
Abstract
A new series of 4-methyl-6-nitro-2-oxo-2H-chroman-7yl-2-(4-(4-fluorophenyl)-6-phenyl-2H-1,3-thiazin-2-yl-amino)acetates 5a–j were synthesized from 6-nitro-4-methyl coumarinyl chloroacetate (5) and 2-amino thiazines (IIIa–j). The structure of the final compounds was adequately confirmed via spectroscopic techniques (IR, 1H NMR, 13C NMR, Mass) and characterization of physical properties. Final compounds were screened for their antimicrobial, antitubercular, and antioxidant activities. Compounds 5c and 5h found to have antibacterial potency against E. coli with MIC values 50 µg/mL compared to standard drugs. Compound 5d demonstrated better antifungal potency (MIC = 200 µg/mL) against C. albicans when compared with griseofulvin. Compounds 5b and 5h found to be encouraging antitubercular (MIC = 62.5 µg/mL with 98–99% inhibition) against M. tuberculosis H37Rv. The newly synthesized 5h and 5b were appeared to have high radical scavenging efficacies as 33.99 ± 0.301 and 35.35 ± 0.470 μg/mL ± SD of IC50 values, respectively, in DPPH and ABTS bioassay.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The natural compound such as coumarin has served as valuable leads for the development of newer biological potent analogs (Kostova et al. 2011; Patel et al. 2017). Coumarins form an elite class of compounds, which exhibit a variety of therapeutic activities including antibacterial (Bhat et al. 2009; Muratovic et al. 2013), antimalarial (Patel et al. 2012), antioxidant (Kostova 2006; Nagamallu et al. 2016), anticancer (Sashidhara et al. 2010; Thakur et al. 2015), antiplatelet (Roma et al. 2003), antithrombotic (Kontogiorgis et al. 2015), analgesic (Keri et al. 2010), antifungal (Al-Amiery et al. 2012; Rehman et al. 2005), antiviral (Hassan et al. 2016), anticoagulant (Rost et al. 2005), anti-inflammatory (Bansal et al. 2013), and antitumor (Chen et al. 2013). On the other hand, the nitrogen and sulfur heterocyclic ring families are very interesting due to their physicochemical properties, especially in the sense of design of new drugs and new materials. The core moiety of 1,3-thiazines has N-C-S linkage have been used as antimicrobial activity (Koketsu et al. 2002), antitumor (Wang et al. 2012), antituberculosis (Tiwari et al. 2016), analgesic and anti-inflammatory (Jupudi et al. 2013), and antioxidant (Jeleń et al. 2015).
The development of coumarins as antioxidant agents has attracted much attention in recent years. Coumarins afford an opportunity for the discovery of new antioxidants with truly novel mechanisms of action. The present article deals with the rational design of coumarin clubbed thiazine motif with an understanding of the mechanisms of existing synthetic and natural coumarins. Antimicrobial activity of 4- and 7-hydroxy and nitrocoumarins has been reviewed extensively (Debeljak et al. 2007; Dekić et al. 2011) and it has been observed that when it has been nitrated, its antimicrobial activities is enhanced. Additionally, a recent Quantitative Structure Activity Relationship (QSAR) study of the antimicrobial activity of some 3-nitrocoumarins has put forward some new arguments in this direction. With this consideration and continuation of our ongoing interest in the synthesis of the thiazine clubbed coumarin derivatives, we have been prompted to synthesize newer, possibly more potent, pharmacologically active compounds. We have condensed chloroacetate of 4-methyl-7-hydroxy-6-nitrocoumarin with substituted amino derivatives of thiazines obtained via chalcone derivatives. The synthesized compounds were assigned on the basis of IR, 1H NMR, 13C NMR and mass spectral data. The in vitro evaluation of these derivatives viz., antimicrobial activity on four different bacteria (Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, and Streptococcus pyogenes) and three different fungi (Candida albicans, Aspergillus niger, and Aspergillus clavatus), antitubercular activity on Mycobacterium tuberculosis (H37Rv) virulent strain is presented. Antioxidant acticity of these derivatives have been evaluated by DPPH (2,2’-diphenyl-1-picrylhydrazyl) and ABTS (2,2’-Azino-bis-3-ethylbenzthiazoline-6-sulfonic acid) radical scavenging assay.
Materials and methods
All solvents, chemicals, and reagents were purchased from Sigma-Aldrich with the highest purity and used without further purification. Melting points were determined with an open capillary method on ‘‘Equiptronics’’ digital melting point apparatus, model no. EQ-730 and are uncorrected. IR spectra were recorded on a Perkin Elmer spectrophotometer (KBr pellets) instrument. 1H and 13C NMR spectra were recorded on Bruker Avance II 400 MHz NMR Spectrometer using DMSO-d6 as solvent and TMS as an internal standard. All chemical shifts were reported as δ values (ppm). Mass spectra were recorded using Expression CMS from Advion, USA using ESI as ion source (mobile phase 0.1% formic acid in 80:20, Methanol: Water). Analytical thin-layer chromatography (TLC) was performed with Merck silica gel plates and visualized with ultraviolet (UV) irradiation (254 nm) or iodine.
Experimental procedure
Chemistry
4-Methyl-7-hydroxy-coumarin (3) has been prepared as described in literature
Yield: 85%, mp: 183 to 185 °C. IR (KBr, cm−1): 3493 (–OH), 3098, 2817 (–CH3, asym, sym), 1735 (>C=O str), 1148 (–C–O–C). 1H NMR (DMSO-d6) δ (ppm): 10.35 (s, 1H, phenolic -OH), 7.48 (d, 1H, aromatic) 6.76 (d,1H,aromatic), 6.69 (s, 1H,aromatic), 2.37 (s, 3H, –CH3) (Furniss et al. 1989).
