
Abstract
A new class of 2-benzoxazolinone derivatives was designed and synthesized for its anti-human immunodeficiency virus-1 activity. The benzoxazolinone scaffold could be replaced with catechol moiety in the potent but toxic integrase strand transfer inhibitors. The biological evaluation of the synthesized compounds revealed that all compounds were active against human immunodeficiency virus-1 at 100 μM. It is also found that most of the compounds presented no significant cytotoxicity at concentration of 100 μM. The most potent compound with thiadiazole ring as the linker inhibited the human immunodeficiency virus-1 with 84% rate. Docking of this structure in the active site of prototype foamy virus integrase indicated that the chelation of two Mg2+ cations might be the probable mechanism of the anti-human immunodeficiency virus-1 activity. Our results indicated that the synthesized compounds can provide a very good basis for the development of new anti-human immunodeficiency virus-1 agents.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Human immunodeficiency virus (HIV-1) life cycle has been interrupted through inhibition of the three essential viral enzymes encoded in the pol gene: reverse transcriptase (RT), protease (PR) and integrase (IN) (Chi et al. 2007; Tanis et al. 2010). The standard AIDS chemotherapy referred to as highly active antiretroviral therapy (HAART) is the combination of RT and PR inhibitors (Dayam et al. 2007). HAART therapy has suffered from important limitations including drug toxicity, patient disinclination, and drug resistance. Regarding to the problems, there is a demand for alternative drug targets (Pawar et al. 2014; Wang and Vince 2008). IN as the third enzyme of the pol gene is a requisite for stable infection and has no host cell equivalent. Therefore, IN has emerged as a novel target for designing safe and potent inhibitors (Billamboz et al. 2011). Integration process consists of two steps termed 3’-processing and strand transfer. The first step initiates after assembly of the viral DNA on IN in cytoplasm (Boros et al. 2006; Quashie et al. 2012). In this reaction, (3’-processing) IN catalyzes the cleavage of a dinucleotide from each 3’ end of the DNA. After translocation of the preintegration complex to the nucleus, strand transfer occurs (Dayam et al. 2007). Here, the viral DNA integrates into the host cell DNA (Al-Mawsawi et al. 2007; Boros et al. 2006).
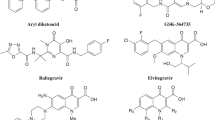
The active site of the IN consists of a DDE motif (D64D116E152) that coordinates two Mg2+ ions (Hassounah et al. 2016). Chelation of these metal cofactors is a promising approach to selective inhibition of strand transfer. As could be depicted in FDA approved IN inhibitors (raltegravir, elvitegravir, and dolutegravir in Fig. 1), the ketoenolcarboxyl group in place of chelating motif and coplanar hydrophobic aryl group are the common pharmacophores for an integrase strand transfer inhibitor (INSTI) (Li et al. 2014). The development of INSTIs has led to different chemical classes including beta-diketoacids, naphthyridinecarboxamides, pyrimidinones, pyridoxine hydroxamic acids, and quinolone carboxylic acids (Quashie et al. 2012; Stranix et al. 2016; Tanis et al. 2010).

FDA approved INSTIs
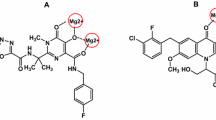
2-Benzoxazolinone is a druggable scaffold with versatile biological activities such as anti-HCMV (human cytomegalovirus) and anti-VZV (varicella zoster virus) effects (Bach et al. 2015; De Clercq and Poupaert 1999; Poupaert et al. 2005). This scaffold serves as a bioisoster for phenol or catechol and amides in many designs (Gerova et al. 2016). This bioisosteric replacement provides a tool to overcome poor pharmacokinetic and unfavorable toxic properties of catechol containing lead compounds (1 in Fig. 2) (Ingale and Bhatia 2011; Poupaert et al. 2005). Apparently, 2-benzoxazolinone with modified local steric and electronic properties, could result in more selectivity and affinity (Poupaert et al. 2005). In this research, we selected the 2-benzoxazolinone as a new chemical scaffold for INST inhibitory potential and joined it with a hydrazide linker that is a part of some potent INSTIs such as compound 2 (Fig. 2). We incorporated the aryl group in these structures to occupy the hydrophobic module of the IN active site.

Lead compounds (1 and 2) and our designed structures
To compare the effect of ring closure in the linker on anti-HIV-1 activity, some derivatives with thiadiazole ring as the linker were also considered (designed molecules in Fig. 2). However, the isosteric relationships between 2-benzoxazolinones and purines (De Clercq and Poupaert 1999), the 3’ end dinucleotide of viral DNA presenting in the IN active site could be stabilized through π–π interactions.
Results and discussion
Chemistry
The title compounds were synthesized according to the pathway outlined in Scheme 1. Benzo[d]oxazol-2(3H)-one (1) was prepared by classical procedure starting from 2-aminophenol and urea. Treatment of (1) with ethyl chloroacetate in the presence of potassium carbonate and acetone as the solvent gave the N-alkylated product (2). This ester intermediate reacted with hydrazine hydrate to afford the acid hydrazide (3). The acylated compounds (4a–f) were obtained by the reaction of hydrazide (3) with substituted benzoyl chlorides in dry dimethyl formamide (DMF) and catalytic amount of anhydrous sodium carbonate. For the synthesis of compounds (5a–c), hydrazide (3) and some phenyl isothiocyanate derivatives were heated under reflux in absolute ethanol. Dehydrocyclization of compounds (5a–c) was performed in conc. sulfuric acid to afford the thiadiazole containing derivatives (6a–c).

