
Abstract
In recent years, a large number of in silico and in vitro assays have been developed for safety assessment in early drug discovery. These methods are usually validated using datasets of known drugs with large chemical diversity, while application to homologous series has been rarely explored. Here we report a case study about phospholipidosis (PLD) risk evaluation for a dataset of nine compounds, designed and synthesized to modulate the physico-chemical properties typical of cationic amphiphilic compounds (CADs), representing the main class of PLD inducers. Our aim was to investigate the effect of structure modification on PLD induction according to a number of standard in silico and in vitro methods. As a result, we found that different in silico methods lead to conflicting results when applied to our series of weak PLD inducers, thus the apparently easy-to-use definition of CADs requires special attention. Moreover, when weak inducers are tested in vitro, the revealed PLD effect may vary based on the purity grade of the tested compound and the features of the selected assay. Finally, we have shown that slight modifications on a chemical scaffold can have an impact on the PLD effect. This study also exemplifies that current in silico methods possibly overestimate the PLD induction effect of cationic amphiphilic compounds compared to the in vitro, with the risk of discarding promising compounds based on incorrect safety liabilities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Drug failure due to safety issues represents a major factor in the productivity gap for pharmaceutical companies (Blomme and Will 2016; Waring et al. 2015). Thus, several strategies have been recently developed to move the safety assessment of new chemical entities early in drug discovery, and new technologies are being applied for this purpose (Waring et al. 2015; DiMasi et al. 2010). An exhaustive review about toxicology strategies in drug discovery has been recently published (Blomme and Will 2016), including in silico and in vitro assays.
Among the toxicity risk assessments moved to an early discovery phase, the evaluation of phospholipidosis (PLD) associated to drugs apparently represents a simple task (Chatman et al. 2009). PLD is a lipid storage disorder characterized by the accumulation of phospholipids in the lysosomes of the affected tissues upon drug treatment, forming microscopic multilamellar inclusion bodies (Chatman et al. 2009; Ikeda et al. 2008; Reasor et al. 2006; Halliwell 1997). In recent years, much effort has been devoted to predict the PLD effect of drugs (Ploemen et al. 2004; Tomizawa et al. 2006; Pelletier et al. 2007; Ivanciuc 2008; Hanumegowda et al. 2010; Lowe et al. 2012; Kruhlak et al. 2008; Goracci et al. 2013), as well as to develop efficient in vitro assays (Kasahara et al. 2006; Bhandari et al. 2008; van de Water et al. 2011) for fast and cost-effective PLD evaluation. Although the mechanism is not yet fully understood, knowledge about this adverse event is rather advanced, and the chemical features of PLD inducers seem quite simple to be described. Indeed, cationic amphiphilic drugs (CADs) are considered the most relevant class responsible for this disorder (Reasor et al. 2006; Kodavanti and Mehendale 1990; Lullmann et al. 1975), although a number of aminoglycoside or macrolide antibiotics are also PLD inducers (PLD+) (Munic et al. 2011; Mingeot-Leclercq and Tulkens 1999). Despite this side-effect, CADs are, for their amphiphilic nature, elected drugs to penetrate the blood brain barrier, and thus are often developed to treat neurological diseases (Muehlbacher et al. 2012). In addition, they are also used in a wide range of therapeutic areas, including the treatment of arrhythmia, allergy, and fungal infections (Kodavanti and Mehendale 1990). The definition of CADs is apparently rather trivial. CADs are usually described as drugs that share two structural features: a rigid hydrophobic moiety (primarily aromatic rings) and a polar “head group” including an amine group, which is mainly in its protonated state under physiological conditions (Reasor et al. 2006). Due to this simple definition and the general PLD/CADs qualitative correlation, a number of rule-based methods for PLD predictions mainly based on CADs chemical features (pKa, CLogP, volume of distribution, and amphiphilic moment) have been developed in the last 15 years (Ploemen et al. 2004; Tomizawa et al. 2006; Pelletier et al. 2007; Hanumegowda et al. 2010; Fischer et al. 2012). Interestingly, depending on the PLD prediction methods, physico-chemical properties defining CADs are quantitatively different. For example, Ploemen et al. (2004) set as preliminary reference values pKa > 8 and ClogP > 1, while Muehlbacher et al. (2012) used pKa > 7.4 and logP > 3. In addition, Katzmi et al. (2013) set a pKa > 6 as reference value, while Tomizawa et al. (2006) introduced the net charge (NC) calculated at pH 4.0 for CADs definition (CLogP > 1 and 1 ≤ NC ≤ 2). Finally, for Nadanaciva et al. (2011) CADs having a basic center with a pKa between 6.5 and 11 are likely to cause lysosomotropism. Thus, depending on the threshold used, new chemical entities (NCE) may or may not be classified as CADs. In this context, the need to improve the definition for CADs inducing PLD has been recently raised (Slavov et al. 2014; Sun et al. 2012; Choi et al. 2013), considering that, as noted by Choi (2013), not all CADs induce PLD. Consequently, the importance of describing the spatial distribution of the chemical groups leading to the amphiphilic nature has been also addressed to refine the CADs definition. To this aim, the peculiar chemical nature of PLD-inducing CADs has been recently summarized by two toxicophore models by Slavov et al. (2014) and by us (Goracci et al. 2015). A deeper understanding of the PLD effect associated with CADs should avoid being too general thereby limiting the exclusion of potentially good drugs in an early phase.
However, if from the chemistry perspective a better definition of CADs inducing PLD is in progress, on the biological side, eventual anomalous effects of CADs towards PLD induction could be related also to the choice of the in vitro assay used for toxicological evaluation.
In the present study, we report a case study to evaluate the effect of chemical features of CADs and the biological assays used on the determination of the PLD induction. Indeed, we realized that focused investigations into this issue are extremely scarce (Quaglino et al. 2004). Our starting point was the N-(2-chlorobenzyl)quinuclidin-3-amine (1) (Fig. 1), which possesses all the chemical features generally attributed to a PLD+ compound. Indeed, compound 1 has a calculated LogP value of 2.5, with the most basic center having a pKa of 9.45, and it is predicted as a PLD+ according to various in silico models for PLD risk assessment (see Table 1). In addition, compound 1 nicely fits the Slavov two-center toxicophore (Slavov et al. 2014), with a distance between the tertiary amino group (the most basic center) and the aromatic ring of 5.3 Å, and our recently published toxicophore model (Goracci et al. 2015). Despite these chemical features, compound 1 recently resulted to be a false positive in vitro, using two different biological assays (Goracci et al. 2015).

Structure of compound 1
Therefore, we decided to further investigate this case, re-synthesizing compound 1 with higher purity (>99%), and synthesizing eight new analogs (compounds 2–9, Table 1). The PLD effect of compounds 1–9 was predicted according to four in silico methods, and then determined by a well-established cell-based in vitro assay using two different fluorescent phospholipids as probes.
Materials and methods
Computational study
PLD effect predicted according to Ploemen (P) (Ploemen et al. 2004) and Tomizawa (T) (Tomizawa et al. 2006) models The rule-based methods P and T are based on Eqs. (1) and (2), respectively:
where pKaMB refers to the pKa of the most basic center in the molecule and NC correspond to the net charge at pH 4.0. Values calculated for compound 1–9 are reported in Table 1. The pKaMB, CLogP, and NC values were calculated using MoKa (Molecular Discovery Ltd, UK) (Cruciani et al. 2009; Milletti et al. 2010).
