
Abstract
In the search of more powerful and versatile antioxidant agents for biomedical applications, a structural modification of the resveratrol scaffold was carried out. Six trans-resveratrol (TR) analogs with a trans-stilbazole framework were obtained. Quantum calculations based on the density functional theory (DFT) were used to study the relationship between the structure of synthesized compounds and antioxidant activity. Calculations have shown that derivatives 2b, 2c, and 2e should exhibit higher antioxidant activity than TR, whereas 2a, 2d, and 3a are less effective antioxidants. The obtained data correlate well with the results of the analysis carried out with diphenyl-1-picrylhydrazyl (DPPH). All compounds showed a lower cytotoxicity compared to TR in the MTT test on mouse fibroblast cells L929.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Natural polyphenols are the subject of research due to the wide range of biological effects associated with their antioxidant activity. Resveratrol (1), the phytoalexin found in grapes and some other plants, is one of the substances from this class (Scalbert et al. 2005). Resveratrol has heart protecting (Kopp 1998) and anticancer (Baur and Sinclair 2006) activities, slows the aging process (Valenzano et al. 2006) and inhibits the neurodegenerative diseases development (Karuppagounder et al. 2009). However, this wide physiological activity potential of polyphenols, particularly resveratrol, is limited by their low bioavailability. Resveratrol has poor water solubility and low absorption from the digestive tract after oral administration. Thus the optimization of plant polyphenols bioavailability is the basis for the full realization of their biomedical potential. The study of the resveratrol antioxidant effect enhancement is important due to the leading role in its physiological activity. According to modern concepts, the antioxidant interaction with active radicals can occur in several alternative mechanisms (Scheme 1) (Mikulski et al. 2010).

Alternative mechanisms of antioxidant action. a Electron transfer from the antioxidant on the active radical lead to a cation radicals and anion and the subsequent proton transfer from the cation radical to the anion. This process named as single-electron transfer followed by proton transfer (SET-PT). b Direct transfer of a hydrogen atom from the antioxidant to the active radical. This process named as hydrogen atom transfer (HAT). c The antioxidant deprotonation followed by the transfer of the electron from the formed anion to the active radical. Then the protonation of anion formed from the active radical occurs. This process named as sequential proton loss electron transfer (SPLET)
The implementation of the first mechanism is determined by the reducing activity of the antioxidant. For the HAT and SPLET processes the stability of the resulting particles (АrO• or АrO–) plays the key role. This stability is determined by the efficient electron density delocalization.
Thus, the most convenient descriptor for the antioxidant activity evaluation is the adiabatic ionization potential (AIP) for the first mechanism and the bond-dissociation enthalpy (BDE) for the second process. Proton affinity (PA) and electron transfer enthalpy (ETE) can be the descriptors for the SPLET mechanism, as the estimation of the electron transfer stage (Urbaniak et al. 2012). In addition, an important descriptor allowing the formed radical АrO• stability evaluation is the spin density and electronic density of HOMO delocalization.
It is known that reducing BDE, which can be achieved by introducing donor substituents into the phenol system (especially in para-position to the OH group), leads to the antioxidant activity increasing. However, the significant decreasing of BDEO-H leads to the decreasing of AIP (by increasing the HOMO energy) to the values when the direct reaction of the compound with molecular oxygen becomes possible. It is difficult to prevent this interaction because it will increase the prooxidant activity of the compounds or even make them unstable in air.
The increasing of prooxidant properties is not a completely negative process. Thus studies of polyhydroxylated resveratrol analogs showed a significant increase in their cytotoxicity to cancer cells (HL-60 and Jurkat human leukemia cells), associated with their prooxidative effects in biological systems (Murias et al. 2005).
At the same time, the instability in the air is a crucial parameter for the practical use. One of the solutions to this problem is the transition from phenol substrates to 3-hydroxy pyridine and pyrimidine derivatives. It was found that this substitution increases the ionization potential of more than 10 kcal/mole, while the O–H bond dissociation energy O–H increasing only 1.1 kcal/mol (Pratt et al. 2001). The presence of the π-deficient pyridine ring allowed to add donor groups (including the most powerful dialkylamino group) to the antioxidant structure. As the result, their inhibitory activity increased significantly without increasing the oxygen oxidation (Nara et al. 2008).
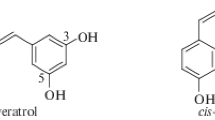
In the present study we carried out the theoretical and experimental research into resveratrol analogs containing 3-pyridinol fragment (2a–2e) (Fig. 1).

Structures of resveratrol (1) and its analogs (2a–2e)
This modification has been motivated by several problems. First, the presence of the pyridine ring increases the water solubility and bioavailability by the quaternary ammonium salts formation option. Second, it allows the synthesis of analogs with greater antioxidant activity compared to the natural prototype.
Materials and methods
Experimental
Except where indicated, materials and reagents were used as supplied by the suppliers without further purification. The 1H and 13C NMR spectra were recorded on a Jeol JNM-ECX400 device (399.78 and 100.53 MHz, respectively) in DMSO-d 6 . Chemical shifts were measured with reference to the residual protons of the solvent (DMSO-d 6 , 1H, 2.49 ppm, 13C, 39.50 ppm). Optical density measurements were carried out in the visible region at 517 nm on the “Shimadzu UV2600 Vis” instrument, the thickness of the absorbing layer was 10 mm. ESI-MS spectra were obtained on a Thermo Scientific MSQ Plus mass spectrometer in positive (negative) ion detection mode (source temperature 350 °C, gas-nebulizer−nitrogen, gas flow 50 l/min, needle voltage 4.5 kV, voltage on cone 75 V, scanning in the range of 100–400 Da with a speed of 2 scans/s). The eluent is a mixture of ACN and 0.1% formic acid in a ratio of 60:40, a flow rate of 0.05 ml/min. Microwave syntheses were carried out in the Monowave 300 reactor in special tightly sealed ampoules made of borosilicate glass.
Synthesis
(E)-2-(4-Methoxystyryl)pyridine (3b)
A mixture of picoline-2 (930 mg, 10 mmol), 4-methoxybenzaldehyde (1.36 g, 10 mmol) and ZnCl2 (1 g, 7.35 mmol) was placed in the vial for the microwave reactor and heated for 0.5 h at 200 °C. After cooling, acetone was added to the reaction mixture, the insoluble precipitate was filtered off, and the filtrate was evaporated. Then 10 mL of water, 25 mL of concentrated ammonia solution were added to the precipitate and boiled. After cooling, the mixture was extracted with methylene chloride, the extract was dried with anhydrous magnesium sulfate, and the solvent was evaporated to yield 3b as a cream-pink solid (269 mg, 12%). The spectral data coincide with those described in the literature (Mao et al. 2014).