Reagents and conditions: a Conc. HCl, reflux, 140–170 °C, 4 h b acetone, K2CO3, reflux, 4 h c abs. ethanol, reflux, 2 h d dry dimethyl formamide (DMF), anhydrous Na2CO3, r.t, 2 h e abs. ethanol, reflux, 4 h f Conc. sulfuric acid, r.t, 1 h
Anti-HIV-1 activity
The activity of all compounds against single cycle replicable HIV NL4-3 was measured by determining their ability to inhibit p24 expression in Hela cells cultures. The well-known nucleoside RT inhibitor (AZT) was assayed in the same cells as a reference. All synthesized compounds were also tested for their cytotoxicity in MTT-based cell viability assay. Results have been summarized in Table 1. All compounds were active at 100 μM against HIV-1 with inhibition rate ranging from 5–84%. The cell viability has been in the range of 37–94%. Our results indicated that the presence of 2-amino-1,3,4-thiadiazole ring as a spacer, significantly improves the antiviral activity in comparison to benzohydrazide or carbothioamide linker. Accordingly, compound 6a having the aminophenyl at C-2 position of 1,3,4-thiadiazole ring showed the highest inhibitory effect with 84% inhibition rate among the synthesized compounds. It seems that the heteroatoms of thiadiazole ring would have been involved in chelation of Mg2+ ions presenting in the IN active site. However, substitution of phenyl ring with either electron-withdrawing or electron-donating group in compounds possessing thiadiazole (6b and 6c) or benzohydrazide (4b–f) linker decreased the potency, whereas cell viability has not meaningfully affected by this replacement. Our results showed that compounds with carbothioamide linker (5a–c) exhibited higher anti HIV-1 activity compared with their analogues having bezohydrazide linker (4a–f). According to these results compound 5b having electron-withdrawing group such as fluorine substituent at para position of N-phenyl ring exhibited the highest anti-HIV activity (81%) among the series of compounds containing acyclic carbothioamide or benzohydrazide linker. This finding conforms to the common 4-flourophenyl pharmacophore of INSTIs. This motif would have been involved in charge transfer interaction with positively charged pocket of the IN active site. It can be concluded that the introduction of 1,3,4-thiadiazol ring to the 2-benzoxazolinone scaffold results in the greater anti-HIV-1 activity and lesser cytotoxicity. In addition, the activity has benefited from the simple hydrophobic phenyl ring Table 2).
Molecular modeling (docking)
In order to explore the binding pose of the synthesized structures in the IN active site, docking studies were performed using AutoDock Tools. The studies were based on the X-ray crystallographic structure of prototype foamy virus (PFV) IN (PDB: 3OYA) in complex with DNA, 2 Mg2+, and raltegravir at 2.65 Å resolution. According to the literature, secondary structures of HIV-IN catalytic core domain and PFV IN are very similar with relative mean square deviations (RMSD) 1.04 Å (Billamboz et al. 2011; Hajimahdi et al. 2013; Yu et al. 2013). According to these data, we undertook docking studies based on the crystallographic structure of PFV IN. This procedure was validated by docking of Raltegravir into the IN active site. Then, the best obtained form was superimposed on the co-crystallized raltegravire using the Pymol Molecular Graphics System, version 0.99rc6 (Calculated RMSD = 1.92) (Billamboz et al. 2011). Docking of the most potent structure into the IN active site (Fig. 3) exhibited two important interactions: (1) chelation of two Mg2+ ions by the thiadiazole ring heteroatoms and the nitrogen of aminophenyl substituent (2) the 2-benzoxazolinone ring and the displaced 3’-end-DA17 interacted via π–π interactions. Furthermore, the superimposition of the most active compound on the crystallographic ligand (raltegravir) revealed the similar pose of these two structures in the IN active site (Fig. 4).

Compound 6a in PFV IN active site

Superimposition of compound 6a on Raltegravir in PFV IN active site
Conclusion
Herein we investigated the anti-HIV-1 activity of a series of 2-benzoxazolinones derivatives as a potential scaffold for INSTI activity. Also we introduced substituted phenyl ring in these structures to insert into the hydrophobic pocket of IN active site. According to the biological evaluations, all compounds were active anti-HIV-1 agents at 100 μM. The 2-aminophenyl-1, 3, 4-thiadozole ring of the most potent compound (6a, 84% at 100 μM) interacts with 2 Mg2+ cations in the IN active site. In accordance with the docking study results, the benzoxazolinone scaffold involves in π–π stacking interactions with viral DNA in the IN active site. Thus, the anti-HIV-1 activity of these compounds may be interpreted as INST inhibitory mechanism.
Experimental
Materials
All reagents purchased from the Aldrich (USA) or Merck (Germany) Chemical Company and were used without further purifications.
