
Abstract
Biochemical analysis of secondary metabolites of marine old-woman octopus Cistopus indicus (family Octopodidae) led to the identification of two sixteen-membered spiro-linked macrocyclic bislactones, named as cistobislactone A and cistobislactone B with unprecedented feature of henicos framework, based on extensive spectroscopic analyses. Cistobislactone B exhibited potential inhibition property against arachidonate 5-lipoxygenase (IC50 2.18 mM) than that demonstrated by cistobislactone A (IC50 2.54 mM) and standard non-steroidal anti-inflammatory agent ibuprofen (IC50 4.50 mM) thus signifying the higher anti-inflammatory activity of the cistobislactone B analogue. The studied macrocyclic bislactones exhibited promising antioxidant potential, in which cistobislactone B exhibited potential radical quenching (IC50 2.33 mM) and hydrogen peroxide scavenging (IC50 1.81 mM) activities that were proximal to the commercial anti-oxidant α-tocopherol (IC50 ~ 1.60 mM). This further reinforced its attenuation property against arachidonate 5-lipoxygenase. Considerably greater electronic properties coupled with balanced hydrophobicity of cistobislactone B could ascribe the superior ligand-receptor interfaces leading to its anti-inflammatory activity. Molecular docking analysis of cistobislactone B with 5-lipoxygenase recorded lesser docking score (−12.24 kcal mol−1) and binding energy (−11.24 kcal mol−1), which further supported its anti-inflammatory activity. Cistobislactone B, with six fold lesser value of inhibition constant (Ki 5.76 nM) towards 5-lipoxygenase than that displayed by cistobislactone A, could describe the superior protein-ligand interactions of the former. The undescribed cistobislactone B might be a potential natural anti-inflammatory lead to moderate the odds of inflammatory pathologies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Biochemistry of the arachidonate 5-lipoxygenase (5-LO) encompasses the biosynthesis of leukotrienes, and catalyzes the primary steps in the biotransformation of arachidonic acid to the mediators of inflammation [1]. Pro-inflammatory profile of leukotrienes suggested that 5-LO antagonists could encompass pharmacological potential in the treatment of various inflammatory disorders [2]. Other pro-inflammatory enzyme cyclooxygenase (COX) exists as two isoforms, an inducible kind (COX-2) and a constitutive type (COX-1), exhibiting important roles to cause inflammation by biosynthesizing inflammatory prostaglandins (PGs) from arachidonic acid [3]. Widely used anti-inflammatory drugs comprise of non-steroidal anti-inflammatory drugs (NSAIDs) that were found to have gastrointestinal ulcerogenic side-effects [4]. In this context, the therapeutic approach to develop dual inhibitors of 5-LO and COX in an attempt to augment their characteristic anti-inflammatory effects with reduced undesirable side-effects is significant.
Macrocyclic lactones comprise a major class in natural products owing to their noticeable role in the chemistry of antibiotics and various bioactive potentials [5]. These structurally diverse and complex organic molecules fascinated medicinal chemistry researchers across the globe over the past six decades on account of their significant role in modern drug discovery and development [6]. Natural macrocycles of marine origin are common of fourteen to sixteen skeletal ring systems with different structure motifs, in which fourteen-membered macrocyclic frameworks are by far the most abundant [7, 8]. As accounted earlier, oxygenated macrocyclic compounds possessing greater than sixteen-membered ring size could possess a lactone moiety, whereas thirteen and fourteen-membered analogous contain ether linkages. Conspicuously, a fourteen-membered bislactone, cordybislactone has been reported from the fungus Cordyceps sp. with anticancer activity along with anti-mycobacterial property [9]. A new class of 25-membered ring marine macrocyclic lactones with tetrahydropyran moieties, named lituarines with antifungal, cytotoxic and anti-neoplastic activities were reported from the sea pen Lituaria australasiae [10]. An unusual tetraquinaneoxa-built bislactone carrying linear R-alkylbutenolide paracaseolide A, displaying anti-tumor activity, was characterized from Sonneratia paracaseolaris [11]. Earlier literature reported the isolation of a tetrahydrofuran attached sixteen-membered macrocyclic bislactone from Amphioctopus neglectus belonging to the family Octopodidae [12].
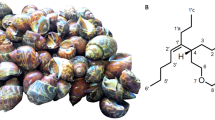
Cephalopods are being a part of various world cuisines for centuries. In India, benthic octopuses form the basis of economically important fisheries [13]. It was reported that old-woman octopus Cistopus indicus forms greater than 90 percent of total octopus landings in the southwest coast of the Indian peninsular region [14, 15]. The marine octopuses belonging to the class Cephalopoda were proved to be the productive sources of various pharamacoactive secondary metabolites and nutritionally rich functional food ingredients. Organic extract from C. indicus was reported to exhibit potent antioxidant and anti-inflammatory properties particularly 5-LO attenuation activity [16, 17]. Although several chemical studies were carried out on bivalve and gastropod mollusks, the order Octopoda has been less investigated so far. The present work describes the isolation and structural characterization of two new sixteen-membered spiro-linked macrocyclic bislactones (cistobislactones A and B) (Fig. 1) from the ethyl acetate-methanol (EtOAc-MeOH, 1:1 v/v) extract of C. indicus (family Octopodidae) harvested from the southwest coast of Indian peninsular expanse. The purified compounds were analyzed for their potential to attenuate 5-LO and inducible form of COX-2, and compared the activities with regard to their potential to attenuate the constitutive form of COX-1. The isolated compounds were characterized by detailed spectroscopic experiments. The targeted bioactivities of the cistobislactones A-B were structurally correlated with various physicochemical parameters, whereas their binding efficiency with active site of 5-LO was evaluated using molecular docking analysis (Fig. S1).

(a) Structural representation of macrocyclic bislactone analogues, cistobislactones A and B purified from the organic extract of (b) marine old-woman octopus C. indicus
Results and discussion
General
Macrocyclic lactones comprise an important chemical class, which are ubiquitously found in the marine organisms, and were demonstrated with potent anti-inflammatory activities [18, 19]. Herein, we have isolated two sixteen-membered spiro-linked macrocyclic bislactones, cistobislactones A and B, from the organic extract of marine octopus C. indicus with potential 5-LO attenuation properties.
