
Abstract
The prevailing COVID-19 pandemic, triggered by the novel coronavirus SARS-CoV-2, stands as the predominant global health crisis of the decade, claiming millions of lives and causing profound disruptions to society. Despite the rapid development of vaccines against COVID-19, the situation remains challenging, necessitating the exploration of new antiviral drugs. In this study, we present the design and synthesis of diphenyl-1H-imidazole derivatives as a potential lead series for inhibiting the SARS-CoV-2 3CLpro enzyme. The synthesized molecules underwent screening for inhibiting the SARS-CoV-2 3CLpro enzyme at a concentration of 20 µM. Compounds 6–14 exhibited inhibition ranging from 88 to 99%. Further assessments were conducted to evaluate the anti-SARS-CoV-2 activity of these compounds against both the ancestral SARS-CoV-2 strain and the Delta variant in virus-infected cells. Compounds such as 4-(4-chlorophenyl)-2-(3,4-dimethoxyphenyl)-1H-imidazole (9), 4-(2,4-dichlorophenyl)-2-(3,4-dimethoxyphenyl)-1H-imidazole (10), and 4-(4-(2,4-dichlorophenyl)-1H-imidazol-2-yl)benzene-1,2-diol (14) exhibited promising activity against both the SARS-CoV-2 strain (with IC50 values of 7.7 µM, 12.6 µM, and 11.8 µM, respectively) and the Delta variant (with IC50 values of 7.4 µM, 13.8 µM, and 12.1 µM, respectively). Moreover, the 3CLpro inhibition IC50 values for these compounds correlated well with the observed antiviral activity, measuring at 5.1 µM (9), 10.9 µM (10), and 7.3 µM (14). These findings underscore the efficacy of diphenyl-1H-imidazole derivatives as promising candidates for further development and optimization in the fight against COVID-19.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
COVID-19 caused by novel coronavirus (SARS-CoV-2) is the major pandemic of the decade claiming approximately over 6.6 million lives, causing severe socio-economic implications globally [1]. Vaccination has helped to get considerable control on the spread and fatality of the condition; however, complete treatment is not available [2,3,4]. Certain repurposed drugs have found benefits; however, they have their own side effects and limitations. FDA has approved some anti-viral drugs to treat mild to moderate COVID-19 condition if detected in early stages. The NIH has suggested few treatment combinations for healthcare workers, which includes Nirmatrelvir with Ritonavir, Remdesivir, Molnupiravir for different age groups. These antiviral drugs have many side effects and may show drug-drug interactions [5]. Despite substantial efforts, there no precise cure for COVID-19, and discovery of new molecules with promising antiviral activity is required.
Viral proteases are the potentially validated target for anti-viral drug discovery over many years. In addition to this, in case of SARS-CoV-2 virus, many variants (SARS-CoV-2, Delta, Omicron) are observed with different virulence capacity and morbidity. In all these variants, the virulence capacity varies mainly because of mutations in spike protein. However, major mutations in the active site of 3CLpro (main protease/Mpro) are not observed. Thus, the antiviral activity of 3CLpro inhibitors is expected to remain unaffected by any of the mutations of spike protein, generally observed in different SARS-CoV-2 variants [3, 6]. Therefore, 3CLpro is a promising target for the treatment of COVID-19.
In the current study, we have identified 2,4-diphenyl-1H-imidazole as a promising scaffold and explored its interactions with the active site of the SARS-CoV-2 3CLpro enzyme. Following the assessment of its enzyme inhibition potential in vitro and its antiviral activity in virus-infected cells, we proceeded to synthesize derivatives and subjected them to screening for both SARS-CoV-2 3CLpro enzyme inhibition and anti-SARS-CoV-2 activity (against both the ancestral SARS-CoV-2 and Delta variants) in virus-infected cells. Dose-response curves were generated for the top three molecules to ascertain their IC50 values. Additionally, an in-silico analysis was conducted to investigate their interactions with the active site of the SARS-CoV-2 3CLpro enzyme. Detailed discussion of all these findings is provided in the subsequent sections.
