
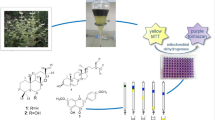
Abstract
A new glyceride, 2,3-dihydroxypropyl-31-hydroxyhentriacontanoate (1), a new glucoside stigmasterol derivative, stigmasterol 3-O-β-D-glucopyranoside 6’-hexadecanoate (2) along with eleven known compounds (3-13) were isolated from the fruits and the stem bark of Tetrapleura tetraptera (Schumach. & Thonn.) Taub. using silica gel vacuum liquid and column chromatography. The structures of the isolated compounds were elucidated based on spectroscopic and spectrometry methods including NMR (1D and 2D), high-resolution mass spectrometry (HRESIMS) and comparison with data reported in the literature. The crude extract and the isolated compounds were evaluated for their antibacterial activity against a panel of bacterial strains using the microdilution technique and for their cytotoxic potential on breast cancer cell lines (MCF-7, MDA-MB-231) using doxorubicin as reference medicine. All the tested compounds exhibited significant antibacterial activity against Klebsiella aerogenes and Bacillus subtilis with an MIC value of 18.5 µg/mL. Furthermore, compounds (3) and (9) were active against all bacterial strains with MICs values ranging from 18.5 to 74 µg/mL. Compounds (12-13) and the crude extract were cytotoxic against MDA-MB-231 cells with their CC50 values ranging between 14.5 and 20.0 µg/mL. These results confirm that T. tetraptera is a potential source of antibacterial agents and exhibits selective toxicity against breast cancer cell lines. Apart from compound (3) and compounds (10-12), all the other compounds were isolated from Tetrapleura genus for the first time. Compounds (1), (2), (6), (7) and (8) were also isolated from the Fabaceae family for the first time.

Graphical abstract
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bacteria are ubiquitous, mostly free-living organisms often consisting of one biological cell. They constitute a large domain of prokaryotic microorganisms and live in symbiotic and parasitic relationships with humans, animals, and plants. All human organs are susceptible to bacterial infections and each bacterial species preferentially infects specific organs. The emergence of multidrug-resistant bacteria has further compromised the accessibility and affordability of many currently prescribed antibiotics worldwide [1, 2]. According to the Centers for Disease Control and Prevention (CDC) report, each year in the United States, at least 2 million people are infected with bacteria that are resistant to one or more of the antibiotics used in the treatment of infections [3,4,5]. This adverse situation is more intricate in developing countries due to lack of finances, laboratory diagnosis and an effective disease surveillance system.
Appropriate actions are thus needed to overcome these drawbacks. These actions include the control of antibiotics use, evaluation of bacterial and antimicrobial resistance mechanisms and more importantly, the development of new drugs either synthetically or naturally. One of the ways new drugs can be developed is through the exploration and investigation of medicinal plants for antibacterial and antimicrobial agents. In the last few years, several studies have been conducted in different countries to prove the efficacy of plants in treating several diseases and their bioactivity has been attributed to the presence of secondary metabolites [6,7,8,9].
Tetrapleura tetraptera (Schumach. & Thonn.) Taub., commonly known as Aridan, is a hardy, single-stemmed perennial tree with dark green leaves, a thick woody base and spreading branches. The plant grows up to 20–25 meters in height and is native to most of tropical Africa, particularly the rainforest belts of West, Central and East Africa (Uganda, Burkina Faso, Mali, Gambia, Nigeria, Cameroon, and Gabon). In Central and West Africa, T. tetrapleura is used as a therapeutic agent for the treatment and management of different ailments; fresh fruits are used as ingredient for the preparation of medicine against stomach gripes, inflammation, febrile convulsion, and rheumatic pains while an infusion of fruits, leaves and stem bark is taken to control diabetes mellitus, jaundice, malaria, asthma, hypertension, and inflammatory conditions [10,11,12,13].
Previous studies done with the extract demonstrated the cytotoxic potential of the dichloromethane-methanol extract from the fruits of T. tetraptera against nine cancer cell lines with IC50 values ranging from 10.27 μg/mL (in CCRF-CEM leukemia cells) to 23.61 μg/mL (against HCT116 p53 − /− colon adenocarcinoma cells) [14]. In another study, streptozotocin (STZ)-induced diabetic rats were treated with aqueous extract (50–800 mg/kg p.o.) of the fruit of T. tetraptera which exhibited hypoglycaemic effects [15].
Phytochemical investigation of the stem bark and fruits of the plant led to the isolation and identification of 3-O-β-D-glucopyranosyl-(1 → 3)-β-D-glucopyranosylolean-12-en-28-oic acid, olean-12-en-3-β-O-D-glucopyranoside, (3 R,4 S)-3,4-dimethyloxetan-2-one, naringenin, luteolin, 3-O-β-D-glucopyranosyl-(1 → 6)-β-D glucopyranosylurs-12-en-28-oic acid [14], aridanin [10] and Tetrapterosides A and B [16]. In addition, gas chromatography-mass spectroscopy of oil from the leaves of T. tetraptera led to the identification of thirty-one volatile compounds mainly unsaturated fatty acids and alkenes [17]. Some of these compounds have been shown to exhibit a wide range of biological activities such as cytotoxicity, anti-leishmanial, antiplasmodial and antidiabetic activities [10, 12, 14]. In the present study, a new glyceride, a new stigmasterol glucoside derivative and other constituents were isolated from the fruits and the stem bark of T. tetraptera. The cytotoxicity and antibacterial potential of both the pure isolated compounds and the crude extract were evaluated on two breast cancer cell lines and a panel of bacterial strains respectively.
