
Abstract
Three antioxidant and anti-inflammatory oxygenated meroterpenoids, 1-(3-methoxypropyl)-2-propylcyclohexane (C13) (1), 3-(methoxymethyl)heptyl 3-(cyclohex-3-enyl) propanoate (C18) (2), and 2-ethyl-6-(4-methoxy-2-((2-oxotetrahydro-2H-pyran-4-yl)methyl)butoxy)-6-oxohexyl 5-ethyloct-4-enoate (C29) (3) were purified from the methanol:ethyl acetate fraction of red seaweed Kappaphycus alvarezii (family Solieriaceae) collected from the Gulf-of-Mannar on the southeast coast of peninsular India. The highly oxygenated C29 meroterpenoid 3 displayed potential antioxidative activities (IC50 < 0.35 mg/mL) as evaluated by 2, 2’-azino-bis (3-ethylbenzothiazoline)-6-sulphonic acid and 1, 1-diphenyl-2-picryl-hydrazil free radical scavenging assays. The compound 3 displayed potential in vitro inhibitory activities towards pro-inflammatory 5-lipoxidase (IC50 1.04 mg/mL), which indicated its potential anti-inflammatory properties against inducible inflammatory mediators causing an inflammatory response. Structure-activity relationship analyses displayed the functional roles of lipophilic-hydrophobic characteristics and electronic parameter to determine its potential anti-inflammatory activity in terms of inhibiting inducible inflammatory cyclooxygenase and lipoxidase.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Terpenoids are structurally diverse secondary metabolites with more than 40,000 reported structural diversity possessing valuable bioactive properties (Gershenzon and Dudareva 2007). Terpenoids were recognized to possess potential pharmacological properties against deadly diseases, such as malaria (Parshikov et al. 2012), cardiovascular ailments (Liebgott et al. 2000) and cancer (Ebada et al. 2010). Seaweeds or marine macroalgae were found to be the potential reservoir of bioactive secondary metabolites including terpenes, sterols, polyphenols, acetogenins, etc. (Reis et al. 2013), and the most prominent among these are meroterpenoid group of compounds (Chakraborty et al. 2016). A rare meroterpenoid (secotaondiol) with potential gastroprotective activity was described in a previous report of literature (Areche et al. 2015). The meroditerpene, 11-hydroxy-11-O-methylamentadione, isolated from the seaweed Cystoseira usneoides showed anti-inflammatory effects in dextran sodium sulphate-persuade colitis in a murine model. The terpenoid compound was found to significantly inhibit the generation of the cytokine (a type of inflammatory signaling molecule) and tumour necrosis factor in lipopolysaccharide-induced human monocytic leukaemia cell line (Zbakh et al. 2016). Three antioxidative aryl meroterpenoids were previously isolated from the red seaweed Hypnea musciformis (Chakraborty et al. 2016).
Among different red seaweeds, Kappaphycus alvarezii (Doty) (family Solieriaceae) is a commercially important and cultivable species that is predominantly abundant in tropical coastal and marine habitats in the southeast Asian countries, especially Malaysia, India, Indonesia, Philippines, China and Taiwan (Chandrasekaran et al. 2008; Ask and Azanza 2002). We have previously described the anti-inflammatory and antioxidant potentials of unprecedented cyclic ether along with pharmacologically active polygalactan from this seaweed species (Makkar and Chakraborty 2017a; Makkar and Chakraborty 2017b, c). The present study aimed to isolate three specialized oxygenated meroterpenoids, 1-(3-methoxypropyl)-2-propylcyclohexane (C13) (1), 3-(methoxymethyl)heptyl 3-(cyclohex-3-enyl)propanoate (C18) (2) and 2-ethyl-6-(4-methoxy-2-((2-oxotetrahydro-2H-pyran-4-yl)methyl)butoxy)-6-oxohexyl 5-ethyloct-4-enoate (C29) (3) of K. alvarezii collected from the shallow marine habitats of the Gulf-of-Mannar on the south-east coast of Peninsular India (Fig. 1). The anti-inflammatory and antioxidative activities of these compounds were evaluated by different in vitro models and their structures were proposed on the basis of 2D-NMR experiments. The physicochemical characteristics of meroterpenoids contributing towards the antioxidant and anti-inflammatory activities were ascertained by structure-activity relationship analyses.

