
Abstract
In this study, we present the synthesis of novel isothiochromenes, thiazolidonone, thiazinone, arylidines, triazoles, and pyrimidinone compounds based on the starting material 3-amino- isothiochromene-4-carbonitrile 3. The chemical structures were confirmed using spectroscopic methods and elemental analyses. These compounds were screened for their in vitro antiviral and antitumor activities. Compounds 10a-c and 22a-b showed activity against herpes simplex virus-1 (HSV-1) and human immunodeficiency virus-1 (HIV-1). Compounds 15 and 21a-b exhibited activity against various types of cancer cell lines.

Compounds 22a-b and 10a-c showed to be active against herpes simplex virus-1 (HSV-1) and virus-1 (HIV-1). Also, the products 21a-b and 15 proved to a wide extent antitumor activity
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Many biological activities in the human body are modulated by heterocyclic compounds such as nucleic acids, hormones, and neurotransmitters, etc. Heterocycles also play a crucial role in drug discovery where they are involved in many therapeutic areas. One of those heterocyclic compounds, the pyrimidine moiety, is an essential component of many classes of drugs, such as antineoplastic (Uramustine), antibacterial (Trimethoprim), antifungal (Flucytosine), and antivirals (Broxuridine) (Selvam et al. 2012; Abu-Hashem and Hussein 2015).
Substituted thiopyrans are heterocycles that exhibit an extensive range of biological activities such as anti-inflammatory (Rogier et al. 2001), antibacterial (Brown et al. 2002), effective antagonists at α1- adrenoreceptor (Quaglia et al. 2002), and dopamine D3 receptor-selective agonists (Van Vliet et al. 2000). Furthermore, isothiochromene-based compounds are reported to exhibit antitubercular, anti-HIV-1 (Bennani et al. 2007), anticancer and antitrypanosomal activities (Kaminskyy et al. 2014)
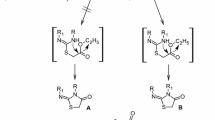
Also, thiazolopyrimidine compounds are shown to have activities as acetylcholinesterase inhibitors (Zhi et al. 2008). Meanwhile, thiazine-based compounds are known to have antimicrobial (Ram et al. 2013), antiviral (Galal et al. 2011), anti-inflammatory, analgesic, and ulcerogenic activities (Vijay 2011). Likewise, 1, 2, 4-triazolopyrimidine derivatives displayed numerous pharmacological activities as antimicrobial (Abu-Hashem et al. 2017), anti-inflammatory, analgesic (Sung and Lee 1992), and antimalarial (Havaldar and Patil 2008) agents. Also, 1, 2, 3-triazine is an interesting class of heterocyclic compounds. They are known to have pharmacological activity and may be considered as lead molecules for the development of future drugs (Kumar et al. 2014) for examples: Tubercidin (a) is used to preventing the growth of bacteria. Toyocamycin (b) is recognized as antibiotic and antineoplastic. Sangivamycin (c) is active against leukemia, lung carcinoma, against colon, and gallbladder cancer in humans. 2-Aza-adenosine (d) exhibits the highest cytotoxicity against epidermoid carcinoma cells (Migawa et al. 2005), (Fig. 1).

Biologically active 1, 2, 3-triazine derivatives
Pyrrolopyrimidines are a valuable group of bioactive compounds showing antivirals, antiprotozoal, antibacterial and anticancer (De Coen et al. 2016), anti-hyperglycemic activities (Mohamed et al. 2014) and anti-cancer agents (Dholakia et al. 2015). Based on the potential biological activities of these heterocycles we synthesized new pyrimidine-, thiazolopyrimidine-, pyrimidothiazine-, triazine-, triazolo- pyrimidine-, pyrrolopyrimidine, and pyridine-based compounds incorporating isothiochromene in order to investigate their antiviral and cytotoxic activities. Thus, it is a real challenge to combine the aforementioned rings together in a molecular framework to make use of the additive effect of these rings toward the antiviral and cytotoxic activities.
Results and discussion
Chemistry
The synthetic strategies, adopted to obtain the poly-functional substituted isothiochromenes, are shown in Schemes 1–4. Compound 3 was prepared via Gewald reaction (Gewald et al. 1966) of 2, 6-bis (pyridin-4-ylmethylene) cyclohexan-1-one 1 (Somers-Edgar et al. 2009) with malononitrile and elemental sulfur in ethanol containing diethylamine. The IR spectrum of compound 3 showed absorption bands at ν 3420 (NH2), 2212 (CN) cm−1. Furthermore, the 1H NMR spectrum of 3 exhibited two singlet signals at δ 4.62, 6.28, and 8.23 ppm for one proton (C1-H) of the thiopyran ring and the amino group (as D2O exchangeable) and the methine proton respectively. Moreover, its mass spectrum revealed the ion peak at m/z 358 (M+, 100 %), which agrees with the molecular formula (C21H18N4S). Compound 3 reacted with carbon disulfide in ethanol and in the presence of aqueous potassium hydroxide according to the reported condition by (Rashad et al. 2005) to afford the pyrimidine-1, 3(2H)-dithione (5) through the intermediate 4. The IR spectrum of compound 5 revealed the absorption bands at ν 3395 (NH), 1620 (C=N) and 1355, 1370 cm−1 (2 C=S). Moreover, its 1H NMR spectrum exposed two singlet signals at δ 9.19 and 9.21 ppm due to (2NH). Subsequently, compound 5 reacted with chloroacetic acid in the presence of Ac2O/AcOH to give the isothiochromeno[3,4-d]thiazolo[3,2-a]pyrimidin-9(10H)-one 8, the formation of 8 can be explained via nucleophilic addition of the thiol group of 1, 3(2H)-dithione 5 to in-situ formed chloroacetic anhydride 6, followed by cyclization of the formed intermediate 7. Knovenagel condensation of compounds 8 with the appropriate aromatic aldehydes derivatives namely, benzaldehyde, or 4-chlorobenzaldehyde or 4-methoxybenzaldehyde afforded the isothiochromeno[3,4-d]thiazolo[3,2-a]pyrimidin-9(10H)-one 10a-c. The IR spectra of compounds 8 displayed a characteristic absorption band at ν 1710 due to the carbonyl group of thiazolone ring respectively. The 1H NMR spectrum of 8 exhibited multiplet signals at δ 1.19–1.69 ppm corresponding to six protons of cyclohexene (3CH2) and two singlet signals 4.18 and 8.07 ppm due to two protons of the thiazolidinone moiety (CH2) and the methine proton of thiopyran ring, respectively. Furthermore, the 1H-NMR spectra of 10a-c displayed the disappearances of the singlet signal for two protons of the thiazolidinone moiety (CH2) (Scheme 1).

Synthesis of thiazolopyrimidines 8 and 10a–c. Reagents and conditions: (i) Et2NH/EtOH, reflux, 80–90 °C, 7–9 h. (ii) KOH, stirring, 13–15 h. (iii) Ac2O/AcOH, reflux, 7–10 h. (iv) AcOH, reflux, 12–14 h

Synthesis of pyrimido[2,1-b][1,3] thiazin-9-ones 13 and, 14a–c. Reagents and conditions: (i) Ac2O/AcOH, reflux, 7–10 h. (ii) AcOH, reflux, 12–14 h

Synthesis of 1-chloro-6H-isothiochromeno[3,4-d][1,2,3]triazine 15, 3H-isothio- chromeno[3,4-d]pyrimidine 18a,b and 8H-isothiochromeno[4,3-e][1,2,4]triazolo[1,5-c] pyrimidine 22a, and 22b. Reagents and conditions: (i) NaNO2, HCl/ AcOH, stirring, 4 h. (ii) pyridine, reflux, 10–12 h. (iii) Ac2O, reflux, 10–12 h. (iv) dioxane, reflux, 6–8 h. (v) reflux, 4 h

Synthesis of pyrrolo[3,2-d]pyrimidine 26 and 32, and isothiochromeno[3,4-b]pyrrole 30 and 31. Reagents and conditions: (i) K2CO3, Acetone, reflux, 10 h. (ii) K2CO3, DMF, reflux, 8 h. (iii) KSCN, HCl, reflux, 7 h. (iv) EtOH/EtONa, reflux, 10 h. (v) EtOH/ EtONa, reflux, 10–12 h
Similarly, 10-(sub.-benzylidene)-5-(pyridin-4-yl)-1-(pyridin-4-ylmethylene)-13-thioxo-1, 3, 4, 5, 10, 11-hexahydro-2H,9H,13H-isothiochromeno[3’,4’:4,5]pyrimido[2,1-b][1,3]thiazin-9-one 14a-c, were prepared via the reaction of 5 with 3-bromopropanoic acid, followed by the condensation of the formed pyrimido[2,1-b][1,3] thiazin-9-one derivative 13 with the same aldehydes 9a–c. The IR spectrum of compound 13 revealed a characteristic absorption bands at ν 1718 cm−1 due to the carbonyl group of the thiazinone ring. Furthermore, the mass spectra of 14a–c showed the molecular ion peaks at m/z 576 (M+, 98 %), 611 (M+, 100 %), 606 (M+, 90 %), respectively (Scheme 2).