7-Hydroxy-4-methyl-6-nitro-2H-chromen-2-one (4) has been prepared as described in literature
Yield: 72%, mp: 193–195 °C (as reported). IR (KBr, cm−1): 3490 (–OH), 3094, 2827 (–CH3, asym, sym), 1745 (>C=O str), 1190 (–C–O–C), 1536, 1357 (–NO2). 1H NMR (DMSO-d6) δ (ppm):13.26 (s, 1H, Phenolic –OH), 8.44 (s, 1H, aromatic), 7.09 (s, 1H, aromatic), 6.35 (s, 1H, aromatic), 2.37 (s, 3H, –CH3) (Ganguly et al. 2001).
4-Methyl-6-nitro-2-oxo-2H-chromen-7-yl 2-chloroacetate (5) has been prepared as described in literature
Yield: 69%, mp: 109–112 °C. IR (KBr, cm−1): 3003, 2799 (–CH3, asym, sym), 1751, 1657 (>C=O str), 1190 (–C–O–C), 1571, 1346 (–NO2), 831 (–C–Cl). 1H NMR (DMSO-d6) δ (ppm): 8.38 (s, 1H, aromatic), 7.27 (s, 1H, aromatic), 6.37 (s, 1H, aromatic), 4.47 (s, 2H, –COCH2), 2.48 (s, 3H, –CH3) (Qandil and Fakhouri 2012).
General method for the synthesis of (E)-1-(4-fluorophenyl)-3-phenylprop-2-en-1-one (IIa)
p-Fluoro acetophenone 6 (0.01 mol) and benzaldehyde Ia (0.01 mol) were dissolved in 15 mL ethanol. NaOH solution (0.02 mol) in ethanol was added slowly and the mixture was stirred at 20 °C for 2 h until the entire mixture becomes very thick. The progress of the reaction was monitored by TLC (toluene: acetone, 80:20). Then the reaction mixture was poured slowly onto 400 mL of water with stirring and kept in refrigerator for 24 h. The precipitate obtained was filtered, washed, and recrystallized from ethanol. The other compounds IIb–j were prepared by the same method using substituted benzaldehydes Ib–j.
General method for the synthesis of 4-(4-Fluorophenyl)-6-phenyl-2H-1,3-thiazin-2-amine (IIIa)
A mixture of (E)-1-(4-fluorophenyl)-3-phenylprop-2-en-1-one IIa (0.01 mol), thiourea (0.01 mol) was dissolved in ethanolic sodium hydroxide (10 mL) was refluxed about 2–3 h. The progress of the reaction was monitored by TLC (ethylacetate: n-hexane (1:3)). The reacting mixture was poured onto 400 mL of cold water and stirred for an hour and then kept in refrigerator for 24 h. The precipitate of 4-(4-fluorophenyl)-6-phenyl-2H-1,3-thiazin-2-amine IIIa obtained was filtered, washed, and recrystallized with ethanol. The other compounds IIIb–j were prepared by the same method using substituted chalcones IIb–j. Compound IIIh: IR (KBr, cm−1): 3182 (–NH2), 2959 (–CH3), 1599 (–C=N), 1170 (C–F); 1H NMR (DMSO-d6) δ (ppm): 8.76 (s, 2H, –NH2), 6.89–7.61 (m, 8H, aromatic), 6.45 (s, 1H, aromatic), 5.28 (s, 1H, aromatic), 2.97 (t, 2H, –CH2CH2CH3), 1.65 (m, 2H, –CH2CH2CH3), 0.94 (t, 2H, –CH2CH2CH3).
General method for the synthesis of (5a–j)
All the reactions were carried out under nitrogen atmosphere. In a round bottom containing compound 5 (0.1 mol) and IIIa–j (0.1 mol), DMF and K2 CO3 (2.4 equivalent) were added under constant stirring. Reaction Mixture was refluxed for 8–9 h. After the completion of the reaction (TLC monitored), reaction mixture was poured over crushed ice, solids that are separated out was filtered, washed with saturated solution of NaHCO3 and dried. The crude product was purified by column chromatography using silica gel 100–200 mesh and gradient (0–80%) ethylacetate in hexane as eluent. The precipitate obtained was filtered, washed, and recrystallized.
4-Methyl-6-nitro-2-oxo-2H-chromen-7yl-2-(4-(4-fluorophenyl)-6-phenyl-2H-1,3-thiazin-2-yl-amino) acetate (5a)
Yellow solid, yield: 59 %, m.p.: 129–131 °C, M.F.: C28H19N3FO6S.
IR (KBr) ν cm−1: 3192 (–NH), 1715, 1671 (–C=O), 1601 (C=N), 2961, 2840 (–CH3), 1508, 1364 (–NO2), 1177(C–F); 1H NMR (400 MHz, DMSO-d6, TMS) δ ppm: 8.55 (s, 1H, –CH), 6.32–7.78 (m, 12H, aromatic), 5.23 (s, 1H, –CH), 3.71 (s, 2H, –CH2NH), 3.23 (s, 1H, –CH2NH), 2.40 (s, 3H, –CH3); 13C NMR (100 MHz, DMSO-d6 TMS) δ ppm: 160.11 (C-2),113.88 (C-3), 152.54 (C-4), 120.68 (C-5), 139.40 (C-6), 135.67 (C-7), 115.79 (C-8), 159.14 (C-9), 117.65 (C-10), 19.40 (C-12), 168.16 (C-14), 43.98 (C-16), 81.13 (C-18),157.35 (C-20), 95.50 (C-21), 153.11 (C-22), 134.18 (C-24), 130.12 (C-25, C-29), 115.57 (C-26, C-28), 165.09 (C-27), 136.32 (C-32), 128.55 (C-33, C-37), 129.04 (C-34, C-36), 126.32 (C-35); m/z: 545.54 (M+).