Results & discussion
Designing aspect
Majority of the antiviral drugs contain heterocyclic moieties with diverse substitutions. Mainly because of its active site interaction abilities, good synthetic feasibility for derivatization, and amphoteric nature, imidazole is one of the important heterocycles in the field of anti-viral drug discovery [7]. Various anti-viral agents like ledipasvir, daclatasvir (anti-hepatitis C virus), capravirine (anti-HIV), enviroxime (anti-rhinovirus and polio virus) contains imidazole scaffold. Various imidazole alkaloids (naamidine, pyronaamidine, leucettaamine) isolated from the marine sponge Pericharax heteroraphis were found effective on H1N1 influenza virus [8,9,10]. Apart from these, various research groups are working on imidazole-based drug discovery for viral infections like COVID-19, Dengue, Zika virus. Considering imidazole as a promising heterocycle, here we initiated imidazole-based lead identification. In the present report, our focus is on substituted 2,4-diphenyl imidazole.
The 3CLpro is cysteine protease enzyme made up of 306 amino acids having three major domains. Domain 1 is 8–101 amino acid residue region, domain 2 is comprised of amino acids from 102 to 184, and the 3rd domain is from 201 to 203 amino acids. The loop region with amino acids from 185 to 200 connects domain 2 with domain 3. The active site/binding site region is present between domains 1 and 2 with a Cys145-His41 catalytic dyad. This active site region is mainly comprised of S1, S1’, S2 and S4 sub-regions. The S1 region is made up of Phe140, Leu141, Asn142, His163, Glu166 and His172 amino acids. The S1’ small region is made up from S1 subsite by intrusion of Asn142 and it consists of Thr25, Thr26, and Leu27. The S2 subsite is hydrophobic and is made up of His41, Met49, Tyr54, Met165 and Asp187. The final S4 region comprises Met165, Leu167, Phe185, Gln189, and Gln192 amino acids [11, 12] (Fig. 1).

Active site of enzyme 3CLpro comprising of S1, S1’, S2 and S4 sub-regions
In virtual interaction studies, the 2-phenyl moiety of 2,4-diphenyl imidazole was observed in S2 binding subsite surrounded by Met49, Tyr54, Arg188, and Gln189. Whereas the 4-phenyl of same scaffold observed in S1 sub-site surrounded by Phe140, Ser144, and His163. The imidazole ring was observed located centrally with free N–H facing towards S4 subsite (Fig. 2).

Interactions of 2,4-diphenyl imidazole (6) with 3CLpro active site
To understand the enzyme inhibition potency of basic scaffold 6, it was tested using FRET based SARS-CoV-2 3CLpro enzyme inhibition assay and at 20 µM concentration, 89.96% enzyme inhibition was observed. Further, this compound was tested against ancestral SARS-CoV-2 and Delta variants of SARS-CoV-2 infected Vero E6 cells at the same concentration and showed 14.1% and 6.9% antiviral activity, respectively. This considerably good enzyme inhibition and low to moderate antiviral activity could be because of poor cell permeability. This suggested the possibility of the scaffold for further development. Thus, we decided to make some hydrophobic and hydrophilic modifications in the scaffold to extemporize the lead activity.
Synthesis of 2, 4-diphenyl-1H-imidazole derivatives
Compounds 6–14 were synthesized as depicted in Scheme 1. The starting material 2-Bromo-1-(substitutedphenyl)ethanone (2a–2d) were synthesized using various acetophenone (1a–1d). Second starting material analogs (5a, 5b) were synthesized from benzaldehyde/3,4-dimethoxybenzaldehyde (3a, 3b) via benzonitrile/3,4-dimethoxybenzonitrile (4a, 4b). The benzimidamide (5a/5b) were treated with different bromoketone (2a–2d) to get final compounds 4-(Substitutedphenyl)-2-(3,4-dimethoxyphenyl)-1H-imidazole (6–10). To improve the polar properties of compounds 6–10, O-demethylation was carried to obtain 4-(4-substitutedphenyl-1H-imidazol-2-yl)benzene-1,2-diol (11–14).