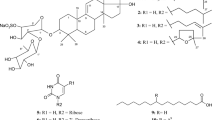
Oxygenated meroterpenoids, 1-(3-methoxypropyl)-2-propylcyclohexane (1), 3-(methoxymethyl)heptyl 3-(cyclohex-3-enyl)propanoate (2) and 2-ethyl-6-(4-methoxy-2-((2-oxotetrahydro-2H-pyran-4-yl)methyl)butoxy)-6-oxohexyl 5-ethyloct-4-enoate (3) isolated from red seaweed K. alvarezii. The thalli of the studied seaweed were displayed as inset
Materials and methods
Chemicals and instrumentation
FTIR spectral data were recorded in Perkin–Elmer Series 2000 (scan range of 400 and 4000 cm−1). A Varian Cary 50 Uv–vis spectrometer (Varian Cary, USA) was utilized to acquire the UV spectral data. Two-dimensional NMR spectral experiments were performed on a Bruker Avance DPX 500 (500 MHz) spectrometer (CDCl3 as aprotic solvent). Standard pulse sequences were used for HMBC, HSQC, 1H–1H COSY, NOESY, and DEPT experiments. GC-MS analyses were carried out with an EI mode {Varian GC (CP-3800) housed in a mass spectrometer (Varian 1200 L)}. ESI-MS data were obtained by using a liquid chromatography-mass spectrometry system (Applied Biosystems QTrap 2000, Germany). The solvents used for analyzing samples were of analytical grade (E-Merck, Germany).
Collection of seaweed samples of K. alvarezii and extraction
The red seaweed K. alvarezii were freshly collected from the Gulf of Mannar in Mandapam region located between 8°48′ N, 78°9′ E and 9°14′ N, 79°14′E on the southeast coast of India. The seaweed thalli were washed in running water for 15 min and shade dried (~36 °C, 36 h). The shade dried seaweed thalli were ground before being extracted (1 kg) with solvent methanol: ethyl acetate (3 h, 1:1, v/v, 60–70 °C) before being dried through anhydrous Na2SO4. The pooled filtrate was concentrated below 50 °C by using a rotary vacuum evaporator (Heidolph, Germany) to dryness to yield a dark brown residue of crude ethyl acetate-methanol (EtOAc: MeOH) fraction of K. alvarezii (45 g, yield on dry basis 4.5%).
Purification of meroterpenoids from the red seaweed K. alvarezii
The crude EtOAc: MeOH fraction of K. alvarezii (20 g) was loaded over a glass column filled with silica gel (60–120 mesh, 600 g), before being fractionated by repeated chromatography to separate various fractions. The initial elution was carried out with n-hexane and the solvent polarity was gradually increased with the addition of EtOAc (3:7 v/v n-hexane: EtOAc) to obtained thirty fractions (20 mL) that were minimized to five homogeneous groups (FN34-FN38), whereas FN35 (320.3 mg) was fractionated with n-hexane: EtOAc (4:1, v/v) to yield compound 1 (120 mg). The purity of compound 1 was ascertained by TLC (silica gel GF254; MeOH: EtOAc, 1:19 v/v, Rf: 0.96) and reverse-phase HPLC (acetonitrile ACN: MeOH, 2:4 v/v) experiments. The fraction FN36 was flash chromatographed (Biotage SP1-B1A, Sweden) on a silica gel column (loaded with 230–400 meshed silica gel) by employing a step gradient of EtOAc/n-hexane (0–10% EtOAc) to yield a total of ninety fractions (10 mL). Following thin layer chromatography (TLC) analyses, the identical fractions were pooled to obtain five fractions {50 mL, FN36-(1-5)}. The fraction FN36(2) was flash chromatographed on a column (230–400 meshed silica gel) with EtOAc/n-hexane (1:9 to 3:7, v/v) to yield compound 2 {FN36(2), 120.5 mg). The purity of compound 2 was ascertained by TLC {silica gel GF254; MeOH/dichloromethane DCM 1:99 v/v, Rf: 0.80) and reverse-phase HPLC (ACN: MeOH, 1:2 v/v) experiments. The fraction FN36(4) was fractionated with silica gel flash chromatography with EtOAc/n-hexane (1:1, v/v), and thereafter with MeOH/DCM (1:9, v/v) to yield fifty fractions (10 mL). Following TLC analyses, the identical fractions were pooled to yield FN45-1 through FN45-11. The fraction FN45-1 was further purified by preparatory silica gel TLC, whereas the plate was eluted with DCM/MeOH (9:1, v/v) to afford compound 3 {FN36(4), 50.5 mg) as major component, and its purity was ascertained by TLC (silica gel GF254; MeOH/ CHCl3 1:19 v/v, Rf: 0.96) and reverse-phase HPLC (ACN: MeOH, 1:2 v/v) experiments.