4-Methyl-6-nitro-2-oxo-2H-chromen-7yl-2-(4-(4-fluorophenyl)-6-2-chlorophenyl-2H-1,3-thiazin-2-yl-amino)acetates (5b)
Cream yellow solid, yield: 59%, mp: 156–158 °C, M.F.: C28H19ClFN3O6S, IR (KBr) ν cm−1: 3185 (–NH), 1720, 1667 (–C=O), 1591 (–C=N-), 2956, 2835 (–CH3), 1514, 1354 (–NO2), 1172 (C–F), 749 (C–Cl); 1H NMR (400 MHz, DMSO-d6, TMS) δ: 8.54 (s, 1H, –CH), 6.31–7.78 (m, 11H, aromatic), 5.39 (s, 1H, –CH), 3.69 (s, 2H, –CH2NH), 3.30 (s, 1H, –CH2NH), 2.39 (s, 3H, –CH3); 13C NMR (100 MHz, DMSO-d6 TMS) δ: 160.12 (C-2),113.94 (C-3), 152.51 (C-4), 120.72 (C-5), 139.44 (C-6), 135.64 (C-7), 115.77 (C-8), 159.12 (C-9), 118.67 (C-10), 19.41 (C-12), 168.17 (C-14), 40.96 (C-16), 111.13 (C-18),157.37 (C-20), 74.16 (C-21), 160.14 (C-22), 134.13 (C-24), 130.16 (C-25, C-29), 115.52 (C-26, C-28), 165.12 (C-27), 131.51 (C-32), 133.10 (C-33), 131.56 (C-37), 127.80 (C-34), 131.25 (C-36), 128.35 (C-35),; m/z: 563.09 (M+), 565.09(M+2).
4-Methyl-6-nitro-2-oxo-2H-chromen-7yl-2-(4-(4-fluorophenyl)-6-4-chlorophenyl-2H-1,3-thiazin-2-yl-amino)acetates (5c)
Pale yellow solid, yield: 65%, mp: 163–165 °C, M.F.: C28H19ClFN3O6S, IR (KBr) ν cm−1: 3238 (–NH), 1740, 1669 (–C=O), 1599 (–C=N–), 3067, 2840 (–CH3), 1538, 1358 (–NO2), 1258, 1057 (–C–O–C–), 1170 (C–F), 758 (C–Cl); 1H NMR (400 MHz, DMSO-d6, TMS) δ: 8.54 (s, 1H, –CH), 6.30–7.77 (m, 11H, aromatic), 5.34 (s, 1H, –CH), 3.70 (s, 2H, –CH2NH), 3.23 (s, 1H, –CH2NH), 2.41 (s, 3H, –CH3); 13C NMR (100 MHz, DMSO-d6 TMS) δ: 160.12 (C-2),113.94 (C-3), 152.51 (C-4), 120.72 (C-5), 139.44 (C-6), 135.64 (C-7), 115.77 (C-8), 159.12 (C-9), 118.67 (C-10), 19.41 (C-12), 168.17 (C-14), 40.96 (C-16), 111.13 (C-18),157.37 (C-20), 74.16 (C-21), 160.14 (C-22), 134.13 (C-24), 130.16 (C-25, C-29), 115.52 (C-26, C-28), 165.12 (C-27), 131.51 (C-32), 133.10 (C-33), 131.56 (C-37), 127.80 (C-34), 131.25 (C-36), 128.35 (C-35); m/z: 563.09 (M+), 565.09(M+2).
4-Methyl-6-nitro-2-oxo-2H-chromen-7yl-2-(4-(4-fluorophenyl)-6-2-hydroxyphenyl-2H-1,3-thiazin-2-yl-amino) acetate (5d)
Pale yellow solid, yield: 61 %, m.p.: 174–176 °C, M.F.: C28H20FN3O7S.
IR (KBr) ν cm−1: 3484 (–OH), 3190 (–NH), 1758, 1664 (–C=O), 1622 (C=N), 2965, 2842 (–CH3), 1523, 1351 (–NO2), 1174 (C–F);1H NMR (400 MHz, DMSO-d6, TMS): δ ppm: 10.11 (s, 1H, –OH), 8.58 (s, 1H, –CH), 6.37–7.77 (m, 11H, aromatic), 5.22 (s, 1H, –CH), 3.71 (s, 2H, –CH2NH), 3.23 (s, 1H, –CH2NH), 2.44 (s, 3H, –CH3); 13C NMR (100 MHz, DMSO-d6, TMS) δ ppm: 160.12 (C-2),113.92 (C-3), 152.50 (C-4), 120.68 (C-5), 139.46 (C-6), 135.67 (C-7), 115.72 (C-8), 159.10 (C-9), 117.64 (C-10), 19.44(C-12), 168.15 (C-14), 43.93 (C-16), 81.12 (C-18),157.35 (C-20), 95.51 (C-21), 153.12 (C-22), 134.11 (C-24), 130.12 (C-25, C-29), 115.59 (C-26, C-28), 165.15 (C-27), 122.32 (C-32), 158.54 (C-33), 117.52 (C-34), 128.35 (C-35), 121,44 (C-36), 125.02 (C-37); m/z: 561.54 (M+).
4-Methyl-6-nitro-2-oxo-2H-chromen-7yl-2-(4-(4-fluorophenyl)-6-4-hydroxyphenyl-2H-1,3-thiazin-2-yl-amino) acetate (5e)
Pale yellow solid, yield: 69 %, m.p.: 184–186 °C, M.F.: C28H20N3FO7S.
