
Abstract
The increase in antibiotic resistance due to multiple factors has warranted the need for search of new compounds which are active against multidrug resistant pathogens. In this context a small focused library of thiosemicarbazide derivatives of 2-arylthiazole-4-carbaldehyde, 4-methyl-2-arylthiazole-5-carbaldehyde and 1-(4-methyl-2-arylthiazol-5-yl) ethanone, (5a–l) has been synthesized. The title compounds were screened for inhibitory activity against Mycobacterium tuberculosis H37Ra (ATCC 25177) and Mycobacterium bovis Bacille Calmette Guerin (ATCC 35743) strains. The synthesized compounds, 5a–l were further assayed for their cytotoxic activity against the two human cancer cell lines, HeLa and human colon carcinoma 116 cell lines and showed no significant cytotoxic activity against these two cell lines at the maximum concentration evaluated. Further, the synthesized compounds were found to have potential antibacterial activity against Gram-negative bacteria, Escherichia coli, Pseudomonas flurescence and Gram-positive bacteria, Staphylococcus aureus, Bacillus subtilis. Most of the synthesized compounds showed moderate activity against fungal strain Candida albicans. This study provides valuable directions to our ongoing endeavor of rationally designing more potent antimycobacterial agent.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tuberculosis (TB), an infection caused by Mycobacterium tuberculosis (MTB) is a huge menace to the human race. To combat with microbial pathogens, different kinds of antimicrobial agents have been developed substantially. However, the emergence of multidrug-resistant bacterial strains, the restricted spectrum of action, toxicity, extensive side effects and drug interactions, create a serious challenge to the scientific community and the need for an effective therapy has led to a search for new therapeutic options (Kunin and Ellis 2000; Cannon et al. 2009; Achkar and Fries 2010; Kathiravan et al. 2012).
The simplest hydrazine derivative of thiocarbamic acid called thiosemicarbazide has received considerable attention because of their variable bonding modes, structural diversity, promising biological activities, and ion sensing ability (Casas et al. 2000; Kumari et al. 2012). During the past few decades, interest has been rapidly growing in gaining insight into the properties and transformations of thiosemicarbazides and their derivatives due to their appreciable pharmacological activities viz. antitubercular activity (Patel et al. 2014; Tan et al. 2012; Kucukguzel et al. 2006; Sriram et al. 2006; Moglea et al. 2016), anti-inflammatory activity (Salgın-Goksen et al. 2007), antibacterial activity (Alagarsamy et al. 2012; Rane et al. 2014; Sheikhy et al. 2012; Umadevi et al. 2012; Plech et al. 2011; Chornous et al. 2014; Pitucha et al. 2016), anticancer activity (Arora et al. 2014; Perkoviisć et al. 2012; Malki et al. 2015; Zhang et al. 2011), cytotoxic and antiproliferative activity (Mavrova et al. 2014), antiprotozoal activity (Leite et al. 2006; Haraguchi et al. 2011), etc. Use of these compounds in organic synthesis has become a classical strategy for the synthesis of several heterocycles. Thiosemicarbazide is a useful structural moiety and optimization of this structure can result in ground breaking discovery of new class of therapeutic agents.
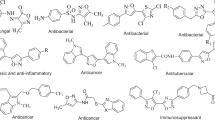
Similarly, Thiazole and its derivatives are also important pharmacophore in medicinal chemistry that could provide a rich spectrum of biological activities (Abhale et al. 2015, 2016, 2017; Johnson et al. 2012; Gaikwad et al. 2012; Tomasic et al. 2015; Urbanek et al. 2013; Inamdar et al. 2013; Lee et al. 2012; Malik et al. 2013; Navale et al. 2013; Mishra et al. 2015; Siddiqui and Ahsan 2010; Yusufzai et al. 2017) (Fig. 1).

Antibacterial and antitubercular agents containing thiosemicarbazone moiety
Considering the importance of thiosemicarbazide and thiazole derivatives, it was thought worthwhile to synthesize thiazole substituted thiosemicarbazide derivatives. In the present work the synthesis and antimycobacterial screening of thiosemicarbazide derivatives of 2-arylthiazole-4-carbaldehyde, 4-methyl-2-arylthiazole-5-carbaldehyde, and 1-(4-methyl-2-arylthiazol-5-yl) ethanone is reported.