(E)-4-(2-(Pyridin-2-yl)vinyl)phenol (3а)
A solution of stilbazole 3b (130 mg, 0.62 mmol) in CH2Cl2 (30 mL) was cooled to −10 °C with constant stirring, and BBr3 (0.5 mL, 5.3 mmol) in CH2Cl2 (30 mL) was slowly added dropwise. At the end of the dropping, the mixture was brought to a room temperature and stirred for 4 h under an argon atmosphere. Then 20 mL of distilled water was added dropwise to the reaction mixture. The aqueous layer was separated, neutralized with a solution of ammonia to pH 7, and extracted with ethyl acetate. The extract was dried over anhydrous magnesium sulfate, and the solution was evaporated to yield 3а as a yellow solid (100 mg, 83%); mp 189–190 °C. The spectral data coincide with those described in the literature (Choi et al. 2010).
1,2-Dimethyl-5-methoxypyridinium iodide (4)
Sodium (280 mg, 12.2 mmol) was dissolved in 4 mL of absolute methanol in a vial for the microwave reactor, and 6-methylpyridin-3-ol (980 mg, 9 mmol) was added, heated for 90 minutes at 100 °C. The mixture was cooled to room temperature and methyl iodide (1.4 mL, 22.5 mmol) was added, heated for 120 minutes at 100 °C. The solution was evaporated, and the residue was recrystallized from ethanol to yield 4 as a cream solid (1.76 g, 74%); mp 130–133 °С;1H NMR (DMSO-d 6 , 400 MHz): δ 2.67 (3H, s, CH3), 3.95 (3H, s, OCH3), 4.19 (3H, s, NCH3), 7.92 (1H, d, J = 8.7 Hz, H-3), 8.13 (1H, dd, J = 8.7, 2.7 Hz, H-4), 8.80 (1H, d, J = 2.7 Hz, H-6); 13C NMR (DMSO-d 6 , 100 MHz): δ 18.7 (CH3, CH3), 45.7 (CH3, NCH3), 57.2 (CH3, OCH3), 129.3 (CH, H-4), 130.9 (CH, H-3), 133.0 (CH, H-6), 147.9 (C, H-2), 155.9 (C, H-5); MS (ESI) m/z calculated for [C8H12NO+] = 138.09 (100.0%), 139.09 (8.7%) found 138.20 (100.0%), 139.20 (9.7%).
General procedure for the condensation of the salt (4) with aromatic aldehydes
Salt 4 (700 mg, 2.64 mmol), the corresponding aldehyde (2.64 mmol) and a few drops of piperidine in butan-1-ol (10 mL) were placed in a round bottom flask equipped with a reflux condenser. The reaction mixtures were boiled for 5–10 h. After the reaction was complete, the mixtures were cooled; the precipitates were filtered off, washed with diethyl ether (with acetone in the case of compound 5a) to yield the products of condensation.
(E)-5-methoxy-1-methyl-2-styrylpyridin-1-ium iodide (5а)
Grey solid (490 mg, 53%); mp 213–215 °С (decomp.); 1H NMR (DMSO-d 6 , 400 MHz): δ 4.01 (3H, s, OCH3), 4.38 (3H, s, NCH3), 7.42–7.50 (3H, m, H-3′, H-4′, H-5′), 7.52 (1H, d, J = 16.0 Hz, H-a), 7.77–7.84 (3H, m, H-2′, H-6′, H-b), 8.20 (1H, dd, J = 9.6, 2.7Hz, H-4), 8.45 (1H, d, J = 9.6 Hz, H-3), 8.79 (1H, d, J = 2.7 Hz, H-6); 13C NMR (DMSO-d 6 , 100 MHz): δ 46.2 (CH3, NCH3), 57.4 (CH3, OCH3), 117.1 (CH, C-a), 125.7 (CH, C-3), 128.1 (2CH, C-2′, C-6′), 128.9 (2CH, C-3′, C-5′), 130.1 (CH, C-4′), 130.6 (CH, C-4), 132.8 (CH, C-6), 135.0 (C, C-1′), 140.5 (CH, C-b), 145.2 (C, C-2), 156.0 (C, C-5); MS (ESI) m/z calculated for [C15H16NO+] = 226.12 (100.0%), 227.13 (16.2%) found 226.07 (100.0%), 227.08 (17.0%).
(E)-5-methoxy-2-(4-methoxystyryl)-1-methylpyridin-1-ium iodide (5b)
Yellow solid (850 mg, 84%); mp 215–218 °С (decomp.); 1H NMR (DMSO-d 6 , 400 MHz): δ 3.82 (3H, s, OCH3), 3.98 (3H, s, OCH3), 4.34 (3H, s, NCH3), 7.04 (2H, d, J = 8.7 Hz, H-3′, H-5′), 7.36 (1H, d, J = 16.0 Hz, H-a), 7.73–7.86 (3H, m, H-2′, H-6′, H-b), 8.15 (1H, dd, J = 9.2, 2.7 Hz, H-4), 8.40 (1H, d, J = 9.2 Hz, H-3), 8.71 (1H, d, J = 2.7 Hz, H-6); 13C NMR (DMSO-d 6 , 100 MHz): 46.1 (CH3, NCH3), 55.4 (CH3, OCH3), 57.3 (CH3, OCH3), 114.5 (2CH, C-3′, C-5′), 114.5 (CH, C-a), 125.2 (CH, C-3), 127.8 (C, C-1′), 130.0 (2CH, C-2′, C-6′), 130.7 (CH, C-4), 132.2 (CH, C-6), 140.5 (CH, C-b), 145.9 (C-2), 155.6 (C, C-5), 161.0 (C, C-4′); MS (ESI) m/z calculated for [C16H18NO2+] = 256.13 (100.0%), 257.14 (17.3%) found 256.09 (100.0%), 257.12 (17.3%).