The treatment of 2-aminoisothiochromene 3 with sodium nitrite solution in the presence of the concentrated hydrochloric acid generated the 1-chloro-6H-isothiochromeno[3,4-d][1,2,3]triazine 15, 1H NMR spectrum of 15 displayed two signals at δ 4.42 (s, 1H, thiopyran), and δ 8.23 (s, 1H, C=CHAr), respectively. Its mass spectrum showed the molecular ion peak at m/z 405 (M+, 100 %), which is in agreement with the molecular formula (C21H16ClN5S). Furthermore, the 3H-isothio- chromeno[3,4-d]pyrimidine-3-thione 18a and the 3H-isothiochromeno[3,4-d]pyrimidin-3-one 18b were prepared via refluxing of compound 3 with phenylisothiocyanate 16a or phenyl isocyanate 16b in of pyridine, respectively through the formation of the intermediate 17a,b. IR spectra of 18a and 18b indicated the presence of three absorption bands at ν 3250, 3220, and 1275 cm−1 corresponding to 2NH and (CS) groups. The 1H NMR spectrum of compound 18a exhibited two characteristic broad singlet signals at δ 10.58 and 10.62 corresponding to the two protons of 2NH groups. Moreover, condensation of 3 with triethyl orthoformate 19a or triethyl orthoacetate 19b in refluxing acetic anhydride gave the ethylformimidate derivative (20a) and the ethylacetimidate (20b), respectively. Compounds 20a or 20b undergo cyclic condensation upon treating with phenylhydrazine in dioxane affording the 1H-isothiochromeno[3,4-d]pyrimidin-2(6H)-amines (21a) and (21b) respectively. The 1H NMR spectrum of compound (21a) displayed two broad singlet signals at δ 9.35 and 10.58 ppm due to (2NH) protons. The mass spectra of 20a, 20b, 21a, and 21b revealed the molecular ion peaks at m/z 414 (M+, 100 %), 428 (M+, 90 %), 476 (M+, 100 %), 490 (M+, 88 %). Subsequently, compounds 21a, and 21b underwent cyclization upon heating under reflux in excess of triethyl orthoformate 19a to achieve the 8H-isothiochromeno[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine 22a, and 22b, respectively. 1H NMR spectrum of 22a showed triplet and quartet signals at δ 1.31 and δ 4.22 ppm due to the ethyl group, and four singlet signals at δ 4.72 (s, 1H, thiopyran), 6.34 (s, 1H, CHOEt, triazole ring), 8.13 (s, 1H, C=CHAr), and 8.84 (s, 1H, N=CH), (Scheme 3)
The cyclocondensation of compound (3) with ethyl bromoacetate in dry acetone and anhydrous potassium carbonate gave the ethyl glycinate 23, which underwent cyclization to the corresponding ethylisothiochromeno[3,4-b]pyrrole-2-carboxylate 24 upon refluxing in dimethylformamide. The structure of 23 and 24 was recognized through the spectral and elemental analysis. The IR spectrum of 24 exhibited three characteristic absorption bands at ν 3405, 3280 and 1735 cm−1 due to the presence of NH2, NH and carbonyl group. Hence, compound 24 reacted with potassium thiocyanate to give the ethylisothiochromeno [3,4-b] pyrrole-2-carboxylate 25, which cyclized via refluxing in ethanol in the presence of sodium ethoxide to achieve the corresponding isothiochromeno[4′,3′:4,5] pyrrolo[3,2-d] pyrimidin-8(2H)-one 26. The 1H-NMR spectra of (26) revealed three broad singlet signals at δ 10.35, 12.32, 13.13 ppm due to the presence of 3NH groups. The mass spectra of compounds 25 and 26 indicated the molecular ion peaks at m/z 503 (M+, 90 %), 457 (M+, 99 %), Finally, the treatment of compound 3 with 2-chloroacetamide or phenacyl bromide or ethyl cyanoacetate was refluxed in sodium ethoxide solution afforded the corresponding isothiochromeno [3,4-b]pyrrole-2-carboxamide 30, isothiochromeno[3,4-b]pyrrol-2-yl) phenyl methanone 31 and isothiochromeno[3,4-b]pyridine-2-carbonitrile 32, respectively. IR spectrum of compound 30 exposed the absorption band at ν 3410-3250 (br) and 1675 cm−1 due to the 2NH2, NH, and the carbonyl groups. Meanwhile, the 1H-NMR spectrum of 31 showed two characteristic singlet signals at δ 6.43 and δ 12.67 ppm corresponding to the two protons of NH2 and the one proton of NH groups. Furthermore, the 13C-NMR of 32 showed signals at δ 15.7, 21.6, 31.2 ppm for three carbon atoms of (3CH2, cyclohexene), 65.6, 88.9 ppm for two carbon atoms of thiopyran ring, 94.1 ppm for one carbon atom of pyridine ring, 102.4 ppm for one carbon atom of cyano group, 111.9 ppm for one carbon atom of (CH), 165.7 ppm for one carbonyl group, and 172.1 ppm for one carbon atom of (C-NH2). The MS of 30, 31, and 32 showed the molecular ion peaks at m/z 415 (M+, 90 %), 476 (M+, 95 %), 425 (M+, 92 %), respectively (Scheme 4).
Biological screening
In vitro anti-Herpes Simplex-1 Virus activity
The in vitro Anti-Herpes Simplex-1 Virus (HSV-1) for the isothiochromene derivatives were evaluated with the method reported by (El-Subbagh et al. 2000; Hufford et al. 1991) where Aphidicolin (0.001 µg/mL) was used as a positive control. The results in (Table 1) revealed that compounds 10a, 10b, 10c, 14b, 22a, and 22b displayed the highest activity among the tested compounds where they decrease the number of viral plaques by 82, 88, 85, 80, 90, and 95 % respectively. The remarkable anti-HSV-1 activity of these compounds may be attributable to the presence of the 3-(4-substitutedbenzylidene)-5-thioxo-3H-thiazolo[3,2-a]pyrimidin-2(5H)-one, 3-(4-chlorobenzylidene)-6-thioxo-pyrimido[2,1-b][1,3]thiazin-2(6H)-one, and 2-ethoxy-5-substituted-3-phenyl-[1,2,4]triazolo[1,5-f]pyrimidine moieties, respectively (El-Subbagh et al. 2000).
In vitro anti-Human Immunodeficiency-1 Virus activity
The effect of the newly synthesized isothiochromene and the azidothymidine (AZT) drug on acutely and persistently infected (T4) cell lines in vitro were evaluated using the method reported by (Chen et al. 1992; Weislow et al. 1989). The method used to estimate the anti-Human Immunodeficiency-1 Virus (HIV-1) effectiveness is intended to discover agents acting at any phase of the virus procreative cycle. The result in (Table 2) displayed that the compounds 10a-c, 22a, and 22b have moderate activity against (HIV-1) cytopathic effect. Whereas; compounds 3, 5, 8, 13, 14a, 14b, 14c, 15, 18a, 18b, 20a, 20b, 21a, 21b, 23, 24, 25, 26, 30, 31, and 32 are inactive.
Antitumor screening (in vitro cytotoxicity)
The isothiochromene derivatives were tested for their in vitro cytotoxicity using the standard MTT method (Mosmann 1983; Denizot and Lang 1986; Thabrew et al.1997) against the human oral carcinoma cells (KB), nasopharyngeal carcinoma cells (CNE2), breast adenocarcinoma cells (MCF-7), and gastric carcinoma cells (MGC-803). The MTT method is based on the reduction of soluble 3-(4, 5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2H-tetrazolium bromide. The result in (Table 3) revealed that the isothiochromenes 15, 21a, 21b, 22a, and 22b exhibited high cytotoxicity against all carcinoma cell lines as follows; the KB (IC50 11.9, 11.7, 11.5, 12.5, and 12.2 µM), the CNE2 (IC50 13.3, 12.9, 12.6, 13.8, and13.5 µM), the MCF-7 (IC50 13.1, 12.8, 12.4, 13.9, and 13.4 µM), the MGC-803 (IC50 12.4, 12.2, 11.9, 13.2, and 12.9 µM), respectively. Also, some compounds as; 5, 18a, 18b, 26, and 32 showed moderate cytotoxicity against the carcinoma cell lines and the rest of the compounds showed weak cytotoxicity activities.
Structural activity relationship
By comparing the observed antiviral and cytotoxic activities of the isothiochromenes obtained in this study to their structures, the (SAR’s) were postulated. (I) the presence of the isothiochromen moiety may be necessary for the broad spectrum of the antiviral and cytotoxicity activity. (II) 8H-isothiochromeno[4,3-e][1,2,4]triazolo[1,5-c] pyrimidines 22a-b exhibited high in vitro anti-herpes simplex -1 virus activity and moderate anti-human immunodeficiency-1 virus activity. The 1, 2, 4 triazole template, either fused with a pyrimidine ring (Abu-Hashem et al. 2017) or not (Sung and Lee 1992; Havaldar and Patil 2008) display remarkable biological activity, thus the noticeable behavior of 22a-b may be attributed to the presence of the triazolo[1,5-c]pyrimidine moiety. (III) Compound 10a–c and 14a–c are more potent than compound 8 and 13: a comparison of the corresponding structures leads to the conclusion that this fact may be attributed to the presence of the arylidene moiety in the more active molecules. (IV) Compounds 15, 21a-b, and 22a-b exhibited high cytotoxicity against all carcinoma cell lines. The 1, 2, 3-triazine scaffold from 15 has a potent biological activity (Kumar et al. 2014). The iminopyrimido moiety from compounds 21 was already identified as presenting in vitro antitumor activity against human breast cancer cells MCF-7 (El-Ashmawy et al. 2013). Thus, the remarkable antineoplastic activity of the compounds may be attributable to the presence of the iminopyrimidine, chlorotriazine, and triazolo-pyrimidine moieties, respectively.
Conclusions
The objective of the present study is to synthesize and evaluate the antiviral and cytotoxic activities of some new isochromene-based compounds with the hope of discovering new structure leads serving as antiviral or antitumor agents. The data revealed that isothiochromenotriazolopyrimidines 22a,b possess promising in vitro antiviral activity against HSV-1 and HIV-1. While isothiochromenotriazine 15 and isothiochromenopyrimidin-2(6H)-amine21a, 21b showed wonderful cytotoxic activity against carcinoma cell lines when compared to the 5-fluorouracil drug.