IR (KBr) ν cm−1: 3478 (–OH), 3186 (–NH), 1765, 1671 (>C=O), 1627 (C=N), 2959, 2851 (–CH3), 1530, 1356 (–NO2), 1180 (C–F);1H NMR (400 MHz, DMSO-d6, TMS): δ ppm: 10.20 (s, 1H, –OH), 8.51 (s, 1H, –CH), 6.32–7.74 (m, 11H, aromatic), 5.27 (s, 1H, –CH), 3.69 (s, 2H, –CH2NH), 3.30 (s, 1H, –CH2NH), 2.51 (s, 3H, –CH3); 13C NMR (100 MHz, DMSO-d6, TMS) δ ppm: 160.09 (C-2),113.88 (C-3), 152.46 (C-4), 120.74 (C-5), 139.41 (C-6), 135.61 (C-7), 115.72 (C-8), 159.10 (C-9), 117.64 (C-10), 19.44(C-12), 168.15 (C-14), 43.93 (C-16), 81.12 (C-18),157.35 (C-20), 95.51 (C-21), 153.12 (C-22), 134.11 (C-24), 130.12 (C-25, C-29), 115.59 (C-26, C-28), 165.15 (C-27), 122.32 (C-32), 158.54 (C-33), 117.52 (C-34), 128.35 (C-35), 121,44 (C-36), 125.02 (C-37); m/z: 561.45 (M+).
4-Methyl-6-nitro-2-oxo-2H-chromen-7yl-2-(4-(4-fluorophenyl)-6-4-fluorophenyl-2H-1,3-thiazin-2-yl-amino) acetate (5f)
Light yellow solid, yield: 64%, m.p.: 158–160 °C, M.F.: C28H19N3F2O6S.
IR (KBr) ν cm−1: 3219 (–NH), 1745, 1669 (–C=O), 1619 (C=N), 2954, 2851 (–CH3), 1168 (aryl-F), 1525, 1358 (–NO2); 1H NMR (400 MHz, DMSO-d6, TMS) δ ppm: 8.53 (s, 1H, –CH), 6.33–7.79 (m, 11H, aromatic), 5.25 (s, 1H, –CH), 3.71 (s, 2H, –CH2NH), 3.22 (s, 1H, –CH2NH), 2.43 (s, 3H, –CH3); 13C NMR (100 MHz, DMSO-d6, TMS) δ ppm: 160.11 (C-2),113.88 (C-3), 152.48 (C-4), 120.69 (C-5), 139.42 (C-6), 135.62 (C-7), 115.76 (C-8), 159.11 (C-9), 117.65 (C-10), 19.40 (C-12), 168.15 (C-14), 43.90 (C-16), 81.12 (C-18),157.33 (C-20), 95.50 (C-21), 153.10 (C-22), 134.14 (C-24), 130.12 (C-25, C-29), 115.52 (C-26, C-28), 165.16 (C-27), 131.33 (C-32), 128.51 (C-33, C-37), 115.06 (C-34, C-36), 161.34 (C-35); m/z: 563.53 (M+).
4-Methyl-6-nitro-2-oxo-2H-chromen-7yl-2-(4-(4-fluorophenyl)-6-p-tolyl-2H-1,3-thiazin-2-yl-amino) acetate (5g)
Yellow solid, yield: 62%, m.p.: 165–167 °C, M.F.: C29H22FN3O6S.
IR (KBr) ν cm−1: 3211(–NH), 1741, 1664 (–C=O), 1629 (C=N), 2941, 2861 (–CH3), 1171 (aryl-F), 1517, 1353 (–NO2); 1H NMR (400 MHz, DMSO-d6, TMS) δ ppm: 8.56 (s, 1H, –CH), 6.38–7.79(m, 11H, aromatic), 5.24 (s, 1H, –CH), 3.70 (s, 2H, –CH2NH), 3.21 (s, 1H, –CH2NH), 2.41 (s, 3H, –CH3), 2.38 (s, 3H, –CH3); 13C NMR (100 MHz, DMSO-d6 TMS) δ ppm: 160.09 (C-2),113.91 (C-3), 152.54 (C-4), 120.66 (C-5), 139.46 (C-6), 135.67 (C-7), 115.79 (C-8), 159.15 (C-9), 117.65 (C-10), 19.47 (C-12), 168.18 (C-14), 43.95 (C-16), 81.11 (C-18),157.32 (C-20), 95.51 (C-21), 153.09 (C-22), 134.16 (C-24), 130.14 (C-25, C-29), 115.55 (C-26, C-28), 165.13 (C-27), 133.34 (C-32), 125.51 (C-33, C-37), 129.04 (C-34, C-36), 136.35 (C-35), 22.37 (C-38); m/z: 559.56 (M+).
4-Methyl-6-nitro-2-oxo-2H-chromen-7yl-2-(4-(4-fluorophenyl)-6-4-propylphenyl-2H-1,3-thiazin-2-yl-amino)acetate (5h)
Dark yellow solid, yield: 68%, m.p.: 140–142 °C, M.F.: C31H26 F N3O6S.
IR (KBr) ν cm−1: 3192 (–NH), 1715, 1671 (>C=O), 1601 (–C=N), 2961, 2840 (–CH3), 1509, 1364 (–NO2), 1233, 1055 (–C–O–C), 1177 (C–F); 1H NMR (400 MHz, DMSO-d6, TMS): δ ppm: 8.57 (s, 1H, –CH), 6.34–7.76 (m, 11H, aromatic), 5.23 (s, 1H, –CH), 3.74 (s, 2H, –CH2NH), 3.22(s, 1H, –CH2NH), 2.42 (s, 3H, –CH3), 2.99 (t, 2H, –CH2CH2CH3), 1.67 (m, 2H, –CH2CH2CH3), 0.96 (t, 2H, –CH2CH2CH3); 13C NMR (100 MHz, DMSO-d6 TMS) δ ppm: 160.10 (C-2),113.90 (C-3), 152.50 (C-4), 120.70 (C-5), 139.44 (C-6), 135.64 (C-7), 115.77 (C-8), 159.13 (C-9), 117.67 (C-10), 19.41(C-12), 168.18 (C-14), 43.94 (C-16), 81.10 (C-18), 157.34 (C-20), 95.50 (C-21), 153.10 (C-22), 134.14 (C-24), 130.14 (C-25, C-29), 115.56 (C-26, C-28), 165.10 (C-27), 133.35 (C-32), 126.52 (C-33, C-37), 128.00 (C-34, C-36), 140.34 (C-35), 34.41 (C-38), 24.01 (C-39), 13.31 (C-40); m/z: 587.15 (M+).
