
Abstract
To investigate the chemical constituents of Eupatorium adenophorum Spreng. (syn. Ageratina adenophora (Spreng.) R.M. King & H. Rob.) growing in Vietnam, the water-soluble fraction from the leaf methanol extract was fractionated by column chromatography. A new sesquiterpenoid, named adenophorone (1), was isolated along with 11 known compounds (2-12). The interpretation of HR-MS and 1D and 2D NMR spectroscopic data together with experimental and theoretical ECD calculations established the absolute stereostructure of compound 1. Two isolated flavonol glucosides 9 and 10 were subjected to enzyme inhibition assays. Gossypetin 5-O-(6′′-(E)-caffeoyl)-β-D-glucopyranoside (9) inhibited α-glycosidase activity with an IC50 value of 24.0 ± 1.61 μg/mL (80% inhibition at 256 μg/mL) and acetylcholinesterase activity with an IC50 value of 217.60 ± 15.47 μg/mL (54% inhibition at 256 μg/mL). Quercetagetin 7-O-β-D-glucopyranoside (10) slightly inhibited the two enzymes (27% and 34% inhibition, respectively) at the maximal tested concentration of 256 μg/mL. The present study is the first report on the chemical constituents of the water-soluble fraction of E. adenophorum. The study also provides some evidence for the α-glycosidase and acetylcholinesterase activities of the rare polyhydroxyflavonol acylglycoside gossypetin 5-O-(6′′-(E)-caffeoyl)-β-D-glucopyranoside.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Eupatorium is a large plant genus of the family Asteraceae, which is distributed throughout tropical America, Europe, Africa, and Asia [1]. The genus Eupatorium has been known for its therapeutic properties for many decades and is a promising bioresource for bioactive substances for drug development [2]. In Vietnam, Nguyen T. B. and Pham H. H. recorded twelve Eupatorium species, including E. odoratum L., E. triplinerve Vahl., E. reesesii Wall., E. quaternum DC., E. cannabium L., E. capillifolium (Lamk.) Small, E. chinense L., E. coelestinum L., E. fortunei Turcz., E. heterophyllum DC., E. japonicum Thunb., and E. lindleyyanum DC. [3, 4]. E. adenophorum Spreng. [syn. Ageratina adenophora (Spreng.) R.M. King & H. Rob.] is a perennial shrub growing 1–2 m in height, which is considered a harmful weed of crops and the natural environment. Originating from Mexico, E. adenophorum has spread to Vietnam, China, and adjacent regions. In Vietnam, E. adenophorum is included in the List of Invasive Plant Species of Vietnam according to Circular 35/2018/TT-BTNM issued by the Ministry of Natural Resources and Environment of Vietnam in 2018. The leaves of E. adenophorum are used in traditional medicine in China, India, and Nigeria [5,6,7,8]. In eastern Himalayas young leaves and shoots of E. adenophorum are given orally against dysentery [5]. In Nigeria the leaves of E. adenophorum are used locally to treat fever, diabetes, and inflammation [6]. The root juice of E. adenophorum is recommended to treat fever [7, 8]. In China, E. adenophorum has been used as a traditional Chinese medicine to treat fever, desinsectization, traumatism, and phyma [8]. Phytochemistry and biological activities of leaf extracts and isolates of E. adenophorum have been described in several reviews and in a growing number of publications. The presence of cadinane sesquiterpenes [9,10,11], quinic acid derivatives [9, 10, 12], flavonoids [9], and phenolic acid derivatives [9, 13, 14] has been reported from the title plant. Diverse biological activities such as antiinflammatory, cytotoxic, antioxidant, antibacterial, antiviral, wound healing, analgesic, acarcidal, and insecticidal activities of the leaf extracts of E. adenophorum have been reported [8,9,10]. In the bioassays of the isolates, cadinene sesquiterpenes and phenolic compounds have been found to be the active principles of E. adenophorum [8]. Thus, 9-oxo-10,11-dehydroageraphorone showed an EC50 value of 0.325 mg/mL against Fusarium oxysporum and 5-O-trans-o-coumaroylquinic acid methyl ester showed an IC50 value of 542.3 μM against Magnaporthe grisea [8]. Chlorogenic acid methyl ester showed scavenging properties against DPPH radicals with an IC50 of 212.2 μM [8]. Thymol derivatives (7,8,9-trihydroxythymol and 7-formyl-9-isobutyryloxy-8-hydroxythymol) showed cytotoxic activity against human cancer cell lines MCF-7, NCI-H460, and HeLa with the respective IC50 values ranging from 44.65 to 83.19 μM [8]. 9-Oxo-10,11-dehydroageraphorone, 9-oxoageraphorone, and 9β-hydroxyageraphorone showed insecticidal effects against Psoroptes cuniculi [8]. So far, most of the previous studies have focused on the extractives in organic solvents; water-soluble fractions from the leaves of E. adenophorum have not been investigated. Most of the reported biological studies have been performed with the extracts, and the bioactive compounds present in the extracts have not been identified. There are not any investigations on the chemical constituents and biological activities of E. adenophorum growing in Vietnam.