Experimental
Materials, equipment’s, and methods
All materials used were obtained from Sigma Aldrich (Saint Lewis, USA). The melting points are in degree centigrade (uncorrected) and were determined on Gallenkamp electric melting point apparatus. TLC analysis was carried out on silica gel 60 F254 precoated aluminum sheets. The IR spectra were recorded (KBr) on a Perkin–Elmer 1430 spectrometer (λ, cm−1) in National Research Center, Egypt. 1H and 13C-NMR spectra were measured on JEOL-ECA 500 and JEOL JNM-LA-400 FT NMR Spectrometers at 500/400, 125/100 MHz, respectively, using tetramethylsilane (TMS) as an internal reference and DMSO-d6 as the solvent at the Microanalytical Center in National Research Center, Egypt. The mass spectra (EI) were recorded on GCMS-QP 1000 EX (Shimadzu) at National Research Center, Egypt. Elemental analyses (C, H, and N) were carried out at the Microanalytical Center in National Research Center, Egypt. The elemental analyses were found to agree favorably with the calculated values. The synthesized compounds were tested in vitro for their antiviral activity using HSV-1, HIV-1 and cytotoxicity activity at the Pharmacology Unit, Department of Pharmacognosy, College of Pharmacy, University of Mansoura, Mansoura, Egypt and National Cancer Institute, Cairo University, Cairo, Egypt.
Synthesis of 3-amino-1-(pyridin-4-yl)-5-(pyridin-4-ylmethylene)-5, 6, 7, 8-tetrahydro-1H-isothiochromene-4- carbonitrile (3)
A mixture of 2, 6-bis(pyridin-4-ylmethylene) cyclohexan-1-one (2.76 g, 0.01 mol), malononitrile (0.66 g, 0.01 mol), sulfur (0.32 g, 0.01 mol), and diethylamine (0.01 mol) in ethanol (30 mL) was heated at 80–90 °C for 7–9 h, then the mixture was left for 36 h at 0 °C. The formed product was collected by filtration, washed with ethanol (25 mL), dried and crystallized from absolute methanol, as yellow crystals (90%), mp 290–292 °C. IR (ν, cm−1) KBr: 3420 (br. NH2), 3050 (CH-aryl), 2952 (CH-aliph), 2212 (CN), 1624 (C=N). 1H NMR (DMSO-d6, ppm) δ 1.57–2.23 (m, 6H, 3CH2, cyclohexene), 4.62(s, 1H, thiopyran), 6.28 (brs, NH2, D2O exchangeable),7.56–7.96 (m, 8H, Ar-H), 8.23 (s, 1H, C=CHAr), 13C NMR (DMSO-d6) δ 15.6, 21.5, 31.2 (3 C, CH2), 65.5, 88.9 (2 C, thiopyran),111.9 (CN), 116.2 (1 C, C=CHAr), 121.4, 124.8, 130.2, 130.8, 130.9, 131.5, 136.1, 147.7, 158.3 (13 C, Ar-C), 165.6 (C-NH2); MS (70 eV, %) m/z 358 (M+, 100 %); Anal. Calc. (Found) for C21H18N4S (358.46): C, 70.36 (70.45); H, 5.06 (5.14); N, 15.63(15.72); S, 8.94(8.51).
Synthesis of 6-(pyridin-4-yl)-10-(pyridin-4-ylmethylene)-4, 6, 7, 8, 9, 10-hexahydro-1H-isothio chromeno[3, 4-d]pyrimidine-1,3 (2H)-dithione (5)
A mixture of compound 3 (3.58 g, 0.01 mol), and carbon disulfide (excess 15 mL) was heated under reflux in ethanolic potassium hydroxide solution for 15 h. The solution was allowed to cool to 0 °C for 5 h; the solid precipitate was filtered off, washed with water (30 mL), dried and crystallized from dimethylformamide to give (5) as a yellow crystals (85%); mp 315–317 °C. IR (ν, cm−1) KBr: 3395 (brs, NH), 3055 (CH-aryl), 2950 (CH-aliph), 1620 (C=N), 1355, 1370 (2 C=S).1H NMR (DMSO-d6, ppm) δ 1.26–2.35 (m, 6H, 3CH2, cyclohexene), 4.29 (s,1H, thiopyran),7.45–8.02 (m, 8H, Ar-H), 8.27 (s, 1H, C=CHAr), 9.19, 9.21 (brs, 2NH, D2O exchangeable), 13C NMR (DMSO-d6) δ 16.8, 22.5, 32.5 (3 C, CH2), 60.2, 90.8 (2 C, thiopyran), 114.7 (1 C, C=CHAr), 122.9, 123.8, 132.5, 133.9, 142.4, 145.5, 146.8, 148.7, 149.1,158.6 (14 C, Ar–C), 168.5, 185.4 (2 C, 2 C=S); MS (70 eV, %) m/z 434 (M+, 100 %); Anal. Calc. (Found) for C22H18N4S3 (434.59): C, 60.80 (60.75); H, 4.17 (4.25); N, 12.89 (12.94); S, 22.13 (22.06).
Synthesis of 5-(pyridin-4-yl)-1-(pyridin-4-ylmethylene)-12-thioxo-1,3,4,5,7,7a-hexa- hydro-2H,12H-isothiochromeno[3,4-d]thiazolo[3,2-a]pyrimidin-9(10H)-one (8) and 5-(pyridin-4-yl)-1-(pyridin-4-ylmethylene)-13-thioxo-1,3,4,5,7,7a,10,11-octahydro-2H,9H,13H-isothiochromeno [3′,4′:4,5] pyrimido[2,1-b][1,3]thiazin-9-one (13)
General method: A mixture of compound 5 (4.34 g, 0.01 mol), and chloroacetic acid (0.93 g, 0.01 mol) or β-bromopropionic acid (1.52 g, 0.01 mol) was heated under reflux in a solution of glacial acetic acid and acetic anhydride (8 ml, 3:1 V) in the presence of sodium acetate anhydrous (0.82 g, 0.01 mol) for 7–10 h. The reaction mixture was into crashed ice water. The formed precipitate was washed with water and filtered off, dried, and recrystallized from the suitable solvent to give 8 and 13 respectively.
Compound 8: Yellowish crystals, yield (75%), mp 328–330 °C, crystallized from EtOH; IR (ν, cm−1) KBr: 3050 (CH-aryl), 2954 (CH-aliph), 1710 (C=O), 1624 (C=N), 1325 (C=S), 1H NMR (DMSO-d6, ppm) δ 1.19–1.69 (m, 6H, 3CH2, cyclohexene), 4.18 (s, 2H, CH2, thiazolidinone), 4.62 (s, 1H, thiopyran), 7.10–7.74 (m, 8H, Ar-H), 8.07 (s, 1H, C=CHAr), 13C-NMR (DMSO-d6) δ 15.7, 21.6, 31.2 (3 C, CH2), 47.7 (1 C, thiopyran), 89.1 (1 C, CH2, thiazole),111.8 (1 C, C=CHAr), 116.2, 121.5, 124.8, 130.3, 131.5, 135.8, 136.1, 136.9, 140.1, 140.2, 140.3, 140.5 (16 C,Ar-C), 165.6 (1 C, C=O), 172.5 (1 C, C=S); MS (70 eV, %) m/z 474 (M+, 100 %); Anal. Calc. (Found) for C24H18N4OS3 (474.62): C, 60.74 (60.78); H, 3.82 (3.75); N, 11.80 (11.90); S, 20.26 (20.20).
Compound 13: Yellow crystals, yield (70%), mp 338–340 °C, crystallized from MeOH; IR (ν, cm−1) KBr: 3058 (CH-aryl), 2960 (CH-aliph), 1718 (C=O), 1621 (C=N), 1322 (C=S). 1H NMR (DMSO-d6, ppm) δ 1.01 (t, 2H, J = 3.70 Hz, CH2CH2S, thiazine), 1.75–2.41 (m, 6H, 3CH2, cyclohexene), 3.13 (t, 2H, J = 3.74 Hz, CH2CH2S, thiazine), 4.51 (s, 1H, thiopyran), 7.05–8.07 (m, 8H, Ar-H), 8.09 (s, 1H, C=CHAr),13C NMR (DMSO-d6) δ 17.9, 23.6, 30.5 (3 C, CH2), 55.8 (1 C, thiopyran), 48.5 CH2CH2S, thiazine, 50.5 (CH2CH2S, thiazine), 113.9 (1 C, C=CHAr), 123.8, 124.5, 128.4, 132.7, 133.9, 143.5, 145.6, 147.8, 149.4, 149.9, 155.7, 158.7 (16 C,Ar-C),169.2 (1 C, C=O), 180.1(1 C, C=S); MS (70 eV, %) m/z 488 (M+, 100 %); Anal. Calc. (Found) for C25H20N4OS3 (488.64): C, 61.45 (61.54); H, 4.13 (4.18); N, 11.47 (11.40); S, 19.68 (19.61).
Synthesis of 10-(4-substitute-benzylidene)-5-(pyridin-4-yl)-1-(pyridin-4-ylmethylene)-12-thioxo-1,3,4,5-tetrahydro -2H,12H-isothiochromeno[3,4-d]thiazolo[3,2-a]pyrimidin-9(10H)-one(10a-c) and 10-(4-substituted-benzylidene)-5-(pyridin-4-yl)-1-(pyridin-4-ylmethylene)-13-thioxo-1,3,4,5,10, 11-hexahydro-2H,9H,13H-isothiochromeno[3’,4’:4,5]pyrimido[2,1-b][1,3] thiazin-9-one (14a-c)
General method: A mixture of compound 8 (4.74 g, 0.01 mol) or compound 13 (4.88 g, 0.01 mol), and aromatic aldehyde derivatives, namely: benzaldehyde (1.06 g, 0.01 mol) or 4-chloro- benzaldehyde (1.40 g, 0.01 mol) or 4-methoxybenzaldehyde (1.36 g, 0.01 mol), respectively was refluxed in a solution of glacial acetic acid (10 mL) and anhydrous sodium acetate anhydrous (0.82 g, 0.01 mol) for 12–14 h. The reaction mixture was into crashed ice water and the formed precipitate product was collected by filtration and crystallized from the appropriate solvent to afford 10a–c and 14a–c, respectively.