(E)-2-(3,4-dimethoxystyryl)-5-methoxy-1-methylpyridin-1-ium iodide (5c)
Yellow solid (870 mg, 80%); mp 206–208 °С (decomp.); 1H NMR (DMSO-d 6 , 400 MHz): δ 3.82 (3H, s, OCH3), 3.85 (3H, s, OCH3), 3.99 (3H, s, OCH3), 4.36 (3H, s, NCH3), 7.05 (1H, d, J = 8.2 Hz, H-2′), 7.34–7.44 (3H, m, H-5′, H-6′, H-a), 7.74 (1H, d, J = 16.0 Hz, H-b), 8.16 (1H, dd, J = 9.2, 2.7 Hz, H-4), 8.39 (1H, d, J = 9.2 Hz, H-3), 8.72 (1H, d, J = 2.7 Hz, H-6); 13C NMR (DMSO-d 6 , 100 MHz): δ 46.2 (CH3, NCH3), 55.6 (CH3, OCH3), 55.8 (CH3, OCH3), 57.3 (CH3, OCH3), 110.6 (CH, C-5′), 111.7 (CH, C-2′), 114.5 (CH, C-a), 122.9 (CH, C-6′), 125.2 (CH, C-3), 127.9 (C, C-1′), 130.7 (CH, C-4), 132.2 (CH, C-6), 140.9 (CH, C-b), 145.8 (C, C-2), 149.0 (C, C-3′), 151.0 (C, C-4′), 155.5 (C, C-5); MS (ESI) m/z calculated for [C17H20NO3+] = 286.14 (100.0%), 287.15 (18.4%) found 286.12 (100.0%), 287.12 (17.2%).
(E)-2-(3,5-dimethoxystyryl)-5-methoxy-1-methylpyridin-1-ium iodide (5d)
Yellow solid (830 mg, 76%); mp 215–218 °С (decomp.); 1H NMR (DMSO-d 6 , 400 MHz): δ 3.81 (6H, s, 2OCH3), 4.00 (3H, s, OCH3), 4.37 (3H, s, NCH3), 6.59 (1H, t, J = 2.1 Hz, H-4′), 6.97 (2H, d, J = 2.3 Hz, H-2′, H-6′), 7.51 (1H, d, J = 16.0 Hz, H-a), 7.70 (1H, d, J = 16.0 Hz, H-b), 8.20 (1H, dd, J = 9.2, 2.7 Hz, H-4), 8.40 (1H, d, J = 9.2 Hz, H-3), 8.77 (1H, d, J = 2.7 Hz, H-6); 13C NMR (DMSO-d 6 , 100 MHz): δ 46.3 (CH3, NCH3), 55.4 (CH3, 2OCH3), 57.4 (CH3, OCH3), 102.1 (CH, C-4′), 106.2 (2 CH, C-2′, C-6′), 117.6 (CH, C-a), 125.7 (CH, C-3), 130.6 (CH, C-4), 132.9 (CH, C-6), 136.9 (C, C-1′), 140.6 (CH, C-b), 145.2 (C, C-2), 156.1 (C, C-5), 160.7 (2C, C-3′, C-5′); MS (ESI) m/z calculated for [C17H20NO3+] = 286.14 (100.0%), 287.15 (18.4%) found 286.12 (100.0%), 287.12 (18.7%).
(E)-5-methoxy-1-methyl-2-(3,4,5-trimethoxystyryl)pyridin-1-ium iodide (5e)
Yellow solid (815 mg, 70%); mp 201–203 °С (decomp.); 1Н NMR (DMSO-d 6 , 400 MHz): δ 3.72 (3H, s, OCH3), 3.86 (6H, s, 2OCH3), 4.00 (3H, s, OCH3), 4.38 (3H, s, NCH3), 7.13 (2H, s, H-2′, H-6′), 7.45 (1H, d, J = 16.0 Hz, H-a), 7.73 (1H, d, J = 16 Hz, H-b), 8.20 (1H, dd, J = 9.2, 2.7 Hz, H-4), 8.38 (1H, d, J = 9.2 Hz, H-3), 8.75 (1H, d, J = 2.7 Hz, H-6); 13С NMR (DMSO-d 6 , 100 MHz): δ 46.3 (CH3, NCH3), 56.2 (2 CH3, 2OCH3), 57.2 (CH3, OCH3), 61.9 (CH3, OCH3), 106.0 (2CH, C-2′, C-6′), 116.2 (CH, C-a), 125.4 (CH, C-3), 130.6 (CH, C-4), 130.8 (C, C-1′), 132.6 (CH, C-6), 139.6 (C, C-4′), 140.9 (CH, C-b), 145.5 (C, C-2), 153.1 (2C, C-3′, C-5′); 155.9 (C, C-5); MS (ESI) m/z calculated for [C18H22NO4+] = 316.15 (100.0%), 317.16 (19.5%) found 316.12 (100.0%), 317.13 (19.7%).
General procedure for the demethylation of salts (5а–e)
Salts 5a–e (0.78 mmol) and pyridinium chloride were added to a round-bottomed flask equipped with a reflux condenser at a rate of 670 mg (5.77 mmol) per CH3 group. The reaction mixture was heated with stirring with a magnetic stirrer at 200–210 °C for 3 h. At the end of the time, the reaction mixture was poured onto the ice; the pH of the mixture was adjusted to 7 with ammonia and then extracted with ethyl acetate. The extracts were dried over anhydrous magnesium sulfate, the solvent was evaporated. The residues were washed with methylene chloride to yield the products.
(E)-6-Styrylpyridin-3-ol (2а)
Cream solid (80 mg, 52%); mp 174–175 °С; 1H NMR (DMSO-d 6 , 400 MHz): δ 7.15 (1H, dd, J = 8.7, 2.7 Hz, H-4), 7.19 (1H, d, J = 16.5 Hz, H-a), 7.25 (1H, t, J = 7.22 Hz, H-4′), 7.34–7.42 (4H, m, H-3′, H-5′, H-3, H-b), 7.58 (2H, d, J = 7.3 Hz, H-2′, H-6′), 8.14 (1H, d, J = 2.7 Hz, H-6), 9.95 (1H, s, OH); 13С NMR (DMSO-d 6 , 100 MHz): δ 122.4 (CH, H-3), 122.9 (CH, C-4), 126.5 (2CH, C-2′, C-6′), 127.5 (CH, C-a), 127.9 (CH, C-4′), 128.6 (2CH, C-3′, C-5′), 128.7 (CH, C-b), 136.8 (C, C-1′), 137.9 (CH, C-6), 146.3 (C, C-2), 152.8 (C, C-5); MS (ESI) m/z calculated for [C13H11NO+H+] = 198.09 (100.0%), 199.10 (14.2%), found 198.15 (100.0%), 199.15 (14.4%); calculated for [C13H11NO–H+] = 196.08 (100.0%), 197.08 (14.2%) found 196.07 (100.0%), 197.07 (14.2%).