General
Melting points (mp) were determined using a Thomas Hoover capillary apparatus (Philadelphia, USA). Infrared spectra were acquired on a Perkin-Elmer 1420 ratio recording spectrometer. A Bruker FT-250 MHz instrument (Brucker Biosciences, USA) was used to acquire 1HNMR and 13CNMR spectra; chloroform-D and DMSO-d6 used as solvents. Coupling constant (J) values are estimated in hertz (Hz) and spin multiples are given as s (singlet), d (double), t (triplet), q (quartet), m (multiplet), and br (broad). The mass spectral measurements were performed on an 6410 Agilent LCMS triple quadrupole mass spectrometer (LCMS) with an electrospray ionization (ESI) interface.
(Koeksal et al. 2005)
(Salgın-Gökşen et al. 2007)
(Salgın-Gökşen et al. 2007)
General procedure for preparation of substituted N’-(2-(2-oxobenzo[d]oxazol-3(2H)-yl)acetyl)benzohydrazide (4a–f)
A mixture of compound 3 (1 mmol), anhydrous sodium carbonate (0.03 g) and dry DMF (3 mL) was prepared in an ice bath. To the reaction mixture, substituted benzoyl chloride (1 mmol) was added under nitrogen atmosphere and stirred at room temperature for 4 h. Then, it was poured onto a beaker containing 30 g ice-cold water. After stirring at room temperature for 1 h, the precipitate thus formed was filtered, washed with water, and crystallized from ethanol.
N’-(2-(2-oxobenzo[d]oxazol-3(2H)-yl) acetyl) benzohydrazide (4a)
Yield 65%; White crystalline powder; mp 223–225 °C; IR (KBr disk): υ (cm−1) 1663–1755 (C=O), 3286 (N–H); 1HNMR (DMSO-d 6 ): δ (ppm) 4.60 (2H, s, N–CH 2 –CO), 7.10–7.13 (1H, t, H5), 7.18–7.23 (2H, m, H4 and H6), 7.33 (1H, d, J = 8.0 Hz, H7), 7.45 (2H, t, J = 7.5 Hz, H3’ and H5’ Ar), 7.54 (1H, t, J = 7.5 Hz, H4’ Ar), 7.82 (2H, d, J = 7.5 Hz, H2’ and H6’ Ar), 10.45 (2H, d, J = 18.0 Hz, –NH–); 13CNMR (DMSO-d 6 ): δ (ppm) 43.4 (CH2, N–CH 2 –CO), 109.6 (CH, C7), 110.1 (CH, C4), 122.9 (CH, C6), 124.3 (CH, C5), 128.05 (CH, C2 and C6 Ar), 129.07 (CH, C3 and C5 Ar), 131.67 (C, C–N), 132.36 (CH, C4 Ar), 132.63 (C, C–CO–NH), 142.4 (C, C–O), 154.4 (C, N–CO–O), 165.9 (2C, CO–NH–NH–CO); LC-MS (ESI) m/z: 334.1 (M+23, 100). Anal. Calcd. for C16H13N3O4: C, 61.73; H, 4.21; N, 13.50. Found: C, 61.96; H, 4.51; N, 13.35.
4-Fluoro-N’-(2-(2-oxobenzo[d]oxazol-3(2H)-yl)acetyl)benzohydrazide (4b)
Yield 50%; White crystalline powder; mp 241–243 °C; IR (KBr disk): υ (cm−1) 1675–1763 (C=O), 3293–3345 (N–H); 1HNMR (DMSO-d 6 ): δ (ppm) 4.63 (2H, s, N–CH 2 –CO), 7.11–7.20 (1H, m, H5), 7.23–7.25 (2H, m, H4 and H6), 7.29–7.43 (3H, m, H3’ and H5’ Ar and H7), 7.91–7.96 (2H, dd, J = 7.5 Hz, H2’ and H6’ Ar), 10.51 (2H, d, J = 15.0 Hz, –NH–); 13CNMR (DMSO-d 6 ): δ (ppm) 43.0 (CH2, N–CH 2 –CO), 109.2 (CH, C7), 109.7 (CH, C4), 115.4, 115.7 (CH, C3 and C5 Ar, J = 18), 122.5 (CH, C6), 123.9 (CH, C5), 128.6 (C, C–CO–NH), 130.1–130.3 (CH, C2 and C6 Ar), 131.3 (C, C–N), 142.0 (C, C–O), 154.0 (C, N–CO–O), 162.3, 166.2 (C, C–F, J = 240), 164.4, 165.5 (2C, CO–NH–NH–CO); LC-MS (ESI) m/z: 352.1 (M+23, 100). Anal. Calcd. for C16H12FN3O4: C, 58.36; H, 3.67; N, 12.76. Found: C, 58.66; H, 3.41; N, 12.96.