Spectral analysis of 1-(3-methoxypropyl)-2-propylcyclohexane (1)
Yellow oil; UV MeOH λmax (log ε): 245 nm (3.26), TLC (Si gel GF254 15 mm; EtOAc/MeOH 19:1, v/v) Rf: 0.96; Rt (HPLC, ACN: MeOH, 2:4 v/v): 12.401 min; IR (vibrational spectra were measured between 4000 to 450 cm−1 for KBr pellets, all frequencies were reported in cm−1; the notations for the various motions of atoms within the normal modes were defined as: ν, stretching; δ, bending; ω, wagging; ρ, rocking; τ, torsion; s, symmetric; as, asymmetric): 728.70 (C-H ρ), 1014.67 (C–O ν), 1256.18 (CH2 ν), 1376.13 (C-H ρ), 1458.14 (C-H δ), 1644.58 (C=C ν), 2857.12, 2923.46 (C–H ν); 1H NMR (500 MHz CDCl3): δH 1.26 (2H, m, H-1), 1.26 (2H, m, H-2), 1.26 (2H, m, H-3), 1.62 (2H, m, H-4), 2.32 (1H, m, H-5), 2.04 (1H, m, H-6), 1.42 (2H, m, H-7), 1.72 (2H, m, H-8), 4.29 (2H, t, 6.95 Hz, H-9), 3.67 (3H, s, H-10), 1.25 (2H, m, H-11), 1.31 (2H, m, H-12), 0.88 (3H, t, 7.53 Hz, H-13); 13C NMR (125 MHz, CDCl3): δC 29.65 (CH2, C-1), 27.23 (CH2, C-2), 29.65 (CH2, C-3), 37.10 (CH2, C-4), 33.86 (CH, C-5), 32.38 (CH, C-6), 29.88 (CH2, C-7), 29.66 (CH2, C-8), 50.87 (CH2, C-9), 51.45 (CH3, C-10), 31.97 (CH2, C-11), 22.7 (CH2, C-12), 14.12 (CH3, C-13); HMBC and 1H-1H-COSY data (Table 1); HR(ESI) MS m/z measured value 198.1988 [M]+, calcd for C13H26O 198.1984 (Fig. S10-S19).
Spectral analysis of 3-(methoxymethyl)heptyl 3-(cyclohex-3-enyl)propanoate (2)
Yellowish oil; UV MeOH/DCM λmax (log ε): 238 nm (2.82), 262 nm (2.40); TLC (Si gel GF254 15 mm; MeOH/DCM 1:99, v/v) Rf: 0.80; Rt (HPLC, ACN: MeOH, 2:4 v/v): 14.2681 min; IR (KBr, expressed in cm−1): 724.66 (C–H ρ), 878.09 (C-H δ), 1018.99 (C–H ρ), 1114.25 (C–H δ), 1169.65 (C=C ν), 1249.94 (C–CO–C ν), 1366.49 (C=O ν), 1458.06 (C–H ν), 1743.11 (C=O ν), 2856.12 (C-H ν), 2925.01 (C-H ν); 1H NMR (500 MHz CDCl3): δH 1.94 (2H, t, 6.13 Hz, H-1), 5.27 (1H, m, H-2), 5.28 (1H, m, H-3), 2.71 (2H, t, 6.69 Hz, H-4), 1.65 (2H, m, H-5), 1.65 (1H, m, H-6), 1.54 (2H, m, H-7), 2.24 (2H, t, 7.34 Hz, H-8), 4.05 (2H, t, 6.85 Hz, H-10), 1.51 (2H, m, H-11), 2.02 (1H, m, H-12), 4.21 (2H, d, 6.78 Hz, H-13), 3.59 (3H, s, H-14), 1.19 (2H, m, H-15), 1.26 (2H, m, H-16), 1.18 (2H, m, H-17), 0.80 (3H, t, 6.77 Hz, H-18); 13C NMR (125 MHz, CDCl3): δC 27.15 (CH2, C-1), 129.93 (CH, C-2), 129.92 (CH, C-3), 25.5 (CH2, C-4), 32.73 (CH2, C-5), 40.19 (CH, C-6), 24.97 (CH2, C-7), 34.13 (CH2, C-8), 174.37 (C-9), 62.04 (CH2, C-10), 37.61 (CH2, C-11), 31.50 (CH, C-12), 62.20 (CH2, C-13), 51.43 (CH3, C-14), 29.56 (CH2, C-15), 28.82 (CH2, C-16), 26.34 (CH2, C-17), 14.11 (CH3, C-18); HMBC and 1H-1H-COSY data (Table 2); HR(ESI) MS m/z measured value 296.2354 [M]+, calcd for C18H32O3 296.2351 (Fig. S20-S29).