(E)-6-(4-Hydroxystyryl)pyridin-3-ol (2b)
Yellow solid (105 mg, 63%); mp 210–211 °С (decomp.); 1H NMR (DMSO-d 6 , 400 MHz): δ 6.76 (2H, d, J = 8.3 Hz, H-3′, H-5′), 6.95 (1H, d, J = 16.0 Hz, H-a), 7.12 (1H, dd, J = 8.7, 2.7 Hz, H-4), 7.22–7.35 (2H, m, H-3, H-b), 7.40 (2H, d, J = 8.7 Hz, H-2′, H-6′), 8.10 (1H, d, J = 2.7 Hz, H-6), 9.66 (1H, br s, OH-Ph), 9.92 (1H, br s, OH-Py); 13CNMR (DMSO-d 6 , 100 MHz): δ 115.5 (2CH, C-3′, C-5′), 122.2 (CH, C-3), 122.5 (CH, C-4), 124.7 (CH, C-a), 127.9 (2CH, C-2′, C-6′), 127.9 (C, C-1′), 128.8 (CH, C-b), 137.7 (CH, C-6), 146.9 (C, C-2), 152.3 (C, C-5), 157.3 (C, C-4′); MS (ESI) m/z calculated for [C13H11NO2+H+] = 214.09 (100.0%), 215.09 (14.1%), found 214.14 (100.0%), 215.14 (14.7%); calculated for [C13H11NO2–H+] = 212.07 (100.0%), 217.08 (14.1%) found 212.08 (100.0%), 213.07 (13.8%).
(E)-4-(2-(5-Hydroxypyridin-2-yl)vinyl)benzo-1,2-diol (2c)
Orange solid (94 mg, 53%); mp 174–175 °С (decomp.); 1H NMR (DMSO-d 6 , 400 MHz): δ 6.72 (1H, d, J = 7.8 Hz, H-5′), 6.79–6.91 (2H, m, H-6′, H-a), 6.97 (1H, d, J = 1.8 Hz, H-2′), 7.12 (1H, dd, J = 8.2, 2.7 Hz, H-4), 7.22 (1H, d, J = 16.0 Hz, H-b), 7.34 (1H, d, J = 8.2 Hz, H-3), 8.10 (1H, d, J = 2.7 Hz, H-6), 8.92 (1H, br s, OH–Ph), 9.01 (1H, br s, OH–Ph), 9.92 (1H, br s, OH–Py); 13C NMR (DMSO-d 6 , 100 MHz): δ 113.3 (CH, C-2′), 115.7 (CH, C-5′), 118.8 (CH, C-6′), 122.2 (CH, C-3), 122.6 (CH, C-4), 124.5 (CH, C-a), 128.4 (CH, C-b), 129.4 (C, C-1′), 137.4 (CH, C-6), 145.4 (C, C-3′), 145.7 (C, C-4′), 146.8 (C, C-2), 152.3 (C, C-5); MS (ESI) m/z calculated for [C13H11NO3+H+] = 230.08 (100.0%), 231.09 (14.1%) found 230.08 (100.0%), 231.10 (14.2 %); calculated for [C13H11NO3–H+] = 228.07 (100.0%), 229.07 (14.1%) found 228.06 (100.0%), 229.08 (14.0%).
(E)-5-(2-(5-Hydroxypyridin-2-yl)vinyl)benzo-1,3-diol (2d)
Grey solid (90 mg, 50%); mp 215–216 °С (decomp.); 1H NMR (DMSO-d 6 , 400 MHz): δ 6.15 (1H, t, J = 2.1 Hz, H-4′), 6.41 (2H, d, J = 1.8 Hz, H-2′, H-6′), 6.97 (1H, d, J = 16.0 Hz, H-a), 7.12 (1H, dd, J = 8.2, 2.7 Hz, H-4), 7.19 (1H, d, J = 16.0 Hz, H-b), 7.38 (1H, d, J = 8.2 Hz, H-3), 8.11 (1H, d, J = 2.7 Hz, H-6), 9.19 (2H, br s, 2OH-Ph), 9.93 (1H, br s, OH-Py); 13C NMR (DMSO-d 6 , 100 MHz): δ 102.4 (CH, C-4′), 104.7 (2 CH, C-2′, C-6′), 122.5 (CH, C-3), 122.7 (CH, C-4), 127.3 (CH, C-a), 129.3 (CH, C-b), 137.7 (CH, C-6), 138.5 (C, C-1′), 146.3 (C, C-2), 152.7 (C, C-5), 158.5 (2C, C-3′, C-5′); MS (ESI) m/z calculated for [C13H11NO3+H+] = 230.08 (100.0%), 231.09 (14.1%) found 230.07 (100.0%), 231.09 (14.7 %); calculated for [C13H11NO3–H+] = 228.07 (100.0%), 229.07 (14.1%) found 228.06 (100.0%), 229.07 (15.0%).
(E)-5-(2-(5-Hydroxypyridin-2-yl)vinyl)benzo-1,2,3-triol (2e)
Orange solid (100 mg, 52%); mp 130–133 °С (decomp.); 1H NMR (DMSO-d 6 , 400 MHz): δ 6.50 (2H, s, H-2′, H-6′), 6.79 (1H, d, J = 16.0 Hz, H-a), 6.95–7.14 (2H, m, H-4, H-b), 7.33 (1H, d, J = 8.2 Hz, H-3), 8.08 (1H, d, J = 2.7 Hz, H-6), 8.81 (3H, br s, 3OH-Ph), 9.70 (1H, br s, OH-Py); 13C NMR (DMSO-d 6 , 100 MHz): δ 105.7 (2 CH, C-2′, C-6′), 122.1 (CH, C-3), 122.4 (CH, C-4), 124.7 (CH, C-a), 127.4 (CH, C-b), 129.5 (C, C-4′), 133.5 (C, C-1′), 137.5 (CH, C-6), 146.1 (2C, C-3′, C-5′), 151.3 (C, C-2), 152.3 (C, C-5); MS (ESI) m/z calculated for [C13H11NO4+H+] = 246.08 (100.0%), 247.08 (14.1%) found 246.08 (100.0%), 247.10 (15.5%); calculated for [C13H11NO4–H+] = 244.06 (100.0%), 245.06 (14.1%) found 244.06 (100.0%), 245.06 (14.0%).
