
Abstract
Despite the significant development of diagnostic procedures and therapeutic options in past few decades, glaucoma is still highly prevalent and represents one of the leading causes of blindness in the world due to progressive and irreversible changes in optic nerve. Detection of carbonic anhydrase as a suitable target for the control of intraocular pressure indicated the beginning of carbonic anhydrase inhibitors application for the antiglaucoma treatment. Considering the multitude of proven and potential therapeutic applications of carbonic anhydrase inhibitors, the discovery of new chemotypes with carbonic anhydrase inhibitory activity will continue to be a significant aim. In this article we review the literature on synthetic chalcones as human carbonic anhydrase inhibitors, discussing their possible application focusing on chemical structure and Ki experimental values. From currently available experimental data and from the results we have obtained performing in silico calculations, we generated data collection on the basis of which we proposed 14 compounds of particular interest for further lead optimization and drug CAI development. Having previously experimentally determined excellent hCA II selectivity and strong inhibition effect and in our study predicted favorable physicochemical, pharmacokinetic, and toxicological profiles, a benzoxazolone chalcone derivative (139) stands out among the selected compounds. To examine potential therapeutic application, this candidate may be taken for further evaluation in in vivo studies.

Graphical abstract
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glaucoma, the second most common eye disease after cataracts [1], is defined as a group of chronic ocular disorders which, if left untreated, lead to blindness as a result of irreversible damage of retinal ganglion cells and optic nerve. The most important risk factor for glaucoma onset and progression is elevated intraocular pressure (IOP) however, the disorder is associated with broader spectrum of conditions and etiologies (age, myopia, first-degree relatives diagnosed with glaucoma, thinner central corneal thickness, racial background, systemic hypertension, pseudoexfoliation etc.) [2,3,4]. This long-term neurodegeneration process usually remains asymptomatic, as a consequence of which glaucoma is diagnosed usually late [5] (Fig. 1).

Normal eye vs eye with glaucoma. In a healthy eye constant flow of aqueous humor, a transparent liquid rich in bicarbonate secreted by the ciliary body, is maintained. An increase in IOP is the result of fluid building up, caused by misfunction of trabecular meshwork cells to maintain a balanced pressure by allowing aqueous humor to flow out. Common types of glaucoma are herein presented, classified based on the appearance of the drainage system within the eye (taken from https://www.glaucomaassociates.com/glaucoma/types-of-glaucoma/ [72])
The main principle of antiglaucoma therapy is based on lowering IOP, currently the only known modifiable risk factor, either by use of medicaments, laser therapy or surgically [6]. IOP-lowering medications function by either preventing the formation or facilitating the swelling of aqueous humor [7]. Among most commonly prescribed agents that prevent the formation of aqueous humor are carbonic anhydrase (carbonate hydro-lyases, EC 4.2.1.1 or CA) inhibitors (CAIs) that are also used as diuretics and anti-epileptics, while novel generation compounds are undergoing clinical investigation as antiobesity and antitumor drugs [8].
Carbonic anhydrases are a superfamily of metalloenzymes that catalyze the reversible hydration of carbon dioxide to bicarbonate ion and proton [9] playing a crucial role in pH regulation and in several metabolic pathways such as lipogenesis, gluconeogenesis and ureagenesis [10]. A large number of α-CA isoforms have been described in vertebrates, with Zn(II) ions at the active site and 12 catalytically active isoforms known to date, widely differing in cellular localizations (Fig. 2), tissue distribution and physiological roles, participating in many biochemical and physiological processes in which bicarbonate or carbon dioxide are substrates [11]. Modulation of CAs seems to be essential for the treatment of many diseases in which the activity of various isoforms is upregulated.

Localization and multimerization of catalytically active hCA isoforms in the cell. Isoforms CA I, CA II, CA III, CA VII, and CA XIII are cytosolic, CA IV is anchored to the membrane via a covalently attached lipid moiety, CA VA and CA VB are found in mitochondria, CA VI is excreted in human saliva and milk, and CA IX (bears a proteoglycan-like (PG) domain), CA XII, and CA XIV are membrane-bound via a single transmembrane α-helix with the catalytic domain outside of the cell. Catalytically inactive isoforms CA VIII, CA X, and CA XI are not shown in the figure. In eye, the cytosolic isoforms CA I, CA II and two membrane-bound isoforms CA IV and CA XII are present [8, 19]
The relationship between glaucoma and CAs was established in 1950s, due to the studies on the chemistry and dynamics of aqueous humor [12,13,14] and to Wistrand’s study [15], who demonstrated that the enzyme (forms involved in the ciliary processes), by promoting bicarbonate formation through hydration of carbon dioxide, is directly involved in production of aqueous humor. Few years after, Becker [16] showed that the sulfonamide based CAI, acetazolamide (AZA), is causing a drop of IOP in experimental animals and humans, whereas Kinsey and Reddy [17] proved that the phenomenon is due to a reduced bicarbonate production and consequent reduction of aqueous humor secretion, all conditioned by the AZA inhibition (sulfonamides tightly bind to the Zn(II) ion within the enzymatic active site, thus interrupting the carbon dioxide hydration cycle [11]). The above findings made CA a suitable target for IOP controlling, indicating the beginning of CAIs applications for the antiglaucoma treatment (Fig. 3). Many decades later, CAIs are still one of the main clinically utilized drugs that, despite the lack of isozyme selectivity (inhibiting most of the catalytically active CA isoforms, a consequence of which manifesting a wide range of side effects [18]) are used both, systemically and as topically in the treatment of glaucoma [for more details on recent advances in the medicinal chemistry of carbonic anhydrase inhibitors see an excellent review by Kumar et al. [19]].

Structures of CA inhibitors in clinical use: (a) the first generation of CAIs, sulfonamide-containing molecules AZA, methazolamide, and dichlorophenamide, agents intended for oral use. In a dose-dependent manner, oral AZA lowers IOP by about 30% and despite a non-selective strong inhibitory effect on hCAs is still considered as one among the most effective CAIs and IOP-lowering agents and (b) dorzolamide and brinzolamide, topically acting CAIs [7, 73]
What limits the development of effective CAIs involved in particular pathology is the lack of isoenzymes selectivity, which could lead to serious side effects [20]. Therefore, the interest is to develop not only potent CAIs, but also those with promising isoform selectivity in order to constitute a valuable therapeutic tool for the treatment of a desired pathology. For antiglaucoma (IOP-lowering hCAIs) therapy this means should be based on the selective CA targeting (i.e., on inhibition of isoforms responsible for aqueous humor secretion: CA II, IV, and XII [8]. Apart of selectivity, developing agents should possess appropriate physicochemical properties: hydrophilicity (due to drug formulation), a balanced lipophilicity (due to plasma membranes penetration ability for reaching the ciliary processes), favorable pharmacokinetics and low toxicity [18]. Not a simple task, is not it? Nevertheless, the quest for novel and more potent CA inhibitors with fewer side effects is a continuous process in context of which various scaffolds have been investigated [see three extensive reviews provided by Kumar et al. [19], Ghorai et al. [21] and Karioti et al. [22]].
Although IOP lowering with medical, laser, and surgical therapies is itself neuroprotective [23], researchers are persistently working on identifying agents that are able to provide neuroprotection independent of IOP reduction. Oxidative stress, excitotoxicity and neuroinflammation are additional factors associated with the pathophysiology of glaucoma, therefore it is assumed the use of agents with antioxidant, antiapoptotic and anti-inflammatory potential could be beneficial in protecting neuronal integrity and function [24].
Considering information presented so far, desirable scenario in quest for novel therapeutic agents for glaucoma and ocular hypertension treatment could be a structure with excellent CAIs properties accompanied by antioxidative and antiinflamatory activity, and favorable toxicological and pharmacokinetic profile.
Chalcones (trans-1,3-diaryl-2-propen-1-ones), the main precursors in flavonoid biosynthesis, represent a group of lipophilic plant-derived polyphenolic compounds [25] consisting of two aromatic rings (A and B) linked by α,β-unsaturated carbonyl unit (Fig. 4). α,β-Unsaturated carbonyl unit is a good Michael acceptor and participates in target selective Michael addition, a specific mechanism reported for post-translational modification (PTM), involving covalent additions to amino acid side chains and regulates protein function in cell signaling pathways and biological processes [26]. As an electrophilic moiety, α,β-unsaturated carbonyl function reacts with amino acids containing nucleophilic side chains (Cys, His, Lys, and Ser) and those covalent interactions are regarded as partial explanation for remarkable spectrum of pharmacological activities [27, 28].

Chemical structure of trans-chalcone
Having high plasticity for chemical modifications, allowing structure building on a rigid platform, chalcones are considered as privileged scaffolds in the design of pharmacological trials [29]. In order to obtain compounds with superior cytotoxic properties, modification of the basic scaffold has been performed by modulating the aromatic residues, replacing aromatic residues with heteroaryl units, and obtaining hybrid molecules [27].