Bioassay-assisted chromatographic fractionation of cistobislactones A-B from C. indicus
Bioactivity-directed size exclusion chromatography of EtOAc-MeOH extract of C. indicus over cross-linked dextran-based resin (lipophilic Sephadex® LH-20) followed by repeated chromatographic purification could result in four fractions (CI1-CI4) based on TLC and RP-HPLC. The fractions were evaluated for bioactivity analysis against the oxidants along with attenuation potential against 5-LO. The fraction CI2 (8.56 g) showed greater antioxidant potential (IC50 1.23 mg mL−1 DPPH scavenging) than the others, and therefore, it was shortlisted for downstream chromatographic purification on a pressurized liquid column (flash chromatography) using n-hexane/EtOAc/MeOH. This could result in the separation of 3 sub-fractions designated as CI2-1-CI2-3 based on TLC and RP-HPLC. These fractions were resubmitted for bioactivities, wherein CI2-2 registered superior bioactive potential, and hence, was purified using preparative RP-HPLC to yield two homogenous metabolites, which were characterized as cistobislactone A and cistobislactone B (Rt 6.35 and 6.76 min, respectively) (Table 1).
Cistobislactone A
The empirical formula of the macrocyclic bislactone, cistobislactone A, was deduced as C26H44O8 {HR(ESI)MS m/z 485.3118 [M + H]+cal. for C26H45O8 485.3114}, and the structure of the compound was elucidated based on detailed NMR experiments (Table 1; Fig. S2–S11). The FTIR spectrum (Fig. S12) displayed strong absorption bands at 3432 and 1751 cm−1 that confirmed the occurrence of hydroxyl and cyclic ester functional groups, respectively. The other prominent infrared stretching bands at 1058 and 2926, 2854 cm−1 suggested the presence of ether bridges (C–O–C) and C-H alkyl stretch in the studied cistobislactone A. Elemental composition of cistobislactone A showed five degrees of unsaturation. 13C NMR spectrum along with 135DEPT showed resonances for 26 carbons, which were classified into four methyls, eleven sp3 methylenes, six sp3 methines, two sp2 methines along with three quaternary carbons. Typical 13C NMR signals recorded at δC 171.5 and 167.9 were assigned to the carbonyl carbons of ester functionality (Fig. S3). Highly deshielded carbon signals at δC 57.8, 74.3, 63.5, 70.2, 73.7, 83.4, and 76.5 were indicative for oxygenated carbons implying three methylenes, three methines, and one methyl carbon, as revealed by the 1H-1H COSY/HMBC data (Fig. S5, S7). Significantly deshielded proton signals at δH 3.31 and 3.28 suggested the attachment of interchangeable hydroxyl groups to the carbons at δC 74.3 and 76.5, respectively that were further ascertained by the disappearance of deuterated hydroxyl resonances in 1H NMR. The absence of resonance peaks corresponding to δC 171.5, 167.9, and 30.2 in 135DEPT depicted their quaternary nature. Previous literature identified a fourteen-membered macrodiolide, clonostachydiol [9] that confined two ester carbonyls, alkene, and hydroxyls, whereas the titled cistobislactone A enclosed two ester functionalities at C-1 and C-5, one olefinic group (C-11/C-12), two hydroxyl groups (in pyran ring at C-4′), and macrocyclic framework (at C-9 position). Deshielded olefinic protons at δH 5.36 and 5.38 appeared as doublet of triplet, which was attached to the carbons at δC 126.3 and 127.9, respectively. Detailed interpretation of the 1H-1H COSY and HMBC spectra established the presence of three isolated spin systems (I, II, and III) as indicated by the bold-faced lines in Fig. 2. The macrocyclic bislactone core enclosed two spin systems of I and II, whereas 1H-1H COSY interactions in spin system III assigned the presence of spiro-fused pyran ring to the bislactone core. These attributions were further confirmed by the HMBC cross-peaks of H-1′ to C-16, C-5′, and H-16 to C-2 and C-14, and the results were corroborated with the previous report of sixteen-membered macrolide with a pyran ring moiety [20]. Considerably deshielded methylene protons at δH 3.96 (H-4) and δH 4.55 (H-16) displayed three-bond HMBCs with lactone carbonyls C-2 (171.5), and C-6 (167.9) from H-4, which reinforced the presence of nearby electronegative oxygen atom of an ester group [18]. HMBC correlation between H-8c (δH 3.45/3.22) and C-8e (δC 57.8) and that between H-3′ (δH 3.87) and C-1′ (δC 73.7) supported the presence of an ether linkage in 2-methoxy methyl-butyl side chain and in the pyran moiety. HMBC cross-peaks at H-4/C-2, H-3/C-4, H-4/C-6, H-7/C-6, H-7/C-8, H-9/C-10, H-11/C-13, H-14/C-15, H-16/C-14, and H-16/C-2 (Fig. S7) along with 1H-1H COSY relations from H-3 to H-4/H-7 through H-14 disclosed the presence of 16-membered macrocyclic bislactonic ring skeleton in the compound. Large coupling constants between H-11 and H-12 (J = 12.5 and 12.3 Hz, respectively) attributed that these olefinic protons were aligned in trans-fashion with respect to each other, and thus, the geometrical configuration was assigned as E. NOEs between δH 3.87 (H-3′) and 3.31 (H-9) showed their proximities, and these protons were lined up in the same plane of symmetry, and thus, assigned as α-oriented. The proton signal at H-3′ showed a correlation with H-4′ (δH 3.28) thus attributing the position in the same symmetry corresponding to α-orientation. Other NOE correlations between δH 1.77 (H-8b)/2.14 (H-8)/1.63 (H-5′) designated their equal plane of symmetry, and were oriented towards the opposite face of the α-protons, and hence, their positions were deduced as β-oriented. In brief, the relative stereochemistry of six chiral centers at C-8, C-8b, C-9, C-3′, C-4′ and C-5′ was assigned based on NOE correlation spectrum (Fig. S8) in which H-3′, H-9′ and H-4′ were aligned in α-orientation, and those at C-8, C-8b and C-5′ were assigned as β-oriented.