4-Methyl-N’-(2-(2-oxobenzo[d]oxazol-3(2H)-yl)acetyl)benzohydrazide (4c)
Yield 45%; White crystalline powder; mp 229–230 °C; IR (KBr disk): υ (cm−1) 1676–1769 (C=O), 3307–3607 (N–H); 1HNMR (DMSO-d 6 ): δ (ppm) 2.38 (3H, s, –CH3), 4.62 (2H, s, N–CH 2 –CO), 7.15–7.20 (1H, m, H5), 7.23–7.25 (2H, m, H4 and H6), 7.28 (2H, d, J = 7.5 Hz, H3’ and H5’ Ar), 7.36 (1H, d, J = 7.5 Hz, H7), 7.76 (2H, d, J = 7.5 Hz, H2’ and H6’ Ar), 10.42 (2H, s, –NH–); 13CNMR (DMSO-d 6 ): δ (ppm) 21.0 (CH3), 43.0 (CH2, N–CH 2 –CO), 109.2 (CH, C7), 109.7 (CH, C4), 122.5 (CH, C6), 123.9 (CH, C5), 127.5 (CH, C2 and C6 Ar), 129.0 (CH, C3 and C5 Ar), 129.4 (C, C–CO–NH), 131.3 (C, C–N), 142.0 (C, C–CH3), 142.5 (C, C–O), 154.0 (C, N–CO–O), 165.3, 165.5 (2C, CO–NH–NH–CO); LC-MS (ESI) m/z: 348.1 (M+23, 100). Anal. Calcd. for C17H15N3O4: C, 62.76; H, 4.65; N, 12.92. Found: C, 62.48; H, 4.31; N, 13.05.
4-Chloro-N’-(2-(2-oxobenzo[d]oxazol-3(2H)-yl) acetyl)benzohydrazide (4d)
Yield 40%; White crystalline powder; mp 249–251 °C; IR (KBr disk): υ (cm−1) 1980–1774 (C=O), 3286–3354 (N–H); 1HNMR (DMSO-d 6 ): δ (ppm) 4.63 (2H, s, N–CH 2 –CO), 7.11–7.19 (1H, m, H5), 7.23–7.25 (2H, m, H4 and H6), 7.36 (1H, d, J = 7.5 Hz, H7), 7.56 (2H, d, J = 7.5 Hz, H3’ and H5’ Ar), 7.87 (2H, d, J = 7.5 Hz, H2’ and H6’ Ar), 10.56 (2H, d, NH); 13CNMR (DMSO-d 6 ): δ (ppm) 43.4 (CH2, N–CH 2 –CO), 110.0 (2CH, C7), 110.1 (CH, C4), 122.9 (CH, C6), 124.3 (CH, C5), 129.1 (CH, C2 and C6 Ar), 129.8 (CH, C3 and C5 Ar), 131.3 (C, C–CO–NH), 131.6 (C, C–N), 137.2 (C, C–Cl), 142.4 (C, C–O), 154.4 (C, N–CO–O), 164.9,165.9 (2C, CO–NH–NH–CO); LC-MS (ESI) m/z: 368.1 (M+23, 100). Anal. Calcd. for C16H12ClN3O4: C, 55.58; H, 3.50; N, 10.25. Found: C, 55.81; H, 3.21; N, 10.44.
3-Methyl-N’-(2-(2-oxobenzo[d]oxazol-3(2H)-yl) acetyl)benzohydrazide (4e)
Yield 40%; White crystalline powder; mp 208–210 °C; IR (KBr disk): υ (cm−1) 1678–1768 (C=O), 3276–3605 (N–H); 1HNMR (DMSO-d 6 ): δ (ppm) 2.35 (3H, s, –CH3), 4.63 (2H, s, N–CH 2 –CO), 7.15–7.20 (1H, m, H5), 7.24–2.25 (2H, m, H4 and H6), 7.36 (1H, d, J = 7.5 Hz, H7), 7.42–7.44 (2H, m, H4’ and H5’ Ar), 7.65–7.68 (2H, m, H2’ and H6’ Ar), 10.47 (2H, s, –NH–); 13CNMR (DMSO-d 6 ): δ (ppm) 21.3 (CH3), 43.4 (CH2, N–CH 2 –CO), 110.0 (2CH, C7), 110.1 (CH, C4), 122.9 (CH, C6), 124.3 (CH, C5), 124.9 (CH, C6 Ar), 128.4 (CH, C2 Ar), 128.8 (CH, C5 Ar), 131.7 (C, C–N), 132.6 (C, C–CO–NH), 132.9 (CH, C4 Ar), 138.0 (C, C–CH3), 142.4 (C, C–O), 154.4 (C, N–CO–O), 165.8, 166.0 (2C, CO–NH–NH–CO); LC-MS (ESI) m/z: 348.1 (M+23, 100). Anal. Calcd. for C17H15N3O4: C, 62.76; H, 4.65; N, 12.92. Found: C, 63.01; H, 4.81; N, 12.77.