10-(benzylidene)-5-(pyridin-4-yl)-1-(pyridin-4-ylmethylene)-12-thioxo-1, 3, 4, 5-tetrahydro-2H, 12H-isothiochromeno[3,4-d]thiazolo[3,2-a]pyrimidin-9(10H)-one(10a)
Yellowish crystals, yield (60%), mp > 340 °C, crystallized from EtOH; IR (ν, cm−1) KBr: 3060 (CH-aryl), 2958 (CH-aliph), 1720 (C=O),1625(C=N), 1328 (C=S), 1H NMR (DMSO-d6, ppm) δ 1.19–1.69 (m, 6H, 3CH2, cyclohexene), 4.72 (s, 1H, thiopyran),7.10–8.06 (m, 13H, Ar-H), 8.13 (s, 1H, C=CHAr), 8.23 (s, 1H, C=CHAr),13C NMR (DMSO-d6) δ 18.7, 23.6, 33.2 (3 C, CH2), 44.9 (1 C, thiopyran), 110.5, 112.8 (2 C, 2 C=CHAr), 123.4, 124.6, 127.2, 127.9, 128.2, 128.5, 133.2, 133.9, 134.9, 144.1, 146.3, 147.5, 149.2, 149.9, 151.5, 155.4, 158.9 (23 C, Ar–C), 179.8 (1 C, C=O), 186.2(1 C, C=S); MS (70 eV, %) m/z 562 (M+, 100 %); Anal. Calc. (Found) for C31H22N4OS3 (562.72): C, 66.17 (66.24); H, 3.94 (3.90); N, 9.96 (9.92); S, 17.09 (17.15).
10-(4-chlorobenzylidene)-5-(pyridin-4-yl)-1-(pyridin-4-ylmethylene)-12-thioxo-1,3,4,5-tetrahydro-2H,12H-isothiochromeno[3,4-d]thiazolo[3,2-a]pyrimidin-9(10H)-one(10b)
Yellow crystals, yield (65%), mp > 340 °C, crystallized from hexane; IR (ν, cm−1) KBr: 3064 (CH-aryl), 2960 (CH-aliph), 1628(C=N), 1330 (C=S), 1724 (C=O).1H NMR (DMSO-d6, ppm) δ 1.21–1.73 (m, 6H, 3CH2, cyclohexene), 4.62 (s, 1H, thiopyran),7.10–7.95(m, 12H, Ar-H), 8.09 (s, 1H, C=CHAr), 8.18 (s, 1H, C=CHAr),13C NMR (DMSO-d6) δ 15.6, 21.6, 31.2 (3 C, CH2), 65.6 (1 C, thiopyran), 107.8, 111.9 (2 C, 2 C=CHAr), 116.8, 118.2, 121.5, 124.8, 130.2, 130.8, 130.9, 131.5, 136.1, 141.7, 143.6, 147.7, 149.5, 153.6, 158.4, 165.7, 168.2 (23 C, Ar–C), 173.8 (1 C, C=O), 177.9 (1 C, C=S); MS (70 eV, %) m/z 597 (M+, 90 %); Anal. Calc. (Found) for C31H21ClN4OS3 (597.17): C, 62.35 (62.40); H, 3.54 (3.59); N, 9.38 (9.33); S, 16.11 (16.18).
10-(4-methoxybenzylidene)-5-(pyridin-4-yl)-1-(pyridin-4-ylmethylene)-12-thioxo-1,3,4,5-tetra hydro-2H,12H-isothiochromeno[3,4-d]thiazolo[3,2-a]pyrimidin-9(10H)-one(10c)
Brownish crystals, yield (62%), mp > 340 °C, crystallized from benzen; IR (ν, cm−1) KBr: 3059 (CH-aryl), 2957 (CH-aliph), 1722 (C=O), 1626 (C=N), 1328 (C=S), 1H NMR (DMSO-d6, ppm) δ 1.22–1.75 (m, 6H, 3CH2, cyclohexene), 3.85 (s,3H, CH3), 4.67 (s, 1H, thiopyran),7.12–7.94 (m, 12H, Ar-H), 8.08 (s, 1H, methine proton), 8.15 (s, 1H, C=CHAr),13C NMR (DMSO-d6) δ 19.1, 23.4, 31.3 (3 C, CH2), 44.9 (1 C, thiopyran), 56.7(1 C,OCH3), 120.8, 121.9 (2 C, 2CH C=CHAr), 122.8, 123.1, 124.2, 127.4, 129.1, 130.4, 133.5, 134.6, 144.2, 146.7, 147.5, 149.5, 149.8, 150.3, 156.7, 158.1, 159.5 (23 C, Ar–C), 169.2 (1 C, C=O), 175.5 (1 C, C=S); MS (70 eV, %) m/z 592 (M+, 95 %); Anal. Calc. (Found) for C32H24N4O2S3 (592.75): C, 64.84 (64.90); H, 4.08 (4.12); N, 9.45 (9.49); S, 16.23 (16.28).
10-(benzylidene)-5-(pyridin-4-yl)-1-(pyridin-4-ylmethylene)-13-thioxo-1,3,4,5,10,11-hexahydro-2H,9H,13H-isothiochromeno[3′,4′:4,5]pyrimido[2,1-b] [1,3]thiazin-9-one (14a)
Yellow crystals, yield (70%), mp > 340 °C, crystallized from dioxane; IR (ν, cm−1) KBr: 3065 (CH-aryl), 2960 (CH-aliph), 1730 (C=O), 1629(C=N), 1335 (C=S), 1H NMR (DMSO-d6, ppm) δ 1.25–2.31 (m, 6H, 3CH2, cyclohexene), 4.29 (s, 2H, CH2N, thiazine), 4.41 (s, 1H, thiopyran), 7.06–7.95 (m, 13H, Ar–H), 8.07 (s, 1H, C=CHAr), 8.15 (s, 1H, C=CHAr),13C NMR (DMSO-d6) δ 20.2, 24.6, 28.8 (3 C, CH2), 45.2 (1 C, thiopyran), 58.2 (1 C, CH2N, thiazine), 121.2, 123.1 (2 C, 2 C=CHAr), 123.1, 124.4, 127.1, 127.9, 128.6, 128.8, 133.3, 133.8, 135.4, 141.2, 144.7, 146.1, 147.3, 149.5, 149.8, 155.6, 159.1 (23 C, Ar–C), 168.5 (1 C, C=O), 177.4 (1 C, C=S); MS (70 eV, %) m/z 576 (M+, 98 %); Anal. Calc. (Found) for C32H24N4OS3 (576.75): C, 66.64 (66.60); H, 4.19 (4.12); N, 9.71 (9.78); S, 16.68 (16.62).
10-(4-chlorobenzylidene)-5-(pyridin-4-yl)-1-(pyridin-4-ylmethylene)-13-thioxo-1,3,4,5,10,11-hexa hydro-2H,9H,13H-isothiochromeno[3′,4′:4,5]pyrimido[2,1-b][1,3]thiazin-9-one (14b)
Yellowish crystals, yield (75%), mp > 340 °C, crystallized from methanol; IR (ν, cm−1) KBr: 3062 (CH-aryl), 2958 (CH-aliph), 1735 (C=O), 1625 (C=N), 1338 (C=S). 1H NMR (DMSO-d6, ppm) δ 1.18–2.25 (m, 6H, 3CH2, cyclohexene), 4.30 (s, 2H, CH2N, thiazine), 4.33 (s, 1H, thiopyran), 7.21–7.99 (m, 12H, Ar–H), 8.01 (s, 1H, C=CHAr), 8.26 (s, 1H, C=CHAr),13C NMR (DMSO-d6) δ 19.8, 24.7, 28.6 (3 C, CH2), 45.5 (1 C, thiopyran), 57.8 (1 C, CH2N, thiazine), 120.6, 123.3 (2 C, C=CHAr), 123.2, 124.5, 127.6, 128.9, 129.1, 133.1, 133.4, 133.8, 135.2, 140.7, 144.4, 146.6, 147.5, 149.6, 149.9, 156.2, 159.5 (23 C, Ar–C),168.2 (1 C, C=O), 178.2 (1 C, C=S); MS (70 eV, %) m/z 611 (M+, 100 %); Anal. Calc. (Found) for C32H23ClN4OS3 (611.19): C, 62.89 (62.80); H, 3.79 (3.72); N, 9.17 (9.11); S, 15.74 (15.78).
10-(4-methoxybenzylidene)-5-(pyridin-4-yl)-1-(pyridin-4-ylmethylene)-13-thioxo-1,3,4,5,10,11-hexahydro-2H,9H,13H-isothiochromeno[3′,4′:4,5]pyrimido[2,1-b][1,3]thiazin-9-one (14c)
Yellow crystals, yield (71%), mp > 350 °C, crystallized from DMF; IR (ν, cm−1) KBr: 3060 (CH-aryl), 2955 (CH-aliph), 1738 (C=O), 1628(C=N), 1340 (C=S).1H NMR (DMSO-d6, ppm) δ 1.20–2.28 (m, 6H, 3CH2, cyclohexene), 3.80 (s,3H, CH3), 4.29 (s, 2H, CH2N, thiazine), 4.41 (s, 1H, thiopyran), 7.22–7.97 (m, 12H, Ar-H), 8.05 (s, 1H, C=CHAr), 8.22 (s, 1H, C=CHAr),13C NMR (DMSO-d6) δ 24.8, 30.8, 30.9 (3 C, CH2), 58.3 (1 C, thiopyran), 58.5 (1 C, OCH3), 58.9 (1 C, CH2N, thiazine), 115.8, 115.9 (2 C, C=CHAr), 116.1, 121.5, 124.8, 130.3, 130.8, 130.9, 131.5, 136.1, 140.1, 140.2, 147.7, 152.6, 152.8, 158.3, 165.5, 165.6 (23 C, Ar–C), 168.5 (1 C, C=O), 180.1 (1 C, C=S); MS (70 eV, %) m/z 606 (M+, 90 %); Anal. Calc. (Found) for C33H26N4O2S3 (606.78): C, 65.32 (65.38); H, 4.32 (4.40); N, 9.23 (9.29); S, 15.85 (15.91).
