
Abstract
The 2022 European Society of Cardiology/European Respiratory Society (ESC/ERS) guidelines for pulmonary hypertension have introduced a refined risk stratification to guide both initial and subsequent treatment of pulmonary arterial hypertension (PAH). The risk stratification at PAH diagnosis still comprises three risk categories (low, intermediate, high) and lists some new parameters. As the estimated 1‑year mortality is more than 20% in high-risk patients after diagnosis, an initial triple-combination therapy including parenteral prostacyclin analogues is recommended for this group. All other patients should receive a dual-combination therapy with an endothelin receptor antagonist and a phosphodiesterase‑5 inhibitor. However, this approach of initial combination therapy is only recommended for classic PAH, while monotherapy followed by regular follow-up and individualized therapy should be used for patients with cardiopulmonary comorbidities. For PAH patients without cardiopulmonary comorbidities, it is recommended to assess their risk at follow-up with a new 4‑strata classification, where the intermediate-risk group is split on the basis of three noninvasive parameters. Importantly, changes from intermediate–high to intermediate–low risk have been shown to be associated with a better prognosis. In addition, the recommendations on treatment escalation became more precise with the addition of a prostacyclin receptor agonist or switching a phosphodiesterase‑5 inhibitor to a soluble guanylate cyclase stimulator for intermediate–low risk and proceeding to triple-combination therapy with parenteral prostacyclin analogues already for intermediate–high risk. With sotatercept, the first non-vasodilator PAH treatment will become available in the near future to further enrich our treatment options for this chronic and still severe disease.
Zusammenfassung
Die neue Risikostratifizierung der pulmonalarteriellen Hypertonie (PAH) wurde in den 2022 erschienenen ESC/ERS-Leitlinien (European Society of Cardiology/European Respiratory Society) für pulmonale Hypertonie vorgestellt. Zum Zeitpunkt der Erstdiagnose erfolgt weiterhin eine Aufteilung in 3 Risikogruppen (niedrig, intermediär, hoch) anhand bekannter und einiger neuer Parameter. Aufgrund der hohen Einjahresmortalität von über 20 % in der Hochrisikogruppe wird bei diesen Patienten eine initiale Dreifachkombinationstherapie unter Einschluss parenteraler Prostazyklin-Analoga empfohlen, während alle anderen Patienten eine duale Therapie aus einem Endothelinrezeptorantagonist und einem Phosphodiesterase-5-Inhibitor erhalten sollen. Die Empfehlung zur initialen Kombinationstherapie wird aber nur bei klassischer PAH ausgesprochen, während bei PAH mit kardiopulmonalen Komorbiditäten eine initiale Monotherapie erfolgen soll – mit individualisierter Therapie im Verlauf. Für die Risikostratifizierung im weiteren Verlauf wird – nur für PAH Patienten ohne kardiopulmonale Komorbiditäten – eine 4‑stufige Klassifikation empfohlen. Bei dieser wurde die Gruppe des intermediären Risikos anhand dreier nichtinvasiver Parameter in intermediär-niedriges und intermediär-hohes Risiko geteilt. Es konnte gezeigt werden, dass Änderungen der Risikogruppen prognostisch bedeutsam sind, zudem wurden die Empfehlungen zur Therapieerweiterung weiter spezifiziert: Bei intermediär-niedrigem Risiko wird die Ergänzung um einen Prostazyklin-Rezeptor-Agonisten oder eine Umstellung vom Phosphodiesterase-5-Inhibitor auf einen löslichen Guanylatzyklase-Stimulator empfohlen. Bereits bei intermediär-hohem Risiko sollte eine Therapieerweiterung um ein parenterales Prostazyklin-Analogon erfolgen. Mit Sotatercept steht in Kürze der erste Nichtvasodilatator zur Therapie der PAH zur Verfügung.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
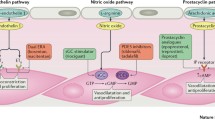
The treatment of pulmonary arterial hypertension (PAH) has evolved considerably over the past 25 years. As early as in 2009, we were able to address all three pathways known to be involved in the development of PAH: the prostacyclin, endothelin, and nitric oxide pathways [1]. In the ensuing years, several new drugs have been licensed, but the major development was the introduction of treatment goals and combination therapy [2]. The 2022 European Society of Cardiology/European Respiratory Society (ESC/ERS) guidelines for pulmonary hypertension present a refined risk stratification and treatment algorithm for PAH, focusing on initial and earlier sequential combination therapy [3]. Importantly, the guidelines define for the first time a phenotype of “PAH with cardiopulmonary comorbidities,” which is not included in the evidence-based treatment algorithm of PAH utilizing the risk stratification and recommendations for combination therapy, as described below. Due to the lack of evidence, these patients should be started on PAH monotherapy and receive individualized treatment during follow-up, which, however, does not exclude treatment escalation. Although the patient group without cardiopulmonary comorbidities is considerably smaller [4], this article focuses on the recommendations for this group of “classic PAH.”