Results
Isolation and structure elucidation
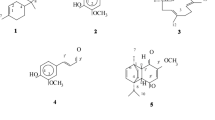
Vacuum liquid chromatography (VLC) and column chromatography (CC) of the dichloromethane-methanol (1:1, v/v) extracts from the fruits and the stem bark of T. tetraptera led to the isolation and characterization of thirteen compounds (1-13). The structures of the isolated compounds are shown in Fig. 1.

Chemical structures of compounds (1-13) isolated from T. tetraptera (Schumach. & Thonn.) Taub
Compound (1) was isolated as a green powder using the mixture of n-hexane/CHCl3 (1/9, v/v) and it is soluble in chloroform. Its HREIMS showed a pseudo molecular ion peak at m/z 592.4722 [M + Cl]− (calcd for C34H68O5Cl, 592.4789) from which the molecular formula was deduced to be C34H68O5, containing one double bond equivalent. The same spectrum also showed a fragment ion peak at m/z 481.3862 attributable to the fragment ion [M-glycerol]− of the molecule. The results of analysis of 1D NMR, DEPT 135 and HSQC together with HREIMS allowed us to deduce that compound (1) was a glyceryl derivative. Its FTIR spectrum showed some important characteristic peaks. A peak at 1732 cm−1 was attributable to a carbonyl group, a broad peak at 3400 cm−1 was attributable to hydroxyl groups. Two peaks at 2916 and 2884 cm−1 were attributable to the aliphatic long chain while a peak at 1257 cm−1 was attributable to the bond C-O. The 1H NMR revealed the presence of three hydrogens geminal to hydroxyl groups at δH 3.85 (1H, q, J = 5.9 Hz, H-2), 3.54 (1H, dd, J = 4.5, 12.0 Hz, H-3a) and 3.56 (1H, dd, J = 4.8, 12.0 Hz, H-3b) and two hydrogens adjacent to a carboxylic acid group at δH 4.14 (1H, dd, J = 6.5, 11.7 Hz, H-1a) and 4.11 (1H, dd, J = 5.8, 11.8 Hz, H-1b). It also revealed the presence of methylene protons at δH 3.55 (2H, m, H-31’), 2.31 (2H, t, J = 7.6 Hz, H-2’), 1.61 (2H, m, H-3’), 1.54 (2H, m, H-30’) and 1.26 (2H, m, H-4’). We observed a broad peak at δ 1.11-1.34 (50H, br s) attributable to an aliphatic chain. The 13C NMR, HSQC and DEPT 135 spectra confirmed the attribution made from 1H NMR by showing a signal at δC 174.0 (C-1’) corresponding to an ester carbonyl, four carbons attached to oxygen at δC 69.4 (C-2), 64.4 (C-1) and 62.5 (C-3) corresponding to glycerol moiety and at δC 61.6 (C-31’) attributable to the terminal methylene attached to the hydroxyl group. It also showed signals of aliphatic methylene carbons at δC 33.5 (C-2’), 25.3 (C-3’), 24.6 (C-4’) and 31.9 (C-30’), and a broad signal was observed at δC 28.5-29.1 (C4’-C28’) integrating for (CH2)25 attributable to the aliphatic long chain. All these data combined are close to those reported in literature and confirmed that the compound is a monoglyceride hydroxyl derivative [18,19,20]. From its HMBC spectrum, some important correlations were observed such as correlations between the ester carbonyl at δC 174.0 (C-1’) and the protons H-1a, H-1b, H-2’ and H-3’ at δH 4.14 (1H, dd, J = 6.5, 11.7 Hz, H-1a), 4.11 (1H, dd, J = 5.8, 11.8 Hz, H-1b), 2.31 (2H, t, J = 7.6 Hz, H-2’) and 1.61 (2H, m, H-3’). There were correlations between C-2 and H-1a, H-1b, between C-1 and H-2, H-3a, H-3b and between C-3 and H-1a, H-1b, H-2. Furthermore, another correlation was observed between the terminal carbon attached to the hydroxyl group at δC 61.9 (C-31’) and the methylene protons at δH 1.54 (m, 2H). Based on these data, compound (1) was assigned the structure of 2,3-dihydroxypropyl-31- hydroxyhentriacontanoate a new derivative to which a trivial name tetrapleurol A was given.
Compound (2) was isolated as a colorless powder using n-hexane/EtOAc (50:50, v/v) and it was soluble in chloroform. Its molecular formula was found on the basis of HR-ESI-MS (m/z 835.6411 [M + Na]+ calcd for 835.6428) to be C51H88O7 indicating 8 double bond equivalents. Its 1H NMR spectrum showed signals at δH 5.17 (dd, J = 15.1, 8.7 Hz, 1H), δH 5.05 (dt, J = 15.0, 6.5 Hz, 1H) and δH 5.37 (dd, J = 7.11, 6.73, 1H) corresponding to H-23, H-22 and H-6 respectively which also corresponded to the four olefinic carbon resonances observed on the 13C NMR spectrum. The two signals at δC 129.4 and 138.2 were typical of C-23 and C-22 of a Δ22 sterol skeleton [21]. The two other signals that appeared at δC 122.1 and 140.4 were characteristic of C-5 and C-6 of a Δ5 sterol frame work [22, 23]. Also, a signal at δC 79.7 corresponding to multiplet at δH 3.57 on the proton spectrum was attributed to an oxymethine carbon which was assigned to C-3 of the steroid moiety. Furthermore, a multiplet was observed at δH 4.36 belonging to the H-6’ of glucose with its anomeric proton at δH 4.39 (H-1’). This suggested that compound (2) was a steroid glycoside derivative. A broad singlet at δH 1.28 together with an ester carbonyl at δC 174.3 indicated the presence of a long ester hydrocarbon chain in the molecule. The COSY and HMBC correlation between the anomeric proton and H-3 confirms the position of the sugar moiety. The HMBC correlation between the ester carbonyl (C-1”) and proton 6’ of the sugar moiety together with correlations between H-1”, H-2” and H-3” confirmed the position of the long hydrocarbon chain. The structure of compound (2) was then found to be stigmasterol 3-O-β-D-glucopyranoside 6’-hexadecanoate Fig. 2.