DPPH radical scavenging activity
DPPH radical scavenging activity was assessed according to the method of Shimada et al. (Shimada et al. 1992). To determine the antiradical activity of the compounds obtained and resveratrol, a series of solutions were prepared by mixing an ethanol solution of DPPH in a final concentration of 45 μM and an ethanol solution of the corresponding polyphenol at a final concentration of 0–1000 μM. The solutions were held for 30 min in darkness at room temperature. The optical density of the solutions was measured at 517 nm. TR was used as a positive control under the same assay conditions. The negative control was without any inhibitor. Lower absorbance at 517 nm represents higher DPPH scavenging activity. The % DPPH radical scavenging activity was calculated from the decrease in absorbance at 517 nm in comparison with negative control. Each series was repeated three times. DPPH radical’s concentration was calculated using the following equation: DPPH scavenging effect (%) = (Ao–Ai)×100/Ao, where Ao was the absorbance of the control and Ai was the absorbance in the presence of the sample.
Cells cultivation
Cells were kept in DMEM medium supplemented with 10% fetal bovine serum (FBS), and confluent monolayers were trypsinized, washed in DMEM, and applied in 96-well microtiter plates (2 × 103 cells/well). Cells were incubated in standard conditions during 48 h (Sanyo incubator, Japan; 37 °C, 5% CO2). Than new compounds and resveratrol were added in final titrated 1:2 concentrations from 800 to 3.125 μM and were incubated with the cells in the same conditions described above during 24 h. At same time control vehicle (solvent used to dissolve the test compounds) was added by using the same procedure, described above, to the control cells.
MTT test procedure
MTT test was carried out by the procedure (Van de Loosdrecht et al. 1994). Mouse fibroblast cells L929 (obtained from the Collections tissue cultures of Ivanovsky Virology Research Institute) were used. After all predetermined incubation time, the medium was replaced with a fresh medium and 30 μl of the MTT solution of 5 mg/ml concentration was added. Colorimetric reaction was developed after incubation with MTT (37 °C, 3 h) followed by the addition of DMSO. The reaction was read spectrophotometrically with a 492 nm filter and a background of 620 nm (microplate ELISA reader, EFOS 9305, Russia). Incubations were tested in triplicate in 3 independent experiments. Cytotoxicity was evaluated through the MTT-derived colorimetric values. Those values were plotted to generate dose–response curves.
All experiments were performed in three replicates. The IC50 calculation was carried out in GraphPad Prizm 7.00. Experimental data are expressed as mean values and standard errors and the significance of differences was analyzed by ANOVA in Microsoft Excel 2010. Differences were considered significant at p < 0.05. The IC50 based upon cell growth in drug-free controls were estimated by curve fitting using GraphPad Prizm 7.00.
Calculation
All computations were carried out by using the GAMESS (US) package (Schmidt et al. 1993). The presented images were generated by the Gabedit visualization program. Calculations were performed for standard conditions at 298.15 K and 1.00 atm in the gas phase by DFT method with hybrid functional B3LYP (Becke 1993). Geometry optimization of the studied molecules was performed in steps. First, the geometry of neutral molecules in their ground states was optimized by a semi-empirical method PM3 and then by using restricted B3LYP/6-31 G level of theory. After that, the selection of the most favorable conformations was carried out. Then the obtained geometry was reoptimized in 6–311 G(d,p) basis set. The calculations for the radicals, anions, and cation radicals were performed from the fully optimized structures of the neutral molecules after H-atom or proton abstraction from the hydroxyl groups or after electron abstraction. Since the radicals, anions, and cation radicals are open-shell systems the unrestricted B3LYP/6-311 G(d,p) level of theory for their full optimization was applied. In optimization of the structure of radicals and cation radicals the spin contamination was monitored. The values of the square of the total spin were the range 0.76–0.78, so the contamination did not exceed 10%. Finally, to verify the stationarity of the energy states obtained, vibrational spectra were calculated for all structures and their evaluation was performed for the absence of negative frequencies. The calculated zero-point energies (ZPE) values were scaled by a factor of 0.9805 (Scott and Radom 1996) and used to correct the total energies.
The solvent (ethanol) effect was estimated on the main descriptors of antioxidant activity since the results of the following studies of antiradical activity in the DPPH test strongly depend on the medium polarity. For this, a conductor polarizable solvation model (CPCM) (Klamt and Schüürmann 1993) was used. In this model ethanol is assumed as a macroscopic continuum characterized a dielectric constant ε = 24.85. The solvent effect was evaluated for the most stable conformations of the molecules, their cation radicals, radicals, and anions, fully optimized in the gas phase. SAR analysis was carried out in Microsoft Excel 2010.
Results and discussion
Chemistry
In this study five resveratrol analogs containing hydroxypyridine fragment (2a–2e) were synthesized. We have proposed the efficient three-step synthesis for derivatives (2a–2e) using more active N-methylpyridinium salt (4) as methylene components on condensation step (Scheme 2).

General synthetic route for synthesis of the resveratrol analogs 2a–2e. Reagents and conditions: (i) 1. CH3ONa, CH3OH, MW. 2. CH3I, MW; (ii) butanol, piperidine; (iii) Py·HCl
The salt (4) was synthesized by methylation of 3-hydroxy-5-methylpyridine under the microwave initiating. The condensation reaction of (4) with appropriate aromatic aldehydes was carried out by boiling in butanol for 5–10 h in the presence of piperidine catalytic amount (Berdnikova et al. 2015) afforded the yields of target N-methylstilbazoles (5a–5e) ranged from 53 to 84%. All products are crystalline compounds with E-configuration. The presence of Z-isomers in the reaction mixtures has not been detected by 1H NMR spectra. The vicinal protons at C = C bond have doublet signals with coupling constants about 16 Hz. Demethylation of compounds (5a–5e) was carried out by heating to 200 °C with freshly prepared anhydrous pyridinium chloride using described protocol (Ruiz et al. 1997). Notably, this process has been effective not only for N- but also for O-demethylation. It allowed converting the compounds (5a–5e) into target phenols (2a–2e) in a one-step process.