Synthesis of 1-chloro-6-(pyridin-4-yl)-10-(pyridin-4-ylmethylene)-7, 8, 9, 10-tetrahydro-6H-isothiochromeno [3, 4- d] [1, 2, 3] triazine (15)
To a cool solution of compound 3 (3.58 g, 0.01 mol) in a mixture of acetic acid (15 mL) and conc. hydrochloric acid (10 mL), a sodium nitrite solution 10% (2 mL) was added with stirring for 15 min. The stirring was continued at (1–5 °C) for 4 h. The formed precipitate was filtered off, dried and crystallized from DMF to give compound 15. Yellow crystals, yield (78%), mp 325–327 °C (dec.); IR (ν, cm−1) KBr: 3065 (CH-aryl), 2952 (CH-aliph), 1625(C=N). 1H NMR (DMSO-d6, ppm) δ 1.57–2.23 (m, 6H, 3CH2, cyclohexene), 4.42 (s,1H, thiopyran), 7.56–7.96 (m, 8H, Ar–H), 8.23 (s, 1H, C=CHAr), 13C NMR (DMSO-d6) δ 15.6, 21.6, 31.2 (3 C, CH2), 65.6 (1 C, thiopyran), 1119 (1 C, C=CHAr), 120.7, 121.5, 124.9, 130.2, 130.8, 130.9, 131.5, 136.1, 143.2, 147.7, 154.1, 158.4 (16 C, Ar–C); MS (70 eV, %) m/z 405 (M+, 100 %); Anal. Calc. (Found) for C21H16ClN5S (405.90): C, 62.14 (62.20); H, 3.97 (3.90); N, 17.25 (17.20); S, 7.90 (7.85).
Synthesis of 1-imino-2-phenyl-6-(pyridin-4-yl)-10-(pyridin-4-ylmethylene)-1, 2, 4, 6, 7, 8, 9, 10-octahydro-3H-isothiochromeno[3,4-d]pyrimidine-3-thione (18a) and 1-imino-2-phenyl-6-(pyridin-4-yl)-10-(pyridin-4-ylmethylene)-1,2,4,6,7,8,9,10-octahydro-3H-isothiochromeno[3,4-d]pyrimidin-3-one (18b)
General method: A mixture of compound 3 (3.58 g, 0.01 mol) and phenylisothiocyanate (1.2 mL, 0.01 mol) or phenylisocyanate (1.1 mL, 0.01 mol) in pyridine (30 mL) was refluxed for 10–12 h. The reaction mixture was left to cool, acidified with dilute hydrochloric acid; the formed product precipitate was filtered off, dried, and recrystallized from the suitable solvent to give 18a and 18b, respectively.
Compound 18a: Recrystallized from THF as yellow crystals in 68% yield, mp > 340 °C (dec.); IR (ν, cm−1) KBr: 3250, 3220 (2NH), 3050 (CH-aryl), 2958 (CH-aliph), 1628 (C=N), 1275 (C=S). 1H NMR (DMSO-d6, ppm) δ 1.19–2.28 (m, 6H, 3CH2, cyclohexene), 4.72 (s,1H, thiopyran), 7.01–8.01 (m, 13H, Ar–H), 8.05 (s, 1H, C=CHAr), 10.58 (br. s, 1H, NH, D2O exchangeable), 10.62 (br. s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ 15.9, 21.8, 31.5 (3 C, 3CH2), 67.4, 91.6 (2 C, thiopyran), 112.8 (1 C, C=CHAr),119.4, 122.2, 128.5, 129.3, 132.7, 133.6, 138.4, 144.6, 146.9, 147.4, 149.1, 149.9, 151.5 (20 C, Ar–C), 159.5 (1 C, C=NH), 181.2 (1 C, C=S); MS (70 eV, %) m/z 493 (M+, 99%); Anal. Calc. (Found) for C28H23N5S2 (493.65): C, 68.13 (68.20); H, 4.70 (4.78); N, 14.19 (14.25); S, 12.99 (12.92).
Compound 18b: Recrystallized from Toluene as yellowish crystals in 66% yield, mp > 350 °C (dec.); IR (ν, cm−1) KBr: 3255, 3222 (2NH), 3051 (CH-aryl), 2962 (CH-aliph), 1670 (C=O), 1629 (C=N).1H NMR (DMSO-d6, ppm) δ 1.20–2.30 (m, 6H, 3CH2, cyclohexene), 4.74 (s,1H, thiopyran), 7.02–7.99 (m, 13H, Ar–H), 8.06 (s, 1H, C=CHAr), 10.60 (br. s, 1H, NH, D2O exchangeable), 10.65 (br. s, 1H,NH, D2O exchangeable); 13C NMR (DMSO-d6) δ 15.6, 21.5, 31.2(3 C, 3CH2), 65.5, 88.9 (2 C, thiopyran),111.9 (1 C, C=CHAr), 116.3, 118.5, 121.6, 124.8, 130.3, 130.8, 130.9, 131.6, 136.2, 139.9, 144.3, 147.8, 152.5, 153.6 (20 C, Ar–C), 158.2 (1 C, C=NH), 160.2 (1 C, C=O); MS (70 eV, %) m/z 477 (M+, 100 %); Anal. Calc. (Found) for C28H23N5OS (477.59): C, 70.42 (70.50); H, 4.85 (4.80); N, 14.66 (14.61); S, 6.71 (6.78).
Synthesis of Ethyl-N-(4-cyano-1-(pyridin-4-yl)-5-(pyridin-4-ylmethylene)-5,6,7,8-tetrahydro-1H-isothio- chromen-3-yl) formimidate (20a) and ethyl-N-(4-cyano-1-(pyridin-4-yl)-5-(pyridin-4-yl methylene)-5, 6, 7, 8-tetrahydro-1H-isothiochromen-3-yl) acetimidate (20b)
General method: A mixture of compound 3 (3.58 g, 0.01 mol) and triethylorthoformate (1.66 mL, 0.01 mol) or triethylorthoacetate (1.83 mL, 0.01 mol) in acetic anhydride (30 mL) was heated under reflux for 10–12 h (TLC control). The solution was cooled overnight to room temperature and then concentrated. The precipitated solid was filtered off and recrystallized to yields 20a and 20b respectively.
Compound 20a: Crystallized from dioxane, yellow crystals in 80% yield, mp 260–262 °C (dec.); IR (ν, cm−1) KBr: 3055 (CH-aryl), 2960 (CH-aliph), 2210(CN), 1622(C=N). 1H NMR (DMSO-d6, ppm) δ 1.29 (t, J = 5.55 Hz, 3H, CH3), 1.32–2.43 (m, 6H, 3CH2, cyclohexene), 4.23 (q, J = 5.60 Hz, 2H, CH2), 4.72 (s,1H, thiopyran), 7.01–7.98 (m, 8H, Ar-H), 8.03 (s, 1H, C=CHAr), 8.05 (s, 1H, N=CHO); 13C NMR (DMSO-d6) δ 20.2 (1 C, CH3), 24.4, 25.9, 28.1 (3 C, CH2), 46.5 (1 C, thiopyran), 66.2 (1 C, OCH2), 98.8 (1 C, thiopyran), 118.7(1 C, CN),121.1 (1 C, C=CHAr), 123.6, 124.8, 133.7, 134.6, 145.1, 146.7, 147.5, 149.4, 149.9, 153.4 (14 C, Ar–C), 155.8(1 C, N=CHOEt); MS (70 eV, %) m/z 414 (M+, 100 %); Anal. Calc. (Found) for C24H22N4OS (414.53): C, 69.54 (69.62); H, 5.35 (5.40); N, 13.52 (13.58); S, 7.73 (7.81).
Compound 20b: Crystallized from benzene, yellowish crystals in 82% yield, mp 273–275 °C (dec.); IR (ν, cm−1) KBr: 3058 (CH-aryl), 2964 (CH-aliph), 2212 (CN), 1620 (C=N). 1H NMR (DMSO-d6, ppm) δ 1.21 (s,1H,CH3),1.33 (t, J = 5.58 Hz, 3H, CH3), 1.36–2.45 (m, 6H, 3CH2, cyclohexene), 4.10 (q, J = 5.62 Hz,2H, OCH2CH3), 4.70 (s,1H, thiopyran), 7.04–7.99 (m, 8H, Ar-H), 8.06 (s, 1H, C=CHAr); 13C NMR (DMSO-d6) δ 19.5 (1 C, CH2CH3), 21.9 (1 C, N=CCH3), 23.6, 25.2, 27.8 (3 C, CH2), 47.1 (1 C, thiopyran), 68.4 (1 C,OCH2), 99.2 (1 C, thiopyran), 118.2 (1 C, CN), 121.5 (1 C, C=CHAr), 123.3, 124.5, 133.2, 134.1, 145.3, 146.6, 147.4, 149.2, 149.6, 152.1 (14 C, Ar–C), 164.5 (1 C, N=COEt); MS (70 eV, %) m/z 428 (M+, 90 %); Anal. Calc. (Found) for C25H24N4OS (428.55): C, 70.07 (70.10); H, 5.65 (5.71); N, 13.07 (13.12); S, 7.48 (7.55).
