
Abstract
In this paper, pyridazin derivatives containing different thiobenzyl moieties were synthesized and screened for their inhibitory activities against rat intestinal α-glucosidase enzyme. The final products were easily obtained without the need to harsh purification steps. The in vitro results revealed that all the synthesized compounds were more potent (IC50s = 26.3–148.9 μM) than the clinically used drug, acarbose. The kinetic study revealed the competitive inhibition behavior of compound 5m (Ki = −56 μM). Docking studies showed imperative interactions such as hydrogen bonding, Pi-Pi T-shaped, and Pi-anion interactions confirming the observed activity. The RMSD value was determined less than 3 Å and validated the study.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus, the third most life-threatening disease in the world, is caused by the increased blood sugar due to either defect in the action or secretion of insulin in the body. Typically, in type 1 diabetes mellitus, beta cells are destructed through autoimmune pathway leading to complete insulin deficiency, while in type 2 diabetes mellitus, because of insulin deficiency the body cannot appropriately use it [1,2,3,4]. It was estimated that 425 million people were suffering from diabetes all over the world in 2017 and it may reach 642 million by 2040 [5, 6].
Type 2 diabetes mellitus (T2DM), also known as non-insulin-dependent diabetes, is a chronic condition accompanied with metabolic disorder which accounts for 90% of diabetic cases. There are several factors such as genetic, sedentary lifestyle, obesity, environmental toxins and advancing age leading to T2DM [7,8,9]. In the long term, diabetes can cause a plethora of complications including cardiovascular disease, stroke, blurred vision, renal failure, peripheral neuropathy and even death [10, 11].
Various medications namely biguanides, dipeptidyl peptidase-4 (DPP-4) inhibitors, GLP-1 agonists and SGLT-2 inhibitors are used to control high glucose levels of blood. The inhibition of α-glucosidase enzyme is an effective method, delaying glucose absorption in the digestive system and thereby suppressing postprandial hyperglycemia [12,13,14]. This hydrolase enzyme is located in jejunum acting through the cleavage of 1,4-α-glycosidic linkage of oligo and polysaccharides [15, 16]. Thus, the inhibition of this enzyme, either by competing for binding or through slowing down the carbohydrate metabolism would reduce the risk of hyperglycemia in diabetic patients. In addition to diabetes, α-glucosidase dysfunction can be attributed to many other diseases such as pompe disease, azoospermia, viral infection, etc. So, it can be deemed as one of the significant targets for drug discovery projects [17, 18].
Despite the fact that most of the α-glucosidase inhibitors (AGIs), used in clinics including miglitol, acarbose, and voglibose can control blood sugar, they are of low efficacy with quite a few side effects like hepatotoxicity, diarrhea, abdominal discomfort, and flatulence [19,20,21,22,23]. It is, therefore, necessary to design and develop novel potential therapeutic agents in an effort to improve efficacy and reduce the side effects by the development of novel bioactive heterocyclic scaffolds [24, 25].
Pyridazine, a six-membered nitrogen-containing heterocyclic core, has found its place between medicinal chemists due to the significant properties [26,27,28,29]. To achieve novel bioactive compounds, several researches have been conducted, focusing on the fused and hybrid forms of pyridazine with different chemical scaffolds and most of them have exhibited a wide range of biological and clinical activities namely MNK1/2, tubulin polymerization inhibitor, antiproliferative, anti-oxidant, anti-inflammatory, and formyl peptide receptor agonist [30,31,32,33,34,35,36,37].
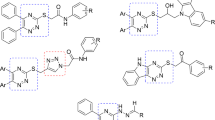
In continuation of our studies [38,39,40,41], in this study, we synthesized a new series of α-glucosidase inhibitors bearing pyridazine as the main core and evaluated their inhibitory activities. Besides, kinetic and docking studies were also performed. Our rational was based on the literature in which triazine-based molecules were significant inhibitors of α-glucosidase enzyme, exemplified in Fig. 1 [42,43,44,45]. Triazine and pyridazine could be considered as bioisosters in medicinal chemistry.

Reported triazine-containing α-glucosidase inhibitors. A triazine-carbazole derivative; B triazine thiazole derivative; C triazine-triazole derivative; D triazine-N-arylacetamides derivative
Results and discussion
Chemistry
Our new derivatives were obtained through a five-step synthetic route as described in Scheme 1. First, compound 2 was obtained through the condensation reaction of benzil 1 with hydrazine in methanol. Then, pyridazine ring was formed by a cyclocondensation reaction of 2-hydrazono-1,2-diphenylethanone 2 with diethyl malonate by using sodium ethoxide as a base after refluxing for three hours. Compound 4 was prepared by the reaction of 1 equiv. of ethyl 3-oxo-5,6-diphenyl-2,3-dihydropyridazine-4-carboxylate 3 with 0.5 equiv. Lawesson’s reagent in refluxing toluene. The nucleophilic substitution reaction occurred in dimethyl formamide (DMF) in the presence of K2CO3. The final compounds were prepared in good yields and in some cases without the need for further purifications.