Syntheses of 2,4-diphenyl-1H-imidazole derivatives. Reagents and conditions: (i) Bromine, ethanol; (ii) Hydroxylamine HCl, DMSO; (iii) (a) dry HCl, ethanol, (b) Ammonium carbonate; (iv) Potassium carbonate, THF, water; (v) BBr3 (1 M in DCM), 0 °C to RT
Antiviral activity of 2,4-diphenyl-1H-imidazole derivatives
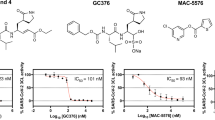
The synthesized compounds were evaluated for 3CLpro enzyme inhibition by quenched fluorescence resonance energy transfer (FRET) assay and antiviral activity against SARS-CoV-2 (ancestral SARS-CoV-2 and the Delta) by plaque assay. The FRET assay showed promising activity by all the synthesized compounds (6–14) against the 3CLpro enzyme with 88–99% inhibition at 20 µM concentration. Considering the observed 3CLpro enzyme inhibition activity, all the synthesized molecules were tested against SARS-CoV-2 (ancestral SARS-CoV-2 and the Delta variant) to understand viral inhibition potency. The initial screening of compounds at 20 µM against SARS-CoV-2 by plaque assay identified 3 hit molecules such as compounds 9, 10, and 14 (Table 1). Further, the IC50 of the hit molecules 9, 10, and 14 was found to be 7.7 µM, 12.6 µM, and 11.08 µM, respectively, against the ancestral SARS-CoV-2 strain, while the IC50 against Delta variant was found to be 7.4 µM, 13.8 µM, and 12.1 µM, respectively. The positive control remdesivir used in the antiviral assay showed an IC50 value of 0.8 µM and 0.7 µM, respectively, against ancestral SARS-CoV-2 and Delta variants (Fig. 3). To correlate the antiviral activity and 3CLpro enzyme inhibition the IC50 of hit compounds 9, 10, and 14 was determined against the 3CLpro enzyme and it was found to be 5.1 µM, 10.9 µM, and 7.3 µM, respectively. A good correlation was observed between enzyme inhibition assay and antiviral activity with hit molecules 9, 10, and 14. The details of enzyme inhibition and antiviral activity is mentioned in Table 1.

Dose Response Curve (DRC) and determination of IC50 against 3CLpro enzyme and SARS-CoV-2 (ancestral SARS-CoV-2 strain and delta variant). (A) Remdesivir; (B) Compound 9; (C) Compound 10; (D) Compound 14
Structure activity relationship
All the synthesized lead compounds (6–14) expressed promising enzyme inhibition activity from 84 to 99% in FRET assay. When these compounds were tested against ancestral SARS-CoV-2 and Delta strain, the results did not correlate with the enzyme activity. The basic amidine intermediate 5a showed 4.7% and 3.4% antiviral activity against ancestral SARS-CoV-2 and delta strains, respectively. Cyclization of this amidine to diaryl imidazole (6) showed improvement in the antiviral activities against both the strains (ancestral SARS-CoV-2: 14.1% & Delta: 6.9%). Introducing 3,4-dimethoxy substitution on B ring of diarylimidazole scaffold as in compound 7 showed drastic improvement in the antiviral activities. This modification improved the electron density of ring B, reduced the percentage of unsaturation of the overall molecule as well as has helped in improvement of lipophilicity of the molecule. The corresponding demethylated compound 11, however, was found to have reduced antiviral activity against ancestral SARS-CoV-2 strain (6.3%) and inactive against delta strain. Considerable enzyme inhibition in FRET assay, but poor activity in cell-based assay could be due to increased polarity and consequently reduced cell permeability of the molecule. This observation was also supported when its activity is compared with the basic diarylimidazole scaffold (compound 6) (Fig. 4).

SAR representation of synthesized lead molecules (6–14)
Introducing methyl substitution on ring A and retaining the dimethoxy substitution on ring B as in compound 8 showed further improvement in the antiviral activity against both the strains (ancestral SARS-CoV-2: 54.7% & Delta: 51.7%). The corresponding dihydroxy derivative (compound 12) showed drastically reduced antiviral activity (ancestral SARS-CoV-2: 1.6% & Delta: 3.4%). Replacement of methyl group (compound 8) with chloro group (compound 9, 10) showed better antiviral activity (compound 9: IC50 values of 7.7 μM and 7.4 μM; Compound 10: 12.6 μM, 13.8 μM against SARS-CoV-2 and delta strain, respectively).
Demethylation of compound 9 leads to compound 13, which showed improved enzyme inhibition, but considerably reduced viral inhibition (ancestral SARS-CoV-2: 28.1% & Delta: 24.1%). Compound 14 is demethylated compound 10, having dichloro substitution on ring A, showed very good viral inhibition activity (ancestral SARS-CoV-2: 11.8 μM & Delta: 12.1 μM). The presence of extra chloro on ring A of compound 14, as compared to compound 13, could be the reason and compensated the additional lipophilicity even after demethylation of compound 14 and supported enhanced activity. Considering molecules 9, 10 and 14 as promising lead molecules, various substitutions, and modifications at free N–H in all three leads are under progress in our lab and planned for separate communications.