4-Methyl-6-nitro-2-oxo-2H-chromen-7yl-2-(4-(4-fluorophenyl)-6-3-bromophenyl-2H-1,3-thiazin-2-yl-amino) acetate (5i)
Pale yellow solid, yield: 62%, m.p.: 125–127 °C., M.F.: C28H19BrFN3O6S.
IR (KBr) ν cm−1: 3326 (–NH), 1749 (–C=O), 1623 (C=N), 2895, 2958 (–CH3), 1514, 1331 (–NO2); 1H NMR (400 MHz, DMSO-d6, TMS) δ: 8.57 (s, 1H, –CH), 6.38–7.77(m, 11H, aromatic), 5.25 (s, 1H, –CH), 3.73 (s, 2H, –CH2NH), 3.22 (s, 1H, –CH2NH), 2.42 (s, 3H, –CH3); 13C NMR (100 MHz, DMSO-d6, TMS) δ ppm: 160.12 (C-2),113.91 (C-3), 152.50 (C-4), 120.89 (C-5), 139.43 (C-6), 135.61 (C-7), 115.72 (C-8), 159.10 (C-9), 117.66 (C-10), 19.40 (C-12), 168.16 (C-14), 43.95 (C-16), 81.12 (C-18),157.33 (C-20), 95.51 (C-21), 153.11 (C-22), 134.14 (C-24), 130.15 (C-25, C-29), 115.55 (C-26, C-28), 165.12 (C-27), 138.37 (C-32), 128.59 (C-33), 127.50 (C-37), 123.06 (C-34), 130.09 (C-36),129.35 (C-35); m/z: 623.32 (M+), 625.18 (M+2), 627.15 (M+4).
4-Methyl-6-nitro-2-oxo-2H-chromen-7yl-2-(4-(4-fluorophenyl)-6-3-phenoxy-2H-1,3-thiazin-2-yl-amino) acetate (5j)
Dark yellow solid, yield: 62%, m.p.: 132–134 °C., M.F.: C34H24FN3O7S.
IR (KBr) ν cm−1: 3318 (–NH), 1749 (–C=O), 1623 (C=N), 2895, 2958 (–CH3), 1514, 1331 (–NO2) 1249, 1050 (–C–O–C–), 1161 (C–F); 1H NMR (400 MHz, DMSO-d6, TMS) δ: 8.57 (s, 1H, –CH), 6.38–7.77(m, 11H, aromatic), 5.25 (s, 1H, –CH), 3.73 (s, 2H, –CH2NH), 3.22 (s, 1H, –CH2NH), 2.42 (s, 3H, –CH3); 13C NMR (100 MHz, DMSO-d6, TMS) δ ppm: 160.23 (C-2),113.88 (C-3), 152.65 (C-4), 120.69 (C-5), 139.51 (C-6), 135.69 (C-7), 115.66 (C-8), 159.54 (C-9), 118.56 (C-10), 19.42 (C-12), 168.21 (C-14), 40.89 (C-16), 111.56 (C-18), 157.24 (C-20), 74.19 (C-21), 160.12 (C-22), 134.18 (C-24), 130.16 (C-25, C-29), 115.62 (C-26, C-28), 165.20 (C-27), 130.66 (C-32), 127.69 (C-33), 142.14 (C-34), 127.39 (C-35), 127.09 (C-36), 126.19 (C-37), 160.61 (C-39), 118.13 (C-40, C-44), 130.11 (C-41, C-43) 122.79 (C-42); m/z: 637.13 (M+).
Biology
In vitro antimicrobial assay
The broth microdilution method has been employed to determine the MICs of synthesized compounds as described in the literature (Patel et al. 2010). Dimethylsulfoxide (DMSO) was used as diluent to get desired concentration of drugs to test upon standard bacterial strains. Prepare a solvent control of a 1:10 dilution of the DMSO used to dissolve the antimicrobial agent being tested. This 1:10 solution is prepared by adding 0.1 ml of solvent to 0.9 ml of the appropriate diluent. The highest dilution showing at least 99% inhibition is taken as MIC. To evaluate the antimicrobial potency of the final derivatives, they were screened against different strains viz., two Gram-positive bacteria S. aureus (MTCC–96) and S. pyogenes (MTCC–442), two Gram-negative bacteria E. coli (MTCC–443) and P. aeruginosa (MTCC–1688), and fungi, C. albicans (MTCC–227), A. niger (MTCC–282), and A. clavatus (MTCC– 1323), and compared with standard drugs, chloramphenicol, ciprofloxacin, and griseofulvin.
In vitro antitubercular assay
Tubercle bacilli are aerobes, grow in specially enriched media containing egg, asparagines, potatoes, serum, and meat extracts. Colonies appear in 2–6 weeks. The drug susceptibility test to determine MIC by LJ Slope method has been employed (Muralidhar and Srivastava 2004). M. tuberculosis H37Rv [acid fast bacilli] (MTCC–200) was used for screening of antitubercular activity. DMSO was used as diluents/vehicle to get desired concentration of drugs to test upon standard bacterial strains. Each synthesized compound was diluted obtaining 2000 μg/mL concentration, as a stock solution and then many dilutions were made shown as in antimicrobial activities (Fig. 1).