Acetylcholinesterase (AChE) is an enzyme that degrades the neurotransmitter acetylcholine in the nerve synapse. AChE inhibitors are candidates for screening agents to treat neurodegenerative diseases such as Alzheimer’s disease. Some flavonoids isolated from E. adenophorum such as quercetagetin-7-O-(6-O-caffeoyl-β-D-glucopyranoside), 5,4’-dihydroxytlavone, and quercetin-3-O-β-D-glucopyranoside showed inhibitory activities against AChE from Caenorhabditis elegans (IC50 values ranged from 12.54 to 89.06 μg/mL) and Spodoptera litura (IC50 values ranged from 12.08 to 86.01 μg/mL) [15]. The acyl glucose moiety in the flavonoid glycosides and the number of hydroxyl groups favoured the AChE inhibitory activity, and among the flavonoids, quercetagetin-7-O-(6-O-caffeoyl-β-D-glucopyranoside) displayed the highest inhibitory effect against AChE with IC50 values of 12.54 μg/mL and 12.58 μg/mL, respectively [15].
Effective control of hyperglycemia in type 2 diabetes mellitus includes the inhibition of carbohydrate hydrolysing enzymes such as α-amylase and α-glycosidase with α-glycosidase being the most important enzyme in carbohydrate digestion. Flavonoids are kown as modulators of the enzyme activity, and flavonoids with two catechol groups of the A- and B-rings, together with a 3-OH group of the C-ring present lower IC50 values [16]. However, the previous study mainly focused on flavonoid aglycones and did not show the influence of the glycosidic or acyl glycosidic moieties of flavonoid glycosides on inhibitory activity. E. adenophorum methanol extract was found to inhibit α-amylase activity with an IC50 value of 1.84 ± 0.007 mg/mL [17]. There are no reports on α-glycosidase inhibitory activity of flavonoid constituents from the extract. The objective of this study was to investigate the water-soluble constituents of a MeOH extract from the leaves of E. adenophorum growing in Vietnam. α-Glycosidase and acetylcholinesterase inhibition assays were also performed to determine antidiabetic and anti-AChE properties of acylated polyhydroxyflavonoid glycosides found in the water-soluble fraction.
Results and discussion
Chemistry
The methanolic extract of the dried leaves of E. adenophorum was subjected to sequential liquid-liquid fractionation with n-hexane and CH2Cl2. The water phase was concentrated and separated by a reversed-phase Diaion HP-20 copolymer column. Repeated column chromatography (CC) led to the isolation of phenolic acids (2, 3, 4, 6, and 7), thymol (5), phenylpropenoic acid (8), flavonols (9 and 10), sterol glucosides (11 and 12), and a new cadinane sesquiterpenoid, adenophorone (1) (Fig. 1). The known compounds from E. adenophorum were identified as 4-hydroxybenzoic acid (2) [18], methyl protocatechuate (3) [18, 19], isovanillic acid (4) [18], 5-O-glucopyranosylthymoquinol (5) [20], o- and p-coumaric acids (6 and 7) [21, 22], 2-O-β-D-glucopyranosylcinnamic acid (8) [23, 24], gossypetin 5-O-(6′′-(E)-caffeoyl)-β-D-glucopyranoside (9) [25], quercetagetin 7-O-β-D-glucopyranoside (10) [23, 26] by comparison of their spectral data (MS and NMR) with those reported in the literature.