Synthesis of 1-imino-N-phenyl-6-(pyridin-4-yl)-10-(pyridin-4-ylmethylene)-7,8,9,10-tetrahydro-1H-isothiochromeno[3,4-d]pyrimidin-2(6H)-amine (21a) and 1-imino-3-methyl-N-phenyl-6-(pyridin-4-yl)-10-(pyridin-4-ylmethylene)-7,8,9,10-tetrahydro-1H-isothiochromeno [3,4-d]pyrimidin-2(6H)-amine (21b)
General method: A suspension of compound 20a (4.14 g, 0.01 mol) or 20b (4.28 g, 0.01 mol) and phenylhydrazine (1.08 g, 0.01 mol) in dioxane (30 mL) was refluxed for 6–8 h. The solid product which formed was filtered off, washed with water, dried in air and recrystallized from the suitable solvent to afford 21a and 21b respectively.
Compound 21a: Crystallized from methanol, yellowish crystals in 78% yield, mp > 340 °C (dec.); IR (ν, cm−1) KBr: 3210, 3190 (2NH), 3054 (CH-aryl), 2956 (CH-aliph), 1626(C=N). 1H NMR (DMSO-d6, ppm) δ 1.30–2.21 (m, 6H, 3CH2, cyclohexene), 4.75 (s, 1H, thiopyran), 7.15–7.95 (m, 13H, Ar-H), 8.05 (s, 1H, C=CHAr), 8.30 (s, 1H, N=CH), 9.35 (br. s, 1H, NH, D2O exchangeable), 10.58 (br. s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ 23.8, 26.2, 28.5 (3 C, 3CH2), 48.1, 108.2 (2 C, thiopyran),120.9 (1 C, C=CHAr), 121.8, 122.5, 123.4, 124.6, 130.5, 133.1, 134.8, 144.1, 145.3, 146.8, 147.5, 147.9, 149.4, 149.7, 154.2 (21 C, Ar-C),159.3 (1 C, C=NH); MS (70 eV, %) m/z 476 (M+, 100 %); Anal. Calc. (Found) for C28H24N6S (476.60): C, 70.56 (70.50); H, 5.08 (5.15); N, 17.63 (17.68); S, 6.73(6.80).
Compound 21b: Crystallized from DMF, yellow crystals in 74 % yield, mp > 350 °C (dec.); IR (ν, cm−1) KBr: 3215, 3195 (2NH), 3052 (CH-aryl), 2954 (CH-aliph), 1628(C=N). 1H NMR (DMSO-d6, ppm) δ 1.29–2.10 (m, 6H, 3CH2, cyclohexene), 2.92(s, 3H, CH3), 4.72 (s,1H, thiopyran), 7.21–8.01 (m, 13H, Ar-H), 8.03 (s, 1H, C=CHAr), 9.28 (br. s, 1H, NH, D2O exchangeable), 10.52 (br. s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ 20.4 (1 C,CH3), 23.5, 26.6, 28.2 (3 C, 3CH2), 46.3,105.5 (2 C,thiopyran), 120.6 (1 C, C=CHAr), 121.5, 122.7, 123.2, 124.4, 129.8, 133.5, 134.7, 144.4, 145.8, 146.5, 147.4, 149.3, 149.6, 153.9, 156.2 (21 C, Ar–C),157.9 (1 C, C=NH); MS (70 eV, %) m/z 490 (M+, 88 %); Anal. Calc. (Found) for C29H26N6S (490.63): C, 70.99 (70.90); H, 5.34 (5.40); N, 17.13 (17.20); S, 6.53(6.61).
Synthesis of 2-ethoxy-3-phenyl-8-(pyridin-4-yl)-12-(pyridin-4-ylmethylene)-2,3,9,10,11,12-hexahydro-8H-isothiochromeno[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine (22a) and 2-ethoxy-5-methyl-3-phenyl-8-(pyridin-4-yl)-12-(pyridin-4-ylmethylene)-2,3,9,10,11,12-hexahydro-8H-isothiochromeno[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine (22b)
General method: A solution of compounds 21a (4.76 g, 0. 01 mol) or 21b (4.90 g, 0. 01 mol) in triethylorthoformate (10 mL) was refluxed for 2–4 h. The reaction mixture was poured into crashed ice water; the formed precipitated was collected via filtration and recrystallized to afford 22a and 22b respectively.
Compound 22a: Crystallized from chloroform, yellow crystals in 75 % yield, mp >340 °C (dec.); IR (ν, cm−1) KBr: 3050 (CH-aryl), 2952 (CH-aliph), 1622(C=N). 1H NMR (DMSO-d6, ppm) δ 1.31 (t, J = 5.60 Hz, 3H, CH2CH3), 1.33–2.43 (m, 6H, 3CH2, cyclohexene), 4.22 (q, J = 5.65 Hz, 2H, OCH2), 4.72 (s,1H, thiopyran), 6.34 (s,1H,CHOEt, triazole ring), 7.01–7.55 (m, 13H, Ar–H), 8.13 (s, 1H, C=CHAr), 8.84 (s, 1H, N=CH); 13C NMR (DMSO-d6) δ 18.5(1 C, CH3), 22.6, 25.8, 27.2 (3 C, 3CH2), 66.5 (1 C, OCH2CH3), 69.7, 190.4 (2C, thiopyran), 112.2 (1 C, C=CHAr), 121.5, 122.4, 123.5, 124.4, 127.2, 130.3, 133.6, 134.5, 142.8, 144.3, 145.9, 146.7, 149.4, 149.7, 150.1, 152.4, 155.2, (23 C, Ar–C); MS (70 eV, %) m/z 532 (M+, 100 %); Anal. Calc. (Found) for C31H28N6OS (532.67): C, 69.90 (69.98); H, 5.30 (5.25); N, 15.78 (15.72); S, 6.02 (6.10).
Compound 22b: Crystallized from ethanol, yellowish crystals in 73% yield, mp >340 °C (dec.); IR (ν, cm−1) KBr: 3053 (CH-aryl), 2954 (CH-aliph), 1621(C=N).1H NMR (DMSO-d6, ppm) δ 1.30 (t, J = 5.64 Hz, 3H, CH3), 1.35–2.44 (m, 6H, 3CH2, cyclohexene), 3.92 (s,3H,CH3), 4.20 (q, J = 5.69 Hz,2H, CH2), 4.70 (s,1H, thiopyran), 6.32 (s,1H, triazole ring), 7.04–7.59 (m, 13H, Ar–H), 8.11 (s, 1H, C=CHAr),13C NMR (DMSO-d6) δ 15.7, 21.6(2 C,2CH3), 31.2, 31.3, 31.9, 38.6 (4 C, 4CH2), 65.6, 88.9 (2 C, thiopyran), 111.9 (1 C, C=CHAr), 116.2, 121.4, 124.8, 130.2, 130.8, 130.9, 131.5, 136.1, 137.2, 138.1, 140.4, 142.5, 144.2, 147.7, 150.1, 152.5, 159.3 (23 C, Ar–C); MS (70 eV, %) m/z 546 (M+, 90 %); Anal. Calc. (Found) for C32H30N6OS (546.69): C, 70.30 (70.38); H, 5.53 (5.59); N, 15.37 (15.32); S, 5.86 (5.80).
Synthesis of Ethyl-(4-cyano-1-(pyridin-4-yl)-5-(pyridin-4-ylmethylene)-5, 6, 7, 8-tetrahydro-1H-isothiochromen-3-yl) glycinate (23)
A mixture of compound 3 (3.58 g, 0.01 mol), ethyl bromoacetate (1.11 mL, 0.01 mol) and anhydrous potassium carbonate (1.38 g, 0.01 mol) in dry acetone (35 mL) was refluxed for 8–10 h. The reaction mixture was allowed to cool to room temperature and water (30 mL) was added, then the solid that separated was filtered off, dried and crystallized from diethyl ether to give 23, yellow crystals in 88 % yield, mp 240–242 °C (dec.); IR (ν, cm−1) KBr: 3250 (NH), 3055 (CH-aryl), 2960 (CH-aliph), 2215 (CN), 1730 (C=O), 1630 (C=N).1H NMR (DMSO-d6, ppm) δ 1.23 (t, 3H, J = 4.81 Hz, CH3), 1.70–2.43 (m, 6H, 3CH2, cyclohexene), 4.03 (s, 2H, NHCOCH2), 4.21 (q, 2H, J = 4.88 Hz, OCH2), 4.72 (s,1H, thiopyran), 7.11–7.96 (m, 8H, Ar–H), 8.24 (s, 1H, C=CHAr), 9.12 (br. s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ 15.2 (1 C, CH3), 23.5, 26.8, 27.9 (3 C, 3CH2), 44.1 (1 C, NHCOCH2), 47.5 (1 C, thiopyran), 64.3 (1 C, OCH2), 78.6 (1 C, thiopyran), 112.2 (1 C, C=CHAr), 115.5 (1 C, CN), 122.4, 124.8, 133.6, 134.8, 144.1, 146.3, 147.7, 149.1, 149.5, 165.2 (14 C, Ar–C), 168.9 (1 C, C=O); MS (70 eV, %) m/z 444 (M+, 100 %); Anal. Calc. (Found) for C25H24N4O2S (444.55): C, 67.55 (67.50); H, 5.44 (5.49); N, 12.60 (12.68); S, 7.21 (7.27).