PLD effect prediction by PLS-DA
An already published PLS-DA model of 331 PLD inducing and non-inducing compounds (accuracy = 0.78, sensitivity = 0.71), was used to predict the PLD risk for compounds in Table 1. Using the VolSurf+ software (Cruciani et al. 2000), compounds 1–9 were imported in their most abundant protonation state at pH 7.4 and all the 128 physico-chemical and ADME related descriptors were used as variables. A complete list of the VolSurf+ descriptors has been reported elsewhere (Mannhold et al. 2006).
PLD effect prediction by PLD-phore
The FLAP software (Molecular Discovery, UK) was used to study the similarity of the tested compounds with the previously defined PLD-phore (Goracci et al. 2015). In particular, the FLAP algorithm (Baroni et al. 2007) was used to align the structures of compounds 1, 3–9 to the PLD-phore previously generated using the FLAPpharm module (Cross et al. 2012a, b). Briefly, the PLD-phore was generated by the automatic alignment of five CADs reported to be PLD inducers, in their protonated state. Subsequently the common interacting features in terms of their GRID molecular interaction fields, pseudo-fields, and atomic points are extracted. An extensive description of the FLAP and FLAPpharm algorithms as well as the applications in defining the pharmacophores have been reported elsewhere (Baroni et al. 2007; Cross et al. 2012a, b; Sirci et al. 2012; Lepri et al. 2014; Tondi et al. 2016; Chapy et al. 2015; Artese et al. 2013). Compounds 1 and 3–9, each one considered in its protonated form and in a maximum of 50 possible conformations to mimic flexibility, were thus aligned on the PLD-phore. During this alignment process a constraint was used for the PLD-phore, defining that its H-bond donor region should be a charged H-bond donor atom. Then, the best alignment in term of the FLAP Glob-Prod descriptor (covering shape, donor, acceptor, and hydrophobic similarity) was selected.
Chemistry
All commercial products were acquired form Sigma Aldrich and used without further purification (≥97% pure). 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded at 400 and 100.6 MHz, respectively, on Bruker Avance II 400 MHz spectrometer at room temperature. Chemical shifts (δ) are given in parts per million (ppm) relative to the internal standard tetramethylsilane. Peak multiplicities are reported as s (singlet), d (doublet), dd (double doublet), t (triplet), dt (double triplet), q (quartet), m (multiplet), or br s (broad singlet). Coupling constants (J) are given in Hz. High resolution mass spectrometry (HRMS) spectra were registered on Agilent Technologies 6540 UHD Accurate Mass Q-TOF liquid chromatography/mass spectrometry (LC/MS) system.
Synthesis of N-substituted 2-chlorobenzylamines (1–7)
A solution of 2-chlorobenzaldehdye (11 mmol) and suitable amine (10 mmol) in MeOH (35 mL) was stirred at r.t. for 24 h. NaBH4 (40 mmol) was carefully added in small portion at 0 °C and the reaction was kept at room temperature for further 4 h. The reaction was carefully quenched with saturated aqueous NaHCO3 solution and evaporated to dryness. The residue was purified by flash chromatography on SiO2 (eluent DCM/MeOH/Et3N 95:5:1, if not stated otherwise) to afford 2-chlorobenzylamines 1–7 (Table 1). The resulting amino compound was dissolved in methanol and HCl 4.0 M in dioxane (40 mmol) was added, the solution was stirred for 30 min and then concentrated again to give the hydrochloric salt in quantitative yield.
N-(2-Chlorobenzyl)quinuclidin-3-amine dihydrochloride (1-syn)
Yield 70%, white solid, mp 141–142 °C; 1H NMR (400 MHz, dimethyl sulphoxide (DMSO)-d6): δ = 10.97 (s, 1H, NH), 10.57 (s, 1H, NH), 9.87 (s, 1H, NH), 7.92 (d, J = 6.1 Hz, 1H, H-6), 7.69–7.54 (m, 1H, H-3), 7.54–7.40 (m, 2H, H-4, H-5), 4.57–4.14 (m, 2H, ArCH 2N), 3.94–3.74 (m, 1H, H-1′), 3.68 (t, J = 11.4 Hz, 1H, H-2′), 3.53–3.40 (m, 1H, H-7′), 3.39–3.33 (m, 1H, H-2′), 3.27–2.96 (m, 3H, H-4′, H-7′), 2.66–2.58 (m, 1H, H-6′), 2.41–2.20 (m, 1H, H-5′), 2.10–1.76 (m, 1H, H-8′), 1.90–1.76 (m, 2H, H-5′, H-8′); 13C NMR (101 MHz, DMSO-d6): δ = 134.3 (C, C-1), 133.1 (CH, C-6), 131.5 (CH, C-4), 130.0 (CH, C-3), 129.9 (C, C-2), 127.9 (CH, C-5), 52.7 (CH, C-1′), 48.6 (CH2, C-2′), 46.2 (CH2, ArCH 2N), 45.8 (CH2, C-7′), 45.3 (CH2, C-4′), 22.0 (CH2, C-8′), 21.9 (CH, C-6′), 17.1 (CH2, C-5′); HRMS-ESI: m/z calcd. for C14H19 35ClN2 251.1315 (M + H+), found 251.1311 (M + H+).
N-(2-Chlorobenzyl)pyridin-3-amine dihydrochloride (2)
Yield 75%, white solid, mp 130–131 °C; purified by column chromatography (eluent DCM/MeOH, 98:2); 1H NMR (400 MHz, MeOD): δ = 8.04 (s, 1H, H-2′), 8.00 (t, J = 3.0 Hz, 1H, H-6′), 7.80–7.69 (m, 2H, H-4′, H-5′), 7.53–7.42 (m, 2H, H-3, H-6), 7.37–7.28 (m, 2H, H-4, H-5), 4.58 (s, 2H, NCH 2Ar); 13C NMR (101 MHz, CD3OD): δ = 147.7 (C, C-3′), 134.2 (C, C-1), 133.3 (C, C-2), 129.5 (CH, C-3), 129.0 (CH, C-4), 128.8 (CH, C-6), 127.9 (CH, C-6′), 127.4 (CH, C-5′), 127.2 (CH, C-4′), 127.1 (CH, C-5), 123.9 (CH, C-2′), 44.0 (CH2, NCH 2Ar); HRMS-ESI: m/z calcd. for C12H11 35ClN2 219.0689 (M + H+), found 219.0689 (M + H+).
N1-(2-Chlorobenzyl)-N2,N2-diethylethane-1,2-diamine dihydrochloride (3)
Yield 87%, white solid, mp 115–116 °C; 1H NMR (400 MHz, MeOD): δ = 7.78 (d, J = 6.3 Hz, 1H, H-6), 7.58 (d, J = 7.3 Hz, 1H, H-3), 7.54–7.37 (m, 2H, H-5, H-4), 4.52 (s, 2H, ArCH 2N), 3.87–3.67 (m, 2H, NCH 2CH2NEt), 3.67–3.53 (m, 2H, NCH2 CH 2NEt), 3.49–3.33 (m, 4H, NCH 2CH3), 1.55–1.26 (m, 6H, NCH2 CH 3); 13C NMR (101 MHz, MeOD): δ = 134.3 (C, C-1), 132.0 (CH, C-6), 131.4 (CH, C-4), 129.9 (CH, C-3), 128.7 (C, C-2), 127.7 (CH, C-5), 48.8 (CH2, ArCH 2N), 48.0 (CH2, NCH 2CH3), 47.5 (CH2, NCH2 CH 2NEt), 42.2 (CH2, NCH 2CH2NEt), 7.9 (CH3, NCH2 CH 3); HRMS-ESI: m/z calcd. for C13H21 35ClN2 241.1472 (M + H+), found 241.1469 (M + H+).
