JL, Cai LM, Chaumeil MM, Eriksson P, Robinson AE, Pieper RO, Phillips JJ, Ronen SM (2014) Glioma cells with the IDH1 mutation modulate metabolic fractional flux through pyruvate carboxylase. PLoS One 9(9):e108289
Cardaci S, Zheng L, MacKay G, van den Broek NJ, MacKenzie ED, Nixon C, Stevenson D, Tumanov S, Bulusu V, Kamphorst JJ et al (2015) Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat Cell Biol 17(10):1317–1326
Lussey-Lepoutre C, Hollinshead KE, Ludwig C, Menara M, Morin A, Castro-Vega LJ, Parker SJ, Janin M, Martinelli C, Ottolenghi C et al (2015) Loss of succinate dehydrogenase activity results in dependency on pyruvate carboxylation for cellular anabolism. Nat Commun 6:8784
Linares JF, Cordes T, Duran A, Reina-Campos M, Valencia T, Ahn CS, Castilla EA, Moscat J, Metallo CM, Diaz-Meco MT (2017) ATF4-induced metabolic reprogramming is a synthetic vulnerability of the p62-deficient tumor stroma. Cell Metab 26(6):817–829
Ma MZ, Zhang Y, Weng MZ, Wang SH, Hu Y, Hou ZY, Qin YY, Gong W, Zhang YJ, Kong X et al (2016) Long noncoding RNA GCASPC, a target of miR-17-3p, negatively regulates pyruvate carboxylase-dependent cell proliferation in gallbladder cancer. Cancer Res 76(18):5361–5371
Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, Osterman AL, Smith JW (2011) Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem 289:42626–42634
Beloueche-Babari M, Wantuch S, Casals Galobart T, Koniordou M, Parkes HG, Arunan V, Chung YL, Eykyn TR, Smith PD, Leach MO (2017) MCT1 inhibitor AZD3965 increases mitochondrial metabolism, facilitating combination therapy and noninvasive magnetic resonance spectroscopy. Cancer Res 77(21):5913–5924
Reed MA, Ludwig C, Bunce CM, Khanim FL, Günther UL (2016) Malonate as a ROS product is associated with pyruvate carboxylase activity in acute myeloid leukaemia cells. Cancer Metab 4(1):15
Dervartanian DV, Veeger C (1964) Studies on succinate dehydrogenase. I. Spectral properties of the purified enzyme and formation of enzyme–competitive inhibitor complexes. Biochim Biophys Acta 92:233–247
Wilmanski T, Buhman K, Donkin SS, Burgess JR, Teegarden D (2017) 1α,25-Dihydroxyvitamin D inhibits de novo fatty acid synthesis and lipid accumulation in metastatic breast cancer cells through down-regulation of pyruvate carboxylase. J Nutr Biochem 40:194–200
Wilmanski T, Zhou X, Zheng W, Shinde A, Donkin SS, Wendt M, Burgess JR, Teegarden D (2017) Inhibition of pyruvate carboxylase by 1α,25-dihydroxyvitamin D promotes oxidative stress in early breast cancer progression. Cancer Lett 411:171–181
Lee SY, Jeon HM, Ju MK, Kim CH, Yoon G, Han SI, Park HG, Kang HS (2012) Wnt/snail signaling regulates cytochrome C oxidase and glucose metabolism. Cancer Res 72(14):3607–3617
Arreola A, Cowey CL, Coloff JL, Rathmell JC, Rathmell WK (2014) HIF1α and HIF2α exert distinct nutrient preferences in renal cells. PLoS One 9(5):e98705
Vastag L, Koyuncu E, Grady SL, Shenk TE, Rabinowitz JD (2011) Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog 7(7):e1002124
Grady SL, Purdy JG, Rabinowitz JD, Shenka T (2013) Argininosuccinate synthetase 1 depletion produces a metabolic state conducive to herpes simplex virus 1 infection. Proc Natl Acad Sci U S A 110(51):E5006–E5015
Janke R, Genzel Y, Wetzel M, Reichl U (2011) Effect of influenza virus infection on key metabolic enzyme activities in MDCK cells. BMC Proc 5(Suppl 8):P129
Yim SA, Lim YS, Kim JW, Hwang SB (2013) Nonstructural 5A protein of hepatitis C virus interacts with pyruvate carboxylase and modulates viral propagation. PLoS One 8(7):e68170
Cao Z, Zhou Y, Zhu S, Feng J, Chen X, Liu S, Peng N, Yang X, Xu G, Zhu Y (2016) Pyruvate carboxylase activates the RIG-I-like receptor-mediated antiviral immune response by targeting the MAVS signalosome. Sci Rep 6(1):22002
Chan YK, Gack MU (2016) Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol 14(6):360–373
Benton BM, Zhang JP, Bond S, Pope C, Christian T, Lee L, Winterberg KM, Schmid MB, Buysse JM (2004) Large-scale identification of genes required for full virulence of Staphylococcus aureus. J Bacteriol 186(24):8478–8489
Sureka K, Choi PH, Precit M, Delince M, Pensinger DA, Huynh TN, Jurado AR, Goo YA, Sadilek M, Iavarone AT et al (2014) The cyclic dinucleotide c-di-AMP is an allosteric regulator of metabolic enzyme function. Cell 158(6):1389–1401
Lasso G, Yu LP, Gil D, Lázaro M, Tong L, Valle M (2014) Functional conformations for pyruvate carboxylase during catalysis explored by cryoelectron microscopy. Structure 22(6):911–922
Menefee AL, Zeczycki TN (2014) Nearly 50 years in the making: defining the catalytic mechanism of the multifunctional enzyme, pyruvate carboxylase. FEBS J 28:1333–1354
Sirithanakorn C, Jitrapakdee S, Attwood PV (2016) Investigation of the roles of allosteric domain arginine, aspartate, and glutamate residues of Rhizobium etli pyruvate carboxylase in relation to its activation by acetyl CoA. Biochemistry 55(30):4220–4228
Westerhold LE, Bridges LC, Shaikh SR, Zeczycki TN (2017) Kinetic and thermodynamic analysis of acetyl-CoA activation of Staphylococcus aureus pyruvate carboxylase. Biochemistry 56(27):3492–3506
Acknowledgements
This work is supported by the International Research Network (IRN) program, contract no. IRN59W0003 from the Thailand Research Fund. U.L. is supported by a royal golden jubilee PhD program, PHD/0103/2557, the Thailand Research Fund. The authors thank Professor John Wallace for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lao-On, U., Attwood, P.V. & Jitrapakdee, S. Roles of pyruvate carboxylase in human diseases: from diabetes to cancers and infection. J Mol Med 96, 237–247 (2018). https://doi.org/10.1007/s00109-018-1622-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-018-1622-0