Molecular docking
To understand the interactions of synthesized molecules with 3CLpro enzyme (PDB code: 6LU7), molecular docking studies were carried out using “flexible docking” protocol within BIOVIA Discovery Studio. The protocol uses a combination of components from other protocols available within BIOVIA Discovery Studio such as CDOCKER to perform the docking and is based on methods within CHARMm to sample sidechain and ligand conformations. Here the molecular interactions of identified potential hits (9, 10, and 14) are discussed (Fig. 5). Unlike compound 6, in all three cases, the 2-phenyl ring was observed to be stabilized in S1 subsite of receptor active site. Whereas the 4-phenyl ring of all three leads was stabilizing receptor-ligand complex by interacting with S4 subsite. The free N–H group is facing the S2 subsite. Considering the linear small molecular nature of synthesized ligands and substantially larger active site cavity, the orientation of ligands within active site can vary while having very stable interactions. The substitutions at N–H of imidazole will decide the final orientation of the ligands in the receptor active site.

Molecular docking interactions of: (a) compound 9; (b) compound 10; (c) compound 14 with 3CLpro (PDB code: 6LU7)
Conclusion
In summary, 2,4-diphenyl-1H-imidazole is a promising lead to inhibit SARS-CoV-2 3CLpro, which is a validated target for developing treatments for COVID-19. Substitution of the phenyl rings with methoxy and chloro groups increases the antiviral potency, probably via increasing the overall lipophilicity and subsequently the cell-permeability. Further these substituents are found to stabilize the molecular interactions with the active sites on SARS-CoV-2 3CLpro in virtual studies. The free NH of the imidazole ring provides additional site for further modifications in order to achieve maximum potency.
Experimental
General
All the chemicals were acquired from S. D. fine chemicals, Spectrochem, Sigma-Aldrich, or Avra chemicals. Pre-coated silica gel TLC plates were used for reaction monitoring. All the mentioned yields are from un-optimized processes. Melting points were determined either using melting point apparatus or by differential scanning calorimetry (DSC) and are uncorrected. Using Perkin-Elmer FT-IR/Bruker spectrophotometer all IR spectra were recorded. The 1H NMR spectra of final lead molecules were recorded on a Bruker Advance-II 500 MHz spectrometer using DMSO-d6 solvents and corresponding chemical shifts (δ) are expressed in parts per million (ppm). LC-MS-, Shimadzu (EI) was used to record the mass of compounds.
Chemistry
2-bromo-1-(substitutedphenyl)ethanone (2a–2d)
Bromine was added drop-wise to a stirred solution of an acetophenone (1 mM) (1a–1d) in ethanol (30 ml) and the solution was stirred at room temperature for 1 h and then poured into water to form a precipitate. This was re-crystallized from ethanol to give pure bromoacetophenone derivatives (2a–2d) with an 85–95% yield.
2-bromo-1-phenylethan-1-one (2a): yield 87%; m.p. 53–56 °C; IR (KBr, cm−1): 3098, 3072, 2939, 1686, 1599, 1572, 748, 687.
2-bromo-1-(p-tolyl)ethan-1-one (2b): yield 92%; m.p. 51–52 °C; IR (KBr, cm−1): 3085, 3038, 2999, 2955, 1691, 1591, 725, 665.
2-bromo-1-(4-chlorophenyl)ethan-1-one (2c): yield 95%; m.p. 94–97 °C; IR (KBr, cm−1): 3083, 3019, 2966, 2940, 1676, 1592, 798, 758.
2-bromo-1-(2,4-dichlorophenyl)ethan-1-one (2d): yield 85%; m.p. 32–34 °C; IR (KBr, cm−1): 3082, 3010, 2965, 2940, 1681, 1585, 868, 797, 769.
3,4-dimethoxybenzonitrile (4a, 4b)
Added a solution of benzaldehyde/3,4-dimethoxybenzaldehyde (3a, 3b) (1 equiv.) in 10 mL dimethylsulfoxide (DMSO) along with hydroxylamine hydrochloride (1.2 equiv.). The reaction mixture was stirred at 110 °C for 10 h. After the reaction completion, the reaction mixture was poured into the ice-cooled water. Filtered the obtained precipitate, washed it with water, and dried it under vacuum to get 4a, 4b.
Benzonitrile (4a): yield 83%; b.p. 188–190 °C; IR (KBr, cm−1): 3078, 3016, 2922, 2223, 1618, 1604, 1488, 812.
3,4-dimethoxybenzonitrile (4b): yield 88%; m.p. 68–70 °C; IR (KBr, cm−1): 3122, 3085, 2962, 2840, 2223, 1596, 1582, 1466, 818.