N-(2-Chlorobenzyl)piperidin-3-amine dihydrochloride (4)
Yield 79%, white solid, mp (dec.) 270 °C; 1H NMR (400 MHz, MeOD): δ = 7.75 (dd, J = 1.5, 7.3 Hz, 1H, H-6), 7.65–7.57 (m, 1H, H-3), 7.56–7.41 (m, 2H, H-4, H-5), 4.63–4.39 (m, 2H, ArCH 2N), 3.87 (d, J = 12.1 Hz, 1H, H-2′), 3.79 (t, J = 10.9 Hz, 1H, H-1′), 3.46 (d, J = 12.8 Hz, 1H, H-4′), 3.24 (t, J = 11.7 Hz, 1H, H-2′), 3.17–3.00 (m, 1H, H-4′), 2.48 (d, J = 10.6 Hz, 1H, H-6′), 2.18 (d, J = 13.9 Hz, 1H, H-5′), 2.03–1.73 (m, 2H, H-5′, H-6′); 13C NMR (101 MHz, MeOD): δ = 134.5 (C, C-1), 132.1 (CH, C-6), 131.5 (CH, C-4), 129.9 (CH, C-3), 128.9 (C, C-2), 127.7 (CH, C-5), 52.1 (CH, C-1′), 46.0 (CH2, ArCH 2N), 43.6 (CH2, C-2′), 43.3 (CH2, C-4′), 24.9 (CH2, C-6′), 20.2 (CH2, C-5′); HRMS-ESI: m/z calcd. for C12H17 35ClN2 225.1159 (M + H+), found 225.1159 (M + H+).
N-(2-Chlorobenzyl)-1-ethylpiperidin-3-amine dihydrochloride (5)
Yield 80%, white solid, mp 110–111 °C; 1H NMR (400 MHz, MeOD): δ = 7.72 (d, J = 6.1 Hz, 1H, H-6), 7.58 (d, J = 7.7 Hz, 1H, H-3), 7.54–7.38 (m, 2H, H-4, H-5), 4.58–4.44 (m, 2H, ArCH 2N), 4.03–3.78 (m, 1H, H-2′), 3.91–3.76 (m, 1H, H-1′), 3.76–3.54 (m, 1H, H-4′), 3.34 (q, J = 6.5 Hz, 2H, NCH 2CH3), 3.24–2.89 (m, 1H, H-2′), 3.07–2.92 (m, 1H, H-4′), 2.56–2.35 (m, 1H, H-6′), 2.25–2.08 (m, 1H, H-5′), 2.07–1.89 (m, 1H, H-6′), 1.90–1.74 (m, 1H, H-5′), 1.43 (t, J = 6.7 Hz, 3H, NCH2 CH 3); 13C NMR (101 MHz, MeOD): δ = 134.5 (C, C-1), 132.1 (CH, C-6), 131.6 (CH, C-4), 129.9 (CH, C-3), 128.8 (C, C-2), 127.8 (CH, C-5), 53.0 (CH2, NCH 2CH3), 52.5 (CH, C-1′), 51.3 (CH2, C-2′), 51.0 (CH2, C-4′), 46.1 (CH2, ArCH 2N), 24.7 (CH2, C-6′), 20.8 (CH2, C-5′), 8.3 (CH3, NCH2 CH 3). HRMS-ESI: m/z calcd. for C14H21 35ClN2 253.1472 (M + H+), found 253.1472 (M + H+).
2-((2-Chlorobenzyl)amino)ethanol hydrochloride (6)
Yield 81%, white solid, mp 134–135 °C; 1H NMR (400 MHz, MeOD): δ = 7.68 (dd, J = 1.9, 7.5 Hz, 1H, H-6), 7.58 (dd, J = 1.4, 7.5 Hz, 1H, H-3), 7.53–7.34 (m, 2H, H-4, H-5), 4.46 (s, 2H, ArCH 2N), 4.02–3.78 (m, 2H, NCH2 CH 2OH), 3.29–3.18 (m, 2H, NCH 2CH2OH); 13C NMR (101 MHz, MeOD): δ = 134.5 (C, C-1), 131.9 (CH, C-6), 131.2 (CH, C-4), 129.8 (CH, C-3), 129.1 (C, C-2), 127.6 (CH, C-5), 56.2 (CH2, NCH2 CH 2OH), 49.2 (CH2, NCH 2CH2OH), 47.7 (CH2, CH2, ArCH 2N); HRMS-ESI: m/z calcd. for C9H12 35ClNO 186.0686 (M + H+), found 186.0677 (M + H+).
N1,N1-Diethyl-N2-(naphthalen-1-ylmethyl)ethane-1,2-diamine dihydrochloride (7)
Yield 77%, white solid, mp 126–127 °C; 1H NMR (400 MHz, MeOD): δ = 8.30 (dd, J = 0.8, 8.6 Hz, 1H, H-10), 8.04 (d, J = 8.3 Hz, 1H, H-4), 8.02–7.98 (m, 1H, H-7), 7.84 (dd, J = 1.0, 7.1 Hz, 1H, H-2), 7.72 (ddd, J = 1.4, 6.9, 8.4 Hz, 1H, H-9), 7.63 (ddd, J = 1.5, 6.9, 8.0 Hz, 1H, H-8), 7.61–7.57 (m, 1H, H-3), 4.87 (s, 2H, ArCH 2N), 3.84–3.73 (m, 2H, NCH 2CH2NEt), 3.69–3.57 (m, 2H, NCH2 CH 2NEt), 3.36 (q, J = 7.0 Hz, 4H, NCH 2CH3), 1.41 (t, J = 7.0 Hz, 6H, NCH2 CH 3); 13C NMR (101 MHz, MeOD): δ = 134.0 (C, C-5), 131.2 (C, C-6), 130.4 (CH, C-4), 129.2 (CH, C-2), 128.7 (CH, C-7), 127.2 (CH, C-9), 126.7 (C, C-1), 126.3 (CH, C-8), 125.1 (CH, C-3), 122.7 (CH, C-10), 48.4 (CH2, ArCH 2N), 47.8 (CH2, NCH 2CH3), 47.4 (CH2, NCH2 CH 2NEt), 41.9 (CH2, NCH 2CH2NEt), 7.8 (CH3, NCH2 CH 3); HRMS-ESI: m/z calcd. for C7H24N2 257.2018 (M + H+), found 257.2018 (M + H+).