Synthesis of Ethyl-1-amino-5-(pyridin-4-yl)-9-(pyridin-4-ylmethylene)-3, 5, 6, 7, 8, 9-hexahydroisothiochromeno [3, 4-b] pyrrole-2-carboxylate (24)
A solution of compound 23 (4.44 g, 0.01 mol), and anhydrous potassium carbonate (1.38 g, 0.01 mol) in dimethylformamide (30 mL) was refluxed for 5–8 h and then left to cool to room temperature and water (25 mL) was added. The formed precipitate was filtered off, dried and crystallized from ethyl acetate to give 24, yellow crystals, yield (85%), mp 250–252 °C (dec.); IR (ν, cm−1) KBr: 3405(NH2), 3280 (NH), 3058 (CH-aryl), 2962 (CH-aliph), 1735 (C=O), 1632 (C=N).1H NMR (DMSO-d6, ppm) δ 1.23 (t, 3H, J = 4.80 Hz, CH3),1.68–2.42 (m, 6H, 3CH2, cyclohexene), 4.22 (q, 2H, J = 4.85 Hz, OCH2), 5.24 (s,1H, thiopyran), 6.27 (br. s, 2H, NH2, D2O exchangeable), 7.10–7.95 (m, 8H, Ar–H), 8.23 (s, 1H, C=CHAr), 10.10 (br. s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ 15.5 (1 C, CH3), 21.8, 25.6, 30.7 (3 C, 3CH2), 52.8 (1 C, OCH2), 65.1, 92.6 (2 C, thiopyran), 111.8 (1 C, C=CHAr), 118.9, 122.6, 124.5, 130.8, 133.7, 135.9,143.5, 144.2, 146.8, 147.6, 149.3,149.9 (16 C, Ar–C), 162.4 (1 C, C=O); MS (70 eV, %) m/z 444 (M+, 92 %); Anal. Calc. (Found) for C25H24N4O2S (444.55): C, 67.55 (67.60); H, 5.44 (5.40); N, 12.60 (12.55); S, 7.21 (7.16).
Synthesis of Ethyl-5-(pyridin-4-yl)-9-(pyridin-4-ylmethylene)-1-thioureido-3, 5, 6, 7, 8, 9-hexahydroisothio- chromeno[3, 4-b] pyrrole-2-carboxylate (25)
A mixture of 24 (4.44 g, 0.01 mol) and 10 % HCl (10 mL) was refluxed with potassium thiocyanate (0.97 g, 0.01 mol) for 5–7 h. The reaction mixture was allowed to cool to room temperature. The formed solid was collected by filtration, washed with water, dried and crystallized from xylene to give 25, Yellow crystals, yield (80%), mp 302–304 °C (dec.); IR (ν, cm−1) KBr: 3412 (NH2), 3300 (NH), 3056 (CH-aryl), 2968 (CH-aliph), 1740 (C=O), 1631(C=N). 1H NMR (DMSO-d6, ppm) δ 1.23 (t, 3H, J = 4.79 Hz, CH3), 1.68–2.42 (m, 6H, 3CH2, cyclohexene), 4.22 (q, 2H, J = 4.87 Hz, CH2), 5.82 (s,1H, thiopyran), 6.28 (br. s, 2H, NH2, D2O exchangeable), 7.10–7.95 (m, 8H, Ar-H), 8.23(s, 1H, C=CHAr), 9.17 (br. s, 1H, NH, D2O exchangeable), 9.32 (br. s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ 15.8 (1 C, CH3), 21.6, 27.5, 31.2 (3 C, 3CH2), 53.9 (1 C,OCH2), 66.3, 90.5 (2 C, thiopyran), 112.5 (1 C, C=CHAr), 119.6, 122.4, 124.7, 131.1, 133.5, 135.7, 143.2, 144.8, 146.7, 147.5, 149.2, 149.6 (16 C, Ar–C), 163.5 (1 C, C=O), 181.2 (1 C, C=S); MS (70 eV, %) m/z 503 (M+, 90 %); Anal. Calc. (Found) for C26H25N5O2S2 (503.64): C, 62.01 (62.10); H, 5.00 (5.09); N, 13.91 (13.97); S, 12.73 (12.80).
Synthesis of 5-(pyridin-4-yl)-1-(pyridin-4-ylmethylene)-10-thioxo-1,3,4,5,7,9,10,11-octahydro isothiochromeno [4′,3′:4,5]pyrrolo[3,2-d]pyrimidin-8 (2H)-one (26)
A solution of 25 (5.03 g, 0.01 mol) in ethanolic sodium ethoxide (0.23 g of sodium metal in 30 mL ethanol) was stirred under reflux for 8–10 h. After cooling, the reaction mixture was neutralized with 10% HCl and the solid formed was collected by filtration, washed with water, dried and then crystallized from dioxane to give 26. Yellow crystals, yield 78%, mp > 340 °C (dec.); IR (ν, cm−1) KBr: 3355 (3NH), 3062 (CH-aryl), 2954 (CH-aliph), 1678 (C=O), 1628 (C=N). 1H NMR (DMSO-d6, ppm) δ 1.75–2.25 (m, 6H, 3CH2, cyclohexene), 4.30 (s,1H, thiopyran), 7.11–7.93 (m, 8H, Ar-H), 8.12(s, 1H, C=CHAr), 10.35 (br. s, 1H, NH, D2O exchangeable), 12.32 (br. s, 1H, NH, D2O exchangeable), 13.13 (br. s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ 15.7, 21.6, 31.2 (3 C, 3CH2), 89.1, 111.8 (2 C, thiopyran), 116.1 (1 C, C=CHAr), 118.1, 121.9,124.8, 128.1, 131.5, 135.8, 136.1, 142.1, 148.2, 152.1, 154.2, 159.8 (16 C, Ar-C), 165.6 (1 C, C=O),172.5 (1 C, C=S); MS (70 eV, %) m/z 457 (M+, 99 %); Anal. Calc. (Found) for C24H19N5OS2 (457.57): C, 63.00 (63.10); H, 4.19 (4.12); N, 15.31 (15.38); S, 14.01 (14.10).
Synthesis of 1-amino-5-(pyridin-4-yl)-9-(pyridin-4-ylmethylene)-3,5,6,7,8,9-hexa- hydroisothio- chromeno[3,4-b]pyrrole-2-carboxamide(30); (1-amino-5-(pyridin-4-yl)-9-(pyridin-4-ylmethylene)-3,5,6,7,8,9-hexahydroisothiochromeno[3,4-b]pyrrol-2-yl)(phenyl)methanone (31) and 1-amino-3-oxo-6-(pyridin-4-yl)-10-(pyridin-4-ylmethylene)-4,6,7,8,9,10-hexahydro-3H -isothiochromeno[3,4-b]pyridine-2-carbonitrile (32)
General method: A mixture of 3 (3.58 g, 0.01 mol), and the appropriate active methylene compounds namely, 2-chloroacetamide (0.93 g, 0.01 mol) or phenacyl bromides (1.99 g, 0.01 mol) or ethyl cyanoacetate (1.13 g, 0.01 mol), was stirred in (40 mL) of anhydrous ethanol containing sodium ethoxide (3%) for 3–4 h, and then was refluxed with stirring for 10–12 h. The reaction mixture was cooled and poured into ice water. The solid formed was filtered off, dried, and recrystallized from suitable solvent to give 30, 31 and 32 respectively.
Compound 30: The compound was obtained from the reaction of 3 (3.58 g, 0.01 mol) and 2-chloroacetamide (0.93 g, 0.01 mol), as a white crystals, crystallized from dioxane (70%), mp 348–350 °C. IR (ν, cm−1) KBr: 3410–3250 broad (2NH2 and NH), 3055 (CH-aryl), 2970 (CH-aliph), 1675 (C=O), 1625 (C=N).1H NMR (DMSO-d6, ppm) δ 1.85–2.27 (m, 6H, 3CH2, cyclohexene), 4.63 (s,1H, thiopyran), 6.42, 6.45 (2 s, 2NH2, D2O exchangeable), 7.25–7.98 (m, 8H, Ar–H), 8.02 (s, 1H, C=CHAr), 12.68 (br., 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ 18.5, 22.4, 30.8 (3 C, 3 CH2), 62.1, 94.3,110.5 (3C, thiopyran), 112.7 (1 C, C=CHAr), 122.7, 124.2, 132.6, 133.7, 135.9, 143.4, 145.6, 147.1, 148.4, 149.1, 149.5 (15 C, Ar–C), 164.6 (1 C, C=O); MS (70 eV, %) m/z 415 (M+, 90 %); Anal. Calc. (Found) for C23H21N5OS (415.52): C, 66.48 (66.40); H, 5.09 (5.15); N, 16.85 (16.77); S, 7.72 (7.65).
Compound 31: The compound was obtained from the reaction of 3 (3.58 g, 0.01 mol) and phenacyl bromides (1.99 g, 0.01 mol), as a yellowish crystals, crystallized from DMF (73%), mp > 340 °C. IR (ν, cm−1) KBr: 3400–3300 broad (NH2 and NH), 3058 (CH-aryl), 2965 (CH-aliph), 1690 (C=O), 1627(C=N).1H NMR (DMSO-d6, ppm) δ 1.80–2.26 (m, 6H, 3CH2, cyclohexene), 4.10 (s,1H, thiopyran), 6.43 (s, NH2, D2O exchangeable), 7.10–7.99 (m, 13H, Ar–H), 8.03 (s, 1H, C=CHAr), 12.67(br., 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ 17.1, 21.4, 30.9 (3 C, 3 CH2), 59.5, 90.4, 98.8 (3C, thiopyran),111.1 (1 C, C=CHAr),122.4, 124.5,128.6, 129.5, 130.8, 132.9, 133.1, 135.2, 136.7, 144.1, 146.2, 147.5, 149.1, 149.5, 149.9 (21 C, Ar–C), 167.1 (1 C, C=O); MS (70 eV, %) m/z 476 (M+, 95 %); Anal. Calc. (Found) for C29H24N4OS (476.60): C, 73.08 (73.14); H, 5.08 (5.14); N, 11.76 (11.70); S, 6.73 (6.78).