Spectral analysis of 2-ethyl-6-(4-methoxy-2-((2-oxotetrahydro-2H-pyran-4-yl)methyl)butoxy)-6-oxohexyl5-ethyloct-4-enoate (3)
Yellow oil; UV MeOH λmax (log ε): 245 nm (3.26); TLC (Si gel GF254 15 mm; MeOH/CHCl3 1:19, v/v) Rf: 0.96; Rt (HPLC, MeOH: ACN, 2:1 v/v): 14.401 min; IR (KBr, expressed in cm−1): 738.89 (C–H ρ), 1073.42 (C–O ν), 1125.95 (CH2 wag), 1170.14 (C–O ν), 1280.43 (CH2 ν), 1369.88 (C–H ρ), 1455.83 (C-H δ), 1589.64 (C=C ν), 1736.56 (C=O ν), 2857.58, 2926.28 (C–H ν); 1H NMR (500 MHz CDCl3): δH 0.87 (3H, t, 6.80 Hz, H-1), 1.30 (2H, m, H-2), 2.02 (2H, m, H-3), 5.35 (1H, t, 5.68 Hz, H-5), 2.02 (2H, m, H-6), 2.32 (2H, t, 7.80 Hz, H-7), 4.16 (2H, d, 5.80 Hz, H-9), 1.73 (1H, m, H-10), 1.27 (2H, m, H-11), 1.62 (2H, m, H-12), 2.32 (2H, t, 7.80 Hz, H-13), 4.26 (2H, d, 9.08 Hz, H-15), 2.49 (1H, m, H-16), 1.50 (2H, m, H-17), 1.69 (1H, m, H-18), 2.32 (2H, t, 7.80 Hz, H-19), 4.19 (2H, t, 9.08 Hz, H-21), 1.69 (2H, m, H-22), 1.73 (2H, m, H-23), 4.30 (2H, t, 7.44 Hz, H-24), 3.67 (3H, s, H-25), 1.42 (2H, m, H-26), 0.85 (3H, t, 6.80 Hz, H-27), 2.13 (2H, m, H-28), 0.87 (3H, t, 7.17 Hz, H-29); 13C NMR (125 MHz, CDCl3): δC 14.20 (CH3, C-1), 25.12 (CH2, C-2), 28.24 (CH2, C-3), 132.45 (C-4), 130.08 (CH, C-5), 34.11 (CH2, C-6), 29.67 (CH2, C-7), 174.56 (C-8), 62.17 (CH2, C-9), 40.19 (CH, C-10), 29.65 (CH2, C-11), 28.99 (CH2, C-12), 24.98 (CH2, C-13), 168.33 (C-14), 68.24 (CH2, C-15), 34.39 (CH, C-16), 29.26 (CH2, C-17), 38.88 (CH, C-18), 32.14 (CH2, C-19), 173.48 (C-20), 60.14 (CH2, C-21), 30.78 (CH2, C-22), 32.73 (CH2, C-23), 65.68 (CH2, C-24), 51.42 (CH3, C-25), 29.85 (CH2, C-26), 19.70 (CH3, C-27), 30.07 (CH2, C-28), 22.80 (CH3, C-29). HMBC and 1H-1H-COSY data (Table 3); HR(ESI) MS m/z measured value 510.3557 [M]+, calcd for C29H50O7 510.3552 (Fig. S30-S39).
Free radical scavenging and anti-inflammatory activities
Radical scavenging potential of the oxygenated meroterpenoids was measured using the 1, 1-diphenyl-2-picrylhydrazyl (DPPH) and 2,2'-azino-bis (3-ethylbenzothiazoline-6-sulphonic acid (ABTS) scavenging assays (Makkar and Chakraborty 2017d). Anti-inflammatory activities were determined by using pro-inflammatory cyclooxygenase-1/2 (COX-1, 2) (Larsen et al. 1996) and 5-lipoxygenase (5-LOX) in vitro inhibition assays, as described previously (Baylac and Racine 2003).
Structure–activity relationship analysis
Structure-activity relationship analysis was carried out by applying distinct structural descriptors (ACD Chemsketch, version 8.0 and ChemDraw Ultra version 8.0), named steric {molar volume}, electronic {topological polar surface area (tPSA) and hydrophobic {octanol-water partition coefficient (log Pow)} molecular descriptor variables.
Statistical analysis
Statistical analysis was performed with the Statistical Program for Social Sciences 10.0 (SPSS Inc, CA, USA). Analyses were performed in triplicate, and the means of all parameters were assessed for significance (p ≤ 0.05) by analysis of variance.
Results and discussion
Spectral analyses of meroterpenoids from K. alvarezii
1-(3-Methoxypropyl)-2-propylcyclohexane (compound 1), a methoxy-substituted C13 meroterpenoid, was purified as yellow oil by extensive column chromatography on adsorbent silica gel. The mass spectrum displayed the molecular ion peak at m/z 198 (Fig. S1) enclosing mono unsaturation (because of the ring system), and the molecular formula as C13H26O based upon combined 1H and 13C NMR spectral data (Table 1). The existence of 13 carbon signals constituting of nine methylenes, two methines and one each of methoxy and methyl carbons was supported by the 13C NMR experiment (Table 1). The deshielded resonance of H-9 (δH 4.29, J = 6.95 Hz) of 1 suggested the C-9 methylene groups remained attached to an electronegative group, possibly of oxygenated origin. The 1H-1H COSY correlations were observed between δH 2.32 (H-5)/ δH 2.04 (H-6); δH 1.62 (H-4)/ δH 2.32 (H-5) were apparent, and were ascribed to the cyclohexane ring framework (Fig. 2, Fig. S2). The methine (–CH) carbon signals were apparent at δC 33.86 (C-5) and C-6 (δC 32.38) that appropriately recognized the junction point of cyclohexane ring system substituted with propane and the methoxypropane moieties, respectively. HMBC correlations from δH 1.25 (assigned as H-11) to δC 22.7 (C-12); δH 1.42 (H-7) to δC 33.86 (C-5)/ δC 31.97 (C-6)/ δC 29.66 (C-8); δH 0.88 (H-13) to δC 31.97 (C-11)/ δC 22.7 (C-12); δH 4.29 (H-9) to δC 29.66 (C-8); δH 3.67 (H-10) to δC 50.87 (C-9) displayed the side chain substitutions of the cyclohexane ring system. In addition, 1H-1H COSY correlations appeared at δH 4.29 (H-9)/ δH 1.72 (H-8), δH 2.04 (H-6)/ δH 1.42 (H-7), which were due to the 1-methoxypropane framework attached to the cyclohexane ring system, and was in accordance with the J1-3 HMBC attributions. Similarly, 1H-1H COSY correlation appeared at δH 1.31 (H-12)/ δH 0.88 (H-13), which was due to the framework attached to the cyclohexane ring system. The combined 1H/ 13C NMR demonstrated highly deshielded oxymethylene protons at δH 4.29 (attributed to H-9) corresponding to the carbon resonance at δC 50.87 (C-9) to assign the propylcyclohexane ring system and side chain substitution in C13 meroterpenoid. The relative stereochemistries of 1 were attributed by NOESY experiments. The reference plane of the compound 1 was arbitrarily chosen as the cyclohexane ring system. NOESY cross peaks between δH 2.32 (H-5)/ δH 1.62 (H-4) suggested their close proximity, and therefore, assigned to align on an identical plane of the cyclohexane ring system with di-equatorial β-faced interaction. Intense NOESY cross peaks between δH 1.62 (H-4)/ δH 2.04 (H-6) appropriately indicated their equi-planer disposition (β-orientation).