(a, b) 1H-1H COSY/HMBC relationships of cistobislactones A-B, wherein the key 1H-1H COSYs were characterized in bold-faced bonds, and HMBCs as double-barbed arrows; (c, d) NOESY relationships (double-sided arrows, pink and blue-colored arrows designated alpha and beta protons, respectively) of cistobislactones A-B, respectively. The NOESY relations were represented by (e, f) computer-generated models of cistobislactone A and B using MM2 force-field calculations
Cistobislactone B
The empirical formula of the second macrocyclic bislactone, cistobislactone B, was deduced as C27H44O7 {HR(ESI)MS m/z 481.3168 [M + H]+cal. for C27H45O7 481.3165}, and the structure of the compound was elucidated by FTIR/NMR experiments (Table 1; Fig. S13–S23). The IR spectrum of cistobislactone B showed the stretching absorptions peaks at 3431, 2924-2856, 1745, 1050 cm−1 that suggested the presence of hydroxyl, alkylic, lactone carbonyl, and ether (C-O-C stretch) functional groups, respectively (Fig. S23). Elemental composition of cistobislactone B showed six degrees of unsaturation. 13C NMR spectrum along with 135DEPT showed resonances for 27 carbons were classified into two carbonyls, four sp2 carbons being for two olefinic bonds of [1,2-disubstituted] and [1,2,2-trisubstituted] nature, respectively, carbons along with five sp3 methines, ten methylenes, four methyls and one sp3 quaternary carbon. 1H-1H COSY spectrum of the compound (Fig. S16) revealed four isolated spin systems, assigning the partial skeletal structure: H-3/H-4 (I), H-7/H-8/H-9/H-10/H-11/H-12/H-13/H-13a/H-13b/H-14 (II), H-8b/H-8c (III), and H-3a′/H-3′/H-4′/H-5′/H-5a′ (IV). HMBC correlations from δH 3.89 (H-4)/δC 171.5 (C-2), δH 2.40 (H-3)/δC 63.1 (C-4), δH 3.89 (H-4)/δC 167.7 (C-6), δH 2.14 (H-7)/δC 167.7 (C-6), δH 2.74 (H-8)/δC 35.9 (C-7), δH 1.99 (H-10)/δC 36.3 (C-8), δH 5.69 (H-12)/δC 25.4 (C-10), δH 5.66 (H-11)/δC 34.0 (C-13), δH 4.50 (H-16)/δC 32.9 (C-14), δH 4.50 (H-16)/δC 27.4 (C-15), and δH 4.50 (H-16)/δC 171.5 (C-2) established the closed 16-membered macrocyclic loop in cistobislactone B. Attachment of a pyran moiety through a spiro linkage to the basic macrocyclic lactone skeleton was assigned by the HMBC correlation between H-16 (δH 4.50) and C-1′ (δC 75.3) and HMBC cross-peak between H-16 (δH 4.50) and C-15 (δC 27.4). Likewise, the side chain of 4-methoxy-but-2-ene-2-yl at C-8 position of the basic lactonic framework was inferred by the HMBC of H-8 (δH 2.74) with C-8a′ (δC 16.2). Similar to cistobislactone A, the olefinic protons positioned at H-11 and H-12 were found to possess larger J values (J = 12.2 and 12.6 Hz, respectively), which attributed that these olefinic protons were aligned in trans-fashion with respect to each other, and thus, the geometrical configuration was assigned as E. Relative stereochemistry of the chiral centers positioned at C-8, C-13, C-3′, C-4′ and C-5′ were inferred from the NOESY experiments, and further corroborated with MM2 force-field calculations. NOEs between the chiral protons at H-3′ (δH 4.03) and H-4′ (δH 3.30) showed their proximities, and were disposed in the same plane of symmetry, and thus, assigned as α-oriented. Likewise, the proton positioned at C-4′ showed NOE correlation with C-13 proton (δH 2.01, H-13′), and was deduced to be α-oriented. NOE coupling between H-8 (δH 2.74) and H-5′ (δH 1.58) designated their equal plane of symmetry, and were oppositely inclined compared to the α-protons, and hence, their positions were deduced as β-oriented.
Bioactive properties of cistobislactones and structure-activity correlation analysis
Organic extract of C. indicus exhibited potential inhibitory activity against 5-LO (IC50 1.14 mg mL−1) and COX-2 (IC50 1.35 mg mL−1). Cistobislactone B demonstrated greater attenuation property against COX-2 (IC50 2.75 mM) than that showed by its cistobislactone A analogue. Greater anti-inflammatory selectivity index (anti-COX-1/anti-COX-2) of cistobislactone B (1.06) compared to the standard anti-inflammatory drug, ibuprofen (selectivity index, 0.43) inferred the higher COX-2 selectivity of the former. It is of note that between the COX isoforms, COX-1 is considered to be a constitutive isoform, which was found to express in most tissues under normal conditions, whereas COX-2 isoenzyme was induced in response to several pro-inflammatory factors [21]. Therefore, the assessment of attenuation potential of bioactive compounds against COX-2 was considered to be an essential tool to encumber inflammatory conditions. Similarly, arachidonate 5-LO inhibitory activity of cistobislactone B (IC50 2.18 mM) was considerably greater than that displayed by cistobislactone A (IC50 2.54 mM) and standard anti-inflammatory drug ibuprofen (IC50 4.50 mM). Arachidonate 5-LO effectively contributes towards the advancement of inflammatory reactions, such as atherosclerosis [22], rheumatoid arthritis [23], and autoimmune diseases [24] owing to its capacity to biosynthesize inflammatory leukotrienes (such as LTB4) and 5-hydroxyeicosatetraenoic acid (HETE). Therefore, 5-LO is a promising target for pharmaceutical applications in several diseases, including inflammation [25]. The selectivity indices (anti-COX-1/anti-COX-2) of cistobislactones towards pro-inflammatory COX isoforms were significantly superior (~1.0) than ibuprofen (0.43). Cistobislactone B exhibited higher antioxidant potentials (DPPH IC50 2.33 mM) and hydrogen peroxide inhibition properties (IC50 1.81 mM) compared to those displayed by cistobislactone A (DPPH IC50 2.80 and H2O2 IC50 2.10 mM) (Table 2).