Since both, natural and synthetic chalcones are known for impressive antioxidative, antiinflamatory and neuroprotective effects [30,31,32], would be rational to assume that these molecules, by preventing neurodegenerative changes in retinal ganglion cells and optic nerve, could express favorable influence in glaucoma pathogenesis. In addition, as many studies shown, chalcones possess a wide variety of cytoprotective and modulatory functions which may have therapeutic potential for multiple diseases [33,34,35,36]. Because of their flexible structure, chalcones can effectively bind to many enzymes and receptors and recently, several studies involving chalcones, reported compounds of the type as potent CA I and II inhibitors [37,38,39,40,41,42,43,44,45,46,47,48,49,50].
For this reason, we aimed our current work towards evaluation of synthetic chalcone derivatives, previously assayed on hCA inhibitory potential, for in silico physicochemical, toxicological, and pharmacokinetic properties, highlighting the most suitable as ligands for the design of novel CAIs with high isoform selectivity and of particular interest for the future development as drug leads. All data related to the chalcone structures involved in CAI experiments were retrieved from the website. The corresponding experimental values were expressed as Ki and/or IC50, depending on the assay type. For the analyses, only the ligands associated to a Ki and/or IC50 value for the hCA isoforms present in the eye/responsible for aqueous humor secretion were taken into account. Whenever was possible, in order to better process the results coming from different laboratories, selectivity ratio (SR Ki hCA I/Ki hCA II, Ki hCA I/Ki AZA and Ki hCA II/Ki AZA) calculations were made on the basis of which the results were also commented. Among the essentials, preclinical data are considered a basic requirement in the development of new drug candidates, and various physicochemical properties can be predicted by in silico approach models, all of which are available from drug evaluation tools. The structures of all ligands in SMILES notation were imported online into drug evaluation tools SwissADME [51] and OSIRIS Property Explorer [52] to predict physicochemical descriptors as well as pharmacokinetic and toxicological properties of the candidate molecules.
Simple chalcone derivatives
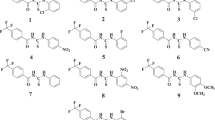
The CA inhibitory potential of the simplest chalcone, (E)-1,3-diphenylprop-2-en-1-one or trans-chalcone (1), and its epoxide (2) (Fig. 5), was tested on hCA I and hCA II isoforms obtained from haemolyzed erythrocytes [53]. Compounds 1 and 2 inhibited both isoforms in a noncompetitive mode with similar Ki values (0.035 and 0.037 µM towards hCA I, respectively; 0.142 and 0.173 µM towards hCA II, respectively), but were less effective compared to the reference inhibitor AZA (Ki = 0.016 and 0.024 µM against hCA I and hCA II, respectively). Compound 2 was subjected to molecular docking study which showed that epoxide oxygen might be responsible for interactions within hCA I active site (hydrogen bond interactions with Gln92). According to the authors [53], this type of interaction was not observed for 2 in the hCA II active site, but hydrogen bonding between the carbonyl oxygen with Gln92 and a water molecule was detected instead. Within hCA I of importance for the structure 2 were π-π stacking interactions between the benzene ring and His94, however inside the hCA II active site this interaction lacked. The authors concluded that aromatic rings are having more important role in enzyme inhibition than α,β‐saturated system [53].

Simple chalcone derivatives as CA inhibitors
Aslan et al. [54] tested a set of seven simple chalcones (compounds 3-9, Table S1) obtained from acetophenones and related benzaldehydes for their inhibitory activity on the isolated hCA I and hCA II. Evaluated derivatives inhibited both isoenzymes in a competitive manner, with Ki values ranging from 1.83 (compound 9) to 7.05 µM for hCA I, and from 0.59 (compound 9) to 5.50 µM for hCA II (Table S1, Fig. 5). The nature and the position of the groups attached to A and B aromatic rings have substantial significance for the enzyme inhibition activity; hCA I potency change in 2′-hydroxy-4-methoxychalcone (3) compared to 4-methoxychalcone (6) was observed as a result of hydroxyl group B ring addition, while by adding fluoro group to 4-fluorochalcone (4) at position 4 on ring B, a new compound (8) with increased inhibitory effects toward hCA I was obtained. Those changes induced selectivity since opposite effect on hCA II was observed (Table S1) [54].
Another group of derivatives (compounds 10–14, Table S1), synthesized by Dizdaroglu et al. [37] were investigated for hCA I and hCA II inhibitory potential and were found to have Ki values in the low nanomolar range. All compounds had better hCA I inhibition activities (Table S1) than AZA (Ki = 250 nM). Among the tested chalcones, 11 (Fig. 5), bearing bromo-substituted A ring and trimethoxy-substituted B ring, had the most potent hCA I inhibition activity (Ki = 8.03 nM). Regarding the hCA II isoform, chalcone with hydroxy-substituted A ring and dimethylamino-substituted B ring (14, Fig. 5), proved to be the most potent and the only among assayed compounds with similar inhibitory activity as AZA (Ki = 11.77 nM and Ki = 12 nM, respectively) [37], based on which can be concluded that N,N-dimethylamino substituent on ring B could have large impact on the activity toward hCA II.
Several p-halogen/p-methoxy A ring-trimethoxy-substituted B ring- chalcones (compounds 15–21, Table S1) were tested against cytosolic isoforms. CA inhibitory activities, measured in nanomolar concentrations (Table S1), were depended on the identity of A ring p-substitution, confirming the known fact that the halogen substituted compounds are effective as CA inhibitors [55]: A ring p-fluoro-substituted 15 (Fig. 5) was the best hCA I inhibitor (Ki = 8.75 nM), while already mentioned A ring p-bromo substituted 11 was the best hCA II inhibitor (Ki = 11.47 nM), both being more effective than reference inhibitor AZA (Ki (hCA I) = 28.75 nM; Ki (hCA II) = 31.67 nM) [38].
Burmaoglu et al. [39] synthesized eight novel halogen-containing chalcone derivatives (22–29, Table S1) with 3′-bromo-2′,4′,6′-trimethoxy-substituted A ring, differing only in type and position of halogen-substituted B ring. The results obtained by evaluating hCA I and hCA II inhibition effect have shown that derivatives have potent inhibitory profiles on both isozymes, with Ki values in the range of low nanomolar concentrations (16.24–40.96 nM for hCA I, 29.61–67.15 nM for hCA II) (Table S1). It could be observed that all compounds expressed slightly weaker hCA II inhibitory activities compared to AZA (Ki = 22.17 nM) however, found to be up to eight times more effective hCA I inhibitors than AZA (Ki = 141.02 nM). With the lowest Ki values, compounds 23 (with ortho-chloro-substituted B ring) and 25 (with para-chloro-substituted B ring) (Fig. 5) were the most effective against hCA I and hCA II isoforms, respectively. The study has indicated pivotal influence of methoxy groups in the A ring for the inhibition of the enzyme receptors: via metal coordination with Zn301 in the catalytic active site of the hCA I or by hydrogen bonding with Try7 and Asn67 residue of hCA II receptor [39]. The confirmation resulted from docking studies which were also part of the research.
Gürdere et al. [40] have investigated inhibition profile of different B ring substituted 4′-aminochalcones (compounds 30–44) towards hCA I and hCA II. Synthetized compounds were more effective inhibitors than AZA (Ki (hCA I) = 83.39; Ki (hCA II) = 104.60 nM), having Ki values ranging from 2.55 to 11.75 for hCA I and from 4.31 to 17.55 nM, with 2-furyl-inserted derivative ((E)-1-(4-aminophenyl)-3-(furan-2-yl)prop-2-en-1-one) 43 as the most potent hCA I inhibitor (Ki = 2.55 nM), and 4′-amino 4-methoxy derivative 36 as the most potent hCA II inhibitor (Ki = 4.31 nM) (Fig. 5) [40].
In an attempt to obtain effective hCA II inhibitors, Mahar et al. [56] performed a synthesis of ten 2′-hydroxy-5′-((trimethylsilyl)ethynyl)chalcones (compounds 45–54, Table S1). According to the results obtained by the enzyme inhibitory assay, all compounds exerted better hCA II inhibition effect than AZA (IC50 = 0.998 μM) (Table S1), with pyridine derivative 47 and compound 49 (Fig. 5) being the most potent (IC50 = 0.033 and 0.054 µM, respectively). For compound 49, which also exhibited strong antioxidant activity and had good binding energy value in in silico study, ligand-protein binding analysis revealed three hydrogen bonds and π–π stacking interactions at the protein active site [56].