N,N-diethyl-2-(isoindolin-2-yl)ethan-1-amine (8)
Yield 50%, white solid, mp (dec.) 238 °C; N,N-diethylethylenediamine (1.88 g, 2.27 mL, 16.2 mmol) was added to a stirred solution of phthalic anhydride (2.00 g, 14.5 mmol) in toluene (100 mL). Catalytic p-TsOH was added and the solution refluxed in a Dean Stark apparatus for 2 h. The solution was concentrated under reduced pressure and the residue was taken up in DCM, washed with NaOH and brine. The organic phase was concentrated to afford 2-(2-(diethylamino)ethyl)isoindoline-1,3-dione (10) as a yellow oil (99% yield), used in the following step without any further purification. 1H NMR (400 MHz, CD3Cl): δ = 7.97–7.78 (m, 2H, Ar–H), 7.78–7.64 (m, 2H, Ar–H), 3.78 (t, J = 7.0 Hz, 2H, NCH 2CH2NEt), 2.72 (t, J = 7.0 Hz, 2H, NCH2 CH 2NEt), 2.59 (q, J = 7.1 Hz, 4H, NCH 2CH3), 1.01 (t, J = 7.1 Hz, 6H, NCH2 CH 3). Phthalimide 10 (1.00 g, 4.06 mmol) was slowly added to a stirred suspension of LiAlH4 (0.77 g, 20.3 mmol) in THF (10 mL) at 0 °C; after the addition, the mixture was refluxed for 24 h. After being cooled to 0 °C, 0.77 mL of water was added, followed by 0.77 mL of 15% NaOH (aq) and 2.31 mL of water. The resulting suspension was stirred for 1 h, the solid was filtered and rinsed with EA. The organic phase was separated and evaporated under vacuum; the residue was treated with HCl in dioxane (2 mL, 4.0 M) and evaporated again to give N,N-Diethyl-2-(isoindolin-2-yl)ethanamine dihydrochloride; 1H NMR (400 MHz, MeOD): δ = 7.45 (s, 4H, H-3, H-4, H-5, H-6), 4.87 (s, 4H, H-7, H-8), 4.27–3.92 (m, 2H, NCH 2CH2NEt), 3.85–3.59 (m, 2H, NCH2 CH 2NEt), 3.40 (q, J = 7.3 Hz, 4H, NCH 2CH3), 1.44 (t, J = 7.3 Hz, 6H, NCH2 CH 3); 13C NMR (101 MHz, MeOD): δ = 133.1 (C, C-1, C-2), 128.9 (CH, C-4, C-5), 122.6 (CH, C-3, C-6), 58.8 (CH2, C-7, C-8), 48.1 (CH2, NCH 2CH2NEt), 47.7 (CH2, NCH 2CH3), 46.8 (CH2, NCH2 CH 2NEt), 7.7 (CH3, NCH2 CH 3); HRMS-ESI: m/z calcd. for C14H22N2 219.1861 (M + H+), found 219.1855 (M + H+).
2-((2-Chlorobenzyl)oxy)-N,N-dimethylethanamine hydrochloride (9)
A solution of 2-dimethylaminoethanol (0.66 g, 7.4 mmol) in anhydrous DMF (2 mL) was carefully added to a stirred suspension of NaH (60% dispersion in mineral oil, 0.29 g, 7.4 mmol) in anhydrous DMF (50 mL). After 30 min, 2-chlorobenzylbromide (1.00 g, 4.9 mmol) in anhydrous DMF (5 mL) was added dropwise, and stirring was continued overnight. The mixture was dropped in water and extracted with DCM. The organic phase was washed with water (three times) and evaporated to give the crude oil, purified by flash chromatography on silica gel (eluent DCM/MeOH/Et3N, 94:5:1). The resulting amino compound was dissolved in methanol and HCl 4.0 M in dioxane (40 mmol) was added, the solution was stirred for 30 min and then concentrated again to give the hydrochloric salt as pale yellow solid (1.2 g, yield 99%); mp 108–109 °C; 1H NMR (400 MHz, MeOD): δ = 7.61–7.51 (m, 1H, H-6), 7.47–7.40 (m, 1H, H-3), 7.40–7.29 (m, 2H, H-4, H-5), 4.74 (s, 2H, ArCH 2O), 3.89 (t, J = 4.7 Hz, 2H, OCH 2CH2N), 3.43 (t, J = 4.7 Hz, 2H, OCH2 CH 2N), 2.94 (s, 6H, NCH 3); 13C NMR (101 MHz, MeOD): δ = 134.8 (C, C-1), 133.3 (C, C-2), 130.0 (CH, C-6), 129.3 (CH, C-4), 129.1 (CH, C-3), 126.8 (CH, C-5), 70.2 (CH2, ArCH 2O), 63.7 (CH2, OCH 2CH2N), 56.7 (CH2, OCH2 CH 2N), 42.3 (CH3, NCH 3); HRMS-ESI: m/z calcd. for C11H16ClNO 214.0993 (M + H+), found 214.0999 (M + H).
Purity analysis
Purities of the final compounds were determined by LC-MS on Agilent Technologies 6540 UHD Accurate Mass Q-TOF LC/MS. The mobile phase was a mixture of water (A) and acetonitrile (B), both containing formic acid at 0.1%. Chromatographic separation was carried out with Acquity UPLC BEH C18 1.7 µm (C18, 150 × 2.1 mm) column, with a flow rate of 0.650 mL/min in a 10 min gradient elution. Gradient elution was as follows: 99.5:0.5 (A/B) to 5:95 (A/B) over 8 min, 5:95 (A/B) for 2 min, and then reversion back to 99.5:0.5 (A/B) over 0.1 min. New synthesized compounds were ≥98% (>99% for compound 1-syn), while purity of 1-com was 96%, according to previous result (Goracci et al. 2015).
Biological assay
Cell culture and treatments
HepG2 cells (human hepatoma cell line) were purchased from the American Type Culture Collection (ATCC® HB-8065™, Manassas, VA, USA) and all culture reagents were purchased from Lonza (Breda, Netherlands). 3-Amino-7-dimethylamino-2-methylphenazine hydrochloride (Neutral Red) and Hoechst 33342 were purchased from Sigma-Aldrich (St Louis, MO, USA). 1-acyl-2-[12-(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-glycero-3-phosphocholine (NBD-PC) was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL) and N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine triethylammonium salt (NBD-PE), was purchased from Molecular Probes (Eugene, OR, USA). HepG2 cells were maintained in modified Eagle’s Medium supplemented with 10% fetal bovine serum, 50 U/mL penicillin, 50 µg/mL streptomycin, 2 mM l-glutamine and 1% non-essential amino acids solution in a humidified atmosphere containing 5% CO2 at 37 °C. Cells were passaged as needed using 0.5% trypsin-EDTA solution and experiments were performed between passages 10 and 25. For experimental purposes, cells collected and suspended at a density of 1 × 105/mL in fresh medium, were seeded on culture plates. Briefly, 2 × 104 cells were plated in 96-well plates for NR uptake and in 96-well black plates for PLD assay with fluorescent phospholipids. After 24 h, culture medium was replaced with fresh medium containing vehicle (0.05% DMSO) or test compounds and then treated cells were incubated for 24 h.
Cytotoxicity assay
Neutral red (NR) uptake: NR uptake into lysosomes of viable cells was performed according to Repetto et al. (2008) with slight modification. Briefly, following exposure to the selected compounds the medium was removed and 50 µL of NR dye (100 µg/mL) was added to each well. After incubation at 37 °C for 90 min, cells were washed with phosphate buffered saline (PBS) and dye was extracted by adding 100 μL of acidified ethanol solution (ethanol: H2O:CH3COOH 50:49:1, v/v/v). Thus, gentle shaking for 10 min complete the dissolution. Optical density (OD) was measured at 550 nm using a microplate reader (Beckman Coulter DTX880) and blanks, which contain no cells as a reference. The relative uptake of NR (%) by viable cells was calculated by comparing absorbance of compound-treated cells and absorbance of control cells exposed to the vehicle (0.05% DMSO) (OD (treated cells)/OD (control vehicle) × 100). Each experiment was performed in triplicate.