a Key 1H-1H COSY, HMBC and NOESY correlations of compound 1. The 1H-1H COSY cross peaks were displayed by bold face bonds, whereas the selected HMBC correlations were shown as double barbed arrows. The β orientation in the NOESY relations was presented as blue coloured arrows
3-(Methoxymethyl)heptyl 3-(cyclohex-3-enyl)propanoate (compound 2), an oxygenated C18 meroterpenoid displayed the molecular ion peak at m/z 296 (Fig. S4) enclosing three degrees of unsaturation {due to the ester carbonyl group (δC 174.37), olefinic carbon at δC 129.92 (C-3), δC 129.93 (C-2) and a ring system}, and the molecular formula as C18H32O3 based upon combined 1H and 13C NMR spectral data (Table 2). The IR-spectrum of 2 displayed the presence of carbonyl group along with olefinic groups due to the bands recorded at 1458 and 2856 cm−1. The 13C NMR spectrum established the existence of 18 carbon signals constituting eleven methylene, two methine, along with one each of carbonyl, olefinic, methyl and methoxy carbons. The 1H NMR in combination with 13C NMR experiments demonstrated highly deshielded oxymethylene protons at H-10 (δH 4.05, J = 6.85 Hz) and H-13 (δH 4.21, J = 6.78 Hz) that were deduced to be correlated with the corresponding carbon signals at C-10 and C-13 methylene groups, and that was further corroborated based on the existence of an ester carbonyl {δC 174.37 (C-9)} and sharp singlet (integral of three) of O–CH3 group in the NMR spectrum. The 1H-1H COSY correlations between δH 1.94 (denoted as H-1)/ δH 5.27 (H-2) and δH 5.28 (assigned to H-3)/ δH 2.71 (H-4) were ascribed to the cyclohexane ring framework (Fig. S5). The J1-3 HMBC correlation between δH 1.94 (denoted as H-1) to δC 129.92 (C-3), δH 5.27 (H-2) to δC 27.15 (C-1) and δH 2.71 (H-4) to δC 129.92 (C-3) attributed the presence of cyclohexene ring system. The methine (–CH) carbon at C-6 (δC 40.19) recognized the junction of cyclohexene ring moiety and was substituted with 3-(methoxymethyl) heptyl butyrate skeleton (Fig. S6). This was corroborated by the 1H–1H COSY and J1-3 HMBC correlations. HMBC cross peaks between δH 1.54 (assigned as H-7) to δC 174.37 (C-9), δH 2.24 (H-8) to δC 174.37 (C-9) appropriately supported the presence of ester carbonyl carbon attached to the cyclohexene ring system. Additional J1-3 HMBC correlations were displayed between δH 1.19 (assigned to H-15) to δC 62.20 (C-13), δH 1.26 (H-16) to δC 29.56 (C-15), δH 1.18 (H-17) to 29.56 (C-15)/ 14.11 (C-18), δH 0.80 (H-18) to δC 28.82 (C-16), which apparently indicated the substitution of 3-(methoxymethyl)heptyl butyrate to the cyclohexene ring system. The methoxy group was found to appear as a singlet at δH 3.59 (attributed to H-14; HSQC δC 51.43 at C-14) to support the presence of 3-(methoxymethyl) heptyl butyrate framework. In addition, the olefinic group was found to appear as the multiplet at δH 5.27–5.28 (H2-H3) {HSQC, δC 129.93 (C-2), δC 129.92 (C-3)}, which attributed to the cyclohexene ring framework. The chemistries of the stereogenic centres bearing protons were derived using NOESY-assisted relative stereochemical assignments . An intense NOESY correlation was displayed between the protons at δH 1.65 (H-6) and δH 1.94 (H-1; J = 6.13 Hz)/ and δH 2.71 (H-4; J = 6.69 Hz), which suggested their equi-planer disposition, and was arbitrarily attributed as β-oriented. Strong NOESY correlation between δH 2.02 (H-12; J = 6.69 Hz) and δH 4.21 (H-13; J = 6.78 Hz) attributed the protons to dispose at the β-side of the reference plane, which suggested their diaxial orientation with reference to the plane of the symmetry (Fig. 3).