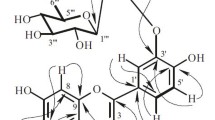
Chemical structures of compounds 1–12
Adenophorone (1) was obtained as a white amorphous powder, \([\alpha]^{23}_{\rm{D}} -\!33.3\, (c\, 0.07, {\rm{CH}}_{3}{\rm{CN}})\). The molecular formula of 1 was established as C15H24O3 from the HR-ESI-MS peak at m/z 275.1621 ([M+Na]+, calcd. 275.1618). The 1H NMR spectrum (CD3OD) showed the presence of three methyl doublets at δH 0.87 (3H, d, J = 6.5 Hz, H-14), 1.01 (3H, d, J = 7.1 Hz, H-13), and 1.02 (3H, d, J = 6.3 Hz, H-15). In addition, two oxymethines at δH 3.88 (1H, d, J = 10.8 Hz, 10.6 Hz, H-4) and 4.20 (1H, td, J = 3.1 Hz, 2.9 Hz, H-7), and an oxymethylene at δH 3.58 (1H, dd, J = 10.7 Hz, 5.4 Hz, H-12a) and 3.97 (1H, dd, J = 11.8 Hz, 10.7 Hz, H-12b) were observed. Based on HSQC correlations, two methylenes at δH 1.26 (1H, ddd, H-8a) and 1.73 (1H, ddd, H-8b), and 2.56 (1H, ddd, H-1a) and 2.60 (1H, dd, H-1b) together with six methines at δH 1.53 (1H, m, H-9), 1.68 (1H, m, H-10), 1.71 (1H, m, H-6), 2.07 (1H, m, H-11), 2.08 (1H, dt, H-5), and 2.38 (1H, dqd, H-3) were identified. The 13C NMR spectroscopic data of 1 aided by HSQC correlations confirmed that the molecule contained 15 carbons, including a ketone carbonyl group at δC 213.2 (C-2), three methyls at δC 13.4 (C-13), 19.8 (C-14), and 10.8 (C-15), two oxymethines at δC 68.4 (C-7) and 80.5 (C-4), an oxymethylene at δC 72.1 (C-12), two methylenes at δC 44.6 (C-1) and 45.3 (C-8), and five methines at δC 26.6 (C-9), 35.9 (C-11), 43.1 (C-6), 45.9 (C-5), 46.0 (C-10), and 52.2 (C-3). By comparing the spectroscopic data of 1 with related sesquiterpenoid skeletons having two methyl doublets and an isopropyl group, it was assumed that 1 had the cadinane skeleton. 1H-1H COSY, HSQC, and HMBC correlations (Fig. 2) were used to verify the suggestion and to place the functional groups and substituents in the planar structure of 1. 1H-1H COSY correlations showed two spin systems: H3-15/H-3/H-4/H-5/H-6/H-11(/H-12)/H3-13 and H-1/H-10/H-9(/H3-14)/H-8/H-7. The two spin systems were connected through 1H-1H COSY interactions between H-5 and H-10 and between H-6 and H-7. The ketone carbonyl group (δC 213.2) was placed at C-2 based on HMBC correlations between H-15 and C-2, and H-15 and C-3. An ether oxygen bridge was identified between C-4 and C-12 based on the HMBC correlation between H-12 and C-4. Finally, the 7-hydroxy group was assigned based on HMBC correlations between H-7 and C-6, and H-7 and C-9. Thus, the planar structure of 1 was unambiguously determined as 4,12-epoxy-7-hydroxycadinan-2-one.

1H-1H COSY and HMBC correlations of 1
The relative stereochemistry of 1 was elucidated based on NOESY correlations (Fig. 3). The same β-orientation of CH3-15 and H-4 was deduced from NOESY correlations between H-4 and C-15 methyl protons. The opposite α-orientation of CH3-14 was determined based on the NOESY correlation between H-4β and H-9. The α-orientation of H-10 was determined based on NOESY correlations between CH3-14 and equatorial H-1a, CH3-14 and H-8a/H-8b, and CH3-14 and H-10, as well as H-10 and H-8a. H-5 was placed on the opposite face from that of H-4β and CH3-15 (α-orientation), since H-3α gave NOE with H-1b and H-1b with H-5. H-6 did not give NOE with CH3-13 and was assigned to possess an opposite α-orientation. H-7 was assigned α-orientated based on NOESY correlations of CH3-13 and H-7. The analysis of NOESY spectroscopic data resulted in the structure of 1 as shown in Fig. 1. Configurational analysis for 1 was performed by comparison of calculated and experimental electronic circular dichroism (ECD) data. The experimental ECD spectrum (solid curve) in Fig. 4 (CD curves) showed a close resemblance to the calculated one (dotted curve) for 1. Therefore, the structure of adenophorone (1) was elucidated to be (3R,4S,5S,6S,7R,9R,10S,11R)-4α,12-epoxy-7β-hydroxycadinan-2-one.

Phase-sensitive NOESY correlations of 1

Experimental and calculated ECD spectra of 1
α-Glycosidase and AchE inhibition assays of compounds 9 and 10
α-Glycosidase aids in the digestion of carbohydrates by cleaving complex carbohydrates to yield glucose, since only monosaccharides such as glucose or fructose can be absorbed into the bloodstream. Two membrane-bound intestinal α-glycosidases, maltase-glucoamylase and sucrase-isomaltase, cleave α-(1,4)-glycosidic linkages to release α-D-glucose in the human digestive system. Inhibiting the function of these enzymes in patients with type 2 diabetes reduces hyperglycemia (high blood sugar). Hyperglycemia mainly affects people with diabetes and can become serious if not treated. α-Glycosidase inhibitors (AGI), such as acarbose and miglitol, are FDA-approved drugs that treat type-2 diabetes [27]. AGIs lower the effect of postprandial sugar in the short term by blocking the breakdown of starchy foods and slowing down the absorption of some sugars. In vitro, the α-glycosidase inhibition assay substrate, p-nitrophenyl α-D-glucopyranoside (pNPG), is hydrolyzed by α-glycosidase to p-nitrophenyl and its absorbance at 410 nm is measured. The colorimetric quantification was used to calculate α-glycosidase inhibitory activity (%) and half-maximal inhibitory concentration (IC50). Table 1 shows the results of α-glycosidase inhibitory activity of compounds 9 and 10. At 256 μg/mL, gossypetin 5-O-(6′′-(E)-caffeoyl)-β-D-glucopyranoside (9) inhibited α-glycosidase activity by 80%. An IC50 of 24.0 ± 1.61 μg/mL was determined, while acarbose showed an inhibitory activity of 134.56 ± 3.02 μg/mL as a positive control. At 256 μg/mL, quercetagetin 7-O-β-D-glucopyranoside (10) inhibited α-glycosidase inhibitory activity by only 27%. According to [16], the aglycones of compounds 9 and 10 satisfy the structural requirements for the α-glycosidase inhibitory activity, including the catechol moiety in the B-ring and 3-OH in the C-ring, however, the loss of 5-OH or 7-OH may reduce the activity. The result of the inhibition assay of 10 reflects the loss of 7-OH since quercetagetin showed the α-glycosidase inhibitory activity with an IC50 value of 180.11 ± 3.68 μM [28]. Regarding the potent activity of compound 9, additional caffeoyl hydroxy groups in the glucose moiety may be involved in binding to the active site of the enzyme.