Various molecular descriptors comprising of electronic, steric and hydrophobic parameters of the isolated cistobislactones were determined and correlated with their bioactive potentials. Electronic parameters, such as topological polar surface area (tPSA 91.29) and molecular polarizability (Pl 52.01) of cistobislactone B were comparable with those of cistobislactone A (tPSA 111.52; Pl 50.90), and greater than ibuprofen (tPSA 37.3; Pl 23.96). The hydrophobicity of cistobislactones (log POW 2.4–3.5) were found within the thresold permissible limit of hydrophobic-lipophilic balance [26], which could be responsible for their potential bioactive properties. An acceptable permeability in cellular network (through inter-membrane barrier) along with the radical scavenging properties of the studied compounds could contribute towards their greater anti-inflammatory activities.
Molecular docking of cistobislactones against inflammatory enzymes and drug-likeness
The attenuation properties of the spiro-linked macrocyclic bislactone derivatives were further corroborated by the in silico molecular docking analysis using arachidonate 5-LO enzyme (Fig. 3; Table 3). Cistobislactones A and B exhibited three hydrogen bonds each in the active site-5-LO complex, whereas ibuprofen displayed two hydrogen bonds in the ligand-enzyme composite (Fig. 3). Three hydrogen bonding interactions with the active site amino acyl residues (Phe304, Leu289, Ile330) (Fig. 3) along with the lesser docking parameters of cistobislactones with arachidonate 5-LO could further attribute their anti-inflammatory potential. Cistobislactone B exhibited lesser binding properties, such as binding energy (−11.24 kcal mol−1), intermolecular energy (−12.42 kcal mol−1), docking score (−12.24 kcal mol−1), and inhibition constant (Ki, 5.76 nM) than those displayed by cistobislactone A (binding energy −10.12 kcal mol−1, intermolecular energy −11.22 kcal mol−1, docking score −11.02 kcal mol−1, and inhibition constant Ki, 38.37 nM), which were accounted for the strong binding affinity of cistobislactone B towards 5-LO enzyme (Table 3). Interestingly, the inhibition constant Ki of cistobislactone B was found to be six-fold lesser than that displayed by cistobislactone A thus attributing the effective binding potential of the former to the enzyme in a non-competitive manner. The molecular docking factors implied that cistobislactone B displayed superior molecular interactions with 5-LO than that exhibited by the anti-inflammatory agent ibuprofen (docking score and binding energy of −3.51 and −3.44 kcal mol−1, respectively). This was further confirmed by higher drug-like attribute of cistobislactone B (drug-likeness score of 0.24) (Fig. 3), and therefore, the compound could be a prospective anti-inflammatory lead attenuating arachidonate 5-LO. Furthermore, superior electronic properties (Pl > 50) could connote its larger electronic rich centers, which could network with the active site of 5-LO resulting in stronger hydrogen bonds with aminoacyl residue as compared to ibuprofen (with lesser polarizability, ~24).

Molecular docking of cistobislactones A and B with 5-LO. (a, b) Nearer view of molecular docking interactions of cistobislactone A and B and hydrogen-bonded amino acid residues in the active catalytic site of 5-LO enzyme; (c, d) Chemdraw representation of hydrogen bond interaction of the amino acyl residues of 5-LO with cistobislactones A and B; (e, f) Drug-likeness score for the cistobislactones with molsoft software. Blue-colored lines illustrated the hydrogen bond interactions
Biosynthetic origin of cistobislactones
Hypothetical biosynthetic pathway of cistobislactones could proceed through the building of intermediate groups and elongation on acyl carrier protein (ACP) and ketosynthase (KS) domain units (Fig. 4). Biosynthesis of cistobislactone A might include the steps involving malonylacetate-ACP intermediates that were decarboxylated followed by sequential removal of acetaldehyde from the S-KS unit. Dehydratase (DH) catalyzed the formation of 3-oxopropyl-1-enyl-4-hydroxy-9-(5-hydroxy-3-(hydroxymethyl)-4,6-dimethyltetrahydro-2H-pyranyl)nonadienoate-S-ACP, which on further cyclization formed macrocyclic bislactonic framework. Addition of 3-methoxypropenol to the olefinic carbon (C-8) facilitated through the nucleophic attack followed by methyl transferase (MT)-catalyzed addition of methyl and subsequent elongation (at C-8b) could lead to the formation of cistobislactone A. Putative biosynthetic pathway to form cistobislactone B might consist of chain extension on both ACP and KS domain through corresponding decarboxylative ketoreduction of malonate S-ACP and sequential flipping of building units between two domains via acyltransferase. A fragment 2, 3-methoxypropenol could be added to the C-8 position of biclactonic macrocyclic skeleton (fragment 1) through a DH-catalyzed reaction. Side chain elongation at C-13 and C-8a could proceed by MT-catalyzed reaction to afford cistobislactone B (Fig. 4).

Scheme of putative biosynthesis of cistobislactone A by consecutive elongation modules through adenylation and condensation. Abbreviations: AT acyltransferase, ACP acyl carrier protein, KR ketoreductase, C condensation, PCP peptidyl carrier protein, KS ketosynthase, DH dehydratase. (A) Condensation reaction between malonate (linked to ACP) and CH3(C=O)- connected with KS; (B) formic-9-(dihydroxy-4,6-dimethyltetrahydro-2H-pyran-3-yl)-4-hydroxy-6-oxonon-2-enoic anhydride-S-KS; (C) 4-hydroxy-9-(5-hydroxymethyl)-dimethyltetrahydro-2H-pyran-3-yl)-6-oxonon-2-enoic 3-oxopropanoic anhydride-S-ACP; (D) 1-hydroxy-3-oxopropyl-4,6-dihydroxy-9-(5-hydroxy-3-(hydroxymethyl)-dimethyltetrahydro-2H-pyranyl)non-2-enoate-S-ASP; (E) 3-oxopropyl-1-enyl-4-hydroxy-9-(5-hydroxy-3-(hydroxymethyl)-dimethyltetrahydro-2H-pyran-3-yl)nona-2,6-dienoate-S-ACP; (F) 3-oxopropyl-4-hydroxy-9-(5-hydroxy-3-(hydroxymethyl)-dimethyltetrahydro-2H-pyranyl)nona-2,6-dienoate-S-ACP; (G) 4,16-dihydroxy-dimethyl-2,8,12-trioxaspiro[5. 15]henicosa-14,18-diene-9,13-dione; (H) methylformate-S-KS; (I) methyl-3-oxopropanoate-S-ACP; (J) 1-methoxypropane-1,3-diol-S-ACP; (K) 3-methoxyprop-2-en-1-ol-S-ACP; (L) 3-methoxyprop-2-en-1-ol; (M) dihydroxy-15-(3-methoxypropyl-dimethyl-trioxaspiro[5.15]henicosa-18-diene-9,13-dione. Scheme of putative biosynthesis of cistobislactone B (steps involving A′-Q′)
Conclusions
Bioassay-guided chromatographic fractionation of the organic extract of the old-woman octopus C. indicus led to the isolation of two unreported sixteen-membered macrocyclic bislactone analogues, named as cistobislactones A and B. Among the isolated analogues, cistobislactone B exhibited greater attenuation properties against arachidonate 5-LO (IC50 2.18 mM) than those displayed by cistobislactone A (IC50 2.54 mM) and standard non-steroidal anti-inflammatory agent ibuprofen (IC50 4.50 mM). Structure-bioactivity correlation analysis established that higher electronic properties (tPSA 91.29 and Pl 52.01) along with optimum hydrophobicity (log POW 3.50) of cistobislactone B could favor the interaction with active sites of 5-LO enzyme, and also predominantly contributed towards greater antioxidant properties, which were corroborated by lesser docking parameters (binding energy,−11.24 kcal mol−1; docking score, −12.24 kcal mol−1). The results illustrated that cistobislactone B could be developed as a promising marine-originated macrocyclic bislactone lead for use against oxidative stress and inflammation.