It is known that resveratrol hydroxyl group in the para-position to the double bond has the highest activity according to its lowest values of BDE and the highest stability of 4′-radical (comparing to 3- and 5-radical). To assess the differences in this activity of OH groups in the benzene and pyridine fragment along with stilbazole (2a) (E)-4-(2-(pyridin-2-yl)vinyl)phenol (3a) was also synthesized. First, (E)-2-(4-methoxystyryl)pyridine (3b) was synthesized by the Knoevenagel condensation method proposed in (Bialon 1902) with some modifications: replacement of convection heating to microwave initiation, which significantly decreased the reaction time and increased the product yield. The product (3b) was demethylated into (3a) by a standard procedure with BBr3.
Calculation
Since the free rotation around the single bond between the pyridine and alkene fragments is possible, the nitrogen atom can occupy two boundary positions relative to the double bond protons (Fig. 2). According to the calculations from two possible options, rotamer (b) with anti-configuration possesses greater stability, which corresponds to the calculation data (Castro et al. 2013) and X-Ray diffraction analysis (Percino et al. 1997) for unsubstituted 2-stilbazole.

Rotamers for compounds 2a–2e
The distance between nitrogen atoms and hydrogen Hb in all structures is in the range 2.477 to 2.504 Å, indicating a certain contribution of weak hydrogen bond (Desiraju and Steiner 2001) to the stabilization of the rotamer (b). Subsequently, all calculations were performed for structures with a skeleton rotamer (b). The most stable conformers of 2c and 2e also have intramolecular hydrogen bonds between hydroxyl groups with lengths in the range 2.125 to 2.197 Å (Fig. 3).

Formation of intramolecular hydrogen bonds for 2c and 2e
The energy values (see Supplementary) analysis showed that all radicals are less stable than the neutral molecules and the corresponding anions, so they can be more reactive in biochemical reactions. The most important structural parameters of the studied molecules and their radicals, such as the C = C and O−H bonds lengths and orders, are given in Supplementary.
It should be noted that the calculated parameters of the central ethylene C = C bonds in the all studied molecules have similar values. However, in the case of 5-O• and 4′-O• radical forms C = C bonds are longer and have a reduced order compared with the neutral molecules. An increasing of length and a decreasing in the order of O−H bonds in the case of an arrangement adjacent to the radical center were noted, indicating the formation of a hydrogen bond between these fragments. A shortening of O3′–H3′ and O5′–H5′ bonds in the case of 5-O• radicals was also indicated.
The calculated torsion angles values between two aromatic cycles and between cycles and O−H fragments indicate the planar geometry of all studied structures, promoting their participation in various stacking interactions. In addition, the planar conformation of free radicals and radical cations should favor the delocalization of spin density and the distribution of the unpaired electron, increasing the stability of radicals and, finally, antioxidant activity.
The energy, distribution of the electron density HOMO in neutral molecules and delocalization of the spin density in the radicals were estimated further. The HOMO energy is an important parameter of the molecules reactivity related to radicals. Molecules with a lower HOMO energy have lower electron-donor ability. The HOMO distribution of the electron density in the molecule allows selecting the regions preferred for the radical attack. More active redox sites of molecules have a high HOMO orbital density. The HOMO distribution analysis (see Supplementary) shows a large range of its delocalization, including both aromatic rings, the double bond between them and hydroxyl groups in the para positions to the double bonds. Thus the presence of a number of active redox sites in the studied molecules was suggested, first of all, 5′-OH- and/or 4-OH-groups. These sites could be attacked by radicals in the real biological systems. In contrast, OH groups in the meta-position should be less sensitive to the interaction with free radicals. This observation for trans-resveratrol is an excellent agreement with the experimental data (Cao et al. 2003).
The spin density distribution in free radicals is a crucial parameter for their stability, which is a fundamental factor of antioxidant activity. The calculations show the spin density delocalization throughout the molecule in all compounds with the para-position of the radical center relatively to ethylene units (see Supplementary). The cyclic fragment directly containing the radical center and the vinyl bond have the maximum localization. In the case of the meta-position of the radical center relative to the central alkene moiety, the spin density is delocalized almost exclusively within the limits of benzene fragment.
Thus, summarizing all given data the formation of strongly resonance stabilized semiquinone structure could be stated for the radicals formed from the hydroxyl located at the para-positions relative to the ethylene moiety. The presence of additional hydroxyl groups adjacent to the radical center contributes to the delocalization and the radical stabilization due to the formation of hydrogen bonds (Fig. 4).

Formation of intramolecular hydrogen bonds for 4′-O-radical of 2e
Based on the obtained data on total energies, the main descriptors for the evaluation of antioxidant activity were calculated (Table 1).
It is known that electron-withdrawing groups (EWG) stabilize the initial molecule and destabilize the corresponding radical and cation radical, while electron-donating groups (EDG) have the opposite effect. Thus, the replacing of the benzene ring with pyridine should increase the AIP and BDE of OH bond, but the addition of the hydroxyl groups should decrease the energy of these processes. This combined result is actually observed. According to data from Table 1, AIP for isomeric stilbazoles 2a and 3a have the highest values; while compounds (2b, 2c, 2e) have the lower AIP compared with trans-resveratrol, i.e., have a high ability for electronic transfer in the interacting with free radicals in biological systems using the SET-PT mechanism. The high AIP value for 2d, comparable to analog 2a with the non-hydroxylated benzene ring, indicates a weak effect of the meta located hydroxyl groups on the cation radical stabilization, resulting in a low degree of π-delocalization according to the spin density distribution in cation radical 2d (see Supplementary).
The BDE values comparison for the pair of isomers 2a and 3a shows that the O−H bond in the pyridine fragment is slightly weaker than in the benzene ring and have the strength similar to the O−H bond of trans-resveratrol, determining its antioxidant properties. The addition of next hydroxyls to the para position decreases the BDE, while the meta-substituted derivative (2d) demonstrates the highest bond strength in the series of compounds studied. The presence of hydroxyl in the adjacent position significantly decreases BDE due to the formation of intramolecular hydrogen bonds.
In contrast to previous descriptors, PA represents the enthalpy of the first-stage reaction in the SPLET mechanism and has the opposite dependence on the electronic effects of the substituents, since EWG stabilizes ArO− and destabilizes the parent structures and EDG have the opposite effect. Indeed, PA for the most easily dissociating O−H bonds in all structures studied is lower than in resveratrol due to the effect of the electron-poor pyridine ring.