Synthesis of final compounds. Reagents and conditions: a hydrazine hydrate, methanol, reflux, 15 min., b Na, EtOH, diethyl malonate, reflux, 3 h., c Lawesson’s reagent, toluene, reflux, 18 h; d benzyl chloride/bromide, DMF, K2CO3, 80 °C
In vitro α-glucosidase inhibitory activity
All of the synthesized compounds were potentially stronger than acarbose as a standard compound. The inhibitory activity was affected by the substitution pattern on phenyl ring. Compound 5m having p-nitrophenyl group as Ar-pendant with IC50 value of 26.3 ± 2.3 μM was considered as the most active and unsubstituted compound 5a was the least active one. Amongst the synthesized derivatives 5a–5m, electron-withdrawing group holders were effective than electron-donating group holders and unsubstituted one. Compounds 5h with 2,6-difluoro, 2,5-dichloro (5i) and 3,4-dichloro (5j) substitution displayed lower activities compared to mono- substituted analogues (5e and 5f). The SAR studies at R groups propounded that the movement of nitro substitution (5k–5m) from ortho to meta and para led to the increment in activities and similar trend was also observed in the case of fluoro substitution (5d and 5e). The change in position of fluorine from ortho to para in compound 5e (IC50 = 50.3 ± 6.5 μM), resulted in increased activity. Considering compounds bearing halogen substituents revealed that among them compounds with the most electronegative atom, fluorine, exhibited the best inhibitory activity. Compound 5h bearing both fluoro groups at ortho position of aryl ring showed almost two folds less activity as compared to compound 5d. In addition, compounds bearing nitro (–NO2) group at meta and para positions of phenyl ring showed good activity when compared with ortho-substituted analogue 5k as well as standard acarbose (IC50 = 173.8 ± 23.9 μM) (Table 1). The cytotoxicity of compound 5m, the most active compound, was investigated against the normal cell line. No toxicity was observed against HDF [41].
α-Glucosidase kinetic study
The mechanism of α-glucosidase activity was studied by using Lineweaver-Burk plot analysis. Four different concentrations of compound 5m were used and p-nitrophenyl-α-D-glucopyranoside (α-PNPG) was used as a substrate. The plot indicated that this compound was competitive inhibitor as judged from Km and Vmax values. Same Vmax values were obtained without the inhibitor at high concentrations of it. To evaluate the binding ability, Ki parameter was detected to be −56 µM (Fig. 2).

The Lineweaver-Burk plot in the absence and presence of four different concentrations of compound 5m
Molecular docking study on α-glucosidase
Molecular docking has found many applications like understanding the mechanism of action [46,47,48,49,50,51,52]. In order to reveal the binding modes of selected compounds 5e, 5l, and 5m to the active site, the crystal structure (PDB ID: 5NN8) was chosen and docking studies were performed by Auto Dock Tools (version 1.5.6). Compound 5m with significant inhibitory activity against α-glucosidase formed a hydrogen bonding between the oxygen of nitro group and HIS674. In case of compound 5l with the nitro group at meta position formed hydrogen bondings with ARG:600 and TRP:613 residues. Compound 5e with fluorine at para formed this important interaction with LEU:405. Conventional hydrogen bonds are shown in green (Fig. 3).

The binding conformations (2D and 3D) of compounds 5e (a, b), 5l (c, d), and 5m (e, f)
One of the phenyl rings in compound 5m formed Pi-Pi T-shaped interaction with PHE649. Compound 5l was also showed the same interaction with this amino acid. In compound 5m, the phenyl group along with ethyl group formed Pi-alkyl interaction with LEU678 and PHE525, respectively. These interactions along with van der Waals interactions explained the observed inhibitory activity against α-glucosidase. Docking studies were validated by re-docking acarbose into the binding site. The root-mean-square distance (RMSD) of co-crystallized and re-docked ligands for acarbose was obtained 0.62 Å (less than 3 Å), which showed high reliability of docking protocol (the related information were inserted in supporting file). The ∆G amounts were determined to be −6.4, −6.1 and −7.1 kcal/mol for compounds 5e, 5l, and 5m, respectively.
Conclusion
In this study, a novel series of substituted pyridazine derivatives were screened against rat small intestine α-glucosidase enzyme. Among all target compounds, compound 5m (IC50 = 26.3 ± 2.3) showed the best inhibitory activity against α-glucosidase. Lineweaver-Burk plot analysis indicated that this compound was competitive α-glucosidase inhibitor. Molecular docking and the binding constant (Ki) revealed the imperative interactions and mechanism of action. Besides, this compound showed no toxicity against HDF line. The antidiabetic potential of the final compounds can serve pyridazin core as promising candidate for further derivatization.
Experimental
Melting points were recorded by the electro thermal 9200 apparatus. The IR spectra were recorded on KBr pellets by using Shimadzu IR470 spectrophotometer. Bruker 500 MHz was used to record 1H NMR spectra. Elemental analysis for C, H and N were performed using a Heraus CHN rapid analyzer.
General procedure for hydrazono-1,2-diphenylethanone 2
Hydrazine hydrate (20 mmol) was added dropwise to a solution of benzil (20 mmol) dissolved in methanol (50 mL). The reaction was refluxed for 15 min. and then the mixture was cooled, and the resultant white solid was filtered and washed with cold methanol [53].
General procedure for the synthesis of compound 3
To the cold solution of sodium (0.05 mol) in 200 mL ethanol, compound 2 was slowly added. After the addition of diethyl malonate (0.075 mol), the reaction mixture was warmed to the room temperature and heated at reflux temperature for 3 h. After removing the solvent under reduced pressure and adjusting the pH to 4 by using HCl (1 N), the solid was collected by filtration and washed with cold water [54].
General procedure for the synthesis of compound 4
To a solution of ethyl 3-oxo-5,6-diphenyl-2,3-dihydropyridazine-4-carboxylate (3, 10 mmol) in 150 mL toluene, Lawesson’s reagent (5 mmol) was added. After refluxing for 18 h and reaction completion, checked by TLC, the reaction was cooled and the solid was separated and used without further purification.