Materials and methods
Chemistry
All the reactions were monitored by thin layer chromatography (TLC). TLC was performed on Merck 60 F-254 silica gel plates. Melting points were determined in capillary tubes in silicon oil bath using a Veego melting point apparatus and are uncorrected. 1H (300 MHz) NMR and 13C (75 MHz) NMR spectra were recorded on Varian mercury XL-300 and Bruker at either 400 MHz (1H NMR) and 100 MHz (13C NMR), spectrometer instruments. Chemical shifts are reported from internal tetramethylsilane standard and are given in δ units. Infrared spectra were recorded on Shimadzu Fourier transform infrared spectroscopy (KBr)-408 in KBr. The LC–MS spectra were recorded on a Shimadzu 2010 LC–MS. Column chromatography was performed on silica gel (100–200 mesh) supplied by Acme Chemical Co. The chemicals and solvents used were laboratory grade and were purified as per literature methods.
General procedure for synthesis of ethyl 2-phenylthiazole-4-carboxylate (2a)
The mixture of ethyl 3-bromo-2-oxopropanoate (40 mmol) and benzothiomide, 1a (40 mmol) in ethanol (30 mL) was refluxed for 3 h. After completion of reaction (TLC) half of the solvent was distilled off and reaction mass was cooled to room temperature. The product was filtered, washed with water and recrystallized from ethanol.
General procedure for synthesis of 2-(2-phenylthiazol-4-yl) methanol (3a)
To a cold solution of lithium aluminum hydride (20 mmol) in diethyl ether (20 mL) 2-phenylthiazole-4-carboxylate, 2a (10 mmol) in diethyl ether (20 mL) was added dropwise over a period of 30 min and the reaction mixture was further stirred for 1 h at 0 °C. After completion of the reaction (TLC), the reaction mixture was quenched by saturated solution of sodium sulphate. The reaction mixture was filtered and the aqueous layer was extracted with diethyl ether (2 × 30 mL), the combined organic layer was washed with water, brine, and dried over sodium sulphate. 2-(2-phenylthiazol-4-yl) methanol was obtained by removing the solvent by distillation.
General procedure for synthesis of 2-phenylthiazole-4-carbaldehyde (4a)
To a solution of 2 (2-phenylthiazol-4-yl) methanol, 3a (14 mmol) in DMSO (30 mL), iodoxy benzoic acid (IBX, 18 mmol) was added and reaction mixture was stirred at 0–20 °C. The progress of reaction was monitored on TLC. After completion of reaction, reaction mixture was filtered. Water (90 mL) was added in the filtrate and the filtrate was extracted with diethyl ether (3 × 30 mL). The organic layer was washed with water, brine and dried over sodium sulphate. Product was obtained by distillation of solvent.
General procedure for synthesis of -1-((2-phenylthiazol-4-yl) methylene) thiosemicarbazide (5a)
The mixture of 2-phenylthiazole-4-carbaldehyde, 4a (2 mmol) and thiosemicarbazide (2.5 mmol) in ethanol (10 mL) was refluxed for 2 h. After completion of reaction (TLC) solvent was cooled to room temperature. The product was filtered, washed with water and recrystallized from ethanol.
1-((2-phenylthiazol-4-yl)methylene)thiosemicarbazide (5a)
Yield 70 %, m.p. 230–232 °C; IR (KBr): 3470, 3430, 3259, 3150, 3018, 2978, 1687, 1645, 1597, 1546, 1491, 1369, 1318, 1284, 1111, 841 cm−1; 1H NMR (DMSO-d6, 500 MHz) δ: 7.46–7.49 (m, 3H, H-3′, H-4′, H-5′), 7.82–7.84 (m, 2H, H-2′,H-6′), 7.87 (s, 1H, Thiazole H-5), 8.10 (s, 1H, NH), 8.25 (s, 1H, NH), 8.26 (s, 1H, NH), 11.45 (s, 1H, N = CH); 13C NMR (DMSO-d6,125 MHz) δ: 118.5 (Thiazole C-5), 126.5 (C-4′), 129.3 (C-3′,-5′), 130.5 (C-2′,-6′), 132.5 (C-1′), 136.4 (C = N), 153.4 (Thiazole C-4), 167.7 (Thiazole C-2), 178.5 (C = S); LC–MS m/z: 263.0 (M + H)+.