Evolution of PAH risk stratification
The 2009 ESC/ERS guidelines for pulmonary hypertension recommended sequential combination therapy in the case of an inadequate clinical response to initial monotherapy [5]. Although there was no detailed risk stratification, determinants of prognosis were listed with cut-off values known to be associated with better or worse prognosis. The 2015 ESC/ERS guidelines for pulmonary hypertension proposed a risk stratification on the basis of the same parameters introduced in 2009, but defined three risk categories, namely, low, intermediate, and high risk [6]. It was admitted that “most of the proposed variables and cut-off values are based on expert opinion.” In addition, the application of the risk stratification to an individual patient was unclear, as not all variables listed could be assessed at every follow-up, and variables could be distributed across the risk groups.
On the basis of registry data, there were two different strategies proposed for the application of risk stratification in clinical practice: The groups from the COMPERA registry and the Swedish PAH registry chose to grade every risk parameter available as low = 1, intermediate = 2, or high = 3 points, according to the cut-offs from the guideline risk table. The sum of those numbers was then divided by the number of parameters and rounded off to the next integer, which then defined the risk group [7, 8]. This classification has been shown to be predictive of survival both at baseline and follow-up. In addition, it was found that the estimated 1‑year mortality according to the risk assessment at baseline was considerably higher than expected, exceeding 20% for patients in the high-risk group [7, 8]. This information has been updated in the current guidelines accordingly [3].
By contrast, the “French approach” counted the number of parameters in the low-risk category only [9]. The four parameters available at baseline and follow-up in a large cohort of 1017 patients with idiopathic, heritable, and drug-induced PAH were World Health Organization (WHO) functional class (FC), 6‑minute walk distance (6-MWD), right atrial pressure, and cardiac index. The number of low-risk criteria (according to the guideline risk table) achieved has been shown to be predictive of survival both at baseline and follow-up, with the best prognosis for patients with all four variables at low risk. Of note, a subgroup of 603 patients also had values for brain natriuretic peptide (BNP) or its N‑terminal form (NT-proBNP) available at the first follow-up. On multivariable analysis with all five parameters, the only significant predictors of survival remaining were WHO-FC, 6‑MWD, and the level of natriuretic peptide, while the hemodynamic parameters no longer offered additional significant prognostic information [9]. This finding is of importance, as invasive follow-up with right heart catheterization is not routinely performed at many European PAH centers [7, 8].
The concept of noninvasive risk stratification at follow-up using WHO-FC, 6‑MWD, and natriuretic peptide levels has been reproduced in the COMPERA registry [10] and finally advanced into a four-strata risk model [11, 12]. Dividing the intermediate-risk group into an intermediate–low- and an intermediate–high-risk group not only reduces the size of this usually largest group, but also carries prognostic information both at baseline and follow-up, and is more sensitive to prognostically relevant changes in risk than the original three-strata model [11]. Therefore, it has been integrated into the 2022 European Guidelines for PAH risk assessment at follow-up including recommendations for sequential combination therapy (Table 1; [3]).