General procedure for the synthesis of target compounds 5a-m
To a solution of ethyl 3-mercapto-5,6-diphenylpyridazine-4-carboxylate (4, 1 mmol) and potassium carbonate (1.5 mmol) in dimethyl formamide (10 mL), benzyl halide derivatives (1.1 mmol) were added. The reaction mixture was heated to 80oC. Upon consumption of starting material, indicated by TLC, the mixture was poured into an ice-water mixture and the resultant solid was collected and washed with water. The crystallization was performed with ethyl acetate/petroleum ether, if needed.
Ethyl 3-(benzylthio)-5,6-diphenylpyridazine-4-carboxylate (5a)
Yield: 67%; m.p. = 165–167 °C; IR (KBr): 1724, 1443, 1327, 1215, 1187, 1024, 766 cm−1; 1HNMR (500 MHz, DMSO-d6) δ: 7.47–7.44 (m, 1H), 7.43–7.27 (m, 9H), 7.15–7.12 (m, 5H), 4.78 (s, 2H), 4.03 (q, J = 7.0 Hz, 2H), 0.86 (t, J = 7.05 Hz, 3H) ppm; 13CNMR (125 MHz, DMSO-d6) δ: 164.09, 157.39, 155.74, 137.06, 136.02, 135.11, 133.63, 130.42, 129.68, 129.23, 129.03, 128.84, 128.64, 128.54, 128.32, 127.91, 127.40, 62.08, 33.85, 13.35 ppm; Anal. Calcd. for C26H22N2O2S: C, 73.21; H, 5.20; N, 6.57%. Found: C, 73.52; H, 4.93; N, 6.24%.
Ethyl 3-((3-methoxybenzyl)thio)-5,6-diphenylpyridazine-4-carboxylate (5b)
Yield: 65% m.p. = 150–152 °C; IR (KBr): 1725, 1593, 1493, 1298, 1211, 1074, 767 cm−1; 1HNMR (500 MHz, DMSO-d6) δ: 7.34–7.24 (m, 9H), 7.13 (d, J = 6.9 Hz, 2H), 7.09–7.06 (m, 2H), 6.85 (d, J = 8.2 Hz, 1H), 4.69 (s, 2H), 4.03 (q, J = 7.2 Hz, 2H), 3.74 (s, 3H), 0.86 (t, J = 7.05 Hz, 3H) ppm; 13CNMR (125 MHz, DMSO-d6) δ: 164.13, 159.31, 157.41, 155.77, 138.57, 136.04, 135.14, 133.66, 130.50, 129.70, 129.04, 128.88, 128.34, 127.93, 121.43, 114.93, 112.83, 62.11, 55.07, 33.87, 13.37 ppm; Anal. Calcd. For C27H24N2O3S: C, 71.03; H, 5.30; N, 6.14%. Found: C, 70.79; H, 5.02; N, 6.39%.
Ethyl 3-((4-methylbenzyl)thio)-5,6-diphenylpyridazine-4-carboxylate (5c)
Yield: 61%; m.p. = 158–160 °C; IR (KBr): 1732, 1538, 1443, 1277, 1375, 1310, 1194, 1005, 764 cm−1; 1HNMR (500 MHz, DMSO-d6) δ: 7.38 (d, J = 7.6 Hz, 2H), 7.34–7.27 (m, 8H), 7.15 (d, J = 8.0 Hz, 2H), 7.12 (d, J = 8.0 Hz, 2H), 4.67 (s, 2H), 4.03 (q, J = 7.1 Hz, 2H), 2.28 (s, 3H), 0.86 (t, J = 7.2 Hz, 3H) ppm; 13CNMR (125 MHz, DMSO-d6) δ: 164.08, 157.32, 155.83, 136.63, 136.03, 135.05, 133.86, 133.63, 130.41, 129.67, 129.15, 129.08, 129.02, 128.84, 128.62, 128.31, 127.90, 62.05, 33.67, 20.71, 13.35 ppm; Anal. Calcd. for C27H24N2O2S: C, 73.61; H, 5.49; N, 6.36%. Found: C, 73.49; H, 5.71; N, 6.05%.
Ethyl 3-((2-fluorobenzyl)thio)-5,6-diphenylpyridazine-4-carboxylate (5d)
Yield: 73%; m.p. = 139–141 °C; IR (KBr): 1723, 1584, 1491, 1310, 1233, 1130, 1080, 763 cm−1; 1HNMR (500 MHz, DMSO-d6) δ: 7.61 (t, J = 7.7 Hz, 1H), 7.37-7.30 (m, 8H), 7.27–7.24 (m, 2H), 7.22–7.20 (m, 2H), 7.17 (t, J = 7.4 Hz, 1H), 4.75 (s, 2H), 4.03 (q, J = 7.1 Hz, 2H), 0.85 (t, J = 7.1 Hz, 3H) ppm; 13CNMR (125 MHz, DMSO-d6) δ: 164.00, 161.55 (d, J = 245 Hz), 155.47, 155.36, 135.95, 135.20, 133.60, 131.54, 130.39, 129.75, 129.65, 128.98, 128.80, 128.59, 128.26, 127.84, 124.46, 123.94 (d, J = 14 Hz), 115.33 (d, J = 21 Hz), 62.05, 27.49, 13.27 ppm; Anal. Calcd. for C26H21FN2O2S: C, 70.25; H, 4.76; N, 6.30%. Found: C, 69.98; H, 4.50; N, 6.02%.