Materials and methods
Instrumentation and materials
Nuclear magnetic resonance (NMR) spectra were obtained with Bruker Avance spectrometer (DPX 500; Bruker, Rheinstetten, Germany, 500 MHz) using dueteriated methanol as solvent and a suitable internal reference (tetramethylsilane, δH 0 ppm). An ultraviolet-visible (UV-VIS) spectrophotometer (Orion™ AquaMate 8000, Thermo Scientific, Waltham, MA) was used to acquire UV spectral data. Gas chromatograph-mass spectrometer (GC-MS) operating with the electron-impact (EI) mode (7890 A GC; 5975 C MS, Agilent Technologies, Inc., Santa Clara, CA) was used for mass spectral analysis, wherein the compounds were fractionated over a medium polar partition-liquid stationary phase (HP-5MS 5% phenylmethyl silox; 30 m length x 0.25 µm film thickness x 250 µm internal diameter). High-resolution electrospray ionization mass spectral (HRESIMS) analysis was performed in ESI positive mode on Agilent 6520 accurate mass Q-TOF LC/MS (Agilent, Santa Clara, CA) coupled with a HPLC (Agilent LC 1200) equipped with a reverse-phase C18 column (ExtendTM, 1.8 µm film thickness, 2.1 × 50 mm). Fourier-transform infrared (FTIR) absorption spectra and optical rotations of the isolated compounds were measured by using an FT-IR spectrophotometer scanning from 4000 to 400 cm−1 (Perkin-Elmer-2000, Waltham, MA) and a polarimeter (ATAGO/AP-300, Atago USA, Inc., Bellevue, WA), respectively. Homogeneity of the purified compounds was analyzed with high performance liquid chromatography (HPLC, Shimadzu LC 20AD, Japan) coupled with Shimadzu LC 2535 binary gradient pump, C18 column (Phenomenex USA, Luna® C18100 Å, 250 mm × 4.6 mm, 5 μm), and photodiode array detector (SPD-M20A, Kyoto, Japan). Finer chromatographic fractionation was carried out with a preparative HPLC connected with a C18-RP (reverse-phase) column (25 cm × 1 cm, 5 μm). Solvents and reagents were of chromatographic/analytical-grade, which were procured from Sigma Aldrich (MO, USA), E-Merck (Darmstadt, Germany), and Spectrochem (Mumbai, India). Cyclooxygenases (COX-1 and COX-2, human recombinant) were procured from Sigma-Aldrich (Missouri, USA) and arachidonate 5-lipoxygenase (5-LO, soybean) was purchased from Sisco Research Laboratories (Mumbai, India). Sephadex LH-20 (lipophilic sephadex) procured from Sigma Aldrich Co. was used for initial purification (gel permeation chromatography) and silica gel plates (GF254) were used for thin layer chromatography (TLC).
Sample collection, pre-treatment, and preparation of organic solvent extracts
Fresh specimens of C. indicus (~5.5 kg) were brought together from the landing centers at Cochin harbor of the Arabian sea (located at Lat 8°48’ N; Long 78°9’ E and Lat 9°14’ N; Long 79°14’E), along the south-western coast of the Indian peninsular region, during the month of January to March, 2019. The octopus sample was unambiguously identified by Dr. K.K. Saji Kumar, Senior Technical Officer of the Molluscan Fisheries Division in Central for Marine Fisheries Research Institute, Kochi, India, based on detailed dorsal and ventral morphological examination of tentacles, suckers, ligula length and mucous pouches. The samples were cleaned with distilled water, and the parts other than the edible portion were carefully removed. The tissue part was further homogenated and lyophilized using a laboratory-scale freeze drier (alpha 1–4 LD plus, Martin Christ, Germany) yielding the freeze-dried powder (1710 g, yield 31.09%). The lyophilized powder (1710 g) was sonicated with EtOAc-MeOH (1:1, v/v; 1500 mL × 2) before being subjected to hot extraction at elevated temperature of about 45–50 °C under N2 atmosphere for about 6 h to avoid the generation of undesired chemical products. The extract was filtered (through Whatman No. 1 filter paper) and passed over anhydrous sodium sulfate before being evaporated (50 °C) to dryness (5–6 h) using a rotary vacuum evaporator (Heidolph Instruments, Schwabach, Germany) under reduced pressure to yield a dark yellow gummy residue of C. indicus crude extract. The extraction was repeated to yield an amount of 52.36 g of crude (extraction yield of dry weight basis 3.06 ± 0.5%).