Acetylcholine (ACh) is a neurotransmitter associated with cognitive, autonomic, and neuromuscular functions. ACh is synthesized from choline and acetyl CoA with the enzymatic catalysis of choline acetyltransferase. ACh has a very short lifetime; it is broken down by the enzyme acetylcholinesterase (AChE) to choline and acetic acid, and thus the neurotransmission process is terminated. Choline is then reentered into nerve cells for the synthesis of ACh. There are lower levels of ACh in the brain of a person with Alzheimer’s disease, harming the communication between nerve cells, and some of the nerve cells that use ACh are also lost. Chemicals that inhibit AChE activity interfere with the hydrolysis process, leading to the accumulation of ACh and a prolonged lifetime of ACh at nerve synapses, and hence easing the symptoms of Alzheimer’s disease. Some natural alkaloids, such as galantamine (galanthamine) from Galanthus, Narcissus, and Leucojum species of the Amaryllidaceae family and huperzine A from Huperzia sp., are available drugs to treat Alzheimer’s disease based on the inhibition of AChE. In the AChE inhibition assay in vitro (Ellman’s method), the substrate acetylthiocholine iodide (ATCI) is hydrolyzed by AChE into thiocholine, which reacts with 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) to form a yellow product, 5-thio-2-nitrobenzoate (TNB). The concentration of TNB is quantified by its absorbance at 412 nm, which reflects AChE activity. The results of the AChE inhibitory activity of compounds 9 and 10 are shown in Table 2. Gossypetin 5-O-(6′′-(E)-caffeoyl)-β-D-glucopyranoside (9) inhibited 54% AChE activity at the maximal concentration of 256 μg/mL. The half-maximal inhibitory concentration (IC50) of 9 was determined to be 217.60 ± 15.47 μg/mL; in the same assay, the IC50 of donepezil was 0.018 ± 0.008 μg/mL. Quercetagetin 7-O-β-D-glucopyranoside (10) inhibited only 34% AChE activity at 256 μg/mL. In comparison with the AChE inhibitory activity of quercetagetin-7-O-(6-O-caffeoyl-β-D-glucopyranoside) [15] the loss of the caffeoyl hydroxy groups in the glucose moiety of compound 10 reduced the activity. Due to the additional binding caffeoyl hydroxy groups in compound 9, the AChE activity of 9, albeit weak, may be explained.
Materials and Methods
General
Optical rotations were measured on a Jasco P-2200 digital polarimeter. IR spectra were measured on a JASCO FT/IR-6100 spectrophotometer. The UV and ECD spectra were obtained with a JASCO J-1100 spectropolarimeter. 1H NMR, 13C NMR, DEPT, COSY, HMQC, HMBC, and NOESY spectra were obtained on a Bruker Avance 500 MHz NMR spectrometer (Billerica, MA, USA) at 500 MHz and 125 MHz or on a Bruker Avance III spectrometer at 600 MHz and 150 MHz. CD3OD and DMSO-d6 were NMR solvents, and TMS (tetramethylsilane) was used as the reference standard. ESI-MS spectra were measured on an Agilent LC-MS Ion Trap LC/MS system (Santa Clara, CA, USA). HR-ESI-MS spectra were measured on a Thermo Fisher Scientific LTQ Orbitrap XL mass spectrometer (Whatham, MA, USA). Thin-layer chromatography (TLC) was performed on Merck pre-coated TLC silica gel 60 F254 sheets (Darmstadt, Germany), and the spots were detected by spraying with 1% vanillin (China)-concentrated H2SO4 (China) or 5% FeCl3 (China)-EtOH (China) and then heating on a hot plate. Column chromatography (CC) was performed on Merck silica gel of 63–200 μm, 40–63 μm, or 15–40 μm (Darmstadt, Germany) particle size and reversed-phase Diaion HP-20 (Mitsubishi Chemicals, Japan). Solvents for column chromatography: n-hexane (Korea), dichloromethane (Taiwan), acetone (Taiwan), ethyl acetate (Singapore), and MeOH (Indonesia), were distilled and dried over Na2SO4 (China) before use.