Ethyl 3-((4-fluorobenzyl)thio)-5,6-diphenylpyridazine-4-carboxylate (5e)
Yield: 67%; m.p. = 110–112 °C; IR (KBr): 1719, 1508, 1303, 1221, 1186, 1078, 841, 700 cm−1; 1HNMR (500 MHz, DMSO-d6) δ: 7.55 (t, J = 5.6 Hz, 2H), 7.33–7.26 (m, 8H), 7.18–7.12 (m, 4H), 4.71 (s, 2H), 4.03 (q, J = 7.1 Hz, 2H), 0.86 (t, J = 7.0 Hz, 3H) ppm; 13CNMR (125 MHz, DMSO-d6) δ: 164.03, 162.37 (d, J = 240 Hz) 157.37, 155.60, 135.98, 135.09, 133.61, 133.37, 131.22, 131.16, 130.38, 129.64, 129.26, 128.98, 128.30 (d, J = 19 Hz), 127.84, 115.21 (d, J = 21 Hz), 62.02, 32.98, 13.30 ppm; Anal. Calcd. for C26H21FN2O2S: C, 70.25; H, 4.76; N, 6.30%. Found: C, 70.49; H, 4.93; N, 5.98%.
Ethyl 3-((4-chlorobenzyl)thio)-5,6-diphenylpyridazine-4-carboxylate (5f)
Yield: 78%; m.p. = 132–134 °C; IR (KBr): 1720, 1489, 1301, 1214, 1186, 844, 701 cm−1; 1HNMR (500 MHz, DMSO-d6) δ: 7.53 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.4 Hz, 2H), 7.34–7.25 (m, 8H), 7.12 (d, J = 8.0 Hz, 2H), 4.70 (s, 2H), 4.04 (q, J = 7.0 Hz, 2H), 0.86 (t, J = 7.1 Hz, 3H) ppm; 13CNMR (125 MHz, DMSO-d6) δ: 164.03, 157.43, 155.43, 136.43, 135.96, 135.14, 133.59, 131.93, 131.05, 130.41, 129.65, 128.99, 128.82, 128.61, 128.41, 128.28, 127.86, 62.06, 32.96, 13.31 ppm; Anal. Calcd. for C26H21ClN2O2S: C, 67.74; H, 4.59; N, 6.08%. Found: C, 68.00; H, 4.28; N, 6.33%.
Ethyl 3-((4-bromobenzyl)thio)-5,6-diphenylpyridazine-4-carboxylate (5g)
Yield: 83%; m.p. = 142–144 °C; IR: (KBr): 1722, 1617, 1326, 1186, 1076, 765, 698 cm−1; 1HNMR (500 MHz, DMSO-d6) δ: 7.53 (d, J = 8.1 Hz, 2H), 7.47 (d, J = 6.7 Hz, 2H), 7.34–7.29 (m, 8H), 7.12 (d, J = 8.1 Hz, 2H), 4.68 (s, 2H), 4.04 (q, J = 7.1 Hz, 2H), 0.86 (t, J = 7.1 Hz, 3H) ppm; 13CNMR (125 MHz, DMSO-d6) δ: 164.00, 157.41, 155.38, 136.86, 135.94, 135.12, 133.58, 131.38, 131.32, 130.38, 129.63, 128.98, 128.80, 128.58, 128.26, 127.83, 120.42, 62.04, 33.01, 13.31 ppm; Anal. Calcd. for C26H21BrN2O2S: C, 61.79; H, 4.19; N, 5.54%. Found: C, 61.52; H, 4.44; N, 5.23%.
Ethyl 3-((2,6-difluorobenzyl)thio)-5,6-diphenylpyridazine-4-carboxylate (5h)
Yield: 61%; m.p. = 97–99 °C; IR (KBr): 1725, 1590, 1470, 1383, 1217, 1079, 792, 700 cm−1; 1HNMR (500 MHz, DMSO-d6) δ: 7.46 (t, J = 5.6 Hz, 1H), 7.43–7.27 (m, 8H), 7.17–7.12 (m, 4H), 4.78 (s, 2H), 4.01 (d, J = 7.0 Hz, 2H), 0.83 (t, J = 6.9 Hz, 3H) ppm; Anal. Calcd. for C26H20F2N2O2S: C, 67.52; H, 4.36; N, 6.06%. Found: C, 67.81; H, 4.03; N, 6.32%.
Ethyl 3-((2,5-dichlorobenzyl)thio)-5,6-diphenylpyridazine-4-carboxylate (5i)
Yield: 75%; m.p.= 123–125 °C; IR (KBr): 1727, 1442, 1365, 1297, 1186, 1079, 828, 701 cm−1; 1HNMR (500 MHz, DMSO-d6) δ: 7.77 (d, J = 1.6 Hz, 1H), 7.34–7.25 (m, 8H), 7.16–7.12 (m, 4H), 4.78 (s, 2H), 4.01 (q, J = 7.0 Hz, 2H), 0.83 (t, J = 7.1 Hz, 3H) ppm; 13CNMR (125 MHz, DMSO-d6) δ: 163.97, 157.66, 155.18, 136.70, 135.91, 135.37, 133.58, 132.25, 131.66, 131.12, 130.49, 130.08, 129.67, 129.23, 128.99, 128.83, 128.63, 128.27, 127.86, 62.11, 31.71, 13.28 ppm; Anal. Calcd. for C26H20Cl2N2O2S: C, 63.03; H, 4.07; N, 5.65%. Found: C, 62.79; H, 4.31; N, 5.91%.