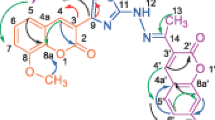
General structure of 4-methyl-6-nitro-2-oxo-2H-chromen-7yl-2-(4-(4-fluorophenyl)-6-substituted phenyl-2H-1,3-thiazin-2-yl -amino) acetate 5a–j
Antioxidant evaluation
DPPH method
Reduction of 2,2-diphenyl-1-picrylhydrazyl (free radical) is the base of the DPPH antioxidant bioassay. It has an odd electron that shows a maximum absorption band of 517 nm (deep violet color) in ethanol. The DPPH bioassay is the widely used and acceptable method for evaluating the free radical scavenging action of the tested compounds. Such substances donate a hydrogen atom when mixed with the DPPH, thereby introducing its reduced congener, diphenylpicrylhydrazine (non-radical) with the loss of violet color. In the present study, DPPH bioassay was adopted to screen the berberine-based compounds for their in vitro antioxidant profiles. The results of this bioassay investigation were introduced in the form of the percentage of radical scavenging antioxidant activity (RSA%) of each substance. The investigation of the DPPH radical scavenging activity was operated according to the methodology described by (Brand-Williams et al. 1995) with some modifications (Mistry et al. 2016). A stable free radical, DPPH, was allowed to react with test compounds in methanol as 20 μg/mL (100, 10, 1, and 0.1) quantities of title compounds were mixed up with 180 μg/mL of DPPH in methanol. Titled compounds donated hydrogen during the mixing thereby introduced the reduction of DPPH and hence a change in the color was observed from deep violet to light yellow at 517 nm after 25 min of reaction in a UV–Visible spectrophotometer (Perkin Elmer). The blank reading was also performed using the mixture of methanol (20 μg/mL) and sample (180 μg/mL of DPPH). Ascorbic acid served as a control drug in this assay, and its solution was prepared by mixing methanol (20 μg/mL) and DPPH radical solution (180 μg/mL). The results of this bioassay, RSA % was determined according to (Mensor et al. 2001) as described in the below equation.
A plot of concentration (Fig. 2) of test compounds and % scavenging introduced IC50 in the presence of an ascorbic acid as standard.

The plot of antioxidant assay result of 5a–j
ABTS method
The ABTS• + radical cation scavenging efficacies of the test compounds was determined according to the method described earlier with some modifications (Mistry et al. 2016). Mixing of an equal amount of 7.0 mM ABTS• + stock solution with 2.45 mM potassium persulfate stock solution produces the ABTS• + cation. The mixture was kept in a dark place at 0 °C temperature for 12 h and ABTS solution was diluted with MeOH so that it gives UV absorption value of 0.700 ( ± 0.200) at the 734 nm. The 1000 μL stock solutions of titled compounds 5a–j were dissolved in methanol and further dilutions of 100, 10, 1, and 0.1 μg/mL taken for test samples. In all 180 μg/mL solutions of compounds to be evaluated and 20 μg/mL of the ABTS solution were mixed in 96-well plates in a dark place and then incubated for 10 min to measure UV absorption at 734 nm. The solution of 180 μg/mL ABTS in 20 μg/mL methanol was used as a control determination, while ascorbic acid was used as a reference drug. The UV absorption data represented the radical scavenging rates that give the corresponding IC50s for the test compounds.
The scavenging capability of ABTS• + radical was calculated using the following equation:
Results and discussion
Chemistry
The synthetic protocol for the lead molecule 4-methyl-6-nitro-2-oxo-2H-chromen-7-yl 2-chloroacetate (5) and final compounds (5a–j) is depicted in Scheme 1. According to the scheme, 4-methyl-7-hydroxy-coumarin (2) has been synthesized by pechmann condentation, which is followed by nitration with nitric acid and acetic acid and then reacting with chloroacetyl chloride to get 4-methyl-3-nitro-2-oxo-2H-chromen-7-yl 2-chloroacetate (5). For the synthesis of final compounds, key intermediate (5) was condensed with amino thiazines derivatives (IIIa–j) obtained from cycloaddition reaction between substituted chalcones (IIa–j) and thiourea. The reaction protocol is illustrated in the following scheme.

a Cooled (5–10 °C), conc. H2SO4; b CAN, 30% H2O2 + 5 mL H2O, stirred; c α-chloroacetyl chloride, CH2Cl2, triethyl amine 1 h stirred; d NaOH, EtOH, 2 -3 h stirred; e thiourea, ethanolic NaOH, 2–3 h refluxed; f 4-methyl-6-nitro-2-oxo-2H-chromen-7-yl 2-chloroacetate (5), DMF, K2CO3, 8-9 h refluxed. Where, R = -H, 2-Cl, 4-Cl, 2-OH, 4-OH, 4-F, 4-CH3, 4-C3H7, 3-Br, 3-OPh
Characterizations of intermediate and final compounds were confirmed by their spectral analysis. The characteristic band of 3192 cm−1 of –NH, while two bands for –C=O appeared at 1715 and 1671 cm−1 and 1523, 1351 cm−1 for –NO2 in IR spectrum confirmed the structure of the final compounds 5a–j. 1H NMR spectrum of final compounds showed singlet at 8.55 of –CH confirmed the neighboring –NO2 group while –CH2NH showed two singlet at 3.71 and 3.23, respectively. 13C NMR spectrum of 5a–j showed peaks nearer at 160.10 and 168.20 for two different C=O and one peak at 19.40 for –CH3 of coumarin. Peaks at 43.90, 81.10, and 111.30 were obtained for –CHNH, –CH of thiazine (C-18) ring, which confirming the structure of final compounds.