Compound 32: The compound was obtained from the reaction of 3 (3.58 g, 0.01 mol) and ethyl cyanoacetate (1.13 g, 0.01 mol), as a yellow crystals, crystallized from methanol (75%), mp > 340 °C. IR (ν, cm−1) KBr: 3405–3280 broad (NH2 and NH), 3054 (CH-aryl), 2960 (CH-aliph), 2210 (CN), 1676 (C=O), 1624(C=N).1H NMR (DMSO-d6, ppm) δ 1.75–2.25 (m, 6H, 3CH2, cyclohexene), 4.25 (s,1H, thiopyran), 6.60 (s, NH2, D2O exchangeable), 7.57–7.98 (m, 8H, Ar-H), 8.12 (s, 1H, C=CHAr),12.32 (br., 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ 15.7, 21.6, 31.2 (3 C, 3CH2), 65.6, 88.9 (2 C, thiopyran), 94.1 (1 C, pyridine),102.4 (1 C, CN), 111.9 (1 C, C=CHAr), 116.3, 121.5, 124.8,130.3,130.8, 130.9,131.5, 136.2, 147.7, 158.4 (14 C, Ar–C), 165.7 (1 C, C=O), 172.1(1 C, C–NH2); MS (70 eV, %) m/z 425 (M+, 92 %); Anal. Calc. (Found) for C24H19N5OS (425.51): C, 67.75 (67.79); H, 4.50 (4.58); N, 16.46 (16.40); S, 7.53 (7.48).
References
Abu-Hashem AA, Hussein HAR (2015) Synthesis, antitumor of new pyrimidine and caffeine derivatives. Lett Drug Des Discov 12:471–478
Abu-Hashem AA, Hussein HAR, Abu-zied KhM (2017) Synthesis of novel 1, 2, 4-triazolopyrimidines and their evaluation as antimicrobial agents. Med Chem Res 26:120–130
Bennani B, Kerbal A, Daoudi M, Baba BF, Houari GA, Jalbout AF, Mimouni M, Benazza M, Demailly G, Akkurt M, Yıldırım SÖ, Hadda TB (2007) Combined drug design of potential mycobacterium tuberculosis and HIV-1 inhibitors: 3Â’,4Â’-di-substituted -4’H-spiro[isothiochromene-3,5’-isoxazol]-4(1H)-one Arkivoc 16:19–40
Brown MJ, Carter PS, Fenwick AE, Fosberry AP, Hamprecht DW, Hibbs MJ, Arvest RL, Mensah L, Milner PH, O’Hanlon PJ, Pope AJ, Richardson CM, West A, Witty DR (2002) The antimicrobial natural product chuangxinmycin and some synthetic analogues are potent and selective inhibitors of bacterial tryptophanyl tRNA synthetase. Bioorg Med Chem Lett 12:3171–3174
Chen MS, Suttman RT, Wu JC, Prisbe EJ (1992) Metabolism of 4-azido thymidine: a compound with potent and selective activity against the human immunodeficiency virus. J Biol Chem 267:257–260
De Coen LM, Heugebaert TSA, García D, Stevens CV (2016) Synthetic entries to and biological activity of pyrrolopyrimidines. Chem Rev 116:80–139
Denizot F, Lang RJ (1986) Rapid colorimetric assay for cell growth and survival. modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. Immunol Methods 22:271–277
Dholakia SP, Dr. Patel MM, Dr. Patel JS (2015) Role of pyrrolopyrimidine derivatives as anticancer agent: mini review Indo Am Pharm Res 5:858–867
El-Ashmawy MB, El-Sherbeny MA, El-Gohary NS (2013) Synthesis and antitumor screening of new series of pyrimido-[4,5-b]quinolines and [1,2,4]triazolo[20,30:3,4]pyrimido[6,5-b] quinolones. Med Chem Res 22:2724–2736
El-Subbagh HI, Abu-Zaid SM, Mahran MA, Badria FA, Al-Obaid AM (2000) Synthesis and biological evaluation of certain α, β-unsaturated ketones and their corresponding fused pyridines as antiviral and cytotoxic agents. J Med Chem 43:2915–2921
Galal SA, El-Naem SI, El-Nezhawy AOH, Ali MA, El-Diwani HI (2011) Novel benzimidazo[2,1-c] [1,4]thiazinone derivatives with potent activity against HSV-1. Arch Pharm 344:255–263
Gewald K, Schinke E, Böttcher H (1966) 2-amino-thiophene aus methylenaktiven nitrilen, carbonylverbindungen und schwefel. Chem Ber 99:94–100
Havaldar FH, Patil AR (2008) Syntheses of 1, 2, 4-triazole derivatives and their biological activity. E-J Chem 5:347–354
Hufford CD, Badria FA, Abou-Karam M, Shier WT, Rogers RD (1991) Preparation, characterization and antiviral activity of microbial metabolites of stemodin. J Nat Prod 54:1543–1552
Kaminskyy D, Kryshchyshyn A, Nektegayev I, Vasylenko O, Grellier P, Lesyk R (2014) Isothiocoumarin-3-carboxylic acid derivatives: synthesis, anticancer and antitrypanosomal activity evaluation. Eur J Med Chem 75:57–66
Kumar R, Singh AD, Singh J, Singh H, Roy RK, Chaudhary A (2014) 1,2,3-triazine scaffold as a potent biologically active moiety: a mini review. Mini Rev Med Chem 14(1):72–83
Migawa MT, Drach JC, Townsend LB (2005) Design, synthesis and antiviral activity of novel 4,5-disubstituted 7-(β-D-Ribofuranosyl) pyrrolo[2,3-d][1,2,3]triazines and the novel 3-amino-5-methyl-1-(β-D-ribofuranosyl)-and 3-amino-5-methyl-1-(2-deoxy-β-dri bofuranosyl)-1,5-dihydro-1,4,5,6,7,8-hexaazaace-naphthylene as analogues of triciribine. J Med Chem 48:3840–3851
Mohamed MS, Ali SA, Abdelaziz DHA, Fathallah SS (2014) Synthesis and evaluation of novel pyrroles and pyrrolopyrimidines as anti-hyperglycemic agents. Bio Med Res Inter 2014:1–13
Mosmann TJ (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Immunol Methods 65:55–63
Quaglia W, Pigini M, Piergentili A, Giannella M, Gentili F, Marucci G, Carrieri A, Carotti A, Poggesi E, Leonardi A, Melchiorre C (2002) Selectivity of 4-phenylchroman analogues for alpha (1)-adrenoreceptor subtypes. J Med Chem 45:1633–1643
Ram SG, Rajesh PG, Parhate VV (2013) Synthesis, characterization and antibacterial activities of some new bromo / nitro 1, 3-thiazines. Rasayan J Chem 6:65–67
Rogier Jr DJ, Carter JS, Talley JJ (2001) Dihydrobenzopyrans, dihydrobenzothiopyrans, and tetrahydroquinolines for the treatment of COX-2-mediated disorders,WO2001049675. Chem Abstr 135:107252
Rashad AE, Sayed HH, Shamroukh AH, Awad HM (2005) Preparation of some fused pyridopyrimidine and pyridothienotriazine derivatives for biological evaluation. Phosphorus, Sulfur, Silicon 180:2767–2777
Selvam TP, James CR, Dniandev PV, Valzita SK (2012) A mini review of pyrimidine and fused pyrimidine marketed drugs. Res Pharm 2(4):01–09
Sung K, Lee AR (1992) Synthesis of [(4, 5-disubstituted-4H-1, 2, 4-triazol-3-yl) thio] alkanoic acids and their analogues as possible anti-inflammatory agents. J Heterocycl Chem 29:1101–1109
Somers-Edgar TJ, Taurin S, Larsen L, Chandramouli A, Nelson MA, Rosengren RJ (2009) Mechanisms for the activity of heterocyclic cyclohexanone curcumin derivatives in estrogen receptor negative human breast cancer cell lines. Invest New Drugs 29:87–97
Thabrew MI, Hughes RD, McFarlane IG (1997) Screening of hepatoprotective plant components using a HepG2 cell cytotoxicity assay. J Pharm Pharmacol 49:1132–1135
Van Vliet LA, Rodenhuis N, Dijkstra D, Wikstrom H, Pugsley TA, Serpa KA, Meltzer LT, Heffner TG, Wise LD, Lajiness ME, Huff RM, Svensson K, Sundell S, Lundmark M (2000) Synthesis and pharmacological evaluation of thiopyran analogues of the dopamine D3 receptor-selective agonist (4aR,10bR)-(+)-trans-3,4,4a,10b-tetrahydro-4-n-propyl-2H,5H[1]benzopyrano [4,3-b]-1,4-oxazin-9-ol (PD 128907). J Med Chem 43:2871–2882
Vijay VD (2011) Synthesis of chalcones.1, 3- thiazines and the biological evaluation for antiinflammatory, analgesic and ulcerogenic activity. Pharma Res 5:127–143
Weislow OS, Kiser R, Fine D, Bader J, Shoemaker RH, Boyd MR (1989) New soluble-formazan assay for HIV-1 cytopathic effects. J Natl Cancer Inst 81:577–586
Zhi H, Chen L, Zhang L, Liu S, Wan DCC, Lin H, Hu C (2008) Design, synthesis, and biological evaluation of 5H-thiazolo[3,2-a]pyrimidine derivatives as a new type of acetylcholinesterase inhibitors. Arkivoc 2008:266–277
Acknowledgements
This work was supported by the Photochemistry Department (Heterocyclic Unit); Chemical Industries Research Division, National Research Center, Dokki, Giza 12622, Egypt.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Abu-Hashem, A.A., Gouda, M.A. & Badria, F.A. Design, synthesis and identification of novel substituted isothiochromene analogs as potential antiviral and cytotoxic agents. Med Chem Res 27, 2297–2311 (2018). https://doi.org/10.1007/s00044-018-2236-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-018-2236-3