Regarding the baseline risk assessment of PAH, the guidelines still recommend a three-strata model, which should be assessed as comprehensively as possible [3]. While the structure of parameters used for risk stratification, reflecting clinical information (e.g., syncope or WHO-FC), exercise capacity (e.g., 6‑MWD, cardiopulmonary exercise testing), and right heart function (e.g., NT-proBNP, echocardiography, hemodynamics), has been maintained, some cut-offs have been slightly modified and some new parameters have been introduced.
For the natriuretic peptide levels, the cut-off dividing the intermediate- and high-risk groups has been modified to fit the four-strata risk assessment at follow-up on the basis of data from the REVEAL registry [3, 13]. High risk is now defined by BNP levels of > 800 ng/L (300 ng/L in 2015) or NT-proBNP levels of > 1100 ng/L (1400 ng/L in 2015). The echocardiography parameters right atrial area and pericardial effusion have remained the same, but the ratio of tricuspid annular plane systolic excursion (TAPSE) to systolic pulmonary arterial pressure (sPAP) has been newly introduced.
The TAPSE/sPAP ratio has been shown to reflect right ventricular–arterial coupling and to be a powerful prognostic marker in PAH independent of other echocardiographic and hemodynamic parameters [14, 15]. However, prospective validation of this new parameter is pending.
Due to increasing evidence in the literature, cardiac magnetic resonance imaging parameters have been introduced in the baseline risk assessment [3]. Regarding hemodynamic parameters, cut-off values for right atrial pressure, cardiac index, and mixed venous oxygen saturation have been kept, while stroke volume index (calculated as cardiac index divided by heart rate) has been newly introduced on the basis of a retrospective study from the French pulmonary hypertension registry [16].
Although the current, refined risk stratification still has some limitations such as missing a prospective evaluation of most of the parameters and application only to PAH patients without cardiopulmonary comorbidities, it facilitates risk stratification especially at follow-up with the opportunity for earlier treatment escalation and more precise information on the prognosis.
Current treatment of pulmonary arterial hypertension
The following treatment recommendations are applicable only to patients with PAH without cardiopulmonary comorbidities (as discussed in the previous section). It should also be kept in mind that all pivotal studies in the field of PAH have been carried out using the hemodynamic criteria as described in the former European Guidelines from 2015, i.e., mean pulmonary arterial pressure (mPAP) ≥ 25 mm Hg, pulmonary arterial wedge pressure ≤ 15 mm Hg, and pulmonary vascular resistance (PVR) > 3 Wood units (WU; [6]), while the current hemodynamic definition has been modified (mPAP > 20 mm Hg and PVR > 2 WU; [3]). Therefore, it should be left to experienced PAH centers to treat and follow up potentially early PAH with only mild hemodynamic impairment (i.e., mPAP 21–24 mm Hg and/or PVR > 2 and < 3 WU) or exercise pulmonary hypertension [3].
General measures, although often recommended on the basis of experience rather than solid evidence, must not be forgotten. There are basic measures that should be applied at any time to all PAH patients, e.g., diuretics if signs of heart failure are present, oxygen when indicated according to local guidelines, vaccination, and psychosocial support. Supervised rehabilitation programs in the meantime are supported by a large body of evidence but should only be prescribed once the PAH treatment is optimized and the patient is in a clinically stable condition. Some drugs such as therapeutic anticoagulation or cardiovascular medication are not recommended in PAH in general unless other indications are present [3].
For patients with idiopathic, heritable, and drug-induced PAH, it is of crucial importance to perform vasoreactivity testing during the diagnostic right heart catheterization in order to identify potential calcium channel blocker responders. Drugs for testing are inhaled nitric oxide, inhaled iloprost, or intravenous epoprostenol. Positive vasoreactivity is defined by a drop in mPAP of ≥ 10 to ≤ 40 mm Hg with increased or unchanged cardiac output. For the first time, the guidelines published an algorithm dedicated to the treatment of vasoreactivity responders including follow-up and treatment goals. After 3–6 months of high-dose calcium channel blockers, these patients should be in WHO-FC I or II, have natriuretic peptide levels in the low-risk category, and have mPAP ≤ 30 mm Hg + PVR ≤ 4 WU on right heart catheterization [3]. Although these criteria are based on consensus, it seems reasonable to add other licensed PAH drugs according to the main treatment algorithm when they are not met.