Plant material
The leaves of E. adenophorum were collected in September 2016 in Sapa, Lao Cai Province, Vietnam. The leaves were shade-dried and the oven-dried at 40 oC. The plant was identified by Dr. Nguyen Thi Kim Thanh (Faculty of Biology, VNU University of Science, VNU Hanoi). A voucher specimen (EA-09-16) was deposited at the same institute.
Extraction and isolation
The dried leaves of E. adenophorum (1.34 kg) were powdered and extracted three times with MeOH (each time for 7 days) at room temperature. The combined MeOH extract was evaporated using a Büchi RType (KRvr 65/45) evaporator under reduced pressure to yield a MeOH extract. The MeOH extract was partitioned between H2O and n-hexane and then dichloromethane successively to afford the corresponding soluble fractions. The water phase was concentrated and separated by using a Diaion HP-20 column (6 cm i.d. × 100 cm length) with a MeOH-H2O solvent gradient (20, 40, 60, and 100%, v/v) to yield four corresponding fractions. The 60% fraction (13.1 g) was chromatographed on a silica gel (63–200 µm) column (3.0 cm i.d. × 30 cm length) and eluted with a stepwise gradient of dichloromethane-MeOH (9:1, 6:1, 1:1, v/v); acetone-MeOH (6:1, 3:1, 1:1, v/v) to yield 9 fractions (F1-F9) on the basis of TLC analysis. Fraction F2 was fractionated on silica gel (40–63 µm) with n-hexane-ethyl acetate (3:1, 1:1, 1:3, v/v) to yield compounds 1 (2.0 mg), 2 (27.5 mg), 3 (3.0 mg), and 4 (17.3 mg). Fraction F4 was separated on a silica gel (40–63 µm) column eluted with n-hexane-acetone (50:1, 12:1, 9:1, 6:1, 3:1, 1:1, v/v) to yield compounds 4 (3.1 mg) and 5 (258.7 mg). Fraction F5 was separated by silica gel (40-63 µm) CC with dichloromethane-ethyl acetate (1:9, 1:15, v/v); ethyl acetate-acetone (6:1, 3:1, 1:1, v/v) to give a mixture of compounds 6 and 7 (5.0 mg). Fraction F6 was separated by silica gel (40–63 µm) CC with ethyl acetate-acetone (9:1, 6:1, 3:1, 1:1, v/v) to yield compound 8 (10.0 mg). Fraction F7 was separated by silica gel (40–63 µm) CC with ethyl acetate-acetone (15:1, 9:1, 6:1, 3:1, 1:1, v/v) to yield compound 9 (51.0 mg). Fraction F8 was recrystallized in MeOH to yield compound 10 (11.7 mg). The 100% fraction (4.55 g) was fractionated on a silica gel (40–63 µm) column (3.0 cm i.d. × 30 cm length) and eluted with a dichloromethane-methanol solvent gradient (9:1, 6:1, 3:1, 1:1, v/v) to yield 9 fractions F1-F9. Fraction F6 was separated by silica gel (40–63 µm) CC with dichloromethane-ethyl acetate (1:3, 1:5, v/v) to yield a mixture of compounds 11 and 12 (3.0 mg).
Adenophorone (1)
White amorphous powder. \([\alpha]^{23}_{\rm{D}} -\!33.3 \,(c\, 0.07, {\rm{CH}}_{3}{\rm{CN}})\). UV λmax (CH3CN) log ε (nm): 2.56 (205), 1.11 (286). IR (KBr) νmax cm−1: 3456, 2946, 2899, 1706, 1458, 1372, 1069, 1034. ECD Δε (nm): −1.52 (204), −0.21 (290) (c 3.97 × 10−3 M, CH3CN). Positive-ion HR-ESI-MS: 275.1621 (calculated for C15H24O3Na: 275.1618). 1H-NMR (CD3OD): δ 0.87 (3H, d, J = 6.5 Hz, H-14), 1.01 (3H, d, J = 7.1 Hz, H-13), 1.02 (3H, d, J = 6.3 Hz, H-15), 1.26 (1H, ddd, J = 13.9 Hz, 12.0 Hz, 2.9 Hz, H-8a), 1.53 (1H, m, H-9), 1.68 (1H, m, H-10), 1.71 (1H, m, H-6), 1.73 (1H, ddd, J = 13.9 Hz, 3.2 Hz, 3.1 Hz, H-8b), 2.07 (1H, m, H-11), 2.08 (1H, dt, J = 10.8 Hz, 4.8 Hz, H-5), 2.38 (1H, dqd, J = 10.6 Hz, 6.3 Hz, 0.9 Hz, H-3), 2.56 (1H, ddd, J = 13.4 Hz, 5.9 Hz, 0.9 Hz, H-1a), 2.60 (1H, dd, J = 13.4 Hz, 2.8 Hz, H-1b), 3.58 (1H, dd, J = 10.7 Hz, 5.4 Hz, H-12a), 3.88 (1H, dd, J = 10.8 Hz, 10.6 Hz, H-4), 3.97 (1H, dd, J = 11.8 Hz, 10.7 Hz, H-12b), 4.20 (1H, td, J = 3.1 Hz, 2.9 Hz, H-7). 13C-NMR (CD3OD): δ 10.8 (C-15), 13.4 (C-13), 19.8 (C-14), 26.6 (C-9), 35.9 (C-11), 43.1 (C-6), 44.6 (C-1), 45.3 (C-8), 45.9 (C-5), 46.0 (C-10), 52.2 (C-3), 68.4 (C-7), 72.1 (C-12), 80.5 (C-4), 213.2 (C-2).