HMBC correlations in compounds (1) and (2)
The eleven known compounds (3-13) were identified as 3-[(2-acetamido-2-deoxy-β-D-glucopyranosyl) oxy]olean-12-ene-28-oic acid (3) [24], sucrose (4) [25], pinitol (5) [26], 4-O-α-D-Glucopyranosyl-D-glucopyranose (6) [25], hexacosanoic acid (7) [27], tetracosanoic acid (8) [27], oleanolic acid (9) [28], betulenic acid (10) [29], stigmasterol (11) [9], stigmasterol 3-O-β-D-glucoside (12) [30] and daucosterol (13) [31]. Compounds (1), (2), (4-9) and (13) are reported for the first time from this plant.
Spectroscopic and spectrometry data of compounds (1) and (2)
Compound (1) 2,3-dihydroxypropyl-31-hydroxyhentriacontanoate. Green powder (11.5 mg); HREIMS, m/z 592.4722 [M + Cl]− (calcd for C34H68O5Cl, 592.4789); IR ʋmax: at 3400, 2916, 2884, 1732, 1257, 1095, 1020 cm−1; 1H-NMR (500 MHz, Chloroform-d) δH 4.14 (1H, dd, J = 6.5, 11.7, H-1a), 4.11 (1H, dd, J = 5.8, 11.8, H-1b), 3.85 (1H, q, J = 5.9, H-2), 3.54 (1H, dd, J = 4.5, 12.0, H-3a), 3.56 (1H, dd, J = 4.8, 12.0, H-3b), 3.55 (2H, m, H-31’), 2.31 (2H, t, J = 7.6, H-2’), 1.61 (2H, m, H-3’), 1.54 (2H, m, H-30’) and 1.26 (2H, m, H-4’); 13C-NMR (126 MHz, Chloroform-d) δC 174.0 (C-1’), 69.4 (C-2), 64.4 (C-1), 62.5 (C-3), 61.6 (C-31’), 33.5 (C-2’), 25.3 (C-3’), 24.6 (C-29’), 31.9 (C-30’) and δC 28.5-29.1 (C4’-C28’) see Table 1.
Compound (2) stigmasterol 3-O-β-D-glucopyranoside 6’-hexadecanoate. Colorless powder (11.3 mg); HREIMS, m/z 835.6411 [M + Na]+ (calcd for C51H88O7Na, 835.6428); 1H-NMR (500 MHz, Chloroform-d) δH 5.17 (1H, dd, J = 15.1, 8.7 Hz, H-23), 5.05 (1H, dt, J = 15.0, 6.5 Hz, H-22), 5.37 (1H, dd, J = 7.11, 6.73, H-6); 13C-NMR (126 MHz, Chloroform-d) δC 174.3 (C-1”), 129.4 (C-22), 138.2 (C-23), 122.1 (C-6), 140.4 (C-5), 101.3 (C-1’), 34.3 (C-2”), 79.7 (C-3), 14.1 (C-16”) see Table 2.
Effects of compounds on bacterial strains
Table 3 shows the MIC values of the extract and the compounds after 24 h of incubation. The crude extract and all the compounds exhibited better bacteriostatic effects with an MIC value of 18.5 µg/mL on Bacillus subtilis (BS) and Klebsiella aerogenes (KA) compared to ampicillin with an MIC of 26 µg/mL on these strains and Streptomycin with MIC of 16 and 512 µg/mL respectively on Bacillus subtilis (BS) and Klebsiella aerogenes (KA). Compound (3) exhibited the highest antibacterial activity amongst all the tested compounds with an MIC value of 18.5 µg/mL on all the bacterial strains and compound (9) showed MICs values ranging from 18.5 to 74 µg/mL on all the bacterial strains.
Cytotoxicity potential of compounds
Table 4 represents the CC50 values of the tested compounds on Vero cells, MCF-7 and MDA-MB-231 cancer cell lines. Compounds (12-13) and the crude extract were cytotoxic against the tested cell lines with CC50 values of 21.2, 18.5 and 17 µg/mL against MCF-7 cell respectively and 20.0, 16.4 and 14.5 µg/mL against MDA-MB-231 cells respectively. However, these compounds were also toxic to normal cells, demonstrating low selectivity index. In general, the other tested compounds inhibited the growth of cancer cells as compared to normal Vero cells. Additionally, the positive control (Doxorubicin) known for its toxic nature on cells, significantly reduced cell viability of all cells showing CC50 values of 2.12, 1.20 and 3.03 µg/mL against Vero, MCF-7 and MDA-MB-231 respectively. From Table 5, compounds (8) and (9) exhibited greater selectivity for normal cells.
Discussion
Bacterial infections have a large impact on public health. Infections can occur in any part of the body either caused by the pathogen itself or by the body’s response to its presence. On the other hand, the burden of bacterial resistance is substantial and is likely to increase in the coming years due to drug resistance among Gram-negative bacteria. In addition, there are very few antibacterial agents with new mechanisms of action under development to meet the challenge of multidrug resistance [32,33,34]. Hence the need to foster research on medicinal plants and their secondary metabolites that could be used as potential pharmaceutical candidates in fighting these infections.