NBD-PC and NBD-PE uptake: Cells were plated in 96-well black plates and allowed to attach for 24 h as described above. Cells were then treated with each of the compound tested together with the fluorescent phospholipid analog NBD-PC, according to Kasahara et al. (2006) or NBD-PE, according to Bhandari et al. (2008). Briefly, the medium in each well was removed, and 50 µL of medium containing 80 µM NBD-PC or 100 µM NBD-PE plus 50 µL of medium containing the tested drugs were added. After 24 h, the cells were washed twice with PBS, and 50 µL of PBS was added to each well. Phospholipid fluorescence was measured with a microplate fluorescence reader (excitation and emission wavelengths of 485/538 nm and 462/536 nm for NBD-PC and NBD-PE, respectively). After the phospholipid fluorescence was measured, cells were incubated for further 20 min at 37 °C with Hoechst 33342 solution (20 µg/mL in PBS). The Hoechst 33342 fluorescence was then measured (excitation and emission wavelengths of 355 and 460 nm, respectively). Values for NBD-fluorescence were normalized to those of Hoechst 33342 fluorescence.
Statistical analysis: Data are expressed as mean ± SD from three independent experiments done at least in triplicate. Statistical comparisons were performed using Student’s t-test. Differences were considered statistically significant when p < 0.05.
Results and discussion
Design and synthesis of analogs of compound 1
As mentioned in the introduction, physico-chemical properties have proved to be useful to evaluate a possible PLD risk effect associated with CAD-like NCE (Ploemen et al. 2004; Tomizawa et al. 2006; Hanumegowda et al. 2010; Fischer et al. 2012). In our previous work, the false-positive result of compound 1 was attributed, among other causes, to the steric hindrance at the most basic center (i.e., the quinuclidine ring). Indeed, the quinuclidine moiety remains barely explored in PLD induction studies, with only three examples reported in the literature database (i.e. quinine, quinidine, and palonosetron) (Ploemen et al. 2004; Tomizawa et al. 2006; Hanumegowda et al. 2010; Orogo et al. 2012). Thus, eight analogs of 1 were designed and synthesized to tune the acid-base properties, the lipophilicity and the steric hindrance at the head-group moiety. Calculated pKa and LogP values are reported in Table 1.
As a first step, the quinuclidine moiety was replaced with pyridine, triethylamino group, piperidine, ethyl piperidine, and hydroxyethyl moiety, to give compounds 2–6, respectively. In compound 2, although the substitution with the pyridine has a minor impact on the LogP, the pKa is significantly reduced by four units. In addition, the second basic center (the benzyl amino group) becomes also irrelevant, being now linked to an aromatic ring. Based on these physico-chemical features, compound 2 was expected to lose the CAD-like features and thus to be a PLD-.
Designing compounds 3–5, the effect of the steric hindrance associated with the quinuclidine ring was investigated. The greatest flexibility was provided in compound 3, maintaining a tertiary amine as the most basic center and keeping the distance between the two basic centers constant. The cyclohexyl ring was retained in compounds 4 and 5, which differ for the absence or the presence of a further alkyl group at amine group, respectively. For these three structures the possible PLD effect was more uncertain, because such fine modulations of a scaffold for PLD studies have been scarcely studied so far (Quaglino et al. 2004).
In compound 6, the most basic center was totally removed and replaced with an alcoholic functional group. Despite this substitution, compound 6 preserves its basic nature while its LogP is significantly reduced. Compounds 7 and 8 are analogs of compound 3, in which the hydrophobic moiety was modified in order to increase or decrease the lipophilicity of the scaffold, respectively. The alkyl-diethylamine moiety of 3 was selected in this study because this group is frequent among CADs (e.g., amiodarone, amiodaquine, chloroquine, and coralgil). Finally, compound 9 was synthesized replacing the benzyl amino basic centre with a benzyl ether; in addition, the tertiary amine head was replaced with a dimethylamino moiety because the reagent was readily available and the dimethylamine is also common in PLD+ drugs (e.g., imipramine, chlorpromazine, and iprindole).
Predicted PLD effect of compounds 1–9
The PLD effect of compound 1 and of its designed analogs 2–9 was predicted using four in silico methods (Table 1): the Ploemen’s method (P) (Ploemen et al. 2004), the Tomizawa’s method (T) (Tomizawa et al. 2006), the projection on a recently published partial least squares discriminant analysis (PLS-DA) model (Goracci et al. 2013), and the similarity towards the toxicophore (PLD-phore) (Cruciani et al. 2009). Additional information about the PLD effect prediction are reported in the Materials and methods section.
As shown in Table 1, compound 2 is the only compound univocally predicted as a PLD non-inducer, in agreement with the lack of a strong basic center. Compounds 8 and 9 are generally predicted as PLD+ compounds, excepting for the P model. On the contrary, compound 6 is generally predicted as PLD- excepting for T model. Finally, compounds 1, 3–5, and 7 are univocally reported as PLD+.
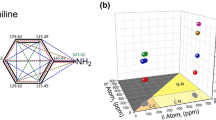
Differently from the P and the T methods, the PLS-DA and the PLD-phore approaches allow a visual inspection of the projection on the model or the compound/toxicophore matching, respectively (Fig. 2). Concerning the projection on the PLS-DA model, compounds 2 and 6 are clearly projected on PLD- region (Fig. 2a, b, red circles), while the remaining compounds are located at edge between PLD+ (blue) and PLD- (red) chemical space. This suggests that, although compounds 1, 3–5, and 7–9 are predicted as PLD+ drugs, their PLD effect is expected rather weak, with compound 7 resulting the one with the highest associated risk of PLD. Concerning the toxicophore similarity, the best alignment for each structure is reported in Fig. 2c, with the exception of compound 2. Indeed, it has already been proven (Slavov et al. 2014; Goracci et al. 2015) that the toxicophore models are generally valid only when applied to strong bases. Figure 2c shows that the alignments of 3–5, 8, and 9 are very similar to that of compound 1, following the Slavov two-center model (Slavov et al. 2014). Compound 7, bearing a naphthalene moiety, shows the best fitting of the hydrophobic region of the toxicophore, also fitting the Slavov three-center model (Slavov et al. 2014). On the contrary, for the hydroxyl compound 6, the alignment to the hydrophobic region of the toxicophore is totally lost, due to the orientation of the remaining basic center to the polar region.

Compound 1 and its analogous external prediction on our already published model (Goracci et al. 2013). PLD+ and PLD- compounds are reported in blue and red, respectively. Yellow projections correspond to compounds in Table 1. In particular, compounds 2 and 6 are highlighted in (a); in b, a detailed inspection of the compounds 1, 3–5, and 7–9 projections, is shown with an enlargement of (a). In c, the alignment of compounds 1, 3–9 on the PLD-phore model generated using FLAP is reported (Goracci et al. 2015; Baroni et al. 2007). Thus, based on physico-chemical properties and fitting to the PLD-phore, compounds 3–5, 7–9 are predicted as likely to induce phospholipid accumulation (PLD+ compounds), while compounds 2 and 6 are predicted as PLD- (color figure online)
Synthesis of compounds 1–9
All tested compounds were synthesized as described in Schemes 1–3. In particular, the compounds 1–7 were obtained by reductive amination of 2-chlorobenzaldehyde and 1-naphthaldeyde with the suitable amine. The reaction was carried out in methanol using sodium borohydride as reducing agent (Schemes 1–2).