Theoretical ECD calculation for the determination of absolute configuration of 1
Configurational analysis was performed with the Spartan′20 V1.1.2. program (Wavefunction, Inc., Irvine, CA, USA) on a commercially available personal computer [operating system: Microsoft 64-bit version of Windows 10 Home edition; 16-core central processing unit: Ryzen 9 5950X processor (Advanced Micro Devices, Inc., Santa Clara, CA, U.S.A.) run at 3.4 GHz; random access memory: 32 GB]. Stable conformers up to 40 kcal/mol for 1 were initially searched using the Merck molecular force field method. Then the aforementioned stable conformers were further optimized using the Hartree-Fock (HF)/3-21 G and ωB97XD/6-31 G* programs. The resulting conformers were subjected to ECD calculations, and the ECD calculations for these conformers were performed with Gaussian 16 (Revision A.03 by Gaussian) [29] on the ChemPark cloud system [30]. The dominant conformers of 1 capable of covering >99% of the population according to Boltzmann’s distribution were selected. Time-dependent density functional theory calculations were conducted at the CAM-B3LYP/TZVP level for these conformers. The resulting rotational strength data were converted to Gaussian curves (bandwidth sigma = 0.6 eV) to obtain the ECD spectra of each conformer, and the spectra were combined after Boltzmann weighting according to their population contributions. The wavelength of the spectra was corrected (+15 nm) based on the absorptions of about 200–250 nm (referring to the experimental and calculated UV spectra) to give the corresponding theoretical ECD spectrum.
α-Glycosidase inhibition assay
The inhibition of α-glycosidase activity was carried out following the method described [31]. A 2.5 mM solution of p-nitrophenyl α-D-glucopyranoside (p-NPG) (Sigma-Aldrich) and 0.2 U/mL α-glycosidase from Saccharomyces cerevisiae (Sigma-Aldrich) were prepared in 100 mM potassium phosphate buffer at pH 6.8. The sample (compound 9 or 10) was prepared in dimethylsulfoxide (DMSO) (Sigma-Aldrich) and serially diluted in the concentrations of 1, 4, 16, 64, and 256 μg/mL. 10 μL of sample was added to a reaction mixture consisting of 40 μL of 100 mM phosphate buffer at pH 6.8, 25 μL of 0.2 U/mL α-glycosidase in a 96-well microplate, and the reaction mixture was incubated for 10 min. at 37 °C. Then, 25 μL of 2.5 mM p-NPG was added, and the reaction mixture was further incubated for 20 min. at 37 °C. After 30 min. 100 mM sodium carbonate solution (100 µL) was added to stop the reaction. The absorbance of the mixture was measured at λ 410 nm on a UV-VIS spectrophotometer (Biotek Instruments, USA). To make a control reaction, the tested sample was replaced by 10 µL of 100 mM phosphate buffer at pH 6.8. Acarbose (Sigma-Aldrich) was used as the reference standard. The α-glycosidase inhibitory activity was calculated using the following equation: α-glycosidase inhibitory activity (%) = (Acontrol − Asample) / Acontrol × 100, where Acontrol is the absorbance of the control and Asample is the absorbance of the sample. IC50 (half-maximal inhibitory concentration) [32] was calculated using Tablecurve software. The experiments were repeated in triplicate. The data were expressed as means of three replicate determinations ± standard error of the mean (SEM).