Bioactivity-guided chromatographic purification of cistobislactones from C. indicus
Organic extract of C. indicus (52.36 g) was fractionated by chromatography over various adsorbent systems. Initial separation was carried out using size-exclusion chromatographic technique. The crude was dissolved in methanol before being loaded onto a packed glass column (5 cm × 150 cm). The fractionation was carried out using MeOH to acquire seven fractions (150–500 mL each) that were pooled to four major fractions (CI1-CI4) based upon RP-C18 HPLC (acetonitrile ACN-MeOH, 50:50 v/v) and TLC (dicholoromethane-MeOH, 9:1 v/v, and n-hexane-EtOAc, 7:3) visualizations. The fraction CI2 (8.56 g) exhibited considerably higher 5-LO enzyme inhibitory potential than other studied fractions, and thus, subjected to chromatographic purification on a pressurized liquid column (flash chromatography, 47 cm × 4 cm) filled with silica gel (230–400 mesh) and eluted with n-hexane/EtOAc/MeOH gradient (30, 70, and 100% EtOAc in n-hexane; and thereafter 30, 70, 100% MeOH in EtOAc) to obtain three sub-fractions (CI2-1-CI2-3) based on TLC/HPLC analyses. Sub-fraction CI2-2 exhibited comparatively greater bioactivities than those recorded with others, and was therefore, chosen for downstream fractionation by preparative HPLC on C18 column (25 cm × 1 cm, 5 μm, flow rate 8 mL/min) fitted with a binary gradient pump (pressure of pump A, 24 Kgf; pressure of pump B 18 Kgf) and photodiode array detector (SPD-M20A, Kyoto, Japan). The solvent system of ACN-MeOH was used as a mobile phase (50:50 v/v) for the preparative HPLC fractionation to afford cistobislactones A-B as yellow oils (98 and 112 mg, respectively).
Bioactivity assessment
Anti-inflammatory potential of cistobislactones were studied by evaluating their inhibition capacities against COX isoforms (COX-1 and 2) [27] and 5-LO [28]. Briefly, the COX enzymes were diluted in Tris buffer (0.1 M, pH 8). Pre-mixed phenol (500 μM), arachidonic acid (50 μM), and dichlorofluorescein (1-DCF, 20 μM) were added to the enzyme mixtures containing cistobislactones with suitable concentrations that were pre-incubated with the enzymes at an ambient temperature for 5 min in the presence of hematin. Absorbance of the reaction product was measured against the reagent blank at 502 nm. 5-LO inhibition assay was performed by adding an aliquot of the stock solution (50 μL, in dimethylsulfoxide and Tween 20 mixture; 29:1 w/w) of the sample with linoleic acid (48 μL) in pre-warmed potassium phosphate buffer (0.1 M, 2.95 mL, pH 6.3). The enzyme 5-LO (100 U) was added to the reaction mixture before being recorded the absorbance at 534 nm.
Antioxidant activities of the cistobislactones were analyzed by using in vitro hydrogen peroxide (H2O2) scavenging and 2, 2-diphenyl-1-picrylhydrazylradical (DPPH) radical quenching experiments [29,30,31]. Briefly, the H2O2 scavenging assay was carried out by mixing the solution of H2O2 (40 mM) in phosphate buffer (pH 7.4) with the freshly prepared sample solutions. The absorbance of the solution was recorded at 230 nm after 10 min against a reagent blank. DPPH scavenging activity was evaluated by adding an equal amount of DPPH solution (in MeOH) to the samples of cistobislactones before being incubated in the dark at room temperature for about 10 min. The decrease in absorbance of the reaction mixture was measured at 517 nm against a reagent blank. The plots of radical scavenging and enzyme inhibition properties were recorded, and the results were expressed as mean inhibitory concentration, IC50 (mg mL−1, mM). Steric bulkiness, hydrophobicity, and electronic parameters were determined by ACD ChemSketch (version 12.0; Advanced Chemistry Development Inc., Toronto, CA) and ChemDraw Ultra (Cambridge Soft Corp., MA; version 12.0) softwares.
In silico molecular modeling
AutoDock 4 (AutoDock Tools, version 1.5.6) software tool was used for in silico molecular docking of cistobislactones with 5-LO (PDB: 1N8Q; resolution 2.1 Å, downloaded from www.pdb.org) and energetically minimized (Swiss-PdbViewer, version 4.1.0) [32]. The cistobislactones were drawn by using ACD/ChemSketch and converted into MDL Molfiles set-up, which was changed into the PDB format with open Babel software. The grid box values were selected as x = 3.699, y = −55.637, z = −30.065 (60 Å X 58 Å X 64 Å) for 5-LO by Auto Grid algorithm. Molecular docking analyses were visualized by USCF Chimera (University of California, San Francisco, version 1.11.2). After the autodocking, root-mean-square deviations of atomic positions were assessed, and the docked conformations were built on their docking scores and binding energies.
Spectral information
Cistobislactone A
Yellowish oil; UV λmax MeOH (log ε): 284 nm (4.02) (Fig. S24); [α]26D -19.2° (MeOH, c = 0.31); Rf = 0.51 (silica gel GF254; dicholoromethane-MeOH, 8:2); Rt = 2.653 min (C18-RP; MeOH-ACN, 1:1 v/v) (Fig. S25); FT-IR (stretching ν, bending δ, rocking ρ) (νmax, cm−1): 895.5 (alkene C-Hδ), 1058.3 (C-O-Cν), 1457.2 (C-Hρ), 1751.6 (C=Oν), 2926.3, 2854.2 (C-Hν), 3432.1 (OHν) (Fig. S12); 1H NMRMeOD (500 MHz, δH in ppm, J in Hz) (Fig. S2); 13C NMRMeOD (125 MHz) (Fig. S3); 135DEPT, 1H-1H COSY, HSQC, HMBC, NOESY data (Fig. S4–S8, Table 1); HRMS (ESI): found m/z 485.3118 [M + H]+, cal. for C26H45O8 485.3114 (∆ = 0.82 ppm) (Fig. S9); GC-MS (EI): found m/z 484.3 [M]+, cal. for C26H44O8 (Fig. S10).
Cistobislactone B
Yellowish oil; UV λmax MeOH (log ε): 287 nm (3.82) (Fig. S26); [α]26D −18.8° (MeOH, c = 0.21); Rf = 0.48 (silica gel GF254; dicholoromethane-MeOH, 8:2); Rt = 2.675 min (C18-RP; MeOH-ACN, 1:1 v/v) (Fig. S27); FT-IR (νmax, cm−1): 898.5 (alkene C-Hδ), 1050.3 (C–O–Cν), 1457.6 (C–Hρ), 1457.6 (C–Hδ), 1745.5 (C=Oν), 2924.3, 2856.2 (C-Hν), 3431.2 (OHν); 1H NMRMeOD (500 MHz, δH in ppm, J in Hz) (Fig. S13); 13C NMRMeOD (125 MHz) (Fig. S14); 135DEPT, 1H-1H COSY, HSQC, HMBC, NOESY data (Fig. S15–S19, Table 1); HRMS (ESI): found m/z 481.3168 [M + H]+, cal. for C27H45O7 481.3165 (∆ = 0.62 ppm) (Fig. S20); GC-MS (EI): found m/z 480.3 [M]+, cal. for C27H44O7 (Fig. S21).