The evidence-based treatment algorithm that will be discussed here is applicable to patients with non-vasoreactive idiopathic, heritable, and drug-induced PAH and PAH associated with connective tissue disease, as these groups are mainly represented in the pivotal PAH trials. For other PAH subgroups such as PAH associated with human immunodeficiency virus, portal hypertension, congenital heart disease, or schistosomiasis, specific recommendations have to be taken into account [3].
While the 2009 European Guidelines recommended initial monotherapy on the basis of the evidence at that time, initial combination therapy became the standard of care in PAH [3, 5, 6]. For patients in the high-risk group at diagnosis (using the three-strata model, see above), a triple-combination therapy including parenteral prostacyclin analogues is recommended (Table 1). Although there are no prospective data on this strategy, a retrospective analysis from the French registry strongly supports this recommendation [17]. Patients treated with initial triple therapy with an endothelin receptor antagonist (ERA), a phosphodiesterase‑5 inhibitor (PDE-5i), and parenteral prostacyclin (epoprostenol) or an analogue (treprostinil) had a significantly better long-term survival of 91% at 5 years compared to 61% in the group of patients on initial mono- or dual-combination therapy, although being at higher risk of mortality at diagnosis.
For PAH patients at intermediate or low risk at baseline, an initial dual-combination therapy with an ERA and a PDE-5i should be started [3]. This recommendation is based mainly on the AMBITION study, which compared an initial combination therapy with the ERA ambrisentan and the PDE-5i tadalafil (50% of patients) with the respective monotherapy (25% of patients each) in a randomized, controlled, double-blind fashion [18]. Although side effects occurred slightly more often in the combination-therapy group, the risk of “first event of clinical failure” was reduced by 50% compared to the pooled monotherapy groups. In addition, a reduction in NT-proBNP and an increase in the 6‑MWD at week 24 significantly favored the initial combination therapy. Another trial, designed to compare initial dual with initial oral triple-combination therapy (ERA, PDE-5i plus the prostacyclin receptor agonist selexipag or placebo; TRITON) in PAH, also showed impressive improvements in PVR (from 12.3 to 6.1 WU, −52%), NT-proBNP (from 1932 to 697 ng/L, −75%), and 6‑MWD (from 347.2 to 407.2 m) after 26 weeks of therapy with the ERA macitentan and the PDE-5i tadalafil [19]. As there was no significant additional effect of selexipag on these endpoints, an initial oral triple therapy is currently not recommended [3]. The strategy of initial dual-combination therapy with an ERA and a PDE-5i in PAH also showed good results in a retrospective series from Italy [20]. Of note, none of the 24 patients (13.3% of the entire group) being at high risk at diagnosis achieved a low-risk profile at follow-up, supporting the strategy of initial triple-combination therapy as proposed by the current guidelines.
The first follow-up after therapy initiation should occur no later than 3–6 months and include at least an assessment of WHO-FC, 6‑MWD, natriuretic peptide level, electrocardiogram, echocardiography (or cardiac magnetic resonance imaging), and an arterial blood gas analysis (or pulse oximetry) to capture response to treatment and potential complications such as desaturations or new cardiac rhythm disturbances [3]. At this time, the four-strata risk stratification should be applied, but the three parameters should be complemented by additional examinations as clinically indicated (Table 1).
For patients in the low-risk category at follow-up, the treatment can be continued without change as they have a good prognosis according to the available data [11, 12]. Of note, especially younger PAH patients can be in a good WHO-FC (I or II) and have a long 6‑MWD in the low-risk category despite showing marked right heart strain on echocardiography or severely impaired hemodynamic parameters on right heart catheterization, which may justify a discussion regarding treatment escalation even with parenteral prostacyclin analogues. Even if there is no prospective evidence for this observation, in a large retrospective series of patients who received treprostinil as add-on therapy or switched from inhaled iloprost or oral selexipag, patients who reached a low-risk profile at follow-up (with an obvious consecutive survival benefit) had significantly longer 6‑MWD (mean of 438 m) and lower natriuretic peptide levels compared to the patients who showed a less favorable course [21]. Taken together with the observation that parenteral prostacyclin analogues are often prescribed late in the disease course with only modest effects on average, these data support an earlier escalation to triple therapy [22,23,24]. Therefore, in the 2022 Guidelines on Pulmonary Hypertension, sequential triple therapy including parenteral prostacyclin analogues is already recommended for patients at intermediate–high risk at follow-up (and to high-risk patients; [3]).