Ethyl 3-((3,4-dichlorobenzyl)thio)-5,6-diphenylpyridazine-4-carboxylate (5j)
Yield: 69%; m.p. = 133–135 °C; IR (KBr): 1719, 1466, 1375, 1186, 1028, 901, 750, 695 cm−1; 1HNMR (500 MHz, DMSO-d6) δ: 7.77 (d, J = 1.9 Hz, 1H), 7.53 (d, J = 8.3 Hz, 1H), 7.42–7.40 (m, 1H), 7.34–7.25 (m, 8H), 7.13 (d, J = 7.1 Hz, 2H), 4.78 (s, 2H), 4.04 (q, J = 7.1 Hz, 2H), 0.86 (t, J = 7.1 Hz, 3H) ppm; 13CNMR (125 MHz, DMSO-d6) δ: 163.97, 157.66, 155.18, 136.70, 135.90, 135.37, 133.58, 132.25, 131.66, 131.12, 130.49 (2 C), 129.67, 129.23, 128.99, 128.83, 128.64, 128.28, 127.87, 62.11, 31.71, 13.29 ppm; Anal. Calcd. for C26H20Cl2N2O2S: C, 63.03; H, 4.07; N, 5.65%. Found: C, 63.41; H, 3.82; N, 5.40%.
Ethyl 3-((2-nitrobenzyl)thio)-5,6-diphenylpyridazine-4-carboxylate (5k)
Yield: 65%; m.p. = 114–116 °C; IR (KBr): 1727, 1524, 1377, 1296, 1192, 1004, 753, 698 cm−1; 1HNMR (500 MHz, DMSO-d6) δ: 8.06 (d, J = 8.9 Hz, 1H), 7.87 (d, J = 7.7 Hz, 1H), 7.73 (t, J = 8.3 Hz, 1H), 7.57 (t, J = 7.9 Hz, 1H), 7.35–7.25 (m, 8H), 7.12 (d, J = 6.7 Hz, 2H), 5.01 (s, 2H), 4.04-3.97 (m, 2H), 0.84 (t, J = 7.1 Hz, 3H) ppm; Anal. Calcd. for C26H21N3O4S: C, 66.23; H, 4.49; N, 8.91%. Found: C, 66.50; H, 4.66; N, 8.69%.
Ethyl 3-((3-nitrobenzyl)thio)-5,6-diphenylpyridazine-4-carboxylate (5l)
Yield: 75%; m.p. = 134–136 °C; IR (KBr): 1726, 1526, 1350, 1295, 1190, 809, 698 cm−1; 1HNMR (500 MHz, DMSO-d6) δ: 8.41 (s, 1H), 8.13 (d, J = 8.95 Hz, 1H), 7.99 (d, J = 7.5 Hz, 1H), 7.64 (t, J = 7.9 Hz, 1H), 7.34–7.25 (m, 8H), 7.12 (d, J = 7.3 Hz, 2H), 4.85 (s, 2H), 4.04 (q, J = 7.1 Hz, 2H), 0.86 (t, J = 7.1 Hz, 3H) ppm; 13CNMR (125 MHz, DMSO-d6) δ: 163.99, 157.56, 147.70, 140.31, 135.93, 135.89, 135.25, 133.55, 130.45, 129.89, 129.60, 128.97, 128.81 (2 C), 128.60, 128.26, 127.81, 123.73, 122.22, 62.09, 32.84, 13.29 ppm; Anal. Calcd. for C26H21N3O4S: C, 66.23; H, 4.49; N, 8.91%. Found: C, 66.50; H, 4.71; N, 8.69%.
Ethyl 3-((4-nitrobenzyl)thio)-5,6-diphenylpyridazine-4-carboxylate (5m)
Yield: 73% m.p. = 144–146 °C; IR (KBr): 1729, 1588, 1493, 1344, 1174, 1087, 699 cm−1; 1HNMR (500 MHz, DMSO-d6) δ: 8.21 (d, J = 8.7 Hz, 2H), 8.00 (d, J = 9.2 Hz, 2H), 7.79 (d, J = 8.7 Hz, 2H), 7.34–7.19 (m, 8H), 4.83 (s, 2H), 4.04 (q, J = 7.1 Hz, 2H), 0.86 (t, J = 7.1 Hz, 3H); 13CNMR (125 MHz, DMSO-d6) δ: 163.98, 157.58, 155.16, 145.81, 137.85, 135.26, 133.56, 130.38, 130.13, 129.64, 128.83, 128.28, 127.85, 126.58, 123.96, 123.54, 122.86, 62.12, 32.90, 13.32 ppm; Anal. Calcd. for C26H21N3O4S: C, 66.23; H, 4.49; N, 8.91%. Found: C, 66.51; H, 4.70; N, 8.66%.
Rat α‑glucosidase assay
According to the reported protocol of Lossow et al., the α-glucosidase (EC 3.2.1.20) was prepared [55]. By the measurement of 4-nitrophenol which was released from para-nitrophenyl α-D glucopyranoside the inhibitory activity was determined [56]. The preparation of 200 μL as the final volume was performed as follows: the enzyme solution (190 μL, 0.15 units/ml), different concentrations of target compounds (5 μL), potassium phosphate buffer. Target compounds were dissolved in DMSO and pre-incubated at 37 °C, the addition of p-nitrophenyl glucopyranoside and then substrate (5 μL, 3 mM), to the enzyme solution was done and then incubated one hour at 37 °C. Finally, with Cytation 3 hybrid microplate reader (BioTek, USA) and the absorbance was measured at 405 nm. By using GraphPadprism 6.0 (SanDiego, California, USA) (https://www.graphpad.com/scientific-software/prism/) was used to obtain IC50 values of tested compounds.
Kinetic analysis
Modes of enzyme inhibition were determined by recording the effect of various concentrations of the substrates 1, 2.5, 5 or 10 mM pNPG on Lineweaver–Burk plots using calculated Vmax and Km. For inhibition test of α-glucosidase: five different concentrations 0, 10, 25, 50, 100 μM of the most active compound were applied. Ki value was determined by depicting the secondary plot of Km against various concentrations of inhibitor.