In this study, the antimicrobial activity of the crude extract and isolated compounds from T. tetraptera were evaluated on a panel of bacterial strains involved in different infections and their cytotoxicity against two cancer cell lines were also investigated. The crude extract and some of the compounds exhibited good inhibitory potential with an MIC value of 18.5 µg/mL against B. subtilis (BS) (Gram-positive) and K. aerogenes (KA) (Gram-negative) bacteria, suggesting that they possess a broad spectrum of activity and can pass through the outer membrane and exert their action. Compound (3) had an MIC value of 18.5 µg/mL against all bacterial strains tested. Compound (9) on the other hand had an MIC values ranging from 18.5 to 74 µg/mL against the tested bacterial strains. This corroborates the study of several authors who demonstrated that oleanolic acid and its analogs are very active against many bacterial strains. They can influence bacterial gene expression, the formation and maintenance of biofilms, cell autolysis and peptidoglycan turnover [35].
Compounds (3) and (9) have the same chemical structure with the only difference of a hydroxyl group attached to C-3 of compound (9) and 2-acetamido-2-deoxy-β-D-glucopyranosyl group attached to C-3 of compound (3). Apart from E. faecalis (EF), B. subtilis (BS) and K. aerogenes (KA) against which compounds (3) and (9) exhibited similar activity, compound (3) had better antibacterial activity than compound (9) against all the other bacterial strains tested. The structure-activity relationship (SAR) of compound (9) and its analogue (3) showed that the introduction of the 2-acetamido-2-deoxy-β-D-glucopyranosyl group at C-3 in (3) considerably increases its antibacterial activity. Likewise, the activities of compounds (3) and (9) were better than the reference antibiotics on a considerable number of the tested microorganisms. The activity of compound (3) against 6 bacterial strains was 6 times better than that of streptomycin. It was also observed that compound (12) differed from compound (13) by the presence of an extra double bond in its structure. However, the activity exhibited by compound (12) against M. smegmatis (MS), K. oxytoca (KO), K. pneumonia (KP) and P. mirabilis (PM) was better than that of compound (13), implying that the presence of a double bond on the aliphatic chain of compound (12) increases its activity. On the other hand, compound (2) differed from (12) by the presence of an hexadecanoate group attached to C-6 of a sugar moiety but its activity against Proteus vulgaris (PV) and Proteus mirabilis (PM) (MIC 4.6 µg/mL) was lower than that of compound (12). Thus, it can be hypothesized that a slight modification of the chemistry of compound (12) could play an important role in the structure-activity relationship (SAR) of this class of compounds. We can also justify the weak activity of compound (2) with the activity of compound (12) which is a fatty acid long chain.
The crude extract of T. tetraptera also exhibited antibacterial activity against E. coli and S. aureus, with an MIC value of 294 µg/mL. This correlates with the study carried out by [36] who demonstrated that T. tetraptera exhibited bacteriostatic activity against E. coli (EC), P. aerogiunosa (PA) and S. aureus (SA) with an MIC value of 250 µg/mL. The mechanism of antibacterial activity of this extract could involve the damage of bacterial cell membranes which resulted in permeability and leakage of macromolecular substances such as DNA, ATP, and proteins [37]. This suggestion is in accordance with [38], who demonstrated that antimicrobial agents could cause irreversible damage that could induce the leakage of proteins and other constituents from bacterial cells. This resulted in the disruption and flux of protons towards the cell exterior, inducing cell death or inhibiting enzymes necessary for amino acid biosynthesis [39, 40].
These results illustrate the potential of T. tetraptera extract and its constituents as an antimicrobial and anticancer lead drug. However, mechanism of cancer cell viability inhibition and structure-activity relationships remain an uncertain gap for prospective investigations. Literature has recorded reports of potential antimicrobial and anticancer properties of triterpenes, phytosterols and fatty acids [41]. Furthermore, [42] postulates that consumption of phytosterol-rich diets may lower the risk of cancer by 20%.
Conclusion
In conclusion, the study of T. tetraptera extract led to the isolation and characterization of thirteen compounds. The crude extract and the isolates showed significant antibacterial activity on the strains tested. Compound (3) showed the highest antibacterial activity against all tested strains. These results suggest the beneficial effect of T. tetraptera and compound (3) in treating bacterial infections. Further studies are needed to determine its mode of action. This work also provides significant chemophenetic contribution on T. tetraptera and to the genus Tetrapleura where very few investigations have been done. Moreover, it has provided further information with regards to possible chemophenetic markers of T. tetraptera and showed the presence of uncommon metabolites in the Fabaceae family.
Experimental
General experimental procedure
Extraction of plant material was done in methanol (MeOH) and dichloromethane (CH2Cl2). Meanwhile, n-hexane, ethyl acetate (EtOAc), chloroform (CHCl3) and methanol were used as pure and binary mixtures at different polarities for fractionation of the extract, isolation, and purification of compounds. Column chromatography (CC) was carried out on silica gel (0.040-0.063 mm). Thin-layer chromatography (TLC) was performed on Merck precoated silica gel 60F254 aluminium. Spots were visualized under UV light (254 and 366 nm) and by spraying the plates with 20% H2SO4 in H2O followed by heating at about 105 °C for 3 min. Using a TOF spectrometer (Bruker, South Africa) equipped with an ESI source, high-resolution mass spectra were obtained. The spectrometer was operated in positive and negative modes (mass range: 50−1500, with a scan rate of 1.00 Hz) with automatic gain control to provide high accuracy mass measurements within 1 ppm deviation using Na formate as calibrant. A spray voltage of 4.5 kV and capillary temperature of 200 °C were used for the experiments with nitrogen as sheath gas (4 L/min). The 1D (1H, 13C, DEPT 135) and 2D (HSQC, HMBC, COSY) NMR spectra were recorded on Bruker Bio Spin GmbH in deuterated solvents (CDCl3, methanol-d4 and DMSO-d6). Chemical shifts were reported in δ (ppm) using tetramethylsilane (TMS) (Sigma Aldrich) as an internal standard, while coupling constants (J) were measured in Hz.