Synthesis of compounds 1–6. Reagents and conditions: (i) 1. MeOH, r.t. 24 h; 2. NaBH4, 4 h; (ii) HCl in dioxane 4.0 M

Synthesis of compounds 7–8. Reagents and conditions: (i) 1. MeOH, r.t. 24 h; 2. NaBH4, 4 h; (ii) HCl in dioxane 4.0 M; (iii) p-TsOH, toluene, reflux; (iv) 1. LiAlH4, THF, reflux, 24 h; 2. HCl in dioxane 4.0 M

Synthesis of compound 9. Reagents and conditions: (i) NaH, DMF, r.t., 16 h
Compound 8 was obtained by condensation of phthalic anhydride with N,N-diethyl-1,2-ethylendiamine (10), followed by reduction (Scheme 2). Finally, benzyl ether derivative 9 was obtained in high yield by reaction of 2-chlorobenzyl bromide with N,N-dimethylethanolamine (Scheme 3).
Biological evaluation
Comparison of cytotoxicity and NBD-phospholipid uptake induction for the commercial and the newly synthesized compound 1
The biological properties of the newly synthesized compound 1 (1-syn, purity >99% by LC-MS; for more details see Materials and methods section) with respect to the commercial sample (AN-465/42886418 from SPECS, named here 1-com, 96% pure by LC-MS) were compared. Viability of HepG2 cells, following 24 h exposure to various concentrations of 1-com or 1-syn, was measured by neutral red (NR) uptake. This assay is based on the ability of viable cells to take up the supravital dye NR by active transport, following its incorporation and binding to lysosomes. Alteration in the cell surface lead to decreased uptake and binding of NR (see Materials and methods section for more details). As shown in Fig. 3, the 1-syn caused a decrease of NR uptake of about 70% at the highest concentration tested, while the 1-com had no effects. The IC50 for the synthesized compound is 124.2 μM.

Effect of 1-com (yellow) and 1-syn (green) compound 1 on HepG2 cell viability. HepG2 were treated for 24 h with selected compounds at the indicated concentrations and viability was assessed by NR uptake assay. Results are expressed as percentage viability of cells exposed to selected compounds with respect to vehicle control (Cveh). Statistical analysis was performed using Student’s t-test. Values are the mean ± SD of three independent experiments done in triplicate (***p < 0.001) (color figure online)
Afterwards, the possible PLD induction by 1-com and 1-syn was evaluated by fluorescence assays, measuring the accumulation of the two fluorescent lipids NBD-PC and NBD-PE (Fig. S1, Supporting Information). As mentioned in the Introduction section, the phosphatidylethanolamine derivative was reported to be a more sensitive probe for PLD risk detection than the phosphatidylcholine one (Bhandari et al. 2008). To take into account cell number, the NBD-PC and NBD-PE fluorescence intensities (Fig. 4a, b) were normalized using Hoechst 33342 (Fig. 4c, d), as described elsewhere (Kasahara et al. 2006; Goracci et al. 2015). Using NBD-PC (Fig. 4e) we could confirm that 1-com did not exhibit phospholipid accumulation at the tested compound concentration. However, when the same assay was performed with 1-syn at 100 µM, an increased uptake of NBD-PC was observed; in fact, the fluorescent ratio between NBD-PC and Hoechst was 1.53 ± 0.24, comparable to the one of amiodarone at 12 µM concentration (1.56 ± 0.27). Thus, the lower purity of 1-com might have negatively influenced the assignment in our previous work (Goracci et al. 2015). When NBD-PE was used instead of NBD-PC, a significant increased accumulation of the fluorescent phospholipid was observed for both 1-syn and 1-com at the highest concentrations tested (Fig. 4f). These results suggest that compound 1 can be classified as a potential weak PLD+, although previously reported as a PLD- one (Goracci et al. 2015) confirming the quality of the two toxicophore models for PLD risk prediction (Slavov et al. 2014; Goracci et al. 2015). It is noteworthy that also the reference compound amiodarone increased the uptake of NBD-PE at a greater extent respect to NBD-PC, suggesting a greater sensitivity of NBD-PE with respect to NBD-PC. In addition, the purity grade of the tested compounds resulted as an important aspect to consider in the case of high-throughput screening campaigns.

Measurement of NBD-PC (a) and NBD-PE (b) uptake in HepG2 cells treated with the commercial (yellow, 1-com) and the newly synthesized (green, 1-syn) compound 1. HepG2 cells were incubated with increasing concentrations of compound 1 in the presence of 40 μM NBD-PC and 50 μM NBD-PE. Amiodarone (AMIO) at 12 µM concentration was used as positive control. After measurement of NBD-PC and NBD-PE fluorescence, nuclei were stained with Hoechst 33342 (c–d). Fluorescence intensities of NBD-PC, NBD-PE, and Hoechst 33342 were measured at 485/538, 462/536, and 355/460 nm (E x/E m) respectively, and results were expressed as percentage of vehicle controls (Cveh). Normalized values were calculated by dividing the NBD-PC or NBD-PE value by the Hoechst 33342 value (e–f). Statistical analysis was performed using Student’s t-test. Results represent the mean ± SD of three independent experiments done in triplicate (*p < 0.05; ***p < 0.001) (color figure online)
Cytotoxicity and NBD-PE uptake induction for the analogs of compound 1
Viability of HepG2 cells for the eight synthesized analogs 2–9 were assayed by NR uptake, as previously described for compound 1. Figure 5 shows the lysosomal uptake of NR by HepG2 cells incubated at various concentrations of compounds 2–9; AMIO and 1-syn are reported as references at 12 and 100 μM, respectively. Among the analogs, only compound 2, bearing the pyridine group, decreased NR uptake at 100 μM (Fig. 5), indicating an impairment of lysosomal function. The other analogs did not significantly affect NR uptake at all the concentration tested. Afterwards, the PLD effect of the analogs of compound 1 was evaluated by the NBD-PE fluorescence assay. Figure 6 shows the level of accumulation of the fluorescent lipid at increasing concentrations of tested compounds, before and after normalization with Hoechst 33342. AMIO and 1-syn were used as positive controls, at 12 and 100 μM, respectively. Among compounds 2–9, only compound 7 displayed a degree of NBD-PE accumulation comparable to that of 100 μM 1-syn with a significant lipid accumulation even at 50 μM. A weak effect was also observed for compound 4, but at 100 μM only. It is noteworthy that compound 7 represents the only tested analog almost fitting the three-centered toxicophore model by Slavov (Slavov et al. 2014). Indeed, the distances connecting the tertiary amino group to the centroids of the two aromatic rings are 6.3 and 6.6 Å (i.e., in the ranges 5.5–7 and 3.5–7 Å, proposed for this model), while the distance between the two centroids of the aromatic rings is slightly lower (3.2 Å) than the proposed range (4–5 Å) (Fig. S2, in the Supporting Information). However, compounds 1, 4, and 7 can be generally classified as weak PLD inducers, compared to the effect exerted by AMIO. Indeed, the fluorescent NBD-PE/Hoechst ratio for compound 1, 4, and 7 at 100 μM was 1.53 ± 0.24, 1.39 ± 0.1, and 1.71 ± 0.12, respectively, while for AMIO a ratio of 2.10 ± 0.09 at 12 μM was observed. Concerning compound 2, although cell viability studies suggested an impairment of lysosomal function, this effect seems to be not associated to lipid accumulation induction.