Docking studies
The compound 5m was docked by using Autodock 4.2.1 software. The structure of the targeted protein α-glucosidase (PDB: 5NN8) was taken from RCSB data bank. Discovery Studio visualizer 4.5 was used to analyze the results.
References
Al-Hassan N. Definition of diabetes mellitus. Br J Gen Pr. 2003;53:567–8.
Ma ZA, Zhao Z, Turk J. Mitochondrial dysfunction and β-cell failure in type 2 diabetes mellitus. Exp Diabetes Res. 2012;703538. https://doi.org/10.1155/2012/703538.
Zaccardi F, Webb DR, Yates T, Davies MJ. Pathophysiology of type 1 and type 2 diabetes mellitus: a 90-year perspective. Postgrad Med J. 2013;92:63–69. https://doi.org/10.1136/postgradmedj-2015-133281.
Ripsin CM, Kang H, Urban RJ. Management of blood glucose in type 2 diabetes mellitus. Am Fam Physician. 2009;79:29–36.
Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, Cho NH. et al. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pr. 2017;128:40–50. https://doi.org/10.1016/j.diabres.2017.03.024.
WHO diabetes statistics report. https://www.who.int/news-room/fact-sheets/detail/diabetes/, 2002 (accessed on 13.06.2022).
Cusi K, DeFronzo RA. Metformin: A review of its metabolic effects. Diabetes Rev. 1998;6:89–131.
Bellary S, Kyrou I, Brown JE, Bailey CJ. Type 2 diabetes mellitus in older adults: clinical considerations and management. Nat Rev Endocrinol. 2021;17:534–48. https://doi.org/10.1038/s41574-021-00512-2.
Cole JB, Florez JC. Genetics of diabetes mellitus and diabetes complications. Nat Rev Nephrol. 2020;16:377–90. https://doi.org/10.1038/s41581-020-0278-5.
Gallagher EJ, LeRoit D. Obesity and diabetes: the increased risk of cancer and cancer related mortality. Physiol Rev. 2015;95:727–48. https://doi.org/10.1152/physrev.00030.2014.
Reddy VP, Zhu X, Perry G, Smith MA. Oxidative stress in diabetes and Alzheimer’s disease. J Alzheimers Dis. 2009;16:763–74. https://doi.org/10.3233/JAD-2009-1013.
Dhameja M, Gupta P. Synthetic heterocyclic candidates as promising α-glucosidase inhibitors: An overview. Eur J Med Chem. 2019;176:343–77. https://doi.org/10.1016/j.ejmech.2019.04.025.
Moorthy NSHN, RamosMJ, Fernandes PA. Studies on α-glucosidase inhibitors development: Magic molecules for the treatment of carbohydrate mediated diseases. Mini Rev Med Chem. 2012;12:713–20. https://doi.org/10.2174/138955712801264837.
Mandal AK. Diabetes, In treating diabetes, what is important? Glucose levels or outcome measures. World J Diabetes. 2015;6:1243–5. https://doi.org/10.4239/wjd.v6.i13.1243
Baron AD. Postprandial hyperglycaemia and α-glucosidase inhibitors. Diabetes Res Clin Pr. 1998;40:S51–S55. https://doi.org/10.1016/s0168-8227(98)00043-6.
Joshi SR, Standl E, Tong N, Shah P, Kalra R. Therapeutic potential ofα-glucosidase inhibitors in type 2 diabetes mellitus: an evidence-based review. Expert Opin Pharmacother. 2015;16:1959–81. https://doi.org/10.1517/14656566.2015.1070827.
Tucci SA, Boyland EJ, Halford JC. The role of lipid and carbohydrate digestive enzyme inhibitors in the management of obesity: a review of current and emerging therapeutic agents. Diabetes Diabetes Metab Syndr Obes Targets Ther. 2010;10:125–43. https://doi.org/10.2147/dmsott.s7005.
Ghani U. Alpha-Glucosidase Inhibitors, Clinically Promising candidates for Anti-Diabetic Drug Discovery. 1st ed. Elsevier; 2019.
Usman B, Sharma N, Satija S, Mehta M, Vyas M, Khatik GL. et al. Recent developments in alpha-glucosidase inhibitors for management of type-2 diabetes: an update. Curr Pharm Des. 2019;25:2510–25. https://doi.org/10.2174/1381612825666190717104547.
Yee HS, Fong NT. A Review of the Safety and Efficacy of Acarbose in Diabetes Mellitus. Pharmacotherapy. 1996;16:792–805.
Kaku K. Efficacy of voglibose in type 2 diabetes. Pharmacotherapy. 2014;15:1181–90. https://doi.org/10.1517/14656566.2014.918956.
Scott LJ, Spencer CM. Miglitol: a review of its therapeutic potential in type 2 diabetes Mellitus. Drugs. 2000;59:521–49. https://doi.org/10.2165/00003495-200059030-00012.
Dhameja M, Gupta P. Synthetic heterocyclic candidates as promising α-glucosidase inhibitors: An overview. Eur J Med Chem. 2019;176:343–77. https://doi.org/10.1016/j.ejmech.2019.04.025.
Kabi AK, Sravani S, Gujjarappa R, Garg A, Vodnala N, Tyagi U, et al. An Overview on Biological Evaluation of Tetrazole Derivatives. In: Swain, BP (eds) Nanostructured Biomaterials. Materials Horizons: From Nature to Nanomaterials. 2022;307-49. Springer, Singapore. https://doi.org/10.1007/978-981-16-8399-2_8.