Aryl-substituted chalcone derivatives
Burmaoglu et al. [41] designed a series of nine novel chalcone derivatives (compounds 55–63, Table S2) and explored their inhibitory activities towards cytosolic hCA I and hCA II isozymes. Tested compounds had the same A ring (3-phenyl-2,4,6-trimethoxy-substituted) structural motif, varying B ring substitution pattern. The synthesized chalcone derivatives showed Ki values in the range of 14.71–62.95 nM against hCA I and 28.93–47.20 nM against hCA II (Table S2). The most effective among compounds was p-chloro substituted 58 showing good selectivity for hCA I isoform, while the most promising inhibitory effect against hCA II was for 56 (o-chloro-substituted B ring) compound (Fig. 6). However, no compound was more effective than the reference AZA (Ki = 8.45 and 4.25 nM for hCA I and hCA II, respectively) [41].

Aryl-substituted chalcone derivatives
N-substituted (amino-/imino-/imido-/diazo-) chalcone derivatives
Yamali et al. [42] designed a series of nine aminoalkylated phenolic chalcones (compounds 64–72, Table S3) via aminomethylation of the corresponding chalcones. The compounds were further screened for CA I and CA II inhibition effect. According to the Ki values (Table S3), all bis-Mannich bases expressed considerable CA inhibitory activities comparable to AZA (Ki (hCA I) = 21.74 nM; Ki (hCA II) = 18.27 nM) with more than half emerging as more potent than this antiglaucoma drug. The lowest Ki values were observed for piperidine derivative 64 (Ki = 11.76 nM) considering hCA I isoform, and morpholine derivative 68 (Ki = 6.08 nM) when it comes to hCA II isoform (Fig. 7) [42].

N-substituted chalcone derivatives
A series of 16 chalcone derivatives (compounds 73–88, Table S3) was prepared by condensation of amino-substituted acetophenones with benzaldehydes in 1:2 molar ratio and subjected to in vitro analysis for hCA I and hCA II inhibition potencies. All of the tested compounds expressed remarkable inhibition activities (Table S3) as compared with standard AZA (Ki = 859.07 and 1022.20 nM for hCA I and hCA II, respectively). According to both Ki and IC50 values, p-methoxy substituted 87 was the most potent hCA I inhibitor (Ki = 141.88 nM; IC50 = 226.10 nM), while compound 74 with m-chloro substituent showed the best inhibition activity against hCA II (Ki = 199.31 nM; IC50 = 295.28 nM) (Fig. 7) [57].
Kocygit et al. [58] synthesized six chalcone-imide derivatives (compounds 89–94, Table S3) and performed biochemical analysis towards both hCA I and hCA II. Cytosolic hCA I form was inhibited by these derivatives with Ki values (426.47–699.58 nM, Table S3) slightly lower than that of the reference inhibitor (977.77 nM). All derivatives were stronger inhibitors of hCA II compared to hCA I, with Ki values (214.92-532.21 nM, Table S3) up to four times lower than that of AZA (904.47 nM). Despite different electron withdrawing and electron donating properties of substituents on B ring of chalcone derivatives, a minimum difference in inhibitory potencies were observed. Among the tested compounds, 91, with m-methyl-substituted B ring, was the most effective hCA I inhibitor (Ki = 426.47 nM), while compound 93, with m-methoxy-substituted B ring was the most effective inhibitor of the hCA II isoform (Ki = 214.92 nM) (Fig. 7) [58].
Considering sulfonamides are among the first discovered CA inhibitors clinically used for decades (AZA), Arslan et al. [43] prepared a series of chalcone derivatives with benzensulfonamide moiety linked to the A ring via diaza bond (compounds 95–100, Table S3). Methyl and methoxy groups were incorporated to the B ring chalcone moiety to induce a slightly lipophilic character to the molecule already contained the hydrophilic phenol and diazo moieties. The derivatives were tested towards cytosolic hCA I and hCA II isoforms, all exhibiting good inhibitory profiles. Isoform hCA I was effectively inhibited by all derivatives with Ki values (9.88–24.40 nM, Table S3) significantly lower than that of the reference AZA (250 nM) with 2,4-dimethoxy derivative 100 (Fig. 7) as the most potent inhibitor. Good inhibitory activities of new compounds towards hCA II with Ki values (18.25–55.43 nM, Table S3) higher than the reference AZA (12.00 nM) were observed. 2-Methyl derivative 95 (Fig. 7) was the most promising among the investigated hCA II, comparable to the clinically used AZA [43].
Phenyl(thio)urea chalcone derivatives
A group of eight phenylurea- (compounds 101–108, Table S4) and four phenylthiourea- (compounds 109–112, Table S4) substituted chalcones was synthesized and evaluated for hCA I and hCA II inhibitory activity. The results showed that all the synthesized compounds inhibited CA isoenzymes activity with IC50 values in micromolar range (Table S4). The most active hCA I inhibitor within phenylurea derivatives was compound 104 (IC50 = 23.06 μM) substituted with a methoxy group in para position of the B ring. When it comes to hCA II inhibition, the addition of a methyl group in para position of both phenylureinyl part and B ring (compound 107) led to the highest inhibitory effect (IC50 = 14.40 μM). Regarding phenylthiourea derivatives, compound 109, bearing a p-chloro-substituted B ring, stood out as the most potent inhibitor of both hCA I and hCA II (IC50 = 36.26 and 23.89 μM, respectively) isoforms (Fig. 8) [59].

Phenyl(thio)urea chalcone derivatives
Heteroaryl chalcone derivatives
Benzoxazolone incorporated chalcone derivatives
Bilginer et al. [60] designed and synthesized a set of eight benzoxazolone chalcone derivatives (113–120, Table S5) evaluating the compounds in hCA I and hCA II inhibitory activity assay. The compounds showed good to moderate CA inhibitory properties, with only compound 120, bearing a p-benzyloxy substituted B ring, having a lower Ki value (28.37 µM) towards hCA I than AZA (30.18 µM), while towards hCA II all compounds had higher Ki values (10.85–37.96 µM) than AZA (4.41 µM) which qualified them as worse inhibitors than the reference (Table S5). However, as the most promising hCA II inhibitor stood out 4-dimethylamino derivative 119 (Ki = 10.85 µM) (Fig. 9) [60].

Benzoxazolone chalcone derivatives
Under the same experimental conditions, Bilginer et al. [61] conducted another study concerning inhibitory properties of benzoxazolone chalcone derivatives towards hCA I and hCA II, this time including halogen-bearing compounds. The tested derivatives (121–132, Table S5) differed in number and position of halogen substituents in the B ring. Similar to the abovementioned series, this group of chalcones also showed moderate inhibition profile, with compound 132 (Fig. 9) being the most active towards both hCA I and hCA II (Ki = 33.5 and 7.3 µM, respectively). However, no compound was more effective CA inhibitor than AZA (Ki = 30.2 and 4.4 µM for hCA I and hCA II, respectively) [61].
Based on the results of both studies, Bilginer et al. [44] designed a series of N-substituted-benzoxazolone chalcone derivatives (compounds 133–139, Table S5) starting from compound 116 which in previous experiments already proved good inhibitory activity on both isoenzymes. Structural modifications were made by aminomethylation of the benzoxazolone ring (Mannich bases). The in vitro inhibition assay results, expressed as Ki values, revealed that aminomethylation significantly improved the inhibition potential of the tested derivatives on cytosolic hCA I and hCA II. Pyrrolidine derivative 136 was found to be the most potent hCA I inhibitor among the tested compounds, with Ki value (0.0123 µM) nearly seven times lower than that of AZA (0.0844 µM). For hCA II the most potent inhibitor was piperazine derivative 139 with a Ki value (0.0086 µM), approximately seven times lower than that of AZA (0.0592 µM) (Fig. 9). Molecular docking emphasized the importance of carbonyl group for the formation of hydrogen bonds with the amino acids present in CAI enzyme active site (thus the prevention of tautomerisation between carbonyl group and nitrogen atom in the benzoxazolone ring by inserting an aminomethyl moiety was crucial for increased CAI potency) and according to the binding pattern of the most potent CAI inhibitor, important interactions were the π-cation interaction (exhibited between the benzoxazolone ring of the compound and the side chain of the amino acid His200), seven hydrophobic interactions (between the compound 136 and the amino acids Phe91, His119, Leu131, Val143, Leu198, and Trp209) and a strong halogen bond-hCA I isoenzyme active site (trifluoromethyl group-Phe91) interaction [44]. Molecular docking study of 139 and hCA II active sites emphasized hydrogen bond interaction between carbonyl group in benzoxazolone with Gln92 once again demonstrating positive contribution of tautomerization prevention on hCA II inhibitory activity. Two additional hydrogen bond interactions were observed between the oxygen atom of the benzoxazolone ring and the amino acids Asn62 and Asn67. The carbonyl group of chalcone participated in a hydrogen bond interaction with Trp5. Indicated also were π-stacking interactions between 139 benzoxazolone ring and the amino acid His94. Four hydrophobic interactions between the molecule 139 and Trp5, Phe20, and Pro202 in the CA II active site were also predicted [44].