3-Fluoro-N’-(2-(2-oxobenzo[d]oxazol-3(2H)-yl) acetyl)benzohydrazide (4f)
Yield 50%; White crystalline powder; mp 219–220 °C; IR (KBr disk): υ (cm−1) 1696–1771 (C=O), 3205–3294 (N–H); 1HNMR (DMSO-d 6 ): δ (ppm) 4.66 (2H, s, N–CH 2 –CO), 7.15–7.20 (1H, m, H5), 7.23–7.25 (2H, m, H4 and H6), 7.36 (1H, d, J = 7.5 Hz, H7), 7.45 (1H, d, J = 7.5 Hz, H4’ Ar), 7.57 (1H, t, J = 7.5 Hz, H5’ Ar), 7.64 (1H, d, J = 10.0 Hz, H2’ Ar), 7.72 (1H, d, J = 7.5 Hz, H6’ Ar), 10.57 (2H, d, J = 22.5 Hz, –NH–); 13CNMR (DMSO-d 6 ): δ (ppm) 47.7 (CH2, N–CH 2 –CO), 114.3 (2CH, C7), 114.4 (CH, C4), 118.8, 119.2 (CH, C2 Ar, J = 24), 123.5, 123.9 (CH, C4 Ar, J = 24), 127.2 (CH, C6), 128.4 (CH, C5), 128.7 (CH, C6 Ar), 135.5, 135.6 (CH, C5 Ar), 136.0 (C, C–N), 139.1, 139.2 (C, C–CO–NH), 146.8 (C, C–O), 158.7 (C, N–CO–O), 164.7, 168.6 (C, C–F, J = 240), 168.9, 170 (2C, CO–NH–NH–CO); LC-MS (ESI) m/z: 352.1 (M+23, 100). Anal. Calcd. for C16H12FN3O4: C, 58.36; H, 3.67; N, 12.76. Found: C, 58.51; H, 3.98; N, 12.64.
General procedure for the preparation of 2-(2-(2-oxobenzo[d]oxazol-3(2H)-yl) acetyl)-N- substituted phenylhydrazinecarbothioamide (5a–c)
A mixture of compound 3 (3 mmol), substituted phenyl isothiocyanate (3.1 mmol) and 10 mL absolute ethanol was refluxed for 4 h. Progress of the reaction was monitored by thin layer chromatography (TLC). After completion of the reaction, it was cooled to room temperature. The precipitated product was filtered and crystallized from ethanol.
2-(2-(2-Oxobenzo[d]oxazol-3(2H)-yl)acetyl)-N-phenylhydrazinecarbothioamide (5a)
Yield 58%; White crystalline powder; mp 190.5–192.5 °C; IR (KBr disk): υ (cm−1) 1204 (C=S), 1666–1746 (C=O), 2959–3251 (N–H); 1HNMR (DMSO-d 6 ): δ (ppm) 4.64 (2H, s, N–CH 2 –CO), 7.16–7.21 (3H, m, H5 and H4 and H6), 7.32–7.44 (5H, m, Ar H), 7.37 (1H, d, J = 7.5 Hz, H7), 9.72 (2H, s, –NH–CS), 10.49 (1H, s, –NH–CO); 13CNMR (DMSO-d 6 ): δ (ppm) 43.1 (CH2, N–CH 2 –CO), 109.7 (2CH, C4 and C7), 122.4, 122.5 (2CH, C5 and C6), 123.9, 124.3 (2CH, C2 and C6 Ar), 125.4, 125.9 (CH, C4 Ar), 128.0, 128.2 (2CH, C3 and C5 Ar), 131.2 (C, C–N), 139.0 (C, C–NH–CS), 142.0 (C, C–O), 154.1 (C, N–CO–O), 165.9 (C, CO–NH–NH), 181.0 (C, CS); LC-MS (ESI) m/z: 343.1 (M+1, 100). Anal. Calcd. for C16H14N4O3S: C, 56.13; H, 4.12; N, 16.36. Found: C, 56.44; H, 4.34; N, 16.10.
N-(4-fluorophenyl)-2-(2-(2-oxobenzo[d]oxazol-3(2H)-yl)acetyl)hydrazinecarbothioamide (5b)
Yield 65%; White crystalline powder; mp 200–202 °C; IR (KBr disk): υ (cm−1) 1218 (C=S), 1675–1755(C=O), 2882–3271 (N–H); 1HNMR (DMSO-d 6 ): δ (ppm) 4.64 (2H, s, N–CH 2 –CO), 7.12–7.20 (5H, m, H3’ & H5’ Ar, H4 and H5 and H6), 7.37 (1H, d, J = 7.5 Hz, H7), 7.47–7.52 (2H, m, H2’ and H6’ Ar), 9.76 (2H, s, –NH–CS), 10.48 (1H, s, –NH–CO); 13CNMR (DMSO-d 6 ): δ (ppm) 43.5 (CH2, N–CH 2 –CO), 110.1 (2CH, C4 and C7), 115.0, 115.2, 115.3, 115.5 (2CH, C3 and C5 Ar), 122.9 (CH, C6), 124.3 (CH, C5), 127.9 (CH, C2 Ar), 131.6 (C, C–N), 135.6,135.7 (CH, C6 Ar), 136.0 (C, C–NH–CS), 142.3 (C, C–O), 154.5 (C, N–CO–O), 158, 162 (C, C–F, J = 240), 166.3 (C, CO–NH–NH), 182.3 (C, CS); LC-MS (ESI) m/z: 361.1 (M+1, 100). Anal. Calcd. for C16H13FN4O3S: C, 53.33; H, 3.64; N, 15.55. Found: C, 53.54; H, 3.75; N, 15.24.