The enthalpies of the second-stage reactions in the SET-PT and SPLET mechanisms − PDE and ETE, respectively, are also important for the overall energy evaluating.
An analysis of the data obtained shows the fine correlation between the PDE and BDE changes, assuming the similar effect of structural and electronic characteristics on these parameters. The data obtained considering the solvent effect are presented in Table 2.
The values of AIP, PDE, and PA are expected to be significantly lower in the ethanol medium compared to the gas phase. It is well known that charged particles are very sensitive to the solvent polarity and their solvation reduces the energy of the processes they participate. At the same time, BDE values analysis (Tables 1 and 2) showed the slight increase of these values in ethanol medium compared to the gas phase. Thus, a more polar ethanol reduces the ability of hydroxyl groups to homolytic dissociation.
Thus, from the thermodynamics viewpoint of the process in the gas phase the HAT mechanism is predominant and in the ethanol medium the process is preferably implemented by the SPLET mechanism, which agrees well with the literature data (Litwinienko and Ingold 2007). It should be noted that the absolute values of the calculated descriptors in the gas phase change upon transition to the ethanol medium, but the relative order of antioxidant activity is generally preserved.
Radical scavenging activity
To study the radical scavenging activity the DPPH scavenging assays were used (Blois 1958). It should be emphasized that there is a big difference between “anti-radical” and “antioxidant” activity. The first one characterizes only compounds ability to react with free radicals (in a particular reaction), while the second shows the ability to inhibit the oxidation process (involving many stages). In addition, some complications in DPPH test may be caused by partial ionization of the testing compounds, which influences the rate of their reaction (Musialik and Litwinienko 2005). However, DPPH test can be used as a simple and rapid method of primary comparative evaluation of the compounds antioxidant activity.
The antioxidant concentration at which the 50% inhibition of DPPH occurs (IC50) was calculated from the graph of the inhibition percentage versus the concentration of the compound (see Supplementary), constructed from the results of three independent measurements. The IC50 values for the testing substances and the resveratrol as a reference compound are shown in Table 3.
A lower IC50 value indicates a higher anti-radical activity of the compound. Analysis of the Table 3 shows that hydroxylstilbazoles (2b), (2c), and (2e), containing hydroxyl groups in the p-position to the double bond, both in the pyridine and benzene fragments, have the highest activity in the interaction with the DPPH radical. Hydroxystilbazoles without OH groups in the p-position to the double bond, or containing OH-groups only in one ring, show a low antiradical activity.
SAR analysis
Comparison of the calculated antioxidant activity descriptors (in ethanol medium) for testing compounds with the experimental data on the interaction with DPPH showed some correlations between them. The data for the thermodynamically more advantageous deprotonation process was used to construct the correlation dependences for the compounds with several hydroxyl groups. A correlation matrix between the experimental values of pIC50 and calculated descriptors in an ethanol medium was obtained (Table 4). The Pearson agreement criterion was used for evaluating the degree of linear dependence between two variables. Correlation coefficients between variables are above 0.70 in most cases. The low correlation between pIC50 and the PDE value characterizing the second step of the SET-PT mechanism is probably due to the fact that the first step is the most important and determining for the mechanisms with several steps from the thermodynamic point of view.
Next, we constructed correlation equations using a multiple linear regression model corresponding to the implementation of three main mechanisms of antioxidant action. The relevance of the models was evaluated by three criteria: the correlation coefficient (R), the value of the Fisher coefficient (F) and the standard deviation (s):
Mechanism SET–PT:
Mechanism HAT:
Mechanism SPLET:
A quantitative estimation shows the good linear correlation between the log IC50 and the descriptors describing the SET-PT mechanism, satisfactory data were also obtained for the SPLET mechanism. Thus, their realization is most likely for the interaction of the testing substances with DPPH, and the fine correlation between the calculated and experimental values confirms the validity of the chosen theoretical model and the calculation results. In the case of the HAT mechanism, the low value of the correlation coefficient indicates its low contribution to the overall process under these experimental conditions. Indeed, the HAT mechanism is the most likely process in the gas phase, but a significant decrease in AIP values, and especially PA, with a certain increase in BDE in the ethanol medium makes the more favorable processes associated with the electronic transfer.
Cytotoxicity
MTT analysis (Mosmann 1983) is a sensitive, quantitative and reliable colorimetric analysis of the viability, proliferation, and activation of cells. Cytotoxicity of synthesized resveratrol analogs was determined using the MTT method and mouse fibroblast cells L929 (obtained from the Collections tissue cultures of Ivanovsky Virology Research Institute).
The results of analysis of the MTT assay data are presented in Fig. 5 (and Table in Supplementary). It was shown that most of the testing compounds reduced the viability of L929 cells to varying degrees. The most significant cytotoxic effect, similar to resveratrol, was demonstrated by analogs 2a and 3a, which reduced the number of fibroblasts in comparison with the drug-free control over the whole range of concentrations. Analogs 2b, 2c, and 2e were less cytotoxic, they exerted a depressing effect on cell growth in concentrations above 50 μM inclusively; at lower concentrations, these compounds did not affect cell growth. Analog 2d was cytotoxic only at maximum concentrations: from 800 to 200–100 μM, otherwise it indices did not differ from the control values, but was significantly less toxic than resveratrol

Comparison of MTT assay in L929 cells; data presented as percentage of control
Compound 2c has the maximum cytotoxic activity in the zone of average concentrations (400–50 μM), whereas in smaller doses there is no difference with the control, but in large doses this compound increases the number and/or viability of the cells by 76%. Therefore, the untypical shape of the survival curve of 2c caused the impossibility of calculating the IC50 by the method of nonlinear regression. The remaining data of half-maximal inhibitory concentrations and confidence intervals are presented in Table 5.
The studied compounds could be arranged as follows in decreasing order of cytotoxicity: resveratrol > analog 3a > 2d > 2e > 2a > 2b. The paradoxical increasing of formazan formation appearing at high concentrations (> 800 μM) of test compounds (especially 2c, which has the lowest IC50 value in the DPPH test) is most likely due to the beginning direct reduction of MTT by the test substances action. The data requires clarification and it is possible to use other tests and models of cell cultures.