Biology
Antimicrobial activity
The minimum inhibitory concentrations (MIC) for the antimicrobial potency of 5a–j were screened against four different bacterial strains and three different fungal strains. The susceptibility of the organisms was determined by the broth microdilution method (Rattan 2000) and compared with standard drugs; chloramphenicol, ciprofloxacin, and griseofulvin. The results of this activity are described in Table 1. Compound 5c with a –Cl group and 5 h with –CH2CH2CH3 at position 4 on benzaldehyde demonstrated remarkable activity (MIC = 50 µg/mL) against E. coli, comparable to chloramphenicol and ciprofloxacin. While other compounds showed poor activity against S. aureus and S. pyogenes. Compounds 5a and 5b, having substituent –H, 2-Cl, respectively, exhibited significant activity with MIC value 250 µg/mL, while compound 5d showed encouraging potency (MIC = 200 µg/mL) against C. albicans compared with griseofulvin. Other compounds with very high MIC values and seem to be poor to moderately active (Table 2).
Antitubercular activity
The MIC of the titled compounds were tested for antituberculer activities (Andrews 2001). The results are mentioned in Table 3. Antituberculer activity results showed that, compound 5b and 5h having 2-Cl and 4-propyl group demonstrated better activity 62.5 µg/mL with 98–99 % inhibition against M. tuberculosis H37Rv (Table 4).
Antioxidant activity
From the antioxidant inspections, molecules 5h and 5b were appeared to have high radical scavenging efficacies as 33.99 ± 0.301 and 35.35 ± 0.470 μg/mL ± SD of IC50 values in DPPH and ABTS bioassay, respectively, and can be comparable to that of control ascorbic acid while other compounds have moderate to poor antioxidant power. The results were summarized in the following graph.
Conclusion
A new series of 4-methyl-6-nitro-2-oxo-2H-chromen-7yl-2-(4-(4-fluorophenyl)-6-substituted-phenyl-2H-1,3-thiazin-2-yl-amino) acetate has been efficiently formulated via coupling 4-methyl-6-nitro-2-oxo-2H-chromen-7-yl 2-chloroacetate with amino thiazine derivatives, which was obtained by treating thiourea with substituted chalcone derivatives at optimum reaction condition. All the synthesized compounds were characterized by spectral techniques. Final compounds were evaluated for their in vitro antioxidant activity using DPPH and ABTS bioassays. The presence of chloro and propyl group on phenyl ring on the chalcone system was essential to exert antioxidant effect and showed excellent free radical scavenging efficacies in DPPH and ABTS bioassays, respectively. Also, a MIC of 5a–j using broth microdilution method towards bacterial and fungal strains was studied and the derivatives 5c and 5h displayed remarkable potency against E. coli with MIC values 50 µg/mL compare to standard drugs. It has been observed that the potent antibacterial and antitubercular candidate proved to possess significant antioxidant activity. The present of propyl group on phenyl ring plays an important role for the potency in above mentioned biological assay.
References
Al-Amiery AA, Kadhum AAH, Mohamad AB (2012) Antifungal activities of new coumarins. Molecules 17:5713–5723
Andrews JM (2001) Determination of minimum inhibitory concentrations. J Antimicrob Chemother 48:5–16
Bansal Y, Sethi P, Bansal G (2013) Coumarin: a potential nucleus for anti-inflammatory molecules. Med Chem Res 22:3049–3060
Bhat MA, Siddiqui N, Khan SA, Mohamed MI (2009) Synthesis of triazolothiazolidinone derivatives of coumarin with antimicrobial activity. Acta Pol Pharm 66:625–632
Brand-Williams W, Cuvelier M-E, Berset C (1995) Use of a free radical method to evaluate antioxidant activity. LWT-Food Sci Tech 28:25–30
Chen H, Li S, Yao Y, Zhou L, Zhao J, Gu Y, Wang K, Li X (2013) Design, synthesis, and anti-tumor activities of novel triphenylethylene–coumarin hybrids, and their interactions with Ct-DNA. Bioorg Med Chem Lett 23:4785–4789
Debeljak Ž, Škrbo A, Jasprica I, Mornar A, Plecko V, Banjanac M, Medić-Šarić M (2007) QSAR study of antimicrobial activity of some 3-nitrocoumarins and related compounds. J Chem Inf Model 47:918–926
Dekić V, Radulović N, Vukićević R, Dekić B, Stojanović-Radić Z, Palić R (2011) Influence of the aryl substituent identity in 4-arylamino-3-nitrocoumarins on their antimicrobial activity. Afr J Pharm Pharmacol 5:371–375
Furniss BS, Hannaford AJ, Smith PW, Tatchell AR (1989) Cognate preparation 4-methyl-7-hydoxycoumarin. In: Vogel AI (Ed) Vogel’s textbook of practical organic chemistry. Longman Scientific & Technical, England, p 1193
Ganguly N, Sukai A, De S (2001) Cerium (IV) ammonium nitrate mediated nitration of coumarins. Synth Commun 31:301–309
Hassan MZ, Osman H, Ali MA, Ahsan MJ (2016) Therapeutic potential of coumarins as antiviral agents. Eur J Med Chem 123:236–255
Jeleń M, Bavavea EI, Pappa M, Kourounakis AP, Morak-Młodawska B, Pluta K (2015) Synthesis of quinoline/naphthalene-containing azaphenothiazines and their potent in vitro antioxidant properties. Med Chem Res 24:1725–1732