Effect of compounds 1-syn, and 2–9 on HepG2 cell viability. HepG2 were treated for 24 h with the selected compounds at the indicated concentrations and viability was assessed by NR uptake assay. Amiodarone (AMIO) at 12 µM concentration was used as positive controls. Results are expressed as percentage viability of cells exposed to selected compounds with respect to vehicle control (Cveh). Statistical analysis was performed using Student’s t-test. Values are the mean ± SD of three independent experiments done in triplicate (**p < 0.01; ***p < 0.001)

Measurement of NBD-PE uptake in HepG2 treated with compounds 2–9. Amiodarone (AMIO) and compound 1-syn at 12 μM and 100 μM concentrations, respectively, were used as positive controls. NBD-PC fluorescence (a) and Hoechst 33342 fluorescence (b) were measured and results are expressed as percentage of vehicle treated cells (Cveh). Normalized values were calculated as ratio between the NBD-PE and the Hoechst 33342 values (c). Statistical analysis was performed using Student's t-test. Results represent the mean ± S.D. of three independent experiments done in triplicate (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 drug-treated HepG2 cells vs. vehicle controls)
A comparison between biological findings for NBD-PE uptake induction and in silico prediction in Table 1 suggests that for this series of compounds, false positive assignments rather than false negative ones are likely to occur, with predictions based on the Ploemen’s rules being the less affected ones. However, Ploemen’s rules were still not found to be a general solution for PLD prediction, performing worse than other methods when applied to a broader chemical space (van de Water et al. 2011; Goracci et al. 2015). Therefore, although the three new PLD inducers 1, 4, 7 were successfully predicted by all the tested in silico methods despite the weakness of their effect, the high number of false positives using in silico tools could lead to discard promising compounds at an early discovery phase.
Conclusions
In summary, we believe that this case study leads to three major considerations. First of all, even though the assumption that CADs are PLD inducers is commonly accepted, CAD-like compounds can be PLD- or weak PLD+ compounds. Therefore, considering the relevant role of CADs in many therapeutic areas, the application of in silico tools tout court without considering uncertainty levels can lead to the discarding of promising compounds. Secondary, the selection of the in vitro assay is determinant when weak CAD-like PLD inducers are tested. Thus, homogeneous data from the same assay should be used for model generation. Finally, when weak PLD inducers or CAD-like non-inducers are included in the dataset used for in silico model generation, they may introduce a serious risk of noise. Indeed, due to the lack of quantitative data on PLD induction, PLD datasets available in the literature are based only on two classes and weak inducers could be cause of discordant PLD effect, as their definition as PLD+ or PLD- might be method dependent. Thus, in our opinion, avoiding the addition of so weak PLD inducers in PLD datasets might be a valuable strategy to work with homogeneous and reliable data, and the limit of prediction for borderline compounds should be taken into account. In this context, the use of the PLS-DA approach could be a valuable strategy to visually inspect borderline compounds.
References
Artese A, Cross S, Costa G, Distinto S, Parrotta L, Alcaro S, Ortuso F, Cruciani G (2013) Molecular interaction fields in drug discovery: recent advances and future perspectives. Wires Comput Mol Sci 3:594–613
Baroni M, Cruciani G, Sciabola S, Perruccio F, Mason JS (2007) A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for ligands and Proteins (FLAP): theory and application. J Chem Inf Model 47:279–294
Bhandari N, Figueroa DJ, Lawrence JW, Gerhold DL (2008) Phospholipidosis assay in HepG2 cells and rat or rhesus hepatocytes using phospholipid probe NBD-PE. Assay Drug Dev Technol 6:407–419
Blomme EA, Will Y (2016) Toxicology strategies for drug discovery: present and future. Chem Res Toxicol 29:473–504
Chapy H, Goracci L, Vayer P, Parmentier Y, Carrupt PA, Decleves X, Scherrmann JM, Cisternino S, Cruciani G (2015) Pharmacophore-based discovery of inhibitors of a novel drug/proton antiporter in human brain endothelial hCMEC/D3 cell line. Br J Pharmacol 172:4888–4904
Chatman LA, Morton D, Johnson TO, Anway SD (2009) A strategy for risk management of drug-induced phospholipidosis. Toxicol Pathol 37:997–1005
Choi SS, Kim JS, Valerio LG, Sadrieh N (2013) In silico modeling to predict drug-induced phospholipidosis. Toxicol Appl Pharmacol 269:195–204
Cross S, Baroni M, Goracci L, Cruciani G (2012a) GRID-based three-dimensional pharmacophores I: FLAPpharm, a novel approach for pharmacophore elucidation. J Chem Inf Model 52:2587–2598
Cross S, Ortuso F, Baroni M, Costa G, Distinto S, Moraca F, Alcaro S, Cruciani G (2012b) GRID-based three-dimensional pharmacophores II: PharmBench, a benchmark data set for evaluating pharmacophore elucidation methods. J Chem Inf Model 52:2599–2608
Cruciani G, Crivori P, Carrupt PA, Testa B (2000) Molecular fields in quantitative structure–permeation relationships: the VolSurf approach. J Mol Struct 503:17–30
Cruciani G, Milletti F, Storchi L, Sforna G, Goracci L (2009) In silico pKa prediction and ADME profiling. Chem Biodivers 6:1812–1821
DiMasi JA, Feldman L, Seckler A, Wilson A (2010) Pharmaceutical innovation in the 21st century: new drug approvals in the first decade, 2000–2009. Clin Pharmacol 87:272–277
Fischer H, Atzpodien EA, Csato M, Doessegger L, Lenz B, Schmitt G, Singer T (2012) In silico assay for assessing phospholipidosis potential of small druglike molecules: training, validation, and refinement using several data sets. J Med Chem 55:126–139
Goracci L, Ceccarelli M, Bonelli D, Cruciani (2013) Modeling phospholipidosis induction: reliability and warnings. J Chem Inf Model 53:1436–1446
Goracci L, Buratta S, Urbanelli L, Ferrara G, Di Guida R, Emiliani C, Cross S (2015) Evaluating the risk of phospholipidosis using a new multidisciplinary pipeline approach. Eur J Med Chem 92:49–63
Halliwell WH (1997) Cationic amphiphilic drug-induced phospholipidosis. Toxicol Pathol 25:53–60
Hanumegowda UM, Wenke G, Regueiro-Ren A, Yordanova, Corradi JP, Adams SP (2010) Phospholipidosis as a function of basicity, lipophilicity, and volume of distribution of compounds. Chem Res Toxicol 23:749–755
Ikeda K, Hirayama M, Hirota Y, Asa E, Seki J, Tanaka Y (2008) Drug-induced phospholipidosis is caused by blockade of mannose 6-phosphate receptor-mediated targeting of lysosomal enzymes. Biochem Biophys Res Commun 377:268–274