(Iso)indole incorporated chalcone derivatives
Indole and methanoisoindole heterochalcone derivatives
Simple indole incorporated chalcone derivatives: Kuday et al. [62] synthesized a group of seven simple indole chalcone derivatives (compounds 140–146, Table S5) in order to evaluate their hCA I and hCA II inhibitory properties. From the obtained results (Table S5), the authors have drawn SAR observations indicating that a presence of one methoxy group at para position of the A ring (compound 141) increased the inhibitory potential of indolyl chalcones towards hCA I (IC50 = 7.42 µM). However, the incorporation of a strong electron-withdrawing group at the same position rendered strong hCA II inhibitory effect, as observed with the p-nitro-substituted derivative 146 (IC50 = 7.22 µM) (Fig. 10) [62].

Simple indole and methanoisoindole chalcone derivatives
Methanoisondole incorporated chalcone derivatives: Kocyigit et al. [45] provided a series of 14 novel methanoisoindole chalcone derivatives (compounds 147–160, Table S5) and evaluated their hCA I and hCA II inhibitory activity. All synthesized compounds elicited remarkable inhibitory activity against both hCA I and hCA II with IC50 (466.4–710.8 pM and 352.5–555.3 pM, respectively) lower or similar to those of AZA (995.7 and 485.0 pM for hCA I and hCA II, respectively). Regarding Ki values, the inhibitory effect of the tested compounds was in the range of 405.3–635.7 pM for hCA I, and 245.4–489.6 pM for hCA II. Taken together, compound 155 with bromo substituent at para position of the B ring stood out as the most promising inhibitor of both CA isoforms (Fig. 10) [45].
By investigating in vitro hCA I and hCA II inhibitory potential, Kocyigit et al. [46] expanded their research to tetrabromo-substituted methanoisoindole chalcone derivatives. Compounds 161–169 expressed superior inhibitory effect towards both isoforms in comparison to AZA (IC50 (hCA I) = 40.45 nM; IC50 (hCA II) = 24.16 nM; Ki (hCA I) = 34.50 nM; Ki (hCA II) = 28.92 nM) in nanomolar concentrations for both IC50 and Ki (Table S5) having the best hCA I inhibitory activity/potency for p-methoxy- and p-substituted B ring linked to 5,6-dibromosubstituted-4,7-methanoisoindole-1,3-dione chalcone derivatives 161 (IC50 = 14.14 and Ki = 11.30 nM) and 164 (IC50 = 13.58 nM and Ki = 13.09 nM). p-Chloro substituted B ring derivative 163 was the most effective inhibitor of hCA II isoform (IC50 = 9.62 nM and Ki = 8.20 nM) (Fig. 10) [46].
Molecular docking studies were performed on both isoenzymes, for compounds 161 and 163 that had the most effective inhibition/binding to the ligand binding site of hCA I and hCA II proteins. The hCA I polar interactions were described with residues His67, His64, His200, Gln92, His94, and Thr199, while hydrophobic interactions were formed by Trp5, Pro202, Pro201, Val207, Trp209, Leu198, Val143, Ala121, Phe91, and Val62 residues. Two different bond type interactions were formed by the residues in the ligand-binding site in hCA I: the hydrogen bond formed by Trp5 and the π–π bond formed by His94. At the hCA II isoenzyme binding site, polar interactions with His64, His94, Asn62, Gln92, and Thr200 and hydrophobic interactions with Trp5, Pro202, Leu198, Val121, and Phe131 residues were observed. A hydrogen bond was formed with Trp5 [46].
Indole incorporated chalcone-sulfonamide hybride derivatives
Compounds belonging to this group were designed as ligands interacting with the middle and outer parts of the active site cavity, known as the most variable regions among the hCA isoforms. This concept was supposed to provide isoform-selective CAIs.
Pyridyl-indole incorporated chalcone-sulfonamide hybride derivatives: Peerzada et al. [63] synthesized a series of several sulfonamide derivatives of pyridyl-indole heteroaryl based chalcones (compounds 170–178, Table S5), screened for in vitro hCA inhibitory activity (hCA II isoenzyme was chosen as off target). Compounds had more inhibition effect on hCA II isoform, having IC50 values (>9.10 µM) higher than that of AZA (0.013 µM) with 2,4-dichlorophenyl-substituted 175 (Fig. 11) as the most active derivative [63].

Indole-sulfonamide chalcone derivatives
Indolylchalcone incorporated- benzensulfonamide triazole linked hybride derivatives: Singh et al. [47] synthesized a series of 17 indolylchalcone derivatives linked to the benzenesulfonamide group via the triazole ring (compounds 179–195, Table S5), and tested their inhibitory activity against cytosolic hCA I and hCA II. Among the assayed derivatives p-bromophenyl and m-methoxyphenyl substituted hybrids (182 and 195, respectively) were the most potent against hCA I isoform (Ki = 18.8 and 38.3 nM, respectively) (Fig. 11), also being more potent than AZA inhibitor (Ki = 250 nM). Molecular docking study performed on two representative (the most active 182 and 195) compounds co-crystalzing at the hCA I active site rationalized the experimental results: elicited potency is for favorable interactions with key residues of the hCA I pocket. By occupying the isoenzyme’s active pocket in a fitting manner, both compounds made several positive interactions with hCA I key residues. For 182 and 195 the O atom from sulfonamide moiety, acting as hydrogen bond acceptor, was responsible for Thr199 and His200 hydrogen bond interactions, while the other O and N atom of sulfonamide moiety established metal ion interactions with Zn(II). Triazole core made an arene-arene (π–π) interaction with Phe91 and several stabilizing hydrophobic interactions were observed with Ile60, Val62, Phe91, Ala121, Leu131, Ala132, Ala135, Leu141, Val143, Leu198, Pro202, and Trp209 (the active site residues stabilizing the binding of 182 and 195 in hCA I active site). The authors speculated the importance of electronic effect of substituents attached to the core, presuming electron donating group (-CH3 (182) -OCH3 (183) and -Br (195)) A ring substituted compounds showed good inhibitory activity as compared to those containing electron withdrawing groups. Those structures, being more potent than the standard AZA, were anticipated as potential lead molecules for the design and development of selective hCA I inhibitors. As far as hCA II isoform was concerned, the most potent against was p-methylphenyl derivative 183 (Fig. 11) (Ki = 36.2 nM), although characterized with weaker inhibition properties when compared to AZA (Ki = 12.1 nM) [47].
Indolylchalcone incorporated -benzensulfonamide amide linked hybride derivatives: Singh et al. [48] similar to structures reported in Singh et al. [47], provided a series of hybrids, indolylchalcone-benzensulfonamide derivatives linked via amide linker (compounds 196–213, Table S5), and tested them against hCA isoforms. Interestingly, amide linker significantly elicited inhibitory effect against hCA II and those derivatives were found to be more selective inhibitors of hCA II when compared to other hCA isoforms. Compounds 204 (Ki = 2.3 nM) and 208 (Fig. 11) (Ki = 2.4 nM) were found to be the most potent within the tested series, more potent against hCA II than AZA (Ki = 12.1 µM) and could be further examined in the design of novel efficient hCA II inhibitors as lead compounds [48].
Oligomeric chalcone derivatives
Bis-chalcone derivatives
Diaza linked 1,4-bis chalcone derivatives
A group of 11 bis-chalcone derivatives (compounds 214–224, Table S6) was prepared by Arslan et al. [64] via diazotization and diazocoupling reactions, and tested for cytosolic hCA I and hCA II inhibitory effect. Monomeric chalcone units were interconnected by -N=N- bond formed between A rings in para- position, generating bis-chalcones differing only in number and position of B ring methoxy groups. Among the investigated bis-chalcones containing single methoxy substituent in the monomeric unit, compound with m-methoxy group in the B ring showed better hCA I inhibitory activity compared to its nearly two times less effective o- and p- analogs (Table S6). The addition of one more methoxy group in the position 5 (compound 222) or two more methoxy groups in positions 4 and 5 (compound 224) of the B ring (Fig. 12) resulted in a significant enhancement of hCA I inhibitory activity, making these two compounds the most potent hCA I inhibitors among the tested derivatives, with Ki values (72.90 and 103.55 nM, respectively) lower than that of AZA (Ki = 250 nM). A rather similar inhibition profile was observed for hCA II, with compound 222 being the most active among the methoxy-substituted derivatives (Ki = 166.54 nM). However, there were no compounds as active as the reference inhibitor was (Ki = 12 nM) [64].