1-((2-(4-chlorophenyl)thiazol-4-yl)methylene)thiosemicarbazide (5b)
Yield 68 %, m.p. 240 °C (dec.); IR (KBr): 3468, 3430, 3261, 3168, 3010, 2976, 1685, 1645, 1597, 1542, 1502, 1368, 1320, 1284, 1122, 1001, 839, 801 cm−1; 1H NMR (500 MHz, DMSO-d 6 ) δ: 7.50 (d, J = 8 Hz, 2H, H-3′, H-5′), 7.92 (d, J = 8 Hz, 2H, H-2′, H-6′), 7.95 (s, 1H, Thiazole H-5), 8.30 (s, 1H, NH), 8.35 (s, 2H, NH2 ), 11.46 (s, 1H, N = CH); 13C NMR (125 MHz, DMSO-d 6 ) δ: 119.0 (Thiazole C-5), 128.8 (C-2′, -6′), 129.9 (C-3′, -5′), 131.9 (C-4′), 135.7 (C-1′), 136.8 (C = N), 152.3 (Thiazole C-4), 165.5 (Thiazole C-2), 178.0 (C = S); LC–MS m/z: 297.0 (M + H)+, m/z: 299.0 (M + H + 2)+.
1-((2-(4-fluorophenyl)thiazol-4-yl)methylene)thiosemicarbazide (5c)
Yield 75 %, m.p. 230 °C (dec.); IR (KBr): 3472, 3433, 3264, 3165, 3010, 2978, 1687, 1645, 1597, 1542, 1502, 1368, 1320, 1284, 1122, 1001, 840, 800 cm−1; 1H NMR (500 MHz, DMSO-d 6 ,) δ: 7.35 (t, J = 6.8 Hz, 2H, H-3′, H-5′), 8.00 (m, 2H, H-2′, H-6′), 8.2 (s, 1H, Thiazole H-5), 8.33 (s, 1H, NH), 8.34 (s, 2H, NH2), 11.58 (s, 1H, N = CH); 13C NMR (125 MHz, DMSO-d 6 ,) δ: 116.8 (C-3′, -5′), 119.5 (Thiazole C-5), 129.4 (C-2′, -6′), 129.9 (C-1′), 136.0 (C = N), 153.6 (Thiazole C-4), 163.5 (C-4′), 166.8 (Thiazole C-2), 178.8 (C = S); LC–MS m/z: 281.0 (M + H)+.
1-((2-p-tolylthiazol-4-yl)methylene)thiosemicarbazide (5d)
Yield 65 %, m.p. 230 °C (dec.); IR (KBr): 3470, 3430, 3260, 3165, 3012, 2970, 1688, 1646, 1600, 1540, 1500, 1368, 1320,1284, 1122, 1001, 839, 799 cm−1; 1H NMR (500 MHz, DMSO-d 6 ) δ: 2.42, (s, 3H, Ar–CH3) 7.35 (d, J = 6.8 Hz, 2H, H-3′, H-5′), 7.84 (d, J = 6.8 Hz, 2H, H-2′, H-6′), 8.00 (s, 1H, Thiazole H-5), 8.22 (s, 1H, NH), 8.32 (s, 2H, NH2), 11.58 (s, 1H, N = CH); 13C NMR (125 MHz, DMSO-d 6 ) δ: 21.2 (Ar–CH3), 119.0 (Thiazole C-5), 127.0 (C-2′, -6′), 130.1 (C-3′, -5′), 130.2 (C-4′), 137.1 (C = N), 140.4 (C-1′), 152.6 (Thiazole C-4), 167.8 (Thiazole C-2), 179.1 (C = S); LC–MS m/z: 277.1 (M + H)+.
General procedure for synthesis of Ethyl 4-methyl