Acetylcholinesterase inhibition assay
The inhibition of acetylcholinesterase (AChE) activity was carried out following the colorimetric method previously described [33,34,35]. The experiment was performed in a 96-well microplate. The sample (compound 9 or 10) and AChE solution (0.25 U/mL) (Sigma-Aldrich) were added to each well, and the solution was mixed and incubated for 15 min. at 25 °C. Then solutions of 2.4 mM 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) reagent (Sigma-Aldrich) and acetylcholine iodide (ATCI) substrate (Sigma-Aldrich) were further incubated for 15 min. at 25 °C. The absorbance of the mixture was measured at λ 410 nm on a UV-VIS spectrophotometer (Biotek Instruments, USA). Donepezil (Sigma-Aldrich) was used as the positive reference standard. The AChE inhibitory activity was calculated using the following equation: AChE inhibitory activity (%) = (Acontrol − Asample) / Acontrol × 100, where Acontrol is the absorbance of the control, Asample is the absorbance of the sample. IC50 (half-maximal inhibitory concentration) [32] was calculated using Tablecurve software. The experiments were repeated in triplicate. The data were expressed as means of three replicate determinations ± standard error of the mean (SEM).
Conclusions
We described the first study of the chemical constituents of the water-soluble fraction from the leaves of E. adenophorum. Twelve compounds were isolated, including a new cadinane sesquiterpenoid, three simple phenolic acids, a thymol derivative, three phenylpropenoic acids, two sterol glucosides, and two polyhydroxyflavonol glycosides. The absolute configuration of the new cadinane sesquiterpenoid was determined by theoretical ECD calculation; and this is also the first example of the determination of the absolute configuration of sesquiterpenoids in E. adenophorum. Two polyhydroxyflavonol glycosides are the rare naturally occurring flavonoids. In the α-glycosidase and acetylcholinesterase inhibition assays, gossypetin 5-O-(6′′-(E)-caffeoyl)-β-D-glucopyranoside exhibited IC50 values of 24.0 ± 1.61 μg/mL and 217.60 ± 15.47 μg/mL, respectively, while quercetagetin 7-O-β-D-glucopyranoside was slightly active. The results are in agreement with the bioactivity tendency of flavonoid glycosides, where the acylated glucopyranosyl moiety is favourable for their inhibitory effects. Since the synergistic properties of extracts from medicinal plants depend on the presence of nonactive and active phytochemicals, the disclosure of the chemical profiles of the water-soluble fraction lays the basis for the possible development of therapeutic herbal formulations from E. adenophorum.
References
Wu ZY, Raven PH, Hong DY, editors. Flora of China. Vol. 20–21 (Asteraceae). Beijing & St. Louis: Science Press & Missouri Botanical Garden Press; 2011.
El-Seedi HR. Antimicrobial activity and chemical composition of essential oil of Eupatorium glutinosum (Lam.). Nat Prod Commun. 2006;1:655–9.
Nguyen TB (ed.). Checklist of Plant Species of Vietnam. Hanoi: Agriculture Publishing House, Vol. 3; 2003.
Pham HH Flora of Vietnam. Ho Chi Minh City: Youth Publishing House, Vol. 3; 2003.
Bantawa P, Rai R. Studies on ethnomedicinal plants used by traditional practitioners, Jhankri, Bijuwa and Phedangma in Darjeeling Himalaya. Nat Prod Radiance. 2009;8:537–41.
Awah FM, Uzoegwu PN, Ifeonu P, Oyugi JO, Rutherford J, Yao XJ, et al. Free radical scavenging activity, phenolic contents and cytotoxicity of selected Nigerian medicinal plants. Food Chem. 2012;131:1279–86.
André R, Catarro J, Freitas D, Pacheco R, Oliveira MC, Serralheiro ML, et al. Action of euptox A from Ageratina adenophora juice on human cell lines: A top-down study using FTIR spectroscopy and protein profiling. Toxicol Vitr. 2019;57:217–25.
Giri S, Sahu R, Paul P, Nandi G, Dua TK. An updated review on Eupatorium adenophorum Spreng. [Ageratina adenophora (Spreng.)]: traditional uses, phytochemistry, pharmacological activities and toxicity. Pharm Res - Mod Chin Med. 2022;2:100068.
Ma QP, Cheng CR, Li XF, Liang XY, Ding J. Chemistry, pharmacological activity and analysis of Ageratina adenophora. Asian J Chem. 2015;27:4311–6.
Tripathi YC, Saini N, Anjum N, Verma PK. A review of ethnomedicinal, phytochemical, pharmacological and toxicological aspects of Eupatorium adenophorum Spreng. Asian J Biomed Pharm Sci. 2018;8:25–35.
Liang X, Yang X, Zhou T, Ma Y, Peng Y, Bahetejiang Y, et al. Three new cadinane-type sesquiterpenes from Eupatorium adenophorum Spreng. Nat Prod Res. 2022;36:4898–905.
Zhang M, Liu W-X, Zheng M-F, Xu Q-L, Wan F-H, Wang J, et al. Bioactive quinic acid derivatives from Ageratina adenophora. Molecules 2013;18:14096–104.
Zheng G, Luo S, Li S, Hua J, Li W, Li S. Specialized metabolites from Ageratina adenophora and their inhibitory activities against pathogenic fungi. Phytochemistry 2018;148:57–62.