Bis- and tris-chalcone derivatives as CA inhibitors
1,3-Bis chalcone derivatives
A group of 19 1,3-bis-chalcone derivatives (225-243, Table S6), with monomeric chalcone units symmetrically condensed, sharing either A (234–242) or B (225–233, and 243) phenyl ring as structure motif, were tested in vitro against hCA I and hCA II. Both isoforms were effectively inhibited by these compounds, with Ki values in the range of 94.33 to 787.38 nM for hCA I, and of 100.37 to 801.76 nM for hCA II, respectively (Table S6) which was in contrast to the higher Ki values for clinical standard AZA (1054.38 nM against hCA I, and 983.78 nM against hCA II). Moreover, IC50 values for the tested derivatives (ranged from 98.63 to 855.44 nM for hCA I and from 77.33 to 717.77 nM for hCA II, Table S6) were lower than those for the reference inhibitor (997.67 and 901.36 nM for hCA I and hCA II, respectively). Among the derivatives “sharing” A ring, a remarkable activity was observed for bromine derivative 239 (Fig. 12) with nearly 11 times lower Ki and IC50 values than AZA for both isoforms. Regarding derivatives “sharing” B ring, the striking feature of the most promising CA inhibitor was bromo substitution (230, Fig. 12) [65]. For the tested 1,3-bis-chalcone derivatives, it could be observed that the presence of a halogen substituent can have a large impact on the hCA inhibitory activity compared to the presence of some other substituents such as, for example, methoxy groups.
Tris-chalcone derivatives
Burmaoglu et al. [49] synthesized via Claisen-Schmidt condensation reaction a group of tris-chalcone derivatives (244–252, Table S6) consisting of identical fluoro-substituted chalcone subunits sharing A ring. The tris-chalcones were further evaluated for their hCA inhibitory activity, and as for the hCA I isoform, all compounds expressed good inhibitory activity with Ki values (19.58–78.73 nM, Table S6) lower than that of the reference AZA (141.02 nM). Derivative 251 (Fig. 12) with two fluorine atoms in m-positions of each subunit expressed the highest potency. Quite similar Ki values were observed for hCA II and were in a range of 12.23 to 41.70 nM (Table S6) with p-fluoro-substituted 246 (Fig. 12) being the most potent among the investigated species and also more potent than AZA (Ki = 22.17 nM) [49].
The same authors [50] tested another group of substituted tris-chalcone derivatives (253–264, Table S6) for CA inhibitory activity. Similar, yet slightly higher activity was observed within the series, with Ki values ranging from 13.6 to 50.0 nM for hCA I and from 9.9 to 39.5 nM for hCA II. p-Chloro-substituted 255 and m-bromo-substituted 257 (Fig. 12) compounds had the best inhibition of hCA I and hCA II, respectively, being even more potent than the reference AZA (Ki = 141.0 nM and 22.2 nM towards hCA I and hCA II, respectively) [50].
Tetra-chalcone substituted phthalocyanines as CA inhibitors
Özen et al. [66] designed two tetra-chalcone substituted phthalocyanine complexes containing Zn(II) or Co(II) ion in their core, and evaluated their hCA I inhibition properties. Compound with Zn(II) ion inhibited hCA I with an IC50 value of 1.69 µM, which was also lower than that of AZA (IC50 = 7.07 µM), while the Co(II)-containing compound showed no inhibition effect, even at 100 µM. As a mechanism of action, the authors suggested that there might be a competition between Zn(II) ion in the complex with Zn(II) ion in the enzyme active site [66].
Arslan [67] synthesized one metal-free and three metallophthalocyanines bearing Co(II), Mn(II), or Cu(II) ion, coordinated with four identical chalcone subunits (Fig. 13). Obtained complexes were tested for their hCA I and hCA II inhibition effect. All derivatives had lower IC50 values (0.5621–0.6413 µM) when compared to that of AZA (0.9857 µM) towards hCA I, but only Mn(II)-coordinating phthalocyanine showed potency towards hCA II inhibition (IC50 = 0.4823 µM), that was comparable to that of the reference inhibitor (IC50 = 0.4894 µM). Since compounds differed only in the presence and type of incorporated metal ion, the author suggested the metal complexing effect changed the electron intensity of the phthalocyanines, resulting in dissimilar interplays via these enzymes [67].

Tetra-chalcone derivatives as CA inhibitors
In silico carbonic anhydrase inhibitors studies
SAR
Inhibition efficacy is a multifactorial process, which is often not easy to rationalize [68], even for compounds with simple chalcone scaffold. By handling the selected chemotype experimental data, focusing our observations primarily on Ki values extracted from the literature [37,38,39,40,41,42,43,44,45,46,47,48,49,50, 53, 54, 56,57,58,59,60,61,62,63,64,65,66,67] and on calculated selectivity ratios (SR), efforts were made for establishing structure–activity relationships. An attempt was directed toward finding useful information about structural elements important for the development of more selective and potent CA inhibitors as an alternative to classical ones.
Previous studies revealed that the modulation of CA inhibitors efficacy strongly depends on the type and position of substituents on both A and B aromatic rings. A general observation regarding the activity is that most of compounds containing electron-donating groups (-CH3, -OH, -NH2, -OCH3, etc.) were having stronger inhibitory effect against hCA I and hCA II [41]. As confirmed by many docking studies, by generating strong electrostatic interactions with the amino acids present in the enzyme active site, these groups are establishing effective connections [41].
Having impact on both hCA I and hCA II isoforms, A ring substituted with -NH2 strongly induces CAI effect [40]. The observed effect is due to aryl amine moiety, which acts as an effective H acceptor, with a tendency to form rather strong H-bonds involving the enzyme active site [37, 40]. The effect of another strong hydrophilic substituent, an -OH group, on inhibition activity was also observed, however not as pronounced as for -NH2. In order to modify and improve the binding characteristics, the design of novel ligands often involves the replacement of functional groups. By replacing hydrophilic substituents (-NH2, -OH) on the A ring with -Br or -CH3 [37], a negligible effect on the activity was achieved. According to the authors [37], the phenomenon can be attributed to newly introduced hydrophobic groups that can also enhance binding affinity, only through hydrophobic interactions. Compounds containing halogen groups are also effective CA inhibitors [55]. In this context, -F group as aromatic ring substituent has particularly good ability for developing strong interactions with the amino acids of active site [41]. Adding polar substituents on B ring affects changes in the inhibitory effect. For example, addition of hydroxyl group at position 2 on ring B in 4-methoxychalcone or fluoro group to 4-fluorochalcone at position 4- on ring B had increased hCA I inhibitory effects. Induced changes however had opposite effect on hCA II [54]. A useful modification to increase the effect on hCA II by lowering the Ki values is the introduction of methoxy (-OCH3) substituents on the B ring. The above consideration is of particular interest, as selective CA targeting is the crucial aspect for the successful pharmacological treatment of diseases in which such enzymes are involved. As can be seen form Table S1, compounds 21, 38, 39, 42, 44 (Ki hCA I/Ki hCA II > 1 and Ki hCA II/Ki AZA < 1) might constitute promising leads for the development of more selective and potent inhibitors as alternatives to the classical CA inhibitors.
What can be noticed only by analyzing numerical data from Table S1, without interpreting the electronic effects or the substitution patterns of the simple chalcones, is that larger number of structures are with pronounced effect on hCA I, with Ki values compatible or often exceeding Ki AZA (except for 19, Table S1). The above observation may suggest the importance of α,β unsaturated carbonyl system as the structural motif interacting with the hCA I active site, but in the absence of sufficient data to confirm this remark, it remains at the level of speculation.
Structural hybridization, which covalently links two or more pharmacophores into a single structural framework, is considered a successful approach for the discovery of new scaffolds with therapeutic applications. Many strategies for the development of selective CAI have been adopted [8, 18, 69] and the entities presented in Table S3 (entries 64–100 compared to Table S1 and Table S2 compounds) perfectly illustrates the purpose of implementing the strategy for the CAI development. As can be also seen from Table S3, chalcone derivatives containing Mannich [42] and Schiff bases [57] and chalcone-imide derivatives [58] are structures with excellent selectivity for the hCA II isoform and strong inhibition effect (Ki values for 74, 93, 68, and 64 are approximately five, four, three, and two times lower than that of AZA) that might be adopted as a promising candidate in drug development. N-aminomethylation by the Mannich reaction proved to be a useful modification in terms of CA inhibitory properties [44] with an interesting example of three 3-[4-(trifluoromethyl)phenyl]acryloyl-2(3H)-benzoxazolones (133, 137, and 139, (Table S5)) with 139 as the best candidate of the series that with the highest hCA II inhibition effect and the lowest Ki hCA II value (8.6 nM) can be regarded as molecule for further investigations [44], representing even a promising scaffold that can be further explored in order to generate chemotype of enhanced CA inhibitory potential and selectivity.