Kabi AK, Sravani S, Gujjarappa R, Garg A, Vodnala N, Tyagi U, et al. An Overview on Biological Activities of 1,2,3-Triazole Derivatives. In: Swain, BP (eds) Nanostructured Biomaterials. Materials Horizons: From Nature to Nanomaterials. 2022;401-23. Springer, Singapore. https://doi.org/10.1007/978-981-16-8399-2_11.
Bansal R, Thota S. Pyridazin-3(2H)-ones: the versatile pharmacophore of medicinal significance. Med Chem Res. 2013;22:2539–52. https://doi.org/10.1007/s00044-012-0261-1.
Sergeev PG, Nenajdenko VG. Recent advances in the chemistry of pyridazine An important representative of six-membered nitrogen heterocycles. Russ Chem Rev. 2020;89:393–429. https://doi.org/10.1070/RCR4922.
Passador K, Thorimbert S, Botuha C. Heteroaromatic Rings of the Future’: Exploration of Unconquered Chemical Space. Synthesis. 2019;51:384–98. https://doi.org/10.1055/s-0037-1611279.
Boraei ATA, Sarhan AAM, Yousuf S, Barakat A. Synthesis of a New Series of Nitrogen/Sulfur Heterocycles by Linking Four Rings: Indole; 1,2,4-Triazole; Pyridazine; and Quinoxaline. Molecules 2020;21:450. https://doi.org/10.3390/molecules25030450.
Tsuboi K, Kimura H, Nakatsuji Y, Kassai M, Deai Y, Isobe Y. Discovery of N-(6-(5-fluoro-2-(piperidin-1-yl)phenyl)pyridazin-3-yl)-1-(tetrahydro-2H-pyran-4-yl)methanesulfonamide as a brain-permeable and metabolically stable kynurenine monooxygenase inhibitor. Bioorg Med Chem Lett. 2021;44:128115. https://doi.org/10.1016/j.bmcl.2021.128115.
Hong B, Yuan X, Wu H, Zhou J, Zhang H. Design, synthesis and biological evaluation of imidazopyridazine derivatives containing isoquinoline group as potent MNK1/2 inhibitors. Bioorg Med Chem. 2021;40:116186. https://doi.org/10.1016/j.bmc.2021.116186.
Abdelbaset MS, Abdelrahman MH, Bukhari SNA, Gouda AM, Abdel-Aziz YM, Abuo-Rahma GEA. Design, synthesis, and biological evaluation of new series of pyrrol-2(3H)-one and pyridazin-3(2H)-one derivatives as tubulin polymerization inhibitors. Bioorg Chem. 2021;107:104522. https://doi.org/10.1016/j.bioorg.2020.104522.
Gaikwad DD, Pawar UD, Chavan SL, Pawar CD, Pansare DN, Shelke RN, et al. Synthesis and anti-proliferative activity studies of 2-(2-(trifluoromethyl)-6-(substituted)imidazo[1,2-b]pyridazin-3-yl)-N-(substituted)acetamide derivatives. J Het Chem. 2020;57:1925–35. https://doi.org/10.1002/jhet.3920.
Mustafa M, Mostafa YA. Antimicrobial Pyridazines: Synthesis, Characterization, Cytotoxicity, Substrate Promiscuity, and Molecular Docking. Chem Biodivers. 2020;17:e2000100. https://doi.org/10.1002/cbdv.202000100.
Qin J, Zhu M, Zhu H, Zhang L, Fu Y, Liu J. et al. Synthesis and antitumor activity of novel pyridazinone derivatives containing 1,3,4-thiadiazole moiety. Phosphorus Sulfur Silicon Relat Elem. 2020;195:592–9. https://doi.org/10.1080/10426507.2020.1737062.
Szczukowski L, Redzicka A, Wiatrak B, Krzyżak E, Marciniak A, Gębczak K, et al. Design, synthesis, biological evaluation and in silico studies of novel pyrrolo[3,4-d]pyridazinone derivatives with promising anti-inflammatory and antioxidant activity. Bioorg Chem. 2020;120:104035. https://doi.org/10.1016/j.bioorg.2020.104035.
Deora GS, Qin CX, Vecchio EA, Debono AJ, Priebbenow RM, Brady J. et al. Substituted Pyridazin-3(2H)-ones as Highly Potent and Biased Formyl Peptide Receptor Agonists. J Med Chem. 2019;62:5242–8. https://doi.org/10.1021/acs.jmedchem.8b01912.
Moghimi S, Salarinejad S, Toolabi M, Firoozpour L, Sadat Ebrahimi SE, Safari F F. et al. Synthesis, in-vitro evaluation, molecular docking, and kinetic studies of pyridazine-triazole hybrid system as novel α-glucosidase inhibitors. Bioorg Chem. 2021;109:104670. https://doi.org/10.1016/j.bioorg.2021.104670.
Peytam F, Takalloobanafshi G, Saadattalab T, Norouzbahari M. et al. Design, synthesis, molecular docking, and in vitro α‑glucosidase inhibitory activities of novel 3-amino‑2,4‑diarylbenzo[4,5]imidazo[1,2‑a]pyrimidines against yeast and rat α‑glucosidase. Sci Rep.2021;11:11911. https://doi.org/10.1038/s41598-021-91473-z.
Peytam F, Adib M, Shourgeshty R, Firoozpour L, Rahmanian-Jazi M. et al. An efficient and targeted synthetic approach towards new highly substituted 6-aminopyrazolo[1,5-a] pyrimidines with α-glucosidase inhibitory activity. Sci Rep.2020;10:2595. https://doi.org/10.1038/s41598-020-59079-z.