Plant material
The fruits and the stem bark of T. tetraptera were collected from Mount Kala (GPS coordinates: Latitude 3◦51′ 32″ N, Longitude 11◦39′ 53′′ E, altitude 1838 m), a locality around Yaoundé, Cameroon, in June 2019 and identified by Mr. Victor Nana, a botanist of the National Herbarium of Cameroon (NHC), Yaoundé, where a voucher specimen (No. 43237) was deposited.
Extraction and isolation
The air-dried fruits of T. tetraptera were milled into fine powder (480.2 g) which was extracted with 5 L (1:10, v/v) of dichloromethane-methanol (1:1, v/v) at room temperature for 72 hours. The plant extract was filtered with Whatman No. 1 filter paper and the brown filtrate was concentrated to dryness using a rotary evaporator. The process was repeated 3 times yielding 80.1 g of crude extract. 70.2 g of this crude extract was subjected to silica gel vacuum liquid chromatography (VLC) and eluted with n-hexane, a mixture of n-hexane/ethyl acetate with increasing polarity and finally with methanol. A total of 100 fractions of 200 mL were collected and combined in 3 series (F1, F2 and F3) based on TLC analysis. The non-polar series F1 (11.2 g) was subjected to column chromatography (CC) and eluted with a binary system n-hexane/EtOAc (from 10:0 to 7:3, v/v) and finally with EtOAc 100%. Fractions 1−10 were obtained upon elution with 100% n-hexane; fractions 11−21 were obtained upon increasing the polarity to 10% EtOAc in n-hexane; fractions 22−30 were obtained upon elution with 15% EtOAc in n-hexane; fractions 31−41 came out when the polarity was increased to 20% EtOAc in n-hexane. The polarity was then increased to 25% EtOAc in n-hexane, leading to the elution of fractions 42−50. Increasing the polarity to 30% gave fractions 51−60. The last fractions 61 to 70 were eluted upon flushing the column with pure ethyl acetate. A total of 70 fractions (200 mL each) were collected from this column. The white precipitates obtained in fractions 15−17 and 20−23 were combined based on TLC analysis and filtered using Whatman filter paper and acetone to afford to compound (7) (10.6 mg) and compound (8) (8.6 mg) respectively. Fractions 43−47 precipitated as a green powder which was filtered using Whatman filter paper and acetone to afford compound (9) (11.3 mg). Fractions 50−54 also precipitated as a green solid and was washed with acetone and filtered to afford to compound (10) (8.3 mg). All the compounds were isolated without further purification. F2 (15.4 g) was also subjected to silica gel CC, eluted with n-hexane/EtOAc (from 9:1 to 5:5, v/v) and the obtained solids were filtered and isolated without further purification giving compounds (11) (9.3 mg), (2) (11.2 mg), (12) (12.4 mg) and (5) (5.7 mg). The methanolic fraction F3 (13.1 g) was also subjected to CC, eluted with a mixture of CHCl3/MeOH (from 9:1 to 6:4, v/v) and the precipitates were washed several times with hexane above Whatman filter paper to afford compounds (13) (11.6 mg), (3) (51.2 mg), (4) (8.1 mg) and (6) (14.6 mg).
The stem bark of the plant was studied following the process stated above with few modifications. In fact, the collected stem bark was air-dried, grinded and the obtained powder was soaked with a mixture of dichloromethane-methanol (1:1, v/v) for 72 hours at room temperature. The brown solution obtained was then concentrated using a rotary evaporator and the process was repeated 3 times leading to 100.7 g of crude extract. A portion of the extract was subjected to silica gel column chromatography and eluted with a mixture of n-hexane/chloroform with increasing polarity, followed by a mixture of chloroform/methanol and finally methanol. A total of 200 fractions of 200 mL each were collected and combined based on TLC analysis. Amongst the collected fractions, some precipitated and were washed and filtered to afford (1), (3), (11) and (13) after spectroscopy analysis and comparison to those obtained from fruits.
Antibacterial assay
Microbial strains
Microbial strains were purchased from Davies Diagnostic, South Africa, and were maintained in glycerol at –8 °C prior to use. It included Gram-positive strains, namely Bacillus subtilis (BS) (ATCC19659), Enterococcus faecalis (EF) (ATCC13047), Staphylococcus epidermidis (SE) (ATCC14990), Staphylococcus aureus (SA) (ATCC25923) and Mycobacterium smegmatis (MS) (MC2155), and Gram-negative bacteria namely Enterobacter cloacae (ECL) (ATCC13047), Proteus vulgaris (PV) (ATCC6380), Klebsiella oxytoca (KO) (ATCC8724), Klebsiella pneumonia (KP) (ATCC13882), Proteus mirabilis (PM) (ATCC7002), Escherichia coli (EC) (ATCC25922) and Klebsiella aerogenes (KA) (ATCC13048).