1,3-Bis and tris-chalcone derivatives were also effective, in particular structures (Table S6) reported by Tutar et al. [65], Burmaouglu et al. [49, 50]. Generally speaking, both isozymes were inhibited, without a particularly pronounced effect in selectivity. Only compound 257 [50], showing four times greater selectivity toward isoform hCA II than for hCA I (Table S6, SRs: Ki hCA I/Ki AZA < 1) and having two times stronger inhibition effect in comparison to AZA (Table S6, SRs: Ki hCA II/Ki AZA < 1), can be considered effective drug candidate. The majority of the investigated structures had better inhibitory profiles compared to AZA (Table S6, SR: Ki hCA I/Ki AZA < 1 and Ki hCA II/Ki AZA < 1).
In silico study of physicochemical, pharmacokinetic, and toxicological properties
Physico-chemical properties
Physico-chemical properties of the evaluated chalcone derivatives (1–264) were calculated using SwissADME [51] in order to predict their oral bioavailability (Table S7). According to “Lipinski’s rule of five” and the “Veber’s rule”, for optimal oral bioavailability it is desirable that the molecule has the following characteristics: molecular weight ≤500, number of hydrogen bond donors ≤5, number of hydrogen bond acceptors ≤10, partition coefficient ≤5, number of rotatable bonds ≤10 and polar surface area ≤140 Å2 [70, 71]. Summarizing the obtained results (Table S7), 30 out of 50 most effective CA inhibitors (Figs. 5–12), including all simple chalcones (1, 2, 9, 11, 14, 15, 23, 25, 36, 43, 47, and 49), both aryl-substituted chalcones (56 and 58), 6 (out of 8) N-substituted chalcones (64, 68, 87, 91, 93, and 95), each representative of phenyl(thio)urea (104, 107 and 109), benzoxazolone (119, 120, 132, 136, and 139), simple indole (141 and 146), methanoisoindole (155) and pyridyl indole-incorporated (175) chalcones, as well as one (out of 3) indole-incorporated chalcone with a triazole-linked benzensulfonamide (183), fulfilled both “rules” predicting their good oral bioavailability. On the other hand, each of the most effective tetrabromo-substituted methanoisoindole chalcones (161, 163 and 164), indole-incorporated chalcones with an amide-linked benzensulfonamide (204 and 208), and bis- and tris-chalcones (222, 224, 230, 239, 251, 246, 255, and 257), as well as AZA, had at least one parameter outside the acceptable values.
Pharmacokinetic properties
According to the pharmacokinetic properties calculated by SwissADME (Table S8) [51], the ability of intestinal absorption was predicted for 35 out of 50 most effective CA inhibitors (Figs. 5–12), including all simple (1, 2, 9, 11, 14, 15, 23, 25, 36, 43, 47, and 49), aryl-substituted (56 and 58), phenyl(thio)urea (104, 107 and 109), benzoxazolone (119, 120, 132, 136 and 139), simple indole (141 and 146), methanoisoindole (155, 161, 163, and 164), and pyridyl indole-incorporated (175) chalcones, as well as 6 (out of 8) N-substituted chalcones (64, 68, 74, 87, 91, and 93). With the exception of 49, 74, 109, 161, 163, 164, and 175, these compounds were also predicted with the ability to pass through BBB. Nine most effective CA inhibitors (49, 56, 58, 64, 68, 74, 107, 230, and 239) are potential P-glycoprotein substrates. The evaluated chalcone derivatives differ sigificantly in their predicted metabolic properties, in terms of whether they are potential CYP inhibitors. Apart from bis-chalcone 222 and tris-chalcones 246, 251, 255, and 257, the most effective CA inhibitors were predicted as inhibitors of at least one CYP isoenzyme (Table S8).
Toxicological properties
Toxicological properties of the evaluated chalcone derivatives (1–264) were predicted using OSIRIS Property Explorer [52] in terms of their potential to cause mutagenic, tumorigenic, irritating and/or reproductive effects (Table S9). Nearly half of the most effective CA inhibitors (compounds 9, 11, 15, 25, 58, 74, 93, 109, 120, 132, 136, 139, 141, 146, 175, 204, 208, 230, 239, 246, 251, 255, and 257) was predicted with no risk for mutagenic, tumorigenic, irritating and reproductive effects. Two compounds (91 and 107) were predicted with medium risk for tumorigenic effects, but with no risk for mutagenic, irritating, and reproductive effects. The other half of the compounds, as well as AZA, was predicted with risk for at least one of the mentioned effects.
Taken together, in silico study of the most effective chalcone CA inhibitors indicates that 14 compounds (9, 11, 15, 25, 58, 93, 109, 120, 132, 136, 139, 141, 146, and 175) were predicted with the most promising physicochemical, pharmacokinetic and toxicological properties, even better than those predicted for AZA.
Conclusion and future perspectives
Lowering intraocular pressure remains the main therapeutic aim in glaucoma therapy and majority of antiglaucoma drugs present on the market are based on this approach. There are evidences that even in patients with well-regulated IOP, glaucomatous changes still progress, indicating that other factors, such as oxidation and inflammation, contribute to neurodegenerative changes in optic nerve. With high plasticity for chemical modifications, impressive antioxidative, antiinflamatory and neuroprotective effects and therapeutic potential for multiple diseases, chalcones are considered privileged scaffolds and potential CA inhibitors. The in silico physico-chemical, toxicological and pharmacokinetic properties evaluation for chalcone derivatives previously assayed on hCA inhibitory activity indicated 9, 11, 15, 25, 58, 93, 109, 120, 132, 136, 139, 141, 146, and 175 as the most promising CA inhibitors. Having better properties than those envisioned for AZA, we hypothesize that these few compounds may be of particular interest for the future development as drug leads. Having insights into Ki and by analyzing SAR, we pointed out structural elements important for the hCA II isoform activity/selectivity. For the inhibition of this isoform mainly implicated in glaucoma, several structural elements on chalcone template are of particular interest: B ring methoxy (-OCH3) substituents, amino groups (-NH2, -NHR, -NR2) and halogens. Data collection related to previous experimental results: (Ki), the SAR and in silico studies, clearly distinguish one candidate from the others. Selected as a compound with an excellent hCA II selectivity and strong inhibition effect, predicted to have favorable physico-chemical, pharmacokinetic and toxicological profiles, 139 may be taken for further evaluation in in vivo studies. Suitable for predicting the active potential of the molecules, for the purpose of drug development, the combined application of in vitro hCA inhibitory potential/in silico tools could be helpful strategy in designing novel and selective CAIs.
References
Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology. 2014;121:2081–90. https://doi.org/10.1016/j.ophtha.2014.05.013.
Boland MV, Quigley HA. Risk factors and open-angle glaucoma: classification and application. J Glaucoma. 2007;16:406–18. https://doi.org/10.1097/ijg.0b013e31806540a1.
Marcus MW, de Vries MM, Junoy Montolio FG, Jansonius NM. Myopia as a risk factor for open-angle glaucoma: a systematic review and meta-analysis. Ophthalmology. 2011;118:1989–94.e2. https://doi.org/10.1016/j.ophtha.2011.03.012.
Ko F, Boland MV, Gupta P, Gadkaree SK, Vitale S, Guallar E, et al. Diabetes, triglyceride levels, and other risk factors for glaucoma in the national health and nutrition examination survey 2005−2008. Invest Ophthalmol Vis Sci. 2016;57:2152–7. https://doi.org/10.1167/iovs.15-18373.
Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA. 2014;311:1901–11. https://doi.org/10.1001/jama.2014.3192.
Carta F, Supuran CT, Scozzafava A. Novel therapies for glaucoma: a patent review 2007–2011. Expert Opin Ther Pat. 2012;22:79–88. https://doi.org/10.1517/13543776.2012.649006.
Scozzafava A, Supuran CT. Glaucoma and the applications of carbonic anhydrase inhibitors. In: Frost S, McKenna R, editors. Carbonic anhydrase: mechanism, regulation, links to disease, and industrial applications. Dordrecht: Springer; 2014. p. 349–59. https://doi.org/10.1007/978-94-007-7359-2_17.
Alterio V, Di Fiore A, D’Ambrosio K, Supuran CT, De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms. Chem Rev. 2012;112:4421–68. https://doi.org/10.1021/cr200176r.
Lindskog S. Structure and mechanism of carbonic anhydrase. Pharmcol Ther. 1997;74:1–20. https://doi.org/10.1016/s0163-7258(96)00198-2.
Supuran CT, Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem. 2007;15:4336–50. https://doi.org/10.1016/j.bmc.2007.04.020.
Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett. 2010;20:3467–74. https://doi.org/10.1016/j.bmcl.2010.05.009.
Friedenwald JS. The formation of the intraocular fluid. Am J Ophthalmol. 1949;32:9–27. https://doi.org/10.1016/s0002-9394(14)78354-0.
Kinsey VE. Comparative chemistry of aqueous humor in posterior and anterior chambers of rabbit eye, its physiologic significance. AMA Arch Ophthalmol. 1953;50:401–17. https://doi.org/10.1001/archopht.1953.00920030409001.