Conclusion
The results of DFT calculations and DPPH tests show that the synthesized derivatives 2b, 2c, and 2e are more active antiradical agents than resveratrol, whereas 3a, 2a, and 2d have a lower ability to interact with free radicals in the media under consideration. Thus, it was shown that the antiradical activity of the obtained compounds strongly depends on the number and position of the hydroxyl groups in the stilbazole framework. At the same time, the medium polarity affects the mechanism of the process essentially but doesn’t affect the relative activity. MTT test data indicate the lower cytotoxicity of the obtained compounds compared to resveratrol, in contrast to which at concentrations <50 μM they didn’t influence the cell growth of mouse fibroblasts L929. Thus, the results obtained contribute to understanding the pharmacological activity of the studied compounds which can be used in food chemistry, medicine and pharmacy as potential antioxidant agents.
References
Baur JA, Sinclair DA (2006) Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 5:493–506
Becke AD (1993) Density‐functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Berdnikova DV, Aliyeu TM, Paululat T, Fedorov YuV, Fedorova OA, Ihmels H (2015) DNA-ligand interactions gained and lost: light-induced ligand redistribution in a supramolecular cascade. Chem Commun 51:4906–4909
Bialon O (1902) Über die Einwirkung von Anisaldehyd auf Chinaldin, α-Picolin und Aldehydcollidin. Ber Dtsch Chem Gessel 35:2786–2790
Blois MS (1958) Antioxidant determinations by the use of a stable free radical. Nature 181:1199–1200
Cao H, Pan X, Li C, Zhou C, Deng F, Li T (2003) Density functional theory calculations for resveratrol. Bioorg Med Chem Lett 13:1869–1871
Castro ME, Percino MJ, Chapela VM, Soriano-Moro G, Ceron M, Melendez FJ (2013) Comparative theoretical study of the UV/Vis absorption spectra of styrylpyridine compounds using TD-DFT calculations. J Mol Model 19:2015–2026
Choi HJ, Song MG, Sim YH, Bae HK, Kim JW, Park LS (2010) Comparative syntheses of arylamine monomer with styrylpyridyl photo-crosslinker of polyarylamine for OLED hole-injection material. Mol Cryst Liq Cryst 531:347–354
Desiraju GR, Steiner T (2001) The weak hydrogen bond in structural chemistry and biology. Oxford University Press, Oxford
Karuppagounder SS, Pinto JT, Xu H, Chen HL, Beal MF, Gibson GE (2009) Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer’s disease. Neurochem Int 54:111–118
Klamt A, Schüürmann G (1993) COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J Chem Soc Perkin Trans 2(2):799–805
Kopp P (1998) Resveratrol, a phytoestrogen found in red wine. A possible explanation for the conundrum of the ‘French paradox’? Eur J Endocrinol 138:619–620
Litwinienko G, Ingold KU (2007) Solvent effects on the rates and mechanisms of reaction of phenols with free radicals. Acc Chem Res 40:222–230
Mao D, Hong G, Wu S, Liu X, Yu J, Wang L (2014) Lewis-acid-catalyzed benzylic reactions of 2-methylazaarenes with aldehydes. Eur J Org Chem 2014:3009–3019
Mikulski D, Szeląg M, Molski M, Górniak R (2010) Quantum-chemical study on the antioxidation mechanisms of trans-resveratrol reactions with free radicals in the gas phase, water and ethanol environment. J Mol Struct 951:37–48
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63
Murias M, Jäger W, Handler N, Erker T, Horvath Z, Szekeres T, Nohl H, Gille L (2005) Antioxidant, prooxidant and cytotoxic activity of hydroxylated resveratrol analogues: structure–activity relationship. Biochem Pharmacol 69:903–912
Musialik M, Litwinienko G (2005) Scavenging of dpph• radicals by vitamin E is accelerated by its partial ionization: the role of sequential proton loss electron transfer. Org Lett 7:4951–4954
Nara SJ, Jha M, Brinkhorst J, Zemanek TJ, Pratt DA (2008) A simple Cu-catalyzed coupling approach to substituted 3-pyridinol and 5-pyrimidinol antioxidants. J Org Chem 73:9326–9333
Percino MJ, Chapela VM, Salmón M, Espinosa-Pérez G, Herrera AM, Flores A (1997) X-ray crystal structure of 2-styrylpyridine. J Chem Crystallogr 27:549–552
Pratt DA, DiLabio GA, Brigati G, Pedulli GF, Valgimigli L (2001) 5-Pyrimidinols: novel chain-breaking antioxidants more effective than phenols. J Am Chem Soc 123:4625–4626
Ruiz A, Rocca P, Marsais F, Godard A, Quéguiner G (1997) Pyridinium chloride: a new reagent for N-demethylation of N-methylazinium derivatives. Tetrahedron Lett 38:6205–6208
Scalbert A, Manach C, Morand C, Rémésy C, Jiménez L (2005) Dietary polyphenols and the prevention of diseases. Crit Rev Food Sci Nutr 45:287–306
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, Windus TL, Dupuis M, Montgomery Jr JA (1993) General atomic and molecular electronic structure system. J Comput Chem 14:1347–1363
Scott AP, Radom L (1996) Harmonic vibrational frequencies:an evaluation of Hartree−Fock, Møller−Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J Phys Chem 100:16502–16513
Shimada K, Fujikawa K, Yahara K, Nakamura T (1992) Antioxidative properties of xanthan on the autoxidation of soybean oil in cyclodextrin emulsion. J Agric Food Chem 40:945–948
Urbaniak A, Molski M, Szeląg M (2012) Quantum-chemical calculations of the antioxidant properties of trans-p-coumaric acid and trans-sinapinic acid. CMST 18:117–128
Valenzano DR, Terzibasi E, Genade T, Cattaneo A, Domenici L, Cellerino A (2006) Resveratrol prolongs lifespan and retards the onset of age-related markers in a short-lived vertebrate. Curr Biol 16:296–300
Van de Loosdrecht AA, Beelen RHJ, Ossenkoppele GJ, Broekhoven MG, Langenhuijsen MMAC (1994) A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J Immunol Methods 174:311–320
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Semenov, A.V., Balakireva, O.I., Tarasova, I.V. et al. Synthesis, theoretical, and experimental study of radical scavenging activity of 3-pyridinol containing trans-resveratrol analogs. Med Chem Res 27, 1298–1308 (2018). https://doi.org/10.1007/s00044-018-2150-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-018-2150-8