The Microdilution method was used to evaluate the minimum inhibitory concentration (MIC) which is defined as the lowest concentration of antimicrobial agent that inhibits microbial growth after 24 h of incubation [43]. In order to achieve this, stock solutions of crude extract and compounds were serially diluted in 100 µL of nutrient broth in a 96 microtiter well plate yielding concentrations of 588, 294, 147, 74, 37, and 18.5 µg/mL. Thereafter, 100 µL of an overnight bacterial culture for each of the bacteria to be tested was brought to 0.5 Mc Farland in nutrient broth, seeded in 96 microtiter plates and incubated at 37 °C for 24 h. Experiments were carried out in duplicate. Streptomycin, ampicillin and nalidixic acid were used as positive control and negative control was prepared to contain 50% nutrient broth in DMSO.
Cell viability assay
Cell lines
Monkey kidney cells (Vero) and breast cancer cell lines MCF-7 (human estrogen receptor (ER)-positive breast adenocarcinoma cells), MDA-MB-231 (human ER-negative breast adenocarcinoma cells) were purchased from Cellonex Separation Scientific SA (Pty) Ltd. (Roodepoort), Johannesburg, South Africa.
Cell culture
Vero, MCF-7 and MDA-MB-231 cells were cultured in sterile Dulbecco’s Minimal Essential Medium (DMEM, Gibco) supplemented with 10% Fetal bovine serum (FBS) and 1% penicillin-streptomycin solution. The cell cultures were maintained at 37 °C in a 5% CO2 humidified atmosphere and pH 7.4. Cell counting was done using handheld automated cell counter (Scapter 3.0™, Merck, Burlington, MA, USA) before every experiment.
The 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay was used to determine the cytotoxic potential of compounds (2 –13) against Monkey kidney (Vero) cells and breast cancer cell lines (MCF-7 and MDA-MB-231). Doxorubicin and untreated cells were served as positive and negative controls respectively. After 24 h incubation at 37 °C in 5% CO2 when the cells have reached 80% confluency, they were harvested using 2% Trypsin-EDTA solution, centrifuged for 5 min at 3000 RCM and re-suspended in Dulbecco’s Modified Eagle Medium (DMEM). Cell counting was done using handheld automated cell counter (Scapter 3.0™, Merck, Burlington, MA, USA) and 1 × 104 cells/well were seeded into 96 well plates. After 24 h incubation, cells were treated with different concentration range (12.5−200 µg/mL) of the test compounds and doxorubicin (12.5−200 µg/mL) prepared in DMEM for 24 h. The MTT solution (20 μL) prepared in PBS (5 mg/mL) was added to all the wells and the plates were incubated for 4 h, followed by 1 h incubation with 100 µL of dimethyl sulphoxide (DMSO) to dissolve the formazan crystals. All experiments were performed in triplicate. The plates were read at 570 nm at a reference wavelength of 630 nm using an ELISA plate reader (Varioskan Flash, ThermoFisher Scientific, Vantaa, Finland).
References
Pitout JDD. Multiresistant Enterobacteriaceae: New threat of an old problem. Expert Rev Anti Infect Ther. 2008;6:657–69. https://doi.org/10.1586/14787210.6.5.657
Falcone M, Paterson D. Spotlight on ceftazidime/avibactam: A new option for MDR Gram-negative infections. J Antimicrob Chemother. 2016;71:2713–22. https://doi.org/10.1093/jac/dkw239
Kebede T, Gadisa E, Tufa A. Antimicrobial activities evaluation and phytochemical screening of some selected medicinal plants: A possible alternative in the treatment of multidrug-resistant microbes. PLoS One. 2021;16:e0249253 https://doi.org/10.1371/journal.pone.0249253
Agyare C, Koffuor GA, Boamah VE, Adu F, Mensah KB, Adu-Amoah L. Antimicrobial and Anti-Inflammatory Activities of Pterygota macrocarpa and Cola gigantea (Sterculiaceae). Evid Based Complement Alternat Med. 2012;902394. https://doi.org/10.1155/2012/902394
Christopher R, Mgani Q, Nyandoro S, Rousseau A, Vuuren S, Isaacs M, et al. Antitrypanosomal, antiplasmodial, and antibacterial activities of extracts from selected Diospyros and Annonaceae species. J Complement Med Res. 2018;7:161 https://doi.org/10.5455/jcmr.20171205011734
Artizzu N, Bonsignore L, Cottiglia F, Loy G. Studies on the diuretic and antimicrobial activity of Cynodon dactylon essential oil. undefined. Fitoterapia 1996;66:174–5
Izzo AA, Carlo G di, Biscardi D, Fusco R de, Mascolo N, Borrelli F, et al. Biological screening of Italian medicinal plants for antibacterial activity. Phytotherapy Research (United Kingdom). 1995
Silihe KK, Zingue S, Winter E, Awounfack CF, Bishayee A, Desai NN, et al. Ficus umbellata Vahl. (Moraceae) Stem Bark Extracts Exert Antitumor Activities In Vitro and In Vivo. Int J Mol Sci. 2017;18:E1073 https://doi.org/10.3390/ijms18061073