2-Ethyl-6-(4-methoxy-2-((2-oxotetrahydro-2H-pyran-4-yl)methyl)butoxy)-6-oxohexyl-5-ethyloct-4-enoate (compound 3), a highly oxygenated C29 meroterpenoid, was purified as yellowish oil with m/z 510 (Fig. S7) bearing five degrees of unsaturation, and its structure was characterized by combined 1H and 13C NMR spectral experiments (Table 3). The IR bending vibration near 1736 cm−1 was associated with the carbonyl group, whereas the olefinic groups were assigned to the absorption bands at 1455 and 2857 cm−1. The 13C NMR spectroscopic data deduced the existence of 29 carbon signals constituting three each of methyl, methylene and ester carbonyl groups along with seventeen methylene and one each of olefinic and methoxy carbons. The 1H NMR in combination with 13C NMR demonstrated highly deshielded oxymethylene protons at δH 4.16 (attributed to H-9, J = 5.80 Hz), 4.19 (H-21, J = 9.08 Hz), 4.26 (H-15, J = 9.08 Hz) and 4.30 (H-24, J = 7.44 Hz) in the proton spectrum suggesting that the C-24, C-21, C-15 and C-9 methylene groups were attached to an electronegative group, possibly of oxygenated origin. This was further corroborated by the presence of ester carbonyl carbons {δC 174.56 (assigned to C-8), δC 168.33 (C-14), δC 173.48 (C-20)} and a singlet (integral of three) of O-CH3 group {δC 51.42 (C-25)}, in the NMR spectrum. The proton-proton connections were apparent between δH 1.73 (ascribed to H-23)/ δH 4.30 (H-24) that assigned the part of a tetrahydro-2H-pyran-2-one ring framework. The J1-3 HMBC correlations between δH 2.32 (ascribed to H-7) to δC 174.56 (C-8), δH 4.26 (H-15) to δC 29.26 (C-17), δH 4.19 (H-21) to δC 173.48 (C-20) and δH 2.32 (H-19) to δC 29.26 (C-17)/ 173.48 (C-20) appropriately deduced the existence of highly deshielded oxymethylene protons and ester carbonyl carbon as part of the tetrahydro-2H-pyran-2-one ring framework. The 1H–1H COSY correlation between δH 1.27 (ascribed to H-11)/ δH 1.62 (H-12)/ δH 2.32 (H-13) were probably attributed to the part of substituted tetrahydro-2H-pyranone ring system (Fig. 3, Fig. S8). In addition, an olefinic group was found to appear as the multiplet at δH 5.35 (H-4/H-5) {HSQC, δC 132.45 (C-4) and δC 130.08 (C-5)}, which was situated at the extended side chain of 3. The 1H-1H correlation between δH 5.35 (denoted as H-5)/ δH 2.02 (H-6) and a HMBC correlation between δH 2.02 (assigned as H-6) to δC 130.08 (C-5) appropriately established the existence of the olefinic group. The HMBC correlation from δH 1.42 (assigned as H-26) to δC 29.85 (C-10) deduced the substitution of 2-ethyl-6-(2-ethyl-4-methoxybutoxy)-6-oxohexyl 5-ethyloct-4-enoate to the pyran-2-one ring system in 3. The methoxy group was found to appear as singlet at δH 3.67 {attributed to H-25; HSQC δC 51.42 (C-25)}, which described the 2-ethyl-6-(2-ethyl-4-methoxybutoxy)-6-oxohexyl 5-ethyloct-4-enoate framework. The relative stereochemistries of the stereogenic centres in 3 were deduced by NOESY experiments. NOESY cross peaks at δH 1.69 (H-18)/ δH 4.19 (H-21; J = 9.08 Hz)/ δH 2.32 (H-19; J = 7.80 Hz) appropriately suggested their close proximity and equi-planer orientation (arbitrarily assigned to β-faced). NOESY correlation between the di-equatorial protons at δH 4.30 (H-24; J = 7.44 Hz)/ δH 2.49 (H-16) apparently attributed to their close spatial arrangements, and therefore, were assigned to be at the β-face with reference to the molecular plane of symmetry. Likewise, an intense NOESY correlation was observed between δH 1.73(H-10)/ δH 1.27 (H-11) that implied their deposition on the same side of the plane with di-axial interaction (Fig. 4, Fig. S8).