For patients at intermediate–low risk on dual therapy with an ERA and a PDE-5i, the guidelines also recommend intensifying PAH treatment. For this purpose, two different options are available: the addition of an (oral) prostacyclin receptor agonist (the only licensed drug currently is selexipag) or the switch from PDE-5i to a soluble guanylate cyclase stimulator (the only licensed drug for PAH currently is riociguat). The first recommendation (Class IIa, level of evidence B) is based on the GRIPHON trial, which tested the effect of selexipag versus placebo in an event-driven, randomized controlled trial on a combined endpoint of clinical deterioration including death in 1156 prevalent PAH patients [25]. In the subgroup of 376 patients (32.5% of the study population) already receiving a combination therapy with an ERA (mainly bosentan) and a PDE-5i (mainly sildenafil), the relative risk for the predefined “morbidity and mortality” endpoint was still significantly reduced by 37% with an even greater risk reduction of 64% in the WHO-FC II group of patients [26]. In addition, other analyses suggest that an earlier compared to a later introduction of selexipag into combination treatment of PAH could be beneficial [27, 28].
The switch from a PDE-5i to riociguat has been investigated in two studies, of which the exploratory RESPITE study showed improvements in 6‑MWD, NT-proBNP, WHO-FC, and hemodynamic parameters measured by right heart catheterization at week 24 [29]. The REPLACE study was randomized and controlled but open label due to technical reasons (multiple doses of two PDE-5i made placebo-control and blinding unfeasible; [30]). The combined endpoint of clinical improvement (at least two out of three using pre-defined cut-offs: WHO-FC, 6‑MWD, NT-proBNP) at week 24 was reached by 41% of patients who were switched to riociguat compared to 20% of patients who continued their PDE-5i. In addition, clinical worsening as a predefined secondary outcome, which was blindly adjudicated, also favored the switch to riociguat (1% vs. 9%, p = 0.0047). Due to the non-blinded fashion of the trial, the guideline recommendation (Class IIb, level of evidence B) for switching PDE-5i to riociguat is somewhat weaker compared to adding selexipag [3]. However, both options are reasonable and should be discussed with patients in a shared-decision process. Due to lack of data, no recommendations can be made for a combination of both steps (switch from PDE-5i and addition of selexipag) or their sequence.
Patients starting on a parenteral prostacyclin analogue should be evaluated for lung transplantation if they are suitable candidates.
New treatments for pulmonary arterial hypertension
Although the therapeutic approach has evolved considerably over time with potential improvement of outcomes by the use of combination treatment, PAH is still a chronic, progressive disease with a high mortality [2, 31, 32]. Therefore, new therapeutics that target the remodeling process in addition to the mainly vasodilatory effects of the current drugs are needed. The first non-vasodilator drug that significantly improved exercise capacity and hemodynamics in a phase‑3 randomized controlled trial in pre-treated, prevalent PAH patients was imatinib [33]. Although the primary endpoint of the IMPRES study was positive, imatinib showed an unsatisfactory safety profile with the occurrence of peripheral edema, gastrointestinal side effects, anemia, and subdural hematoma in patients on therapeutic anticoagulation. Therefore, although having been prescribed at PAH expert centers on an individual basis as compassionate use, the drug was not licensed for the treatment of PAH [34, 35]. With the aim of improving tolerability, current clinical trials follow a more cautious approach of dosing imatinib with close monitoring [36] or an inhaled formulation [37]. The multi-kinase inhibitor seralutinib, which is given twice daily as dry powder inhalation using a commercially available device, has been shown to improve PVR at week 24 in pretreated, prevalent PAH patients compared to placebo in the phase‑2 TORREY study [38, 39]. Other drugs, although thoroughly examined pre-clinically, did not show any effect on hemodynamics in PAH patients after 24 weeks, e.g., the apoptosis signal-regulating kinase 1‑inhibitor selonsertib [40].