Jupudi S, Talari S, Karunakaram D, Govindarajan R (2013) Screening of in vitro antiinflammatory activity of some newly synthesized 1, 3-thiazine derivatives. Int J Res Pharm Chem 3:213–220
Keri RS, Hosamani KM, Shingalapur RV, Hugar MH (2010) Analgesic, anti-pyretic and DNA cleavage studies of novel pyrimidine derivatives of coumarin moiety. Eur J Med Chem 45:2597–2605
Koketsu M, Tanaka K, Takenaka Y, Kwong CD, Ishihara H (2002) Synthesis of 1, 3-thiazine derivatives and their evaluation as potential antimycobacterial agents. Eur J Pharm Sci 15:307–310
Kontogiorgis C, Nicolotti O, Mangiatordi GF, Tognolini M, Karalaki F, Giorgio C, Patsilinakos A, Carotti A, Hadjipavlou-Litina D, Barocelli E (2015) Studies on the antiplatelet and antithrombotic profile of anti-inflammatory coumarin derivatives. J Enzym Inhib Med Chem 30:925–933
Kostova I (2006) Synthetic and natural coumarins as antioxidants. Mini Rev Med Chem 6:365–374
Kostova I, Bhatia S, Grigorov P, Balkansky S, Parmar VS, Prasad AK, Saso L (2011) Coumarins as antioxidants. Curr Med Chem 18:3929–3951
Mistry BM, Patel RV, Keum Y-S, Hwan Kim D (2016) Synthesis of 9-O-3-(1-piperazinyl/morpholinyl/piperidinyl) pentyl-berberines as potential antioxidant and cytotoxic agents. Anticancer Agents Med Chem (Former Med Chem Anticancer Agents) 16:713–721
Mensor LL, Menezes FS, Leitão GG, Reis AS, Santos TCd, Coube CS, Leitão SG (2001) Screening of Brazilian plant extracts for antioxidant activity by the use of DPPH free radical method. Phytother Res 15:127–130
Muralidhar S, Srivastava L (2004) Evaluation of three methods to determine the antimicrobial susceptibility of Mycobacterium tuberculosis. Indian J Med Res 120:463–467
Muratovic S, Duric K, Veljovic E, Osmanovic A, Softic D, Zavrsnik D (2013) Synthesis of biscoumarin derivatives as antimicrobial agents. Asian J Pharm Clin Res 6:132–134
Nagamallu R, Srinivasan B, Ningappa MB, Kariyappa AK (2016) Synthesis of novel coumarin appended bis (formylpyrazole) derivatives: Studies on their antimicrobial and antioxidant activities. Bioorg Med Chem Lett 26:690–694
Patel D, Kumari P, Patel NB (2017) Synthesis and biological evaluation of coumarin based isoxazoles, pyrimidinthiones and pyrimidin-2-ones. Arab J Chem 10:S3990–S4001
Patel K, Karthikeyan C, Moorthy NHN, Deora GS, Solomon VR, Lee H, Trivedi P (2012) Design, synthesis and biological evaluation of some novel 3-cinnamoyl-4-hydroxy-2H-chromen-2-ones as antimalarial agents. Med Chem Res 21:1780–1784
Patel NB, Khan IH, Rajani SD (2010) Pharmacological evaluation and characterizations of newly synthesized 1, 2, 4-triazoles. Eur J Med Chem 45:4293–4299
Qandil AM, Fakhouri LI (2012) a-Anilinoketones, esters and amides: A chemical study. Pharmaceuticals 5:591–612
Rattan A (2000) Antimicrobials in laboratory medicine. Churchill BI, Livingstone, New Delhi, 85
Rehman SU, Chohan ZH, Gulnaz F, Supuran CT (2005) In-vitro antibacterial, antifungal and cytotoxic activities of some coumarins and their metal complexes. J Enzym Inhib Med Chem 20:333–340
Roma G, Di Braccio M, Carrieri A, Grossi G, Leoncini G, Signorello MG, Carotti A (2003) Coumarin, chromone, and 4 (3H)-pyrimidinone novel bicyclic and tricyclic derivatives as antiplatelet agents: synthesis, biological evaluation, and comparative molecular field analysis. Bioorg Med Chem 11:123–138
Rost S, Fregin A, Hünerberg M, Bevans CG, Müller CR, Oldenburg J (2005) Site-directed mutagenesis of coumarin-type anticoagulant-sensitive VKORC1. Thromb Haemost 93:780–786
Sashidhara KV, Kumar A, Kumar M, Sarkar J, Sinha S (2010) Synthesis and in vitro evaluation of novel coumarin–chalcone hybrids as potential anticancer agents. Bioorg Med Chem Lett 20:7205–7211
Thakur A, Singla R, Jaitak V (2015) Coumarins as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Eur J Med Chem 101:476–495
Tiwari R, Miller PA, Chiarelli LR, Mori G, Šarkan M, Centárová I, Cho S, Mikušová K, Franzblau SG, Oliver AG (2016) Design, syntheses, and anti-TB activity of 1, 3-benzothiazinone azide and click chemistry products inspired by BTZ043. ACS Med Chem Lett 7:266–270
Wang W, Zhao B, Xu C, Wu W (2012) Synthesis and antitumor activity of the thiazoline and thiazine multithioether. Int J Org Chem 2:117–120
Acknowledgements
We are thankful to Veer Narmad South Gujarat University for providing necessary facilities, D. Rajani, Microcare Laboratory, Surat, for antimicrobial and antitubercular activity. Nilesh Chauhan is highly obliged to University Grants Commission, New Delhi for awarding Teacher Fellowship Award under Faculty Improvement Program. Vatsal M. Patel is grateful to University Grant Commission for providing BSR-SAP fellowship (F.7-233/2009(BSR)).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Chauhan, N.B., Patel, N.B., Patel, V.M. et al. Synthesis and biological evaluation of coumarin clubbed thiazines scaffolds as antimicrobial and antioxidant. Med Chem Res 27, 2141–2149 (2018). https://doi.org/10.1007/s00044-018-2222-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-018-2222-9