2-(2-(2-Oxobenzo[d]oxazol-3(2H)-yl)acetyl)-N-(p-tolyl) hydrazinecarbothioamide (5c)
Yield 50%; White crystalline powder; mp 185–187 °C; IR (KBr disk): υ (cm−1) 1203 (C=S), 1703–1787 (C=O), 2940–3284 (N–H); 1HNMR (DMSO-d 6 ): δ (ppm) 2.29 (3H, s, –CH3), 4.64 (2H, s, N–CH 2 –CO), 7.14–7.21 (5H, m, H3’ and H5’ Ar, H4 & H5 and H6), 7.28 (2H, d, J = 7.5 Hz, H2’ and H6’ Ar), 7.36 (2H, d, J = 7.5 Hz, H7), 9.65 (2H, s, –NH–CS), 10.46 (1H, s, –NH–CO); 13CNMR (DMSO-d 6 ): δ (ppm) 21.0 (CH3), 43.5 (CH2, N–CH 2 –CO), 110.1 (2CH, C4 and C7), 122.9 (CH, C6), 124.3 (CH, C5), 126.2 (2CH, C2 and C6 Ar), 129.1 (2CH, C3 and C5 Ar), 131.6 (C, C–N), 135.0 (C, C–NH–CS), 136.7 (CH, C–CH3), 142.3 (C, C–O), 154.5 (C, N–CO–O), 166.3 (C, CO–NH–NH), 181.1 (C, CS); LC-MS (ESI) m/z: 357.1 (M+1, 100). Anal. Calcd. for C16H12FN3O4: C, 58.36; H, 3.67; N, 12.76. Found: C, 58.45; H, 3.88; N, 12.61.
General procedure for the preparation of 3-((5-(phenylamino)-1, 3, 4-thiadiazol-2-yl) methyl)benzo[d]oxazol-2(3H)-one (6a–c)
To a cold conc. sulfuric acid (2 mL), compound 5a–c (1 mmol) was added gradually and stirred at the same temperature until dissolution. This solution was left at room temperature for 2 h and then poured onto ice-cold water (5 mL). The formed precipitate was filtered, washed with water and crystallized from ethanol.
3-((5-(Phenylamino)-1, 3, 4-thiadiazol-2-yl) methyl)benzo[d]oxazol-2(3H)-one (6a)
Yield 25%; Yellow crystalline powder; mp 206 °C (decomposed); IR (KBr disk): υ (cm−1) 1480–1500 (C=N), 1750 (C=O), 3300 (N–H); 1HNMR (DMSO-d 6 ): δ (ppm) 5.37 (2H, s, N–CH 2 –CO), 6.96–7.08 (1H, m, H5), 7.13–7.40 (6H, m, H3’ and H4’ and H5’ Ar, H4 and H6 and H7), 7.55 (2H, d, J = 7.5 Hz, H2’ and H6’ Ar), 10.39 (1H, s, –NH); 13CNMR (DMSO-d 6 ): δ (ppm) 109.7 (CH, C7), 109.9 (CH, C4), 117.5 (2CH, C2 and C6 Ar), 117.9 (CH2, N–CH 2 –CO), 122.1 (CH, C4 Ar), 122.8 (CH, C6), 124.1 (CH, C5), 129.1, 129.3 (2CH, C3 and C5 Ar), 130.4 (C, C–N), 140.4 (C, C–NH–thiadiazole), 142.0 (C, C–O), 153.1 (C, C4 thiadiazole), 153.5 (C, N–CO–O), 165.5 (C, C1 thiadiazole); LC-MS (ESI) m/z: 325.1 (M+1, 100). Anal. Calcd. for C16H12N4O2S: C, 59.25; H, 3.73; N, 17.27. Found: C, 59.44; H, 3.96; N, 17.02.
3-((5-((4-Fluorophenyl) amino)-1, 3, 4-thiadiazol-2-yl) methyl)benzo[d]oxazol-2(3H)-one (6b)
Yield 30%; Yellow crystalline powder; mp 204 °C (decomposed); IR (KBr disk): υ (cm−1) 1480–1503 (C=N), 1755 (C=O), 3284 (N–H); 1HNMR (DMSO-d 6 ): δ (ppm) 5.38 (2H, s, N–CH 2 –CO), 7.17 (3H, m, H3’ and H5’ Ar, H5), 7.23–7.27 (1H, m, H6), 7.33 (1H, d, J = 7.7 Hz, H4), 7.39 (1H, d, J = 8.0 Hz, H7), 7.60 (2H, d, J = 8.0 Hz, H2’ and H6’ Ar), 10.44 (1H, s, –NH); 13CNMR (DMSO-d 6 ): δ (ppm) 110.0 (CH, C7), 110.3 (CH, C4), 115.9, 116.2 (CH, C3 and C5 Ar), 119.6, 119.7 (CH, C2 and C6 Ar), 122.0 (CH2, N–CH 2 –CO), 123.2 (CH, C6), 124.5 (CH, C5), 130.7 (C, C–N), 137.3 (C, C–NH-thiadiazole), 142.4 (C, C–O), 153.5 (C, C4 thiadiazole), 153.8 (C, N–CO–O), 155.9, 159.6 (C, C–F), 166.0 (C, C1 thiadiazole); LC-MS (ESI) m/z: 343.1 (M+1, 100). Anal. Calcd. for C16H11FN4O2S: C, 56.13; H, 3.24; N, 16.37. Found: C, 56.41; H, 3.41; N, 16.54.