3,4-dimethoxybenzimidamide (5a, 5b)
To a solution of benzonitrile/3,4-dimethoxybenzonitrile (1 equiv.) in ethanol, dry HCl gas was purged to saturation. The resulted solution was stirred for 10 h. Excess HCl gas was removed and ammonium carbonate (3 equivalent) was added to it. The resulted solution was stirred further for 10 h and concentrated on rotary evaporator to obtain the product 5a, 5b.
Benzimidamide (5a): yield 87%; m.p. 78–80 °C; IR (KBr, cm−1): 3333 (broad), 3053, 2968, 2903, 2842, 1615, 1598, 852, 808.
3,4-dimethoxybenzimidamide (5b): yield 85%; m.p. 110–112 °C; IR (KBr, cm−1): 3305 (broad), 3210, 3089, 2996, 2835, 1644, 1606, 1591, 814.
4-(substitutedphenyl)-2-(3,4-dimethoxyphenyl)-1H-imidazole (6–10)
A solution of benzimidamide (5a)/3,4-dimethoxybenzimidamide (5b) (3 mmol), potassium bicarbonate (12 mmol) in THF (16 ml) and water (4 ml) was heated vigorously at reflux. Various bromoketone (2a–2d) (3 mmol) in THF (4 ml) was added over period of 30 mins and reflux further maintained for 2 h. THF was then recovered, and product was washed with water and recrystallized with ethanol to get pure products 6-10 respectively.
2,4-diphenyl-1H-imidazole (6): yield 67%; m.p. 168–170 °C; IR (KBr, cm−1): 3065, 3032, 1607, 1583, 1459, 714; 1H NMR (DMSO-d6): δ 12.63 (s, 1H, –NH), 8.02–8.01 (d, 2H, ArH), 7.86–7.85 (d, 2H, ArH), 7.72 (s, 1H, ArH), 7.49–7.46 (t, 2H, –SCH2), 7.40–7.35 (m, 3H, ArH), 7.24–7.21 (t, 1H, ArH); MS (m/z): 221.20 (M + H)+.
2-(3,4-dimethoxyphenyl)-4-phenyl-1H-imidazole (7): yield 71%; m.p. 120–122 °C; IR (KBr, cm−1): 3455, 3078, 3002, 2938, 2840, 1606, 1504, 765; 1H NMR (DMSO-d6): δ 12.48 (s, 1H, –NH), 7.86–7.85 (d, 2H, ArH), 7.72 (s, 1H, ArH), 7.61 (s, 1H, ArH), 7.56–7.54 (d, 1H, ArH), 7.38–7.35 (t, 2H, ArH), 7.21–7.18 (t, 1H, ArH), 7.06–7.04 (d, 1H, ArH), 3.86 (s, 3H, –OCH3), 3.81 (s, 3H, -OCH3); MS (m/z): 281.40 (M + H)+.
2-(3,4-dimethoxyphenyl)-4-(p-tolyl)-1H-imidazole (8): yield 68%; m.p. 105–108 °C; IR (KBr, cm−1): 3460, 3060, 3002, 2915, 2835, 1606, 1505, 764; 1H NMR (DMSO-d6): δ 12.42 (s, 1H, –NH), 7.73 (br, 2H, ArH), 7.61 (s, 1H, ArH), 7.57–7.56 (d, 2H, ArH), 7.20–7.19 (d, 2H, ArH), 7.05–7.04 (d, 1H, ArH), 3.85 (s, 3H, –OCH3), 3.80 (s, 3H, –OCH3), 2.31 (s, 3H, –CH3); MS (m/z): 295.20 (M + H)+.
4-(4-chlorophenyl)-2-(3,4-dimethoxyphenyl)-1H-imidazole (9): yield 70%; m.p. 107–110 °C; IR (KBr, cm−1): 3457, 3000, 2920, 2835, 1590, 1495, 765; 1H NMR (DMSO-d6): δ 12.53 (s, 1H, –NH), 7.87–7.86 (d, 2H, ArH), 7.77 (s, 1H, ArH), 7.60 (s, 1H, ArH), 7.56–7.54 (d, 1H, ArH), 7.43–7.42 (d, 2H, ArH), 7.06–7.04 (d, 1H, ArH), 3.85 (s, 3H, –OCH3), 3.81 (s, 3H, –OCH3); MS (m/z): 315.20 (M + H)+, 317.20 (M + H + 2)+.