a Key 1H-1H COSY, HMBC and NOESY correlations of compound 2. The 1H-1H COSY cross peaks were displayed by bold face bonds, whereas the selected HMBC correlations were shown as double barbed arrows. The β orientation in the NOESY relations was presented as blue coloured arrows
Structure–activity relationship analysis
The radical scavenging and anti-inflammatory properties of the oxygenated meroterpenoids isolated from K. alvarezii were compared with commercially available synthetic standards (Table 4). The highly oxygenated C29 meroterpenoid 3 displayed potential antioxidative activities as determined by ABTS and 1, 1-diphenyl-2-picryl-hydrazil (DPPH) free radical scavenging potential (IC50 < 0.35 mg/mL), and was comparable with those exhibited by α-tocopherol (IC50 0.6–0.7 mg/mL, P < 0.05). The electron-delocalization between the carbonyl, methoxy, and olefinic bonds in the molecular structure of these compounds might probably contribute towards the potential free radical scavenging properties (Pietta 2000; Cai et al. 2006). These title meroterpenoid derivatives showed significantly greater inhibition towards the inducible COX-2 than its constitutive cyclooxygenase isoform, and accordingly, their anti-inflammatory selectivity index (SI, anti-COX-1IC50/anti-COX-2IC50) were lower (1.06–1.10) than synthetic NSAIDs (ibuprofen and aspirin, SI: 0.44 and 0.02, respectively; P < 0.05). In particular, no significant variation in the in vitro inhibitory activities towards pro-inflammatory 5-lipoxidase (IC50 1.04–1.14 mg/mL) and cycloxygenase-2 (IC50 1.05–1.09 mg/mL) of compound 3 indicated its potential anti-inflammatory properties against inducible inflammatory mediators causing an inflammatory response. Notably, sodium salicylate appeared to be a weaker inhibitor of the COX isoforms (anti-COX-2 IC50 2.65 mg/mL, anti-COX-1 IC50 1.93 mg/mL), and exhibited significantly lesser activity against 5-LOX (anti-LOX-5 IC50 1.75 mg/mL) (Table 4).
The radical quenching along with cyclooxygenase and lipoxygenase inhibitory activities of the meroterpenoids were determined by lipophilic (log Pow, octanol-water partition-coefficient), steric (molar refractivity, MR) and electronic (tPSA, topological polar surface area) parameters. The radical quenching and anti-inflammatory properties of the studied compounds were found to be directly related to their hydrophobic characters as determined by hydrophobicity-lipophilicity balance (log Pow). A greater value of log Pow indicated the higher molecular hydrophobicity. The compound 1 showed lesser hydrophobicity (log Pow 4) than those displayed by 2 (log Pow 4.26) and 3 (log Pow 5.46). The hydrophobic property was deduced to ascribe the inter-membrane permeability of a compound, the optimal range being 2–5 for appropriate lipophilic–hydrophobic characteristics (Lipinski and Hopkins 2004). The decreased activity of compound 1 might be corroborated with the lesser hydrophobicity and reduced membrane permeability. Resultantly, the lipophilic DPPH radical might easily be associated with meroterpenoids possessing greater hydrophobicity (greater log Pow value) and displaying higher radical scavenging property. On the basis of above attribution, it might be ascribed that the electronic and hydrophobic factors play significant roles to narrate the bioactive potential of the studied compounds. The electron-rich centers were found to constitute the methoxy-substituted side chain, hydroxyl and aryl substituents in the ring framework. These groups might possibly function as the centre of unsaturations, and were attributed to potential anti-inflammatory and radical quenching properties of the meroterpenoids. The aggregate number of electronegative centres and centre of unsaturation were lesser in 1, thereby resulting in lesser activity than those recorded in compounds 2 and 3. The optimum log Pow of the highly oxygenated C29 meroterpenoid 3 (~5.46) along with greater topological polar surface area (tPSA 88.13) might result in its potential anti-inflammatory activity in terms of inhibiting COX-2 (IC50 1.05 mg/mL) and 5-LOX (IC50 1.04 mg/mL) (Table 4).

Key 1H-1H COSY, HMBC and NOESY correlations of compound 3. The 1H-1H COSY cross peaks were displayed by bold face bonds, whereas the selected HMBC correlations were shown as double barbed arrows. The β orientation in the NOESY relations was presented as blue coloured arrows
Conclusions
Three oxygenated meroterpenoids, characterized as 1-(3-methoxypropyl)-2-propylcyclohexane (1), 3-(methoxymethyl)heptyl 3-(cyclohex-3-enyl)propanoate (2) and 2-ethyl-6-(4-methoxy-2-((2-oxotetrahydro-2H-pyran-4-yl)methyl)butoxy)-6-oxohexyl 5-ethyloct-4-enoate (3) were purified from the ethyl acetate-methanol fraction of the intertidal red seaweed K. alvarezii. The highly oxygenated C29 meroterpenoid 3 with potential radical quenching and anti-inflammatory potential might qualify this compound as candidate pharmacological lead against oxidative stress and inflammation.