Kinsey VE, Barany E. The rate flow of aqueous humor. II. Derivation of rate of flow and its physiologic significance. Am J Ophthalmol. 1949;32:189–202. https://doi.org/10.1016/S0002-9394(14)78372-2.
Wistrand PJ. Carbonic anhydrase in the anterior uvea of the rabbit. Acta Physiol Scand. 1951;24:145–8. https://doi.org/10.1111/j.1748-1716.1951.tb00833.x.
Becker B. The mechanism of the fall in intraocular pressure induced by the carbonic anhydrase inhibitor, diamox. Am J Ophthalmol. 1955;39:177–84. https://doi.org/10.1016/0002-9394(55)90022-2.
Kinsey VE, Reddy DV. Turnover of total carbon dioxide in the aqueous humors and the effect thereon of acetazolamide. AMA Arch Ophthalmol. 1959;62:78–83. https://doi.org/10.1001/archopht.1959.04220010082009.
Supuran CT. How many carbonic anhydrase inhibition mechanisms exist. J Enzyme Inhib Med Chem. 2016;31:345–60. https://doi.org/10.3109/14756366.2015.1122001.
Kumar S, Rulhania S, Jaswal S, Monga V. Recent advances in the medicinal chemistry of carbonic anhydrase inhibitors. Eur J Med Chem. 2021;209:112923 https://doi.org/10.1016/j.ejmech.2020.112923
Carta F, Supuran CT, Scozzafava A. Sulfonamides and their isosters as carbonic anhydrase inhibitors. Future Med Chem. 2014;6:1149–65. https://doi.org/10.4155/fmc.14.68.
Ghorai S, Pulya S, Ghosh K, Panda P, Ghosh B, Gayen S. Structure-activity relationship of human carbonic anhydrase-II inhibitors: Detailed insight for future development as anti-glaucoma agents. Bioorg Chem. 2020;95:103557 https://doi.org/10.1016/j.bioorg.2019.103557.
Karioti A, Carta F, Supuran CT. Phenols and polyphenols as carbonic anhydrase inhibitors. Molecules. 2016;21:1649 https://doi.org/10.3390/molecules21121649.
Tsai JC. Innovative IOP-independent neuroprotection and neuroregeneration strategies in the pipeline for glaucoma. J Ophthalmol. 2020;2020:9329310 https://doi.org/10.1155/2020/9329310.
Boia R, Ruzafa N, Aires ID, Pereiro X, Ambrósio AF, Vecino E, et al. Neuroprotective strategies for retinal ganglion cell degeneration: current status and challenges ahead. Int J Mol Sci. 2020;21:2262 https://doi.org/10.3390/ijms21072262.
Nowakowska Z. A review of anti-infective and anti-inflammatory chalcones. Eur J Med Chem. 2007;42:125–37. https://doi.org/10.1016/j.ejmech.2006.09.019.
Chu HW, Sethy B, Hsieh PW, Horng JT. Identification of potential drug targets of broad-spectrum inhibitors with a Michael acceptor moiety using shotgun proteomics. Viruses. 2021;13:1756 https://doi.org/10.3390/v13091756.
Constantinescu T, Lungu CN. Anticancer activity of natural and synthetic chalcones. Int J Mol Sci. 2021;22:11306 https://doi.org/10.3390/ijms222111306.
Salehi B, Quispe C, Chamkhi I, El Omari N, Balahbib A, Sharifi-Rad J, et al. Pharmacological properties of chalcones: A review of preclinical including molecular mechanisms and clinical evidence. Front Pharmcol. 2021;11:592654 https://doi.org/10.3389/fphar.2020.592654.
Zhuang C, Zhang W, Sheng C, Zhang W, Xing C, Miao Z. Chalcone: a privileged structure in medicinal chemistry. Chem Rev. 2017;117:7762–810. https://doi.org/10.1021/acs.chemrev.7b00020.
de Freitas Silva M, Pruccoli L, Morroni F, Sita G, Seghetti F, Viegas C, et al. The Keap1/Nrf2-ARE pathway as a pharmacological target for chalcones. Molecules. 2018;23:1803 https://doi.org/10.3390/molecules23071803.
Ur Rashid H, Xu Y, Ahmad N, Muhammad Y, Wang L. Promising anti-inflammatory effects of chalcones via inhibition of cyclooxygenase, prostaglandin E2, inducible NO synthase and nuclear factor κb activities. Bioorg Chem. 2019;87:335–65. https://doi.org/10.1016/j.bioorg.2019.03.033.
Adelusi TI, Akinbolaji GR, Yin X, Ayinde KS, Olaoba OT. Neurotrophic, anti-neuroinflammatory, and redox balance mechanisms of chalcones. Eur J Pharmcol. 2021;891:173695 https://doi.org/10.1016/j.ejphar.2020.173695.
Kontogiorgis C, Mantzanidou M, Hadjipavlou-Litina D. Chalcones and their potential role in inflammation. Mini Rev Med Chem. 2008;8:1224–42. https://doi.org/10.2174/138955708786141034.
Katsori AM, Hadjipavlou-Litina D. Recent progress in therapeutic applications of chalcones. Expert Opin Ther Pat. 2011;21:1575–96. https://doi.org/10.1517/13543776.2011.596529.
Zhou B, Xing C. Diverse molecular targets for chalcones with varied bioactivities. Med Chem. 2015;5:388–404. https://doi.org/10.4172/2161-0444.1000291.
Jasim HA, Nahar L, Jasim MA, Moore SA, Ritchie KJ, Sarker SD. Chalcones: synthetic chemistry follows where nature leads. Biomolecules. 2021;11:1203 https://doi.org/10.3390/biom11081203.
Dizdaroglu Y, Albay C, Arslan T, Ece A, Turkoglu EA, Efe A, et al. Design, synthesis and molecular modelling studies of some pyrazole derivatives as carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2020;35:289–97. https://doi.org/10.1080/14756366.2019.1695791.
Tuğrak M, Yamalı C, Gül HI, Demir Y. Inhibitory effects of the chalcones towards carbonic anhydrase I, II and acetylcholinesterase enzymes. Erzincan Univ J Sci Technol. 2020;13:1138–46. https://doi.org/10.18185/erzifbed.748798.
Burmaoglu S, Kazancioglu EA, Kaya R, Kazancıoglu M, Karaman M, Algul O, et al. Synthesis of novel organohalogen chalcone derivatives and screening of their molecular docking study and some enzymes inhibition effects. J Mol Struct. 2020;1208:127868 https://doi.org/10.1016/j.molstruc.2020.127868.
Gürdere MB, Budak Y, Kocyigit UM, Taslimi P, Tüzün B, Ceylan M. ADME properties, bioactivity and molecular docking studies of 4-amino-chalcone derivatives: new analogues for the treatment of Alzheimer, glaucoma and epileptic diseases. Silico Pharmcol. 2021;9:34 https://doi.org/10.1007/s40203-021-00094-x.
Burmaoglu S, Kazancioglu EA, Kazancioglu MZ, Sağlamtaş R, Yalcin G, Gulcin I, et al. Synthesis, molecular docking and some metabolic enzyme inhibition properties of biphenyl-substituted chalcone derivatives. J Mol Struct. 2022;1254:132358 https://doi.org/10.1016/j.molstruc.2022.132358.
Yamali C, Gul H, Çakır T, Demir Y, Gülçin I. Aminoalkylated phenolic chalcones: Investigation of biological effects on acetylcholinesterase and carbonic anhydrase I and II as potential lead enzyme inhibitors. Lett Drug Des Discov. 2020;17:1283–92. https://doi.org/10.2174/1570180817999200520123510.
Arslan T, Türkoğlu EA, Şentürk M, Supuran CT. Synthesis and carbonic anhydrase inhibitory properties of novel chalcone substituted benzenesulfonamides. Bioorg Med Chem Lett. 2016;26:5867–70. https://doi.org/10.1016/j.bmcl.2016.11.017.
Bilginer S, Anil B, Koca M, Demir Y, Gülçin İ. Novel Mannich bases with strong carbonic anhydrases and acetylcholinesterase inhibition effects: 3-(aminomethyl)-6-{3-[4-(trifluoromethyl)phenyl]acryloyl}-2(3H)-benzoxazolones. Turk J Chem. 2021;45:805–18. https://doi.org/10.3906/kim-2101-25.
Kocyigit UM, Budak Y, Gürdere MB, Tekin Ş, Köprülü TK, Ertürk F, et al. Synthesis, characterization, anticancer, antimicrobial and carbonic anhydrase inhibition profiles of novel (3aR,4S,7R,7aS)-2-(4-((E)-3-(3-aryl)acryloyl) phenyl)-3a,4,7,7a-tetrahydro-1H-4,7-methanoisoindole-1,3(2H)-dione derivatives. Bioorg Chem. 2017;70:118–25. https://doi.org/10.1016/j.bioorg.2016.12.001.