4-(2,4-dichlorophenyl)-2-(3,4-dimethoxyphenyl)-1H-imidazole (10): yield 73%; m.p. 189–190 °C; IR (KBr, cm−1): 3459, 3035, 3002, 2957, 2836, 1590, 1498, 765; 1H NMR (DMSO-d6): δ 12.71 (s, 1H, –NH), 8.27–8.25 (d, 1H, ArH), 7.87 (s, 1H, ArH), 7.62 (s, 2H, ArH), 7.59–7.57 (d, 1H, ArH), 7.49–7.47 (d, 1H, ArH), 7.07–7.06 (d, 1H, ArH), 3.85 (s, 3H, –OCH3), 3.81 (s, 3H, –OCH3); MS (m/z): 349.25 (M)+, 351.20 (M + 2)+, 353.25 (M + 4)+.
4-(4-substitutedphenyl-1H-imidazol-2-yl)benzene-1,2-diol (11–14)
To obtain products 11–14, A solution of compounds 6–10, respectively (1 equiv.) in dry DCM (36 mL) at 0 °C under N2 was treated drop wise with BBr3 (1 M in DCM, 4 equiv.). The resulting mixture was allowed to attain the RT and stirred overnight, then it was drop wise poured to a stirring ice water (50 mL). The mixture was stirred for 30 min at RT then filtered and dried to obtain the products 11–14 as a yellow solid.
4-(4-phenyl-1H-imidazol-2-yl)benzene-1,2-diol (11): yield 73%; m.p. 98–100 °C; IR (KBr, cm−1): 3426, 3369, 3035, 2799, 1643, 1607, 1518, 686; 1H NMR (DMSO-d6): δ 14.33 (s, 1H, –NH), 10.13 (s, 1H, broad, variable –OH proton peaks), 9.53 (s, 1H, broad, variable –OH proton peaks), 8.20–8.18 (s, 1H, ArH), 7.91–7.90 (d, 2H, ArH), 7.58–7.39 (m, 5H, ArH), 7.02–7.00 (d, 1H, ArH); MS (m/z): 253.25 (M + H)+.
4-(4-(p-tolyl)-1H-imidazol-2-yl)benzene-1,2-diol (12): yield 69%; m.p. 118–120 °C; IR (KBr, cm−1): 3366, 3167, 2967, 1642, 1601, 1520, 715; 1H NMR (DMSO-d6): δ 14.15 (s, 1H, –NH), 10.12 (s, 1H, broad, variable –OH proton peaks), 9.51 (s, 1H, broad, variable –OH proton peaks), 8.13 (s, 1H, ArH), 7.80–7.78 (d, 2H, ArH), 7.48 (s, 1H, ArH), 7.44–7.42 (d, 1H, ArH), 7.36–7.35 (d, 2H, ArH), 7.01–6.99 (d, 1H, ArH), 2.37 (s, 3H, –CH3); MS (m/z): 267.25 (M + H)+.
4-(4-(4-chlorophenyl)-1H-imidazol-2-yl)benzene-1,2-diol (13): yield 72%; m.p. 305–307 °C; IR (KBr, cm−1): 3301, 3276, 3135, 2972, 2888, 1639, 1601, 1510, 808, 704, 647; 1H NMR (DMSO-d6): δ 14.35 (s, 1H, –NH), 10.13 (s, 1H, broad, variable –OH proton peaks), 9.53 (s, 1H, broad, variable –OH proton peaks), 8.23 (s, 1H, ArH), 7.95–7.93 (d, 2H, ArH), 7.65–7.63 (d, 2H, ArH), 7.49–7.48 (d, 1H, ArH), 7.45–7.43 (d, 1H, ArH), 7.01–6.99 (d, 1H, ArH); MS (m/z): 287.20 (M + H)+, 289.20 (M + H + 2)+.
4-(4-(2,4-dichlorophenyl)-1H-imidazol-2-yl)benzene-1,2-diol (14): yield 70%; m.p. 269–271 °C; IR (KBr, cm−1): 3377, 3140, 2987, 1635, 1603, 849, 808, 705; 1H NMR (DMSO-d6): δ 14.55 (s, 1H, –NH), 10.07 (s, 1H, broad, variable –OH proton peaks), 9.51 (s, 1H, broad, variable –OH proton peaks), 8.04 (s, 1H, ArH), 7.89–7.88 (d, 1H, ArH), 7.85–7.83 (d, 1H, ArH), 7.69–7.67 ((d)d, 1H, ArH), 7.46–7.45 (d, 1H, ArH), 7.41–7.39 ((d)d, 1H, ArH), 6.99–6.97 (s, 1H, ArH); MS (m/z): 321.15 (M)+, 323.15 (M + 2)+, 325.15 (M + 4)+.