Statistical analysis
Statistical analysis was performed using the software tool, Statistical Program for Social Sciences 10.0 (SPSS Inc, CA). Triplicate analyses of the independent experiments were performed, and the means were calculated for significant differences (p ≤ 0.05) using analysis of variance (ANOVA).
Data availability
The chromatographic and spectroscopic spectral data are included as supplementary information.
Abbreviations
- 13C NMR:
-
Carbon-13 nuclear magnetic resonance
- 1H NMR:
-
Proton nuclear magnetic resonance
- 2D-NMR:
-
Two-dimensional nuclear magnetic resonance spectroscopy
- 5-LO:
-
5-lipoxygenase
- ACN:
-
Acetonitrile
- ACP:
-
Acyl carrier protein
- CI:
-
Cistopus indicus
- COSY:
-
Correlation spectroscopy
- COX:
-
Cyclooxygenase
- DEPT:
-
Distortionless enhancement by polarization transfer
- DH:
-
Dehydratase
- DPPH:
-
2,2-diphenyl-1-picrylhydrazyl
- EtOAc:
-
Ethyl acetate
- FTIR:
-
Fourier-transform infrared spectroscopy
- GC(EI)MS:
-
Gas chromatography-mass spectrometry (electron ionization)
- HETE:
-
5-hydroxyeicosatetraenoic acid
- HMBC:
-
Heteronuclear multiple bond correlation
- HR(ESI)MS:
-
High resolution mass spectrometry (electrospray ionization)
- HSQC:
-
Heteronuclear single quantum coherence
- Ile:
-
Isoleucine
- Ki :
-
Inhibition constant
- KS:
-
Ketosynthase
- Leu:
-
Leucine
- log POW :
-
Hydrophobic parameter
- LTB4:
-
Leukotriene B4
- LTs:
-
Leukotrienes
- MeOH:
-
Methanol
- MM2:
-
Molecular mechanics 2
- MT:
-
Methyl transferase
- NOEs:
-
Nuclear overhauser effects
- NOESY:
-
Nuclear overhauser effect spectroscopy
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- PGs:
-
Prostaglandins
- Phe:
-
Phenylalanine
- Pl:
-
Polarizability
- Q-TOF LC/MS:
-
Quadruple Time of Flight Liquid Chromatography Mass Spectrometry
- RP-HPLC:
-
Reverse-phase high pressure liquid chromatography
- TLC:
-
Thin layer chromatography
- tPSA:
-
Topological polar surface area
- UV-VIS:
-
Ultraviolet Visible
References
Haeggström JZ, Funk CD. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem Rev. 2011;111:5866–98. https://doi.org/10.1021/cr200246d.
Rainsford KD, Ying C, Smith F. Effects of 5-lipoxygenase inhibitors on interleukin production by human synovial tissues in organ culture: Comparison with interleukin-1-synthesis inhibitors. J Pharm Pharmacol. 1996;48:46–52. https://doi.org/10.1111/j.2042-7158.1996.tb05875.x.
Rouzer CA, Marnett LJ. Cyclooxygenases: Structural and functional insights. J Lipid Res. 2009;50(Suppl):S29–S34. https://doi.org/10.1194/jlr.R800042-JLR200.
Fiorucci S, Meli R, Bucci M, Cirino G. Dual inhibitors of cyclooxygenase and 5-lipoxygenase. A new avenue in anti-inflammatory therapy? Biochem Pharmacol. 2001;62:1433–8. https://doi.org/10.1016/s0006-2952(01)00747-x.
Campbell WC. History of avermectin and ivermectin, with notes on the history of other macrocyclic lactone antiparasitic agents. Curr Pharm Biotechnol. 2012;13:853–65. https://doi.org/10.2174/138920112800399095.
Vardanyan R, Hruby V. Synthesis of best-seller drugs. United States: Academic press, Elsevier; 2016.
Frank AT, Farina NS, Sawwan N, Wauchope OR, Qi M, Brzostowska EM, et al. Natural macrocyclic molecules have a possible limited structural diversity. Mol Divers. 2007;11:115–8. https://doi.org/10.1007/s11030-007-9065-5.
Wessjohann LA, Ruijter E, Garcia-Rivera D, Brandt W. What can a chemist learn from nature’s macrocycles? A brief, conceptual view. Mol Divers. 2005;9:171–86. https://doi.org/10.1007/s11030-005-1314-x.
Ojima K-I, Yangchum A, Laksanacharoen P, Tasanathai K, Thanakitpipattana D, Tokuyama H, et al. Cordybislactone, a stereoisomer of the 14-membered bislactone clonostachydiol, from the hopper pathogenic fungus Cordyceps sp. BCC 49294: Revision of the absolute configuration of clonostachydiol. J Antibiot. 2018;71:351–8. https://doi.org/10.1038/s41429-017-0008-9.
Vidal JP, Escale R, Girard JP, Rossi JC, Chantraine JM, Aumelas A. Lituarines A, B, and C: a new class of macrocyclic lactones from the New Caledonian sea pen Lituaria australasiae. J Org Chem. 1992;57:5857–60. https://doi.org/10.1021/jo00048a017.
Chen XL, Liu HL, Li J, Xin GR, Guo YW. Paracaseolide A, first α-alkylbutenolide dimer with an unusual tetraquinane oxa-cage bislactone skeleton from Chinese mangrove Sonneratia paracaseolaris. Org Lett. 2011;13:5032–5. https://doi.org/10.1021/ol201809q.
Chakraborty K, Krishnan S, Joy M. Macrocyclic lactones from seafood Amphioctopus neglectus: Newly described natural leads to attenuate angiotensin-II induced cardiac hypertrophy. Biomed Pharmacother. 2019;110:155–67. https://doi.org/10.1016/j.biopha.2018.11.034.