The most promising new non-vasodilator PAH drug is sotatercept, a novel fusion protein that binds activins and growth differentiation factors in the attempt to restore balance between growth-promoting and growth-inhibiting signaling pathways in PAH [41]. An impressive, dose-dependent reduction of PVR compared to placebo at week 24 was shown in the phase‑2 PULSAR study, which included 106 prevalent, pre-treated PAH patients [42]. Beneficial effects on secondary endpoints (6-MWD, NT-proBNP) persisted in the open-label extension phase and were transferred to former placebo patients [43]. Sotatercept is given as a subcutaneous injection every 3 weeks. The phase‑3 STELLAR study has met its primary outcome of a significant increase in 6‑MWD at week 24 compared to placebo and the majority of secondary endpoints [44]. Therefore, it is expected that sotatercept will be licensed for the treatment of PAH in the near future.
Conclusion
The 2022 ESC/ERS Guidelines for pulmonary hypertension introduced a refined risk stratification for the treatment of pulmonary arterial hypertension (PAH). For high-risk patients, an initial triple-combination therapy including parenteral prostacyclin analogues is recommended. All other patients should receive a dual-combination therapy with an endothelin receptor antagonist and a phosphodiesterase‑5 inhibitor. For patients with cardiopulmonary comorbidities, monotherapy is recommended with regular follow-up and individualized treatment. Patients without cardiopulmonary comorbidities should be assessed at follow-up with the new 4‑strata classification, where the intermediate-risk group is subdivided on the basis of three noninvasive parameters. The new recommendations include more precise guidance for treatment escalation in patients of intermediate–low risk and intermediate–high risk. Sotatercept, the first non-vasodilator, will become available soon to enhance PAH treatment options.
References
Barst RJ, Gibbs JSR, Ghofrani HA et al (2009) Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol 54:S78–S84
Hoeper MM, Markevych I, Spiekerkoetter E et al (2005) Goal-oriented treatment and combination therapy for pulmonary arterial hypertension. Eur Respir J 26:858–863
Humbert M, Kovacs G, Hoeper MM et al (2022) 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. https://doi.org/10.1093/eurheartj/ehac237
Hoeper MM, Pausch C, Grünig E et al (2020) Idiopathic pulmonary arterial hypertension phenotypes determined by cluster analysis from the COMPERA registry. J Heart Lung Transplant. https://doi.org/10.1016/j.healun.2020.09.011
Galiè N, Hoeper MM, Humbert M et al (2009) Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 30:2493–2537
Galiè N, Humbert M, Vachiery J‑L et al (2015) 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J 46:903–975
Hoeper MM, Kramer T, Pan Z et al (2017) Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J 50:1700740. https://doi.org/10.1183/13993003.00740-2017
Kylhammar D, Kjellström B, Hjalmarsson C et al (2018) A comprehensive risk stratification at early follow-up determines prognosis in pulmonary arterial hypertension. Eur Heart J 39:4175–4181. https://doi.org/10.1093/eurheartj/ehx257
Boucly A, Weatherald J, Savale L et al (2017) Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur Respir J 50:1700889. https://doi.org/10.1183/13993003.00889-2017
Hoeper MM, Pittrow D, Opitz C et al (2018) Risk assessment in pulmonary arterial hypertension. Eur Respir J 51:1702606. https://doi.org/10.1183/13993003.02606-2017
Hoeper MM, Pausch C, Olsson KM et al (2022) COMPERA 2.0: a refined four-stratum risk assessment model for pulmonary arterial hypertension. Eur Respir J 60:2102311. https://doi.org/10.1183/13993003.02311-2021
Boucly A, Weatherald J, Savale L et al (2022) External validation of a refined four-stratum risk assessment score from the French pulmonary hypertension registry. Eur Respir J 59:2102419. https://doi.org/10.1183/13993003.02419-2021