3-((5-(p-Tolylamino)-1, 3, 4-thiadiazol-2-yl)methyl)benzo[d]oxazol-2(3H)-one (6c)
Yield 20%; Yellow crystalline powder; mp 228 °C (decomposed); IR (KBr disk): υ (cm−1) 1482–1546 (C=N), 1750 (C=O), 3257–3298 (N–H); 1HNMR (DMSO-d 6 ): δ (ppm) 2.24 (3H, s, –CH3), 5.36 (2H, s, N–CH 2 –CO), 7.12 (2H, d, J = 7.5 Hz, H3’ and H5’ Ar), 7.14–7.23 (2H, m, H5 and H6), 7.24 (1H, d, J = 7.5 Hz, H4), 7.31 (1H, d, J = 7.5 Hz, H7), 7.43 (2H, d, J = 7.5 Hz, H2’ & H6’ Ar), 10.28 (1H, s, –NH); 13CNMR (DMSO-d 6 ): δ (ppm) 20.8 (CH3), 110.0 (CH, C7), 110.3 (CH, C4), 118.0 (CH, C2 and C6 Ar), 118.4 (CH2, N–CH 2 –CO), 123.2 (CH, C6), 124.5 (CH, C5), 129.9 (CH, C3 and C5 Ar), 130.8 (C, C–N), 131.5 (C, C–CH3), 138.4 (C, C–NH–thiadiazole), 142 (C, C–O), 153.1 (C, C4 thiadiazole), 153.5 (C, N–CO–O), 166.1 (C, C1 thiadiazole); LC-MS (ESI) m/z: 339.2 (M+1, 100). Anal. Calcd. for C17H14N4O2S: C, 60.34; H, 4.17; N, 16.56. Found: C, 60.53; H, 4.35; N, 16.77.
In vitro anti-HIV and cytotoxicity assays
This test was reported earlier in the reference (Zabihollahi et al. 2010). The compounds were added in different concentrations concurrent with inoculation of single-cycle replicable HIV-1NL4-3 virions (200 ng P24) to the Hela cells. The inhibition rate (%) of P24 expression was calculated 72 h post infection by capture enzyme-linked immunosorbent assay (Biomerieux, France). The XTT (sodium3-[1(phenylaminocarbonyl)-3,4-tetrazolium]-bis(4-methoxy-6-nitro)benzene sulfonic acid)proliferation assay was conducted to evaluate the cellular toxicity according to kit instructions (Lin et al. 2002; Scudiero et al. 1988). Cytotoxicity test was performed straight after the P24 assay.
Molecular modeling studies
The most potent compound and the X-ray crystallographic structure of PFV IN (3OYA) were selected for docking studies. The ligand was created and minimized using HyperChem8.0. The co-crystallized ligand and water molecules were extracted, Kollman charges were added nonpolar hydrogens were merged and AutoDock4 atom type assigned to the protein structure. Docking was performed by Auto Dock 4.0 program, using the implemented empirical free energy function and the Lamarckian Genetic Algorithm (LGA) (Hajimahdi et al. 2013; Morris et al. 1998). The active site was defined as a Grid box around the crystallographic ligand raltegravir in (20 × 20 × 20) dimensions. Lamarckian genetic search algorithm was employed and docking run was set to 50.
References
Al-Mawsawi LQ, Dayam R, Taheri L, Witvrouw M, Debyser Z, Neamati N (2007) Discovery of novel non-cytotoxic salicylhydrazide containing HIV-1 integrase inhibitors. Bioorg Med Chem Lett 17:6472–6475
Bach A, Pizzirani D, Realini N, Vozella V, Russo D, Penna I, Melzig L, Scarpelli R, Piomelli D (2015) Benzoxazolone carboxamides as potent acid ceramidase inhibitors: synthesis and structure–activity relationship (SAR) studies. J Med Chem 58:9258–9272. doi:10.1021/acs.jmedchem.5b01188
Billamboz M, Bailly F, Lion C, Calmels C, Andréola M-L, Witvrouw M, Christ F, Debyser Z, De Luca L, Chimirri A, Cotelle P (2011) 2-Hydroxyisoquinoline-1,3(2H,4H)-diones as inhibitors of HIV-1 integrase and reverse transcriptase RNase H domain: Influence of the alkylation of position 4. Eur J Med Chem 46:535–546. doi:10.1016/j.ejmech.2010.11.033
Boros EE, Johns BA, Garvey EP, Koble CS, Miller WH (2006) Synthesis and HIV-integrase strand transfer inhibition activity of 7-hydroxy [1, 3] thiazolo [5, 4-b] pyridin-5 (4H)-ones. Bioorg Med Chem Lett 16:5668–5672
Chi G, Nair V, Semenova E, Pommier Y (2007) A novel diketo phosphonic acid that exhibits specific, strand-transfer inhibition of HIV integrase and anti-HIV activity. Bioorg Med Chem Lett 17:1266–1269
Dayam R, Al-Mawsawi LQ, Neamati N (2007) Substituted 2-pyrrolinone inhibitors of HIV-1 integrase. Bioorg Med Chem Lett 17:6155–6159
De Clercq E, Poupaert JH (1999) Synthesis and antiviral activity of 6-benzoyl-benzoxazolin-2-one and 6-benzoyl-benzothiazolin-2-one derivatives. Antivir Chem Chemother 10:87–97
Gerova MS, Stateva SR, Radonova EM, Kalenderska RB, Rusew RI, Nikolova RP, Chanev CD, Shivachev BL, Apostolova MD, Petrov OI (2016) Combretastatin A-4 analogues with benzoxazolone scaffold: Synthesis, structure and biological activity. Eur J Med Chem 120:121–133. doi:10.1016/j.ejmech.2016.05.012