Enzyme inhibition (3CLpro) assay
The synthesized compounds were screened as inhibitors of SARS-CoV-2 3CLpro at 20 µM using an in vitro quenched fluorescence resonance energy transfer (FRET) assay using a fluorogenic substrate to measure the residual activity. For the study, the MBP-tagged 3CL Protease (SARS-CoV-2) Assay Kit (BPS Bioscience, San Diego, CA, USA) was used according to the manufacturer’s instructions [13, 14]. For determination of the 3CLpro activity, 10 µL of the compounds was pre-incubated with 30 µl of the 3CLpro for 30 min. Subsequently, the fluorogenic substrate was added to a final concentration of 50 µM and the reaction was incubated for 4 h in the dark in the presence of 1 mM 1,4-dithio-D, L-threitol (DTT). The fluorescence intensity was recorded at 460 nm/360 nm. A positive control was included to measure the maximum activity of the protease in the absence of potential inhibitors. Moreover, an inhibition control GC 376 at 20 µM was included in the study. All the compounds were tested initially at 20 µM to establish the enzyme inhibition and antiviral activity. The hit molecules identified from the antiviral screening against SARS-CoV-2 (ancestral SARS-CoV-2 and Delta) were subjected to IC50 determination against SARS-CoV-2 variants by plaque assay and 3CLpro inhibition by FRET assay.
Cell lines and viruses
The African green monkey kidney epithelial cell (Vero E6) was cultured in a humidified CO2 (5%) incubator at 37 °C, in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (100 units/mL), streptomycin (100 µg/mL) and Amphotericin B (0.25 µg/mL).
SARS-CoV-2 isolate USA-WA1/2020 (ancestral SARS-CoV-2 strain) and B.1.617.2 (Delta) were obtained from bei Resources, USA. The virus stocks were prepared by propagating in Vero E6 cells by following the standard protocol [15, 16]. The virus stocks were quantified by the gold standard plaque assay [15]. The SARS-CoV-2 infection study was carried out in high containment (BSL-3) facility.
Screening and determination of IC50 value through dose response curve generation
The compounds were solubilized in dimethyl sulfoxide (DMSO) and screened against SARS-CoV-2 (ancestral SARS-CoV-2 and Delta) by plaque assay. For the initial antiviral screening, Vero E6 cells were seeded at a density of ~30,000 cells per well, in 96 well flat bottom tissue culture plate in 200 µL of complete DMEM. The plate was incubated for 18–24 h, at 37° C in a humidified CO2 (5%) incubator. Next day, the medium was removed from the wells and the test compounds were added in duplicate to respective wells at a final concentration of 20 µM, followed by infection with SARS-CoV-2 isolates at approximately 30 plaque-forming units per well. The plate was incubated at 37° C for 1 h, in a humidified CO2 (5%) incubator for adsorption of virus. The DMEM supplemented with 2.5% FBS (infection medium) was used to dilute the test compounds and virus. The final volume of infection medium containing test compounds and virus was maintained at 40 µL per well to maximize the virus adsorption. After 1 h of incubation, the infection medium was removed from the wells and overlayed with DMEM-CMC, and incubated for 72 h and then the plates were processed to score the plaques. The controls including virus-only wells (with infection and without test compound) and cell-only wells (without infection and test compound) were maintained as positive and negative controls, respectively. The percentage reduction of virus in test compound treated wells were calculated in comparison to positive control (virus-only well).
The hit molecules identified in the initial screen were subjected to six-point dose-response curve (DRC) generation (20 µM, 10 µM, 5 µM, 2.5 µM, 1.25 µM, and 0.625 µM) and IC50 determination in Vero E6 cells. The IC50 of the test compounds was calculated by non-regression analysis using GraphPad prism version 9.2.0.
Molecular docking
The interactions of molecules under study with 3CLpro enzyme (PDB Code: 6LU7) were analyzed using “flexible docking” protocol within BIOVIA Discovery Studio software [17]. To perform the flexible docking, residues (THR25, LEU27, HIS41, VAL42, CYS44, SER46, MET49, LEU50, TYR54, PHE140, LEU141, ASN142, SER144, CYS145, HIS163, HIS164, MET165, GLU166, LEU167, HIS172, ALA173, PHE181, VAL186, ASP187, ARG188, GLN189, THR190 and GLN192) were considered flexible in study. In the study, these residues were considered in creating flexible protein conformations using ChiFlex and side-chain refinement in the presence of the ligand using ChiRotor. In the process, generating protein confirmation was kept true with maximum number to 100. Ligand conformation generation was allowed using BEST method with maximum 255 conformations and energy threshold value to 20 for each ligand under study. Docking protocol was run with 100 number of hotspots and docking refinement was carried using simulated annealing with 2000 heating and 5000 cooling steps. The x, y, z coordinates for docking was set as –11.859595, 13.885757, 69.446622, respectively.