Benza RL, Kanwar MK, Raina A et al (2021) Development and validation of an abridged version of the REVEAL 2.0 risk score calculator, REVEAL lite 2, for use in patients with pulmonary arterial hypertension. Chest 159:337–346. https://doi.org/10.1016/j.chest.2020.08.2069
Tello K, Wan J, Dalmer A et al (2019) Validation of the tricuspid annular plane systolic excursion/systolic pulmonary artery pressure ratio for the assessment of right ventricular-arterial coupling in severe pulmonary hypertension. Circ Cardiovasc Imaging 12:e9047. https://doi.org/10.1161/CIRCIMAGING.119.009047
Tello K, Axmann J, Ghofrani HA et al (2018) Relevance of the TAPSE/PASP ratio in pulmonary arterial hypertension. Int J Cardiol 266:229–235. https://doi.org/10.1016/j.ijcard.2018.01.053
Weatherald J, Boucly A, Chemla D et al (2018) Prognostic value of follow-up hemodynamic variables after initial management in pulmonary arterial hypertension. Circulation 137:693–704. https://doi.org/10.1161/CIRCULATIONAHA.117.029254
Boucly A, Savale L, Jaïs X et al (2021) Association between initial treatment strategy and long-term survival in pulmonary arterial hypertension. Am J Respir Crit Care Med 204:842–854. https://doi.org/10.1164/rccm.202009-3698OC
Galiè N, Barberà JA, Frost AE et al (2015) Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med 373:834–844. https://doi.org/10.1056/NEJMoa1413687
Chin KM, Sitbon O, Doelberg M et al (2021) Three- versus two-drug therapy for patients with newly diagnosed pulmonary arterial hypertension. J Am Coll Cardiol 78:1393–1403. https://doi.org/10.1016/j.jacc.2021.07.057
Badagliacca R, D’Alto M, Ghio S et al (2021) Risk reduction and hemodynamics with initial combination therapy in pulmonary arterial hypertension. Am J Respir Crit Care Med 203:484–492. https://doi.org/10.1164/rccm.202004-1006OC
Olsson KM, Richter MJ, Kamp JC et al (2019) Intravenous treprostinil as an add-on therapy in patients with pulmonary arterial hypertension. J Heart Lung Transplant 38:748–756. https://doi.org/10.1016/j.healun.2019.05.002
Hoeper MM, Gall H, Seyfarth HJ et al (2009) Long-term outcome with intravenous iloprost in pulmonary arterial hypertension. Eur Respir J 34:132–137
Farber HW, Miller DP, Meltzer LA, McGoon MD (2013) Treatment of patients with pulmonary arterial hypertension at the time of death or deterioration to functional class IV: insights from the REVEAL Registry. J Heart Lung Transplant 32:1114–1122
Bartolome SD, Sood N, Shah TG et al (2018) Mortality in patients with pulmonary arterial hypertension treated with continuous prostanoids. Chest. https://doi.org/10.1016/j.chest.2018.03.050
Sitbon O, Channick R, Chin KM et al (2015) Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med 373:2522–2533. https://doi.org/10.1056/NEJMoa1503184
Coghlan JG, Channick R, Chin K et al (2018) Targeting the prostacyclin pathway with selexipag in patients with pulmonary arterial hypertension receiving double combination therapy: insights from the randomized controlled GRIPHON study. Am J Cardiovasc Drugs 18:37–47. https://doi.org/10.1007/s40256-017-0262-z
Gaine S, Sitbon O, Channick RN et al (2021) Relationship between time from diagnosis and morbidity/mortality in pulmonary arterial hypertension: results from the phase III GRIPHON study. Chest 160:277–286. https://doi.org/10.1016/j.chest.2021.01.066
Coghlan JG, Gaine S, Channick R et al (2023) Early selexipag initiation and long-term outcomes: insights from randomised controlled trials in pulmonary arterial hypertension. ERJ Open Res 9:456–2022. https://doi.org/10.1183/23120541.00456-2022
Hoeper MM, Simonneau G, Corris PA et al (2017) RESPITE: switching to riociguat in pulmonary arterial hypertension patients with inadequate response to phosphodiesterase‑5 inhibitors. Eur Respir J 50:1602425. https://doi.org/10.1183/13993003.02425-2016