Kamdem MHK, Ojo O, Kemkuignou BM, Talla RM, Fonkui TY, Silihe KK, et al. Pentacyclic Triterpenoids, Phytosteroids and Fatty Acid Isolated from the Stem-bark of Cola lateritia K. Schum. (Sterculiaceae) of Cameroon origin; Evaluation of Their Antibacterial Activity. Arab J Chem. 2022;15:103506 https://doi.org/10.1016/j.arabjc.2021.103506
Adesina SK, Reisch J. A triterpenoid glycoside from Tetrapleura tetraptera fruit. Phytochemistry. 1985;24:3003–6. https://doi.org/10.1016/0031-9422(85)80044-3
Adesina SK, Iwalewa EO, Johnny II. Tetrapleura tetraptera Taub- Ethnopharmacology, Chemistry, Medicinal and Nutritional Values- A Review. Journal of Pharmaceutical Research International. 2016;1–22. https://doi.org/10.9734/BJPR/2016/26554
Adewunmi CO, Furu P, Marquis BB, Fagbola M, Olatunji OA. Molluscicidal trials and correlation between the presence of Tetrapleura tetraptera in an area and the absence of the intermediate hosts of schistosomiasis and fascioliasis in Southwest Nigeria. J Ethnopharmacol. 1990;30:169–83. https://doi.org/10.1016/0378-8741(90)90006-F
Aladesanmi AJ, Iwalewa EO, Adebajo AC, Akinkunmi EO, Taiwo BJ, Olorunmola FO, et al. Antimicrobial and Antioxidant Activities of Some Nigerian Medicinal Plants. Afr J Tradit Complement Alter Med. 2006;4:173–84. https://doi.org/10.4314/ajtcam.v4i2.31206
Mbaveng AT, Chi GF, Bonsou IN, Ombito JO, Yeboah SO, Kuete V, et al. Cytotoxic phytochemicals from the crude extract of Tetrapleura tetraptera fruits towards multi-factorial drug resistant cancer cells. J Ethnopharmacol. 2021;267:113632 https://doi.org/10.1016/j.jep.2020.113632
Ojewole JAO, Adewunmi CO. Anti-inflammatory and hypoglycaemic effects of Tetrapleura tetraptera (Taub) [Fabaceae] fruit aqueous extract in rats. J Ethnopharmacol. 2004;95:177–82. https://doi.org/10.1016/j.jep.2004.06.026
Noté OP, Mitaine-Offer A-C, Miyamoto T, Paululat T, Pegnyemb DE, Lacaille-Dubois M-A. Tetrapterosides A and B, two new oleanane-type saponins from Tetrapleura tetraptera. Magn Reson Chem. 2009;47:277–82. https://doi.org/10.1002/mrc.2381
Enema OJ, Adesina SK, Umoh UF, Eseyin OA. Gas chromatography-mass spectroscopy (GC-MS) studies of fixed oil of leaf of Tetrapleura tetraptera Taub. (Mimosaceae). J Pharmacogn. Phytochem 2019;8:1237–41
Fotie J, Nkengfack AE, Peter MG, Heydenreich M, Fomum ZT Chemical constituents of the ethyl acetate extracts of the stem bark and fruits of Dichrostachys cinerea and the roots of Parkia bicolor. Bulletin of the Chemical Society of Ethiopia. 2004;18. https://doi.org/10.4314/bcse.v18i1.61646
Mbouangouere R, Tane P, Ngamga D, Ngamga D, Djemgou P, Choudhary M, et al. Piptaderol From Piptadenia africana. Afr J Trad Compl Alt Med. 2008;4:294 https://doi.org/10.4314/ajtcam.v4i3.31222
Sultana N, Armstrong JA, Waterman PG. Benzopyran derivatives from the aerial parts of Eriostemon rhomboideus. Phytochemistry. 1999;52:895–900. https://doi.org/10.1016/S0031-9422(99)00338-6
Rubinstein I, Goad LJ, Clague ADH, Mulheirn LJ. The 220 MHz NMR spectra of phytosterols. Phytochemistry. 1976;15:195–200. https://doi.org/10.1016/S0031-9422(00)89083-4
Marliyana SD, Wibowo FR, Handayani DS, Kusumaningsih T, Suryanti V, Firdaus M, et al. Stigmasterol and Stigmasterone from Methanol Extract of Calophyllum soulattri Burm. F. Stem Bark. J Kim Sains dan Aplikasi. 2021;24:108–13. https://doi.org/10.14710/jksa.24.4.108-113
Erwin, Pusparohmana WR, Safitry RD, Marliana E, Usman Kusuma IW. Isolation and characterization of stigmasterol and β-sitosterol from wood bark extract of Baccaurea macrocarpa Miq. Mull. Arg Rjc 2020;13:2552–8. https://doi.org/10.31788/RJC.2020.1345652
Taiwo BJ, Olubiyi OO, Wang X, Fisusi FA, Akinniyi GA, Van Heerden FR, et al. Schistosomiasis: Snail-vector control, molecular modelling and dynamic studies of bioactive N-acetylglycoside saponins from Tetrapleura tetraptera. Comput Biol Chem. 2018;77:363–72. https://doi.org/10.1016/j.compbiolchem.2018.09.011
Soyler A, Bouillaud D, Farjon J, Giraudeau P, Oztop MH. Real-time benchtop NMR spectroscopy for the online monitoring of sucrose hydrolysis. LWT. 2020;118:108832 https://doi.org/10.1016/j.lwt.2019.108832
Raya-Gonzalez D, Pamatz-Bolaños T, del Rio-Torres RE, Martinez-Muñoz RE, Ron-Echeverria O, Martinez-Pacheco MM. D-(+)-pinitol, a component of the heartwood of Enterolobium cyclocarpum (Jacq.) Griseb. Z Naturforsch C J Biosci. 2008;63:922–4. https://doi.org/10.1515/znc-2008-11-1225
Rehan M, Ansari FA, Singh O. Isolation, Identification, Antibacterial Activity and Docking of Fatty acid and Fatty Alcohol from Rumex dentatus Leaf Extract. Int J Pharm Sci Rev Res. 2020;5:7–11. https://doi.org/10.47583/ijpsrr.2020.v64i01.002
Vyas N, Argal A. Isolation and characterization of oleanolic acid from roots of lantana camara. Asian J Pharm Clin Res. 2014;3:189–91