Sreeja V, Biju Kumar A, Norman MD. First report of Cistopus taiwanicus Liao and Lu. 2009 (Cephalopoda: Octopodidae) from the Indian coast. J Aquat Biol Fish. 2015;3:98–104.
Central Marine Fisheries Research Institute. Annual Report 2019. http://eprints.cmfri.org.in/id/eprint/14753.
FRAD, CMFRI, 2020. Marine Fish Landings in India 2019. Technical Report. ICAR-Central Marine Fisheries Research Institute, Kochi.
Chakraborty K, Joy M. Anti-diabetic and anti-inflammatory activities of commonly available cephalopods. Int J Food Prop. 2017;20:1655–65. https://doi.org/10.1080/10942912.2016.1217008.
Chakraborty K, Joy M, Raola VK, Makkar F. Angiotensin-1 converting enzyme inhibitory activities of common edible cephalopods and their antioxidative effects using different in vitro models. J Food Biochem. 2017;41:e12268. https://doi.org/10.1111/jfbc.12268.
Salas S, Chakraborty K. An unreported polyether macrocyclic lactone with antioxidative and anti-lipoxygenase activities from the Babylonidae gastropod mollusc Babylonia spirata. Med Chem Res. 2018;27:2446–53. https://doi.org/10.1007/s00044-018-2248-z.
Chakraborty K, Francis P. Stomopneulactone D from long-spined sea urchin Stomopneustes variolaris: Anti-inflammatory macrocyclic lactone attenuates cyclooxygenase-2 expression in lipopolysaccharide-activated macrophages. Bioorg Chem. 2020;103:104140. https://doi.org/10.1016/j.bioorg.2020.104140.
Li J, Zhang S, Zhang H, Wang H, Zhang J, Chen A, et al. Isolation and identification of new macrocyclic lactones from a genetically engineered strain Streptomyces bingchenggensis BCJ60. J Antibiot. 2017;70:297–300. https://doi.org/10.1038/ja.2016.130.
Zidar N, Odar K, Glavac D, Jerse M, Zupanc T, Stajer D. Cyclooxygenase in normal human tissues-is COX-1 really a constitutive isoform, and COX-2 an inducible isoform? J Cell Mol Med. 2009;13:3753–63. https://doi.org/10.1111/j.1582-4934.2008.00430.x.
Subbarao K, Jala VR, Mathis S, Suttles J, Zacharias W, Ahamed J, et al. Role of leukotriene B4 receptors in the development of atherosclerosis: Potential mechanisms. Arterioscler Thromb Vasc Biol. 2004;24:369–75. https://doi.org/10.1161/01.ATV.0000110503.16605.15.
Griffiths RJ, Pettipher ER, Koch K, Farrell CA, Breslow R, Conklyn MJ, et al. Leukotriene B4 plays a critical role in the progression of collagen-induced arthritis. Proc Natl Acad Sci U S A. 1995;92:517–21. https://doi.org/10.1073/pnas.92.2.517.
Lordan R, Tsoupras A, Zabetakis I. Inflammation (chapter 2). In: Zabetakis I, Lordan R, Tsoupras A, editors. The Impact of Nutrition and Statins on Cardiovascular Diseases. Ireland: Academic Press; 2019; ISBN 9780128137925:23-51. https://doi.org/10.1016/B978-0-12-813792-5.00002-1.
Basil MC, Levy BD. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat Rev Immunol. 2016;16:51–67. https://doi.org/10.1038/nri.2015.4.
Lipinski C, Hopkins A. Navigating chemical space for biology and medicine. Nature. 2004;432:855–61. https://doi.org/10.1038/nature03193.
Larsen LN, Dahl E, Bremer J. Peroxidative oxidation of leucodichloroluorescein by prostaglandin H synthase in prostaglandin biosynthesis from polyunsaturated fatty acids. Biochim Biophys Acta. 1996;1299:47–53. https://doi.org/10.1016/0005-2760(95)00188-3.
Maneesh A, Chakraborty K, Makkar F. Pharmacological activities of brown seaweed Sargassum wightii (family Sargassaceae) using different in vitro models. Int J Food Prop. 2017;20:931–45. https://doi.org/10.1080/10942912.2016.1189434.
Gulcin I. Comparison of in vitro antioxidant and antiradical activities of L-tyrosine and L-dopa. Amino Acids. 2007;32:431–8. https://doi.org/10.1007/s00726-006-0379-x.
Chew YL, Lim YY, Omar M, Khoo KS. Antioxidant activity of three edible seaweeds from two areas in South East Asia. LWT. J Food Sci Technol. 2008;41:1067–72. https://doi.org/10.1016/j.lwt.2007.06.013.
Maneesh A, Chakraborty K. Previously undescribed fridooleanenes and oxygenated labdanes from the brown seaweed Sargassum wightii and their protein tyrosine phosphatase-1B inhibitory activity. Phytochemistry. 2017;144:19–32. https://doi.org/10.1016/j.phytochem.2017.08.011.
Borbulevych OY, Jankun J, Selman SH, Skrzypczak-Jankun E. Lipoxygenase interactions with natural flavonoid, quercetin, reveal a complex with protocatechuic acid in its X-ray structure at 2.1 A resolution. Proteins. 2004;54:13–19. https://doi.org/10.1002/prot.10579.
Acknowledgements
The authors gratefully acknowledge the funding by the Indian Council of Agricultural Research (ICAR, New Delhi, India) (Grant number MBT/HLT/SUB23). The authors are grateful to the Director, ICAR-CMFRI, and Head, Marine Biotechnology Division of ICAR-CMFRI for facilitating the research activities. The authors are thankful to the Chairman, Department of Chemistry, Mangalore University (Karnataka, India) for providing the necessary support.
Author contributions
SKP and KC designed research, conducted experiments, and analyzed data. KC acquired funds and conceptualized the work. SKP drafted the manuscript. KC reviewed and edited the manuscript. All authors read and approved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
About this article

Cite this article
Paulose, S.K., Chakraborty, K. Cistobislactones A-B, two sixteen-membered spiro-linked macrocylic bislactones from marine octopus Cistopus indicus: new anti-inflammatory agents attenuate arachidonate 5-lipoxygenase. Med Chem Res 30, 2042–2054 (2021). https://doi.org/10.1007/s00044-021-02790-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-021-02790-x