References
https://www.who.int/publications/m/item/covid-19-weekly-epidemiological-update---21-december-2022 (cited on 02-Jan-2023)
Pelly S, Liotta D. Potent SARS-CoV-2 direct-acting antivirals provide an important complement to COVID-19 vaccines. ACS Cent Sci. 2021;7:396–99.
Unoh Y, Uehara S, Nakahara K, Nobori H, Yamatsu Y, Yamamoto S, et al. Discovery of S‑217622, a noncovalent oral SARS-CoV‑2 3CL protease inhibitor clinical candidate for treating COVID-19. J Med Chem. 2022;65:6499–512.
Fan H, Lou F, Fan J, Li M, Tong Y. The emergence of powerful oral anti-COVID-19 drugs in the post-vaccine era. Lancet. 2022;3:e91.
https://www.cdc.gov/coronavirus/2019-ncov/your-health/treatments-for-severe-illness.html (cited on 02-Jan-2023)
Shahanshah MFH, Jain S, Sharma B, Grewall A, Swami S. Comparative analysis of B.1.617.2 (Delta) variant of SARS-CoV-2. JMID. 2022;12:38–51.
DeSimone RW, Currie KS, Mitchell SA, Darrow JW, Pippin DA. Privileged structures: applications in drug discovery. Comb Chem High Throughput Screen. 2004;7:473–93.
Fei F, Zhou Z. New substituted benzimidazole derivatives: a patent review (2010−2012). Expert Opin Ther Pat. 2013;23:1157–79.
Ingle RG, Magar DD. Heterocyclic chemistry of benzimidazoles and potential activities of derivatives. Int J Drug Res Technol. 2011;1:26–32.
Gong KK, Tang XL, Liu YS, Li PL, Li GQ. Imidazole alkaloids from the South China Sea sponge Pericharax heteroraphis and their cytotoxic and antiviral activities. Molecules. 2016;21:150.
Kanhed AM, Patel DV, Teli DM, Patel NR, Chhabria MT, Yadav MR. Identification of potential Mpro inhibitors for the treatment of COVID-19 by using systematic virtual screening approach. Mol Divers. 2021;25:383–401.
Patel DV, Teli DM, Kanhed AM, Patel NR, BS Shah, A Vora, et al. Identification of potential Mpro inhibitors for the treatment of COVID-19 by targeted covalent inhibition: an in silico approach. IJQSPR. 2021;6:58–77
Morse JS, Lalonde T, Xu S, Ray Liu W. Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. ChemBioChem. 2020;21:730–8.
Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–12.
Case JB, Bailey AL, Kim AS, Chen RE, Diamond MS. Growth, detection, quantification, and inactivation of SARS-CoV-2. Virology. 2020;548:39–48.
Jureka AS, Silvas JA, Basler CF. Propagation, inactivation, and safety testing of SARS-CoV-2. Viruses. 2020;12:622.
Dassault Systèmes BIOVIA, Discovery studio modeling environment, release 2022. San Diego: Dassault Systèmes; 2022.
Acknowledgements
Authors (AMK & AV) are thankful to SVKM’s NMIMS University for providing financial support under ‘University Seed Grant’ scheme to carry out this work. The following reagent was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52281. The following reagent was obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, Isolate hCoV-19/USA/PHC658/2021 (Lineage B.1.617.2; Delta Variant), NR-55611, con-tributed by Dr. Richard Webby and Dr. Anami Patel.
Author information
Authors and Affiliations
Contributions
AMK & AV: Designing of project, in-silico studies, synthesis of the compounds and drafting and editing the manuscript; AT: Preliminary screening of compounds; GRR, RKS, MY, RH, MS, SN: 3CLpro inhibition assay and anti-SARS-CoV-2 activity, drafting and editing the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
Authors (AMK and AV) received financial support from SVKM’s NMIMS under ‘University Seed Grant’ scheme for this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kanhed, A.M., Vora, A., Thakkar, A. et al. Design and synthesis of diphenyl-1H-imidazole analogs targeting Mpro/3CLpro enzyme of SARS-CoV-2. Med Chem Res (2024). https://doi.org/10.1007/s00044-024-03263-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00044-024-03263-7