Hajimahdi Z, Zarghi A, Zabihollahi R, Aghasadeghi M (2013) Synthesis, biological evaluation, and molecular modeling studies of new 1, 3, 4-oxadiazole-and 1, 3, 4-thiadiazole-substituted 4-oxo-4H-pyrido [1, 2-a] pyrimidines as anti-HIV-1 agents. Med Chem Res 22:2467–2475
Hassounah SA, Mesplède T, Wainberg MA (2016) Nonhuman primates and humanized mice for studies of HIV-1 integrase inhibitors: a review. Pathogens Immunity 1:41–67
Ingale KB, Bhatia MS (2011) HIV-1 integrase inhibitors: a review of their chemical development. Antivir Chem Chemother 22:95–105
Koeksal M, Goekhan N, Kuepeli E, Yesilada E, Erdoğan H (2005) Synthesis, Analgesic and Antiinflammatory properties of certain 5‐/6‐Acyl‐3‐(4‐substituted‐1‐piperazinylmethyl)‐2‐benzoxazolinones Derivatives. Arch Pharm (Weinheim) 338:117–125
Li B-W, Zhang F-H, Serrao E, Chen H, Sanchez TW, Yang L-M, Neamati N, Zheng Y-T, Wang H, Long Y-Q (2014) Design and discovery of flavonoid-based HIV-1 integrase inhibitors targeting both the active site and the interaction with LEDGF/p75. Bioorg Med Chem 22:3146–3158
Lin C-C, Cheng H-Y, Yang C-M, Lin T-C (2002) Antioxidant and antiviral activities ofEuphorbia thymifolia L. J Biomed Sci 9:656–664
Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ (1998) Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 19:1639–1662
Pawar R, Das T, Mishra S, Pancholi B, Gupta SK, Bhat SV (2014) Synthesis, anti-HIV activity, integrase enzyme inhibition and molecular modeling of catechol, hydroquinone and quinol labdane analogs. Bioorg Med Chem Lett 24:302–307
Poupaert J, Carato P, Colacino E (2005) 2 (3H)-benzoxazolone and bioisosters as “privileged scaffold” in the design of pharmacological probes. Curr Med Chem 12:877–885
Quashie PK, Sloan RD, Wainberg MA (2012) Novel therapeutic strategies targeting HIV integrase. BMC Med 10:1
Salgın-Gökşen U, Gökhan-Kelekçi N, Göktaş Ö, Köysal Y, Kılıç E, Işık Ş, Aktay G, Özalp M (2007) 1-Acylthiosemicarbazides, 1, 2, 4-triazole-5 (4H)-thiones, 1, 3, 4-thiadiazoles and hydrazones containing 5-methyl-2-benzoxazolinones: synthesis, analgesic-anti-inflammatory and antimicrobial activities. Bioorg Med Chem 15:5738–5751
Scudiero DA, Shoemaker RH, Paull KD, Monks A, Tierney S, Nofziger TH, Currens MJ, Seniff D, Boyd MR (1988) Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res 48:4827–4833
Stranix BR, Wu JJ, Milot G, Beaulieu F, Bouchard JE, Gouveia K, Forte A, Garde S, Wang Z, Mouscadet JF, Delelis O, Xiao Y (2016) Pyridoxine hydroxamic acids as novel HIV-integrase inhibitors. Bioorg Med Chem Lett 26:1233–1236. doi:10.1016/j.bmcl.2016.01.028
Tanis SP, Plewe MB, Johnson TW, Butler SL, Dalvie D, DeLisle D, Dress KR, Hu Q, Huang B, Kuehler JE (2010) Azaindole N-methyl hydroxamic acids as HIV-1 integrase inhibitors-II. The impact of physicochemical properties on ADME and PK. Bioorg Med Chem Lett 20:7429–7434
Wang Z, Vince R (2008) Synthesis of pyrimidine and quinolone conjugates as a scaffold for dual inhibitors of HIV reverse transcriptase and integrase. Bioorg Med Chem Lett 18:1293–1296
Yu S, Sanchez TW, Liu Y, Yin Y, Neamati N, Zhao G (2013) Design and synthesis of novel pyrimidone analogues as HIV-1 integrase inhibitors. Bioorg Med Chem Lett 23:6134–6137. doi:10.1016/j.bmcl.2013.09.018
Zabihollahi R, Sadat S, Vahabpour R, Aghasadeghi M, Memarnejadian A, Ghazanfari T, Salehi M, Rezaei A, Azadmanesh K (2010) Development of single-cycle replicable human immunodeficiency virus 1 mutants. Acta Virol 55:15–22. doi:10.4149/av_2011_01_15
Acknowledgements
This study was financially supported by Research Deputy of Shahid Beheshti University of Medical sciences as part of Ph. D thesis of M. Safakish.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Safakish, M., Hajimahdi, Z., Zabihollahi, R. et al. Design, synthesis, and docking studies of new 2-benzoxazolinone derivatives as anti-HIV-1 agents. Med Chem Res 26, 2718–2726 (2017). https://doi.org/10.1007/s00044-017-1969-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-017-1969-8