Moghimi S, Toolabi M, Salarinejad S, Firoozpour L, Sadat Ebrahimi SE, Safari F. et al. Design and synthesis of novel pyridazine N-aryl acetamides: In-vitro evaluation of α-glucosidase inhibition,docking, and kinetic studies. Bioorg Chem. 2020;102:14071. https://doi.org/10.1016/j.bioorg.2020.104071.
Wang G, Wang J, He D, Li X, Li J, Peng Z. Synthesis and biological evaluation of novel 1,2,4-triazine derivatives bearing carbazole moiety as potent α-glucosidase inhibitors. Bioorg Med Chem Lett. 2016;26:2806–9. https://doi.org/10.1016/j.bmcl.2016.04.071.
Wang G, Peng Z, Wang J, Li X, Li J. Synthesis, in vitro evaluation and molecular docking studies of novel triazine-triazole derivatives as potential a-glucosidase inhibitors. Eur J Med Chem. 2017;125:423–9. https://doi.org/10.1016/j.ejmech.2016.09.067.
Wang G, Li X, Wang J, Xie Z, Li L, Chen M. et al. Synthesis, molecular docking and α-glucosidase inhibition of 2-((5,6-diphenyl-1,2,4-triazin-3-yl)thio)-N-arylacetamides. Bioorg Med Chem Lett. 2017;27:1115–8. https://doi.org/10.1016/j.bmcl.2017.01.094.
Wang G, Peng Z, Gong Z, Li Y. Synthesis, biological evaluation, and docking studies of novel 5,6-diaryl-1,2,4-triazine thiazole derivatives as a new class of α-glucosidase inhibitors. Bioorg Chem. 2018;78:195–200. https://doi.org/10.1016/j.bioorg.2018.03.015.
da Silva Júnior OS, Franco CJP, de Moraes AAB, Cruz JN, da Costa KS, do Nascimento LD. et al. In silico analyses of toxicity of the major constituents of essential oils from two Ipomoea L. species. Toxicon. 2021;195:111–8. https://doi.org/10.1016/j.toxicon.2021.02.015.
Lima AM, Siqueira AS, Moller MLS, de Souza RC, Cruz JN, Lima ARJ. et al. In silico improvement of the cyanobacterial lectin microvirin and mannose interaction. J Biomol Struct Dyn. 2020;40:1064–73. https://doi.org/10.1080/07391102.2020.1821782.
Almeida VM, Dias ER, Souza BC, Cruz JN, Santos CBR, Leite FHA. et al. Methoxylated flavonols from Vellozia dasypus Seub ethyl acetate active myeloperoxidase extract: in vitro and in silico assays. J Biomol Struct Dyn. 2020;40:7574–83. https://doi.org/10.1080/07391102.2021.1900916.
Galucio NCR, Moysés DA, Pina JRS, Marinho PSB, Júnior PCG, Cruz JN, et al. Antiproliferative, genotoxic activities and quantification of extracts and cucurbitacin B obtained from Luffa operculata (L.) Cogn. Arab J Chem. 2022;15:103589. https://doi.org/10.1016/j.arabjc.2021.103589.
Rego CMA, Francisco AF, Boeno CN, Paloschi MV, Lopes JA, Silva MDS. et al. Inflammasome NLRP3 activation induced by Convulxin, a C-type lectin-like isolated from Crotalus durissus terrificus snake venom. Sci Rep. 2022;12:4706. https://doi.org/10.1038/s41598-022-08735-7.
Garg A, Kant K, Roy KK, Sahoo A, Malakar CC, Gupta S. Docking-based evaluation against Human Tankyrase-1 and Tankyrase-2 enzyme. Mater Today: Proc. 2022;57:300–6. https://doi.org/10.1016/j.matpr.2022.03.095.
Reetu R, Garg A, Roy KK, Roy A, Gupta S, Malakar CC. In-silico studies for targeting PPARγ for the Type II Diabetes Mellitus. Mater Today: Proc. 2022;57:44–48. https://doi.org/10.1016/j.matpr.2022.01.299.
Al-kahraman MSAY, Al-kahraman YM, Singh GS. Evaluation of some classical hydrazones of ketones and 1,2-diketones as antileishmanial, antibacterial and antifungal agents. Arch Pharm Res. 2012;35:1009–13. https://doi.org/10.1007/s12272-012-0608-7.
Schmidt P, Druey J. Heilmittelchemische Studien in der heterocyclischen Reihe. Mitteilung. Pyridazine II. Eine neue Pyridazinsynthese. Helv Chim Acta. 1954;15:134–40. https://doi.org/10.1002/hlca.19540370116.
Lossow WJ, Migliorini RH, Brot N, Chaikoff IL. Effect of total exclusion of the exocrine pancreas in the rat upon in vitro esterification of C14–labeled cholesterol by the intestine and upon lymphatic absorption of C14–labeled cholesterol. J Lipid Res. 1964;5:198–202.
Kim JH, Cho CW, Kim HY, Kim KT, Choi GS, Kim HH. et al. α-Glycosidase inhibition by prenylated and lavandulyl compounds from Sophora flavescens roots and in silico analysis. Int J Biol Macromol. 2017;102:960–9. https://doi.org/10.1016/j.ijbiomac.2017.04.092.
Acknowledgements
This work was supported and funded by a grant from international campus, School of Pharmacy, Tehran University of Medical Sciences, Grant no. 99-1-220-46947.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Firoozpour, L., Kazemzadeh Arasi, F., Toolabi, M. et al. Design, synthesis and α-glucosidase inhibition study of novel pyridazin-based derivatives. Med Chem Res 32, 713–722 (2023). https://doi.org/10.1007/s00044-023-03027-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-023-03027-9