Ivanciuc O (2008) Weka machine learning for predicting the phospholipidosis inducing potential. Curr Top Med Chem 8:1691–1709
Kasahara T, Tomita K, Murano H, Harada, Tsubakimoto K, Ogihara T, Ohnishi S, Kakinuma C (2006) Establishment of an in vitro high-throughput screening assay for detecting phospholipidosis-inducing potential. Toxicol Sci 90:133–141
Kazmi F, Hensley T, Pope C, Funk RS, Loewen GJ, Buckley DB, Parkinson A (2013) Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells). Drug Metab Dispos 41:897–905
Kodavanti UP, Mehendale HM (1990) Cationic amphiphilic drugs and phospholipid storage disorder. Pharmacol Rev 42:327–354
Kruhlak NL, Choi SS, Contrera JF, Weaver JL, Willard JM, Hastings KL, Sancilio LF (2008) Development of a phospholipidosis database and predictive quantitative structure-activity relationship (QSAR) models. Toxicol Mech Methods 18:217–227
Lepri S, Nannetti G, Muratore G, Cruciani G, Ruzziconi R, Mercorelli B, Palu G, Loregian A, Goracci L (2014) Optimization of small-molecule inhibitors of influenza virus polymerase: from thiophene-3-carboxamide to polyamido scaffolds. J Med Chem 57:4337–4350
Lowe R, Mussa HY, Nigsch F, Glen RC, Mitchell JB (2012) Predicting the mechanism of phospholipidosis. J Cheminform 4:2
Lullmann H, Lullmann-Rauch R, Wassermann O (1975) Drug-induced phospholipidoses. II. Tissue distribution of the amphiphilic drug chlorphentermine. Crit Rev Toxicol 4:185–218
Mannhold R, Berellini G, Carosati E, Benedetti P (2006) AOME properties and their prediction major causes for failure in drug development are unsuitable pharmacokinetic properties of drug candidates including absorption, distribution, metabolism, and excretion (ADME), which were traditionally measured at rather late stages of drug development. Nowadays, the testing of ADME properties is done much earlieri. In: Cruciani G (ed) Molecular interaction fields: applications in drug discovery and ADME prediction. Wiley-VCH, New York, 2006, vol. 27 173–196
Milletti F, Storchi L, Goracci L, Bendels S, Wagner B, Kansy M, Cruciani G (2010) Extending pKa prediction accuracy: high-throughput pKa measurements to understand pKa modulation of new chemical series. Eur J Med Chem 45:4270–4279
Mingeot-Leclercq MP, Tulkens PM (1999) Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother 43:1003–1012
Molecular Discovery Ltd. http://www.moldiscovery.com/. Accessed 11 Feb 2016
Muehlbacher M, Tripal P, Roas F, Kornhuber J (2012) Identification of drugs inducing phospholipidosis by novel in vitro data. ChemMedChem 7:1925–1934
Munic V, Banjanac M, Kostrun S, Nujic K, Bosnar M, Marjanovic N, Ralic J, Matijasic M, Hlevnjak M, Erakovic Haber V (2011) Intensity of macrolide anti-inflammatory activity in J774A.1 cells positively correlates with cellular accumulation and phospholipidosis. Pharmacol Res 64:298–307
Nadanaciva S, Lu S, Gebhard DF, Jessen BA, Pennie WD, Will Y (2011) A high content screening assay for identifying lysosomotropic compounds. Toxicol In Vitro 25:715–723
Nonoyama T, Fukuda R (2008) Drug-induced phospholipidosis: Pathological aspects and its prediction. J Toxicol Pathol 21:9–24
Orogo AM, Choi SS, Minnier BL, Kruhlak NL (2012) Construction and consensus performance of (Q)SAR models for predicting phospholipidosis using a dataset of 743 compounds. Mol Inf 31:725–739
Pelletier DJ, Gehlhaar D, Tilloy-Ellul, Johnson TO, Greene (2007) Evaluation of a published in silico model and construction of a novel Bayesian model for predicting phospholipidosis inducing potential. J Chem Inf Model 47:1196–1205
Ploemen JP, Kelder J, Hafmans T, van de Sandt H, van Burgsteden JA, Salemink PJ, van Esch E (2004) Use of physicochemical calculation of pKa and CLogP to predict phospholipidosis-inducing potential: a case study with structurally related piperazines. Exp Toxicol Pathol 55:347–355
Quaglino D, Ha HR, Duner E, Bruttomesso D, Bigler L, Follath F, Realdi G, Pettenazzo A, Baritussio A (2004) Effects of metabolites and analogs of amiodarone on alveolar macrophages: structure-activity relationship. Am J Physiol Lung Cell Mol Physiol 287:L438–L447
Reasor MJ, Hastings KL, Ulrich RG (2006) Drug-induced phospholipidosis: issues and future directions. Expert Opin Drug Saf 5:567–583
Repetto G, del Peso A, Zurita JL (2008) Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat Protoc 3:1125–1131
Sirci F, Goracci L, Rodriguez D, an Muijlwijk-Koezen J, Gutierrez-de-Teran H, Mannhold R (2012) Ligand-, structure- and pharmacophore-based molecular fingerprints: a case study on adenosine A(1), A (2A), A (2B), and A (3) receptor antagonists. J Comput Aided Mol Des 26:1247–1266
Slavov SH, Wilkes JG, Buzatu DA, Kruhlak NL, Willard JM, Hanig JP, Beger RD (2014) Computational identification of a phospholipidosis toxicophore using (13)C and (15)N NMR-distance based fingerprints. Bioorg Med Chem 22:6706–6714
SPECS. http://www.specs.net/. Accessed 18 Dec 2015
Sun H, Shahane S, Xia M, Austin CP, Huang R (2012) Structure based model for the prediction of phospholipidosis induction potential of small molecules. J Chem Inf Model 52:1798–1805
Tomizawa K, Sugano K, Yamada H, Horii I (2006) Physicochemical and cell-based approach for early screening of phospholipidosis-inducing potential. J Toxicol Sci 31:315–324
Tondi D, Cross S, Venturelli A, Costi M, Cruciani G, Spyrakis F (2016) Decoding the structural basis for carbapenem hydrolysis by class A β-lactamases: fishing for a pharmacophore. Curr Drug Targets 17:983–1005
van de Water FM, Havinga, Ravesloot WT, Horbach GJ, Schoonen WG (2011) High content screening analysis of phospholipidosis: validation of a 96-well assay with CHO-K1 and HepG2 cells for the prediction of in vivo based phospholipidosis. Toxicol In Vitro 25:1870–1882
Waring MJ, Arrowsmith J, Leach AR, Leeson PD, Mandrell S, Owen RM, Pairaudeau G, Pennie WD, Pickett SD, Wang J, Wallace O, Weir A (2015) An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov 14:475–486
Acknowledgements
The authors would like to thank Prof. Francesco Galli for generously sharing his laboratory facilities for biological analysis, and Dr. Simon Cross for helpful comments and for English revision. Financial support from the Italian MIUR within the “FIRB-Futuro in Ricerca 2010” Program—Project RBFR10×500 is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Susan Lepri and Aurora Valeri contributed equally to this work.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Lepri, S., Valeri, A., Buratta, S. et al. Synthesis and phospholipidosis effect of a series of cationic amphiphilic compounds: a case study to evaluate in silico and in vitro assays. Med Chem Res 27, 679–692 (2018). https://doi.org/10.1007/s00044-017-2093-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-017-2093-5