Donfack JH, Fotso GW, Ngameni B, Tsofack FN, Tchoukoua A, Ambassa P, et al. In vitro hepatoprotective and antioxidant activities of the crude extract and isolated compounds from Irvingia gabonensis. 亚洲传统医药. 2010;5:79–88
Téné DG, Tih AE, Kamdem MHK, Talla RM, Diboue PHB, Melongo YKD, et al. Antibacterial and antioxidant activities of compounds isolated from the leaves of Symphonia globulifera (Clusiaceae) and their chemophenetic significance. Biochemical Syst Ecol. 2021;99:104345
Prachayasittikul S, Saraban P, Cherdtrakulkiat R, Ruchirawat S, Prachayasittikul V. New bioactive triterpenoids and antimalarial activity of Diospyros rubra Lec. EXCLI J. 2010;9:1–10
Pop-Vicas A, Tacconelli E, Gravenstein S, Lu B, D’Agata EMC. Influx of multidrug-resistant, gram-negative bacteria in the hospital setting and the role of elderly patients with bacterial bloodstream infection. Infect Control Hosp Epidemiol. 2009;30:325–31. https://doi.org/10.1086/596608
Garnacho-Montero J, Corcia-Palomo Y, Amaya-Villar R, Martin-Villen L. How to treat VAP due to MDR pathogens in ICU patients. BMC Infect Dis. 2014;14:13 https://doi.org/10.1186/1471-2334-14-135
Vargiu AV, Ruggerone P, Opperman TJ, Nguyen ST, Nikaido H. Molecular Mechanism of MBX2319 Inhibition of Escherichia coli AcrB Multidrug Efflux Pump and Comparison with Other Inhibitors. Antimicrob Agents Chemother. 2014;58:6224–34. https://doi.org/10.1128/AAC.03283-14
Wolska K, Grudniak A, Fiecek B, Kraczkiewicz-Dowjat A, Kurek A. Antibacterial activity of oleanolic and ursolic acids and their derivatives. Open Life Sci. 2010;5:543–53. https://doi.org/10.2478/s11535-010-0045-x
Achi OK. Composition and antibacterial activities of Tetrapleura tetraptera Taub. pod extracts. Res J Microbiol. 2006;5:146–422. https://doi.org/10.3923/JM.2006.416.422
Lin L, Agyemang K, Abdel-Samie MAS, Cui H. Antibacterial mechanism of Tetrapleura tetraptera extract against Escherichia coli and Staphylococcus aureus and its application in pork. J Food Saf. 2019;39:e12693 https://doi.org/10.1111/jfs.12693
Cui H, Bai M, Sun Y, Abdel-Samie MAS, Lin L. Antibacterial activity and mechanism of Chuzhou chrysanthemum essential oil. J Funct Foods. 2018;48:159–66. https://doi.org/10.1016/j.jff.2018.07.021
Burt S. Essential oils: their antibacterial properties and potential applications in foods–a review. International Journal. Food Microbiol. 2004;94:223–53. https://doi.org/10.1016/j.ijfoodmicro.2004.03.022
Gill AO, Holley RA. Disruption of Escherichia coli, Listeria monocytogenes and Lactobacillus sakei cellular membranes by plant oil aromatics. Int J Food Microbiol. 2006;108:1–9. https://doi.org/10.1016/j.ijfoodmicro.2005.10.009
Miras-Moreno B, Sabater-Jara AB, Pedreño MA, Almagro L. Bioactivity of Phytosterols and Their Production in Plant in Vitro Cultures. J Agric Food Chem. 2016;64:7049–58. https://doi.org/10.1021/acs.jafc.6b02345
Ramprasath VR, Awad AB. Role of Phytosterols in Cancer Prevention and Treatment. J AOAC Int. 2015;98:735–8. https://doi.org/10.5740/jaoacint.SGERamprasath
Balouiri M, Sadiki M, Ibnsouda SK. Methods for in vitro evaluating antimicrobial activity: A review. J Pharm Anal. 2016;6:71–79. https://doi.org/10.1016/j.jpha.2015.11.005
Acknowledgements
This research was supported by the South African Medical Research Council with funds received from the South African National Department of Health, and the UK Medical Research Council with funds received from the UK Government’s Newton Fund. The authors are also thankful to the Department of Chemical Sciences and the Department of Biotechnology and Food Technology of the University of Johannesburg for providing laboratory space, chemicals and equipment that allowed us to carry on this work.
Author contribution
CRediT authorship contribution statement. MHKK: Conceptualization, Investigation, Methodology, Writing - original draft. GLFM: Review-writing & review. KKS: Writing - review & editing. GKM: Cytotoxic assay, Review-editing. MPM: Review & editing. TAM: Formal analysis, Review-editing. CMT: Writing - review & editing. JLT: Formal analysis, Methodology and review. OO: Review & editing. TYF: Antibacterial analysis. EMM: Supervision, Writing - review & editing. DTN: Supervision, Writing - review & editing.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kamdem, M.H.K., Melacheu, G.L.F., Silihe, K.K. et al. Cytotoxic and antibacterial activities of compounds isolated from the fruits and stem-bark of Tetrapleura tetraptera (Schumach. & Thonn.) Taub. (Fabaceae). Med Chem Res 31, 1948–1958 (2022). https://doi.org/10.1007/s00044-022-02956-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-022-02956-1