References
Areche C, Benites J, Cornejo A, Ruiz LM, García-Beltrán O, Simirgiotis MJ, Sepúlveda B (2015) Seco-taondiol, an unusual meroterpenoid from the Chilean seaweed Stypopodium flabelliforme and its gastroprotective effect in mouse model. Mar Drugs 13:1726–1738
Ask EI, Azanza RV (2002) Advances in cultivation technology of commercial eucheumatoid species: a review with suggestions for future research. Aquacult 206:257–277
Baylac S, Racine P (2003) Inhibition of 5-lipoxygenase by essential oils and other natural fragment extracts. Int J Aromather 13:138–142
Cai YZ, Mei S, Jie X, Luo Q, Corke H (2006) Structure-radical scavenging activity relationships of phenolic compounds from traditional Chinese medicinal plants. Life Sci 78:2872–2888
Chakraborty K, Joseph D, Joy M, Raola VK (2016) Characterization of substituted aryl meroterpenoids from red seaweed Hypnea musciformis as potential antioxidants. Food Chem 212:778–788
Chandrasekaran S, Nagendran AN, Pandiaraja D, Krishnankutty N, Kamalakannan B (2008) Bioinvasion of Kappaphycus alvarezii on corals in the Gulf of Mannar, India. Curr Sci 94:1167–1172
Ebada SS, Lin WH, Proksch P (2010) Bioactive sesterterpenes and triterpenes from marine sponges: occurrence and pharmacological significance. Mar Drugs 8:313–346
Gershenzon J, Dudareva N (2007) The function of terpene natural products in the natural world. Nat Chem Biol 3:408–414
Larsen LN, Dahl E, Bremer J (1996) Peroxidative oxidation of leuco-dichloroluorescein by prostaglandin H synthase in prostaglandin biosynthesis from polyunsaturated fatty acids. BBA Lipid Met 1299:47–53
Liebgott T, Miollan M, Berchadsky Y, Drieu K, Culcasi M, Pietri S (2000) Complementary cardioprotective effects of flavonoid metabolites and terpenoid constituents of Ginkgo biloba extract (EGb 761) during ischemia and reperfusion. Basic Res Cardiol 95:368–377
Lipinski C, Hopkins A (2004) Navigating chemical space for biology and medicine. Nature 432:855–861
Makkar F, Chakraborty K (2017a) Unprecedented antioxidative cyclic ether from the red seaweed Kappaphycus alvarezii with anti-cyclooxygenase and lipoxidase activities Nat Prod Res 31(10):1131–1141
Makkar F, Chakraborty K (2017b) Antidiabetic and anti-inflammatory potential of sulphated polygalactans from red seaweeds Kappaphycus alvarezii and Gracilaria opuntia Int J Food Prop 20(6):1326–1337
Makkar F, Chakraborty K (2017c) Antioxidative sulfated polygalactans from marine macroalgae as angiotensin-I converting enzyme inhibitors. Nat Prod Res. https://doi.org/10.1080/14786419.2017.1363756.
Makkar F, Chakraborty K (2017d) Previously undescribed antioxidative azonicyl morpholinone alkaloid from red seaweed Gracilaria opuntia with anti-cyclooxygenase and lipoxidase properties. Nat Prod Res 32(10):1150–1160
Parshikov IA, Netrusov AI, Sutherland JB (2012) Microbial transformation of antimalarial terpenoids. Biotechnol Adv 30:1516–1523
Pietta PG (2000) Flavonoids as antioxidants. J Nat Prod 63:1035–1042
Reis VM, Oliveira LS, Passos RMF, Viana NB, Mermelstein C, Sant’Anna C, Pereira RC, Paradas WC, Thompson FL, Amado-Filho GM, Salgado LT (2013) Traffic of secondary metabolites to cell surface in the red alga Laurencia dendroidea depends on a two-step transport by the cytoskeleton. PLoS One 8:63929
Zbakh H, Talero E, Avila J, Alcaide A, de los Reyes C, Zubía E, Motilva V (2016) The algal meroterpene 11-hydroxy-11-O-methylamentadione ameloriates dextran sulfate sodium-induced colitis in mice. Mar Drugs 14:149
Acknowledgements
Financial support from the Science and Engineering Research Board (SERB) of Department of Science and Technology, Ministry of Science and Technology in the form of research grant (SR/S1/OC-96/2012 SERB) is gratefully acknowledged. The authors thank the Director, Central Marine Fisheries Research Institute, for his guidance and support. Thanks are due to the Head, Marine Biotechnology Division, Central Marine Fisheries Research Institute for facilitating the research activity. FM acknowledges Department of Science and Technology, for the award of fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
These authors contributed equally: Fasina Makkar and Kajal Chakraborty
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Makkar, F., Chakraborty, K. Antioxidant and anti-inflammatory oxygenated meroterpenoids from the thalli of red seaweed Kappaphycus alvarezii. Med Chem Res 27, 2016–2026 (2018). https://doi.org/10.1007/s00044-018-2210-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-018-2210-0