Zhang M, Ouyang JK, Xu QL, Liu SB, Qian T, Dong LM, et al. Thymol derivatives with antibacterial and cytotoxic activity from the aerial parts of Ageratina adenophora. RSC Adv. 2021;11:5755–61.
Li M, Gao X, Lan M, Liao X, Su F, Fan L, et al. Inhibitory activities of flavonoids from Eupatorium adenophorum against acetylcholinesterase. Pestic Biochem Phys. 2020;170:e104701.
Proença C, Freitas M, Ribeiro D, Oliveira EFT, Sousa JLC, Tomé SM, et al. α-Glucosidase inhibition by flavonoids: an in vitro and in silico structure-activity relationship study. J Enzym Inhib Med Chem. 2017;32:1216–28.
Kapali J, Sharma KR. Estimation of phytochemicals, antioxidant, antidiabetic and brine shrimp lethality activities of some medicinal plants growing in Nepal. J Med Plants. 2021;20:102–16.
Shaikh A, Makhmoor T, Choudhary M. Radical scavenging potential of compounds isolated from Vitex agnus-castus. Turkish J Chem. 2010;34:e119.
Ha T, Pham C, Tra N, Le A, Cham B, Ninh TS. Chemical constituents from methanolic extract of Garcinia mackeaniana leaves and their antioxidant activity. Vietnam J Sci Tech. 2020;58:411–8.
Cho AS, Jeon S, Kim MJ, Yeo J, Seo KI, Choi MS, et al. Chlorogenic acid exhibits anti-obesity property and improves lipid metabolism in high-fat diet-induced-obese mice. Food Chem Toxicol. 2010;48:937–43.
Zheng G, Jia Y, Zhao X, Zhang F, Luo S, Li S, et al. o-Coumaric acid from invasive Eupatorium adenophorum is a potent phytotoxin. Chemoecology .2012;22:131–8.
Ramadoss K, Chapala D, Puttagunta SB. Isolation, characterization, and RP-HPLC estimation of p-coumaric acid from methanolic extract of Durva Grass (Cynodon dactylon Linn.) (Pers.). Int J Anal Chem. 2015;2015:1–7.
Li R, Ding Z, Ding J. Chemical constituents from Eupatorium adenpphorum. Acta Bot Yunnan. 1997;19:196–200.
Giang PM, Thuy NT, Nam DH, Thu TTH, Huong DTV. Phenolic compounds, terpenoids, and sterols from Eupatorium japonicum Thunb. In Vietnam. Vietnam J Chem. 2019;57:243–7.
Wang C, Yang R, Song L, Ning B, Ou-yang C, Cao A, et al. Two new highly-oxygenated flavonoid glycosides from Eupatorium adenophorum Spreng. Phytochem Lett. 2016;16:245–8.
Schmeda-Hirschmann G, Tapia A, Theoduloz C, Rodríguez J, López S, Feresin GE. Free radical scavengers and antioxidants from Tagetes mendocina. Z Naturforsch. 2004;59c:345–53.
Benalla W, Bellahcen S, Bnouham M. Antidiabetic medicinal plants as a source of alpha-glucosidase inhibitors. Curr Diabetes Rev. 2010;6:247–54.
Wang W, Xu H, Chen H, Tai K, Liu F, Gao Y. In vitro antioxidant, anti-diabetic and antilipemic potentials of quercetagetin extracted from marigold (Tagetes erecta L.) inflorescence residues. J Food Sci Technol. 2016;53:2614–24.
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian 16, Revision A.03, Gaussian Inc., Wallingford, CT, USA. 2016.
ChemPark Cloud System. 2022. https://www.chempark.jp/login-chempark.php. Accessed 21 October 2022.
Li T, Zhang XD, Song YW, Liu JW. A microplate-based screening method for alpha-glucosidase inhibitors. Chin J Clin Pharm Ther. 2005;10:1128–34.
Kiziltaş H. Comprehensive evaluation of Reseda lutea L. (Wild Mignonette) and 7 isolated flavonol glycosides: determination of antioxidant activity, anti-Alzheimer, antidiabetic and cytotoxic effects with in vitro and in silico methods. Turk J Chem. 2022;46:1185–98.
Ellman GL, Courtney KD, Andres V, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharm. 1961;7:88–95.
Min BS, To DC, Lee J-S. Cholinesterase inhibitors from Cleistocalyx operculatus Buds. Arch Pharm Res. 2010;33:1665–70.
Somani G, Kulkarni C, Shinde P, Shelke R, Laddha K, Sathaye S. In vitro acetylcholinesterase inhibition by psoralen using molecular docking and enzymatic studies. J Pharm Bioallied Sci. 2015;7:32–36.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Phan, M.G., Dong, N.P., Do, T.V.H. et al. A new cadinane sesquiterpenoid from Eupatorium adenophorum and α-glycosidase and AChE inhibitory activities of a gossypetin acylglucoside. Med Chem Res 32, 2168–2175 (2023). https://doi.org/10.1007/s00044-023-03110-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-023-03110-1