Hoeper MM, Al-Hiti H, Benza RL et al (2021) Switching to riociguat versus maintenance therapy with phosphodiesterase‑5 inhibitors in patients with pulmonary arterial hypertension (REPLACE): a multicentre, open-label, randomised controlled trial. Lancet Respir Med 9:573–584. https://doi.org/10.1016/S2213-2600(20)30532-4
Pitre T, Su J, Cui S et al (2022) Medications for the treatment of pulmonary arterial hypertension: a systematic review and network meta-analysis. Eur Respir Rev 31:220036. https://doi.org/10.1183/16000617.0036-2022
Hoeper MM, Pausch C, Grünig E et al (2022) Temporal trends in pulmonary arterial hypertension: results from the COMPERA registry. Eur Respir J 59:2102024. https://doi.org/10.1183/13993003.02024-2021
Hoeper MM, Barst RJ, Bourge RC et al (2013) Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 127:1128–1138
Speich R, Ulrich S, Domenighetti G et al (2015) Efficacy and safety of long-term Imatinib therapy for pulmonary arterial hypertension. Respiration 89:515–524. https://doi.org/10.1159/000381923
Hoeper MM, Opitz C, Olschewski H et al (2014) Imatinib for pulmonary arterial hypertension. Dtsch Med Wochenschr 139(Suppl 4):S151–S154. https://doi.org/10.1055/s-0034-1387457
Imperial College London (2021) Positioning Imatinib for pulmonary arterial hypertension. clinicaltrials.gov
Aerovate Therapeutics (2023) IMPAHCT: a phase 2b/3, randomized, double-blind, placebo-controlled, 24-week dose ranging and confirmatory study to evaluate the safety and efficacy of AV-101 in patients with pulmonary arterial hypertension (PAH). clinicaltrials.gov
Frantz RP, Benza RL, Channick RN et al (2021) TORREY, a phase 2 study to evaluate the efficacy and safety of inhaled seralutinib for the treatment of pulmonary arterial hypertension. Pulm Circ 11:20458940211057070. https://doi.org/10.1177/20458940211057071
Presentations | Gossamer Bio. https://ir.gossamerbio.com/events-and-presentations/presentations/. Accessed 25 Feb 2023
Rosenkranz S, Feldman J, McLaughlin VV et al (2021) Selonsertib in adults with pulmonary arterial hypertension (ARROW): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir Med. https://doi.org/10.1016/S2213-2600(21)00032-1
Yung L‑M, Yang P, Joshi S et al (2020) ACTRIIA-Fc rebalances activin/GDF versus BMP signaling in pulmonary hypertension. Sci Transl Med 12:eaaz5660. https://doi.org/10.1126/scitranslmed.aaz5660
Humbert M, McLaughlin V, Gibbs JSR et al (2021) Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med 384:1204–1215. https://doi.org/10.1056/NEJMoa2024277
Humbert M, McLaughlin V, Gibbs JSR et al (2023) Sotatercept for the treatment of pulmonary arterial hypertension: PULSAR open-label extension. Eur Respir J 61:2201347. https://doi.org/10.1183/13993003.01347-2022
Hoeper MM, Badesch DB, Ghofrani HA et al (2023) Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension. N Engl J Med 388(16):1478–1490. https://doi.org/10.1056/NEJMoa2213558
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
T.J. Lange has received honoraria for consultation and/or speaker activities and/or support for participation in educational activities from Acceleron Pharma, AstraZeneca, Bayer, Böhringer Ingelheim, Ferrer, Gossamer Bio, Janssen Cilag, MSD, Orphacare, and Pfizer.
For this article no studies with human participants or animals were performed by any of the authors. All studies mentioned were in accordance with the ethical standards indicated in each case.
Rights and permissions
About this article

Cite this article
Lange, T.J. Refined risk stratification, current treatment, and new therapeutic approaches in pulmonary arterial hypertension. Herz 48, 259–265 (2023). https://doi.org/10.1007/s00059-023-05179-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00059-023-05179-1