Kocyigit UM, Budak Y, Eligüzel F, Taslimi P, Kılıç D, Gulçin İ, et al. Synthesis and carbonic anhydrase inhibition of tetrabromo chalcone derivatives. Arch Pharmcol. 2017;350:e1700198 https://doi.org/10.1002/ardp.201700198.
Singh P, Swain B, Thacker PS, Sigalapalli DK, Purnachander Yadav P, Angeli A, et al. Synthesis and carbonic anhydrase inhibition studies of sulfonamide based indole-1,2,3-triazole chalcone hybrids. Bioorg Chem. 2020;99:103839 https://doi.org/10.1016/j.bioorg.2020.103839.
Singh P, Purnachander Yadav P, Swain B, Thacker PS, Angeli A, Supuran CT, et al. Discovery of a novel series of indolylchalcone-benzenesulfonamide hybrids acting as selective carbonic anhydrase II inhibitors. Bioorg Chem. 2021;108:104647 https://doi.org/10.1016/j.bioorg.2021.104647.
Burmaoglu S, Yilmaz AO, Polat MF, Kaya R, Gulcin İ, Algul O. Synthesis and biological evaluation of novel tris-chalcones as potent carbonic anhydrase, acetylcholinesterase, butyrylcholinesterase and α-glycosidase inhibitors. Bioorg Chem. 2019;85:191–7. https://doi.org/10.1016/j.bioorg.2018.12.035.
Burmaoglu S, Yilmaz AO, Polat MF, Kaya R, Gulcin İ, Algul O. Synthesis of novel tris-chalcones and determination of their inhibition profiles against some metabolic enzymes. Arch Physiol Biochem. 2021;127:153–61. https://doi.org/10.1080/13813455.2019.1623265.
SwissADME, http://www.swissadme.ch/. Accessed Jun 2022.
OSIRIS Property Explorer, http://www.organic-chemistry.org/prog/peo/. Accessed June 2022.
Stellenboom N. Comparison of the inhibitory potential towards carbonic anhydrase, acetylcholinesterase and butyrylcholinesterase of chalcone and chalcone epoxide. J Biochem Mol Toxicol. 2019;33:e22240 https://doi.org/10.1002/jbt.22240.
Aslan HE, Demir Y, Özaslan MS, Türkan F, Beydemir Ş, Küfrevioğlu ÖI. The behavior of some chalcones on acetylcholinesterase and carbonic anhydrase activity. Drug Chem Toxicol. 2019;42:634–40. https://doi.org/10.1080/01480545.2018.1463242.
Bayrak Ç, Taslimi P, Gülçin İ, Menzek A. The first synthesis of 4-phenylbutenone derivative bromophenols including natural products and their inhibition profiles for carbonic anhydrase, acetylcholinesterase and butyrylcholinesterase enzymes. Bioorg Chem. 2017;72:359–66. https://doi.org/10.1016/j.bioorg.2017.03.001.
Mahar J, Saeed A, Belfield KD, Ali Larik F, Ali Channar P, Ali Kazi M, et al. 1-(2-Hydroxy-5-((trimethylsilyl)ethynyl)phenyl)ethanone based α,β-unsaturated derivatives an alternate to non-sulfonamide carbonic anhydrase II inhibitors, synthesis via Sonogashira coupling, binding analysis, Lipinsk’s rule validation. Bioorg Chem. 2019;84:170–6. https://doi.org/10.1016/j.bioorg.2018.11.031.
Koçyiğit ÜM, Gezegen H, Taslimi P. Synthesis, characterization, and biological studies of chalcone derivatives containing Schiff bases: Synthetic derivatives for the treatment of epilepsy and Alzheimer’s disease. Arch Pharmcol. 2020;353:e2000202 https://doi.org/10.1002/ardp.202000202.
Kocyigit UM, Budak Y, Gürdere MB, Ertürk F, Yencilek B, Taslimi P, et al. Synthesis of chalcone-imide derivatives and investigation of their anticancer and antimicrobial activities, carbonic anhydrase and acetylcholinesterase enzymes inhibition profiles. Arch Physiol Biochem. 2018;124:61–8. https://doi.org/10.1080/13813455.2017.1360914.
Gençer N, Bilen Ç, Demir D, Atahan A, Ceylan M, Küçükislamoğlu M. In vitro inhibition effect of some chalcones on erythrocyte carbonic anhydrase I and II. Artif Cells Nanomed Biotechnol. 2013;41:384–8. https://doi.org/10.3109/21691401.2012.761226.
Bilginer S, Gul HI, Erdal FS, Sakagami H, Levent S, Gulcin I, et al. Synthesis, cytotoxicities, and carbonic anhydrase inhibition potential of 6-(3-aryl-2-propenoyl)-2(3H)-benzoxazolones. J Enzyme Inhib Med Chem. 2019;34:1722–9. https://doi.org/10.1080/14756366.2019.1670657.
Bilginer S, Gul HI, Erdal FS, Sakagami H, Gulcin I. New halogenated chalcones with cytotoxic and carbonic anhydrase inhibitory properties: 6-(3-Halogenated phenyl-2-propen-1-oyl)-2(3H)-benzoxazolones. Arch Pharmcol. 2020;353:e1900384 https://doi.org/10.1002/ardp.201900384.
Kuday H, Sonmez F, Bilen C, Yavuz E, Gençer N, Kucukislamoglu M. Synthesis and in vitro inhibition effect of new pyrido[2,3-d]pyrimidine derivatives on erythrocyte carbonic anhydrase I and II. Biomed Res Int. 2014;2014:594879 https://doi.org/10.1155/2014/594879.
Peerzada MN, Khan P, Ahmad K, Hassan MI, Azam A. Synthesis, characterization and biological evaluation of tertiary sulfonamide derivatives of pyridyl-indole based heteroaryl chalcone as potential carbonic anhydrase IX inhibitors and anticancer agents. Eur J Med Chem. 2018;155:13–23. https://doi.org/10.1016/j.ejmech.2018.05.034.
Arslan T, Çelik G, Çelik H, Şentürk M, Yaylı N, Ekinci D. Synthesis and biological evaluation of novel bischalcone derivatives as carbonic anhydrase inhibitors. Arch Pharmcol. 2016;349:741–8. https://doi.org/10.1002/ardp.201600122.
Tutar U, Koçyiğit ÜM, Gezegen H. Evaluation of antimicrobial, antibiofilm and carbonic anhydrase inhibition profiles of 1,3-bis-chalcone derivatives. J Biochem Mol Toxicol. 2019;33:e22281 https://doi.org/10.1002/jbt.22281.
Özen F, Günel A, Baran A. DNA-binding, enzyme inhibition, and photochemical properties of chalcone-containing metallophthalocyanine compounds. Bioorg Chem. 2018;81:71–8. https://doi.org/10.1016/j.bioorg.2018.08.002.
Arslan T. Design, synthesis of novel peripherally tetra-chalcone substituted phthalocyanines and their inhibitory effects on acetylcholinesterase andcarbonic anhydrases (hCA I and II). J Organomet Chem. 2021;951:122021. https://doi.org/10.1016/j.jorganchem.2021.122021.
Supuran CT. Carbon- versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2018;33:485–95. https://doi.org/10.1080/14756366.2018.1428572.
Bonardi A, Nocentini A, Bua S, Combs J, Lomelino C, Andring J, et al. Sulfonamide inhibitors of human carbonic anhydrases designed through a three-tails approach: improving ligand/isoform matching and selectivity of action. J Med Chem. 2020;63:7422–44. https://doi.org/10.1021/acs.jmedchem.0c00733.
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. https://doi.org/10.1016/s0169-409x(00)00129-0.
Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–23. https://doi.org/10.1021/jm020017n.
https://www.glaucomaassociates.com/glaucoma/types-of-glaucoma/ Accessed 26 Jul 2022.
Mincione F, Scozzafava A, Supuran CT. The development of topically acting carbonic anhydrase inhibitors as antiglaucoma agents. Curr Pharmcol Des. 2008;14:649–54. https://doi.org/10.2174/138161208783877866.
Funding
The work was funded by the Ministry of Science and Technological Development of Serbia (Project 451-03-68/2022-14/200113) and Faculty of Medicine, University of Niš Internal project No. 40.
Author information
Authors and Affiliations
Contributions
All authors contributed to each stage of the manuscript preparation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
Authors know of no conflict of interest associated with this publication and there has been no financial support of this work that could have influenced its outcome.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gocić, V., Marković, A. & Lazarević, J. The potential of chalcone derivatives as human carbonic anhydrase inhibitors in the therapy of glaucoma. Med Chem Res 31, 2103–2118 (2022). https://doi.org/10.1007/s00044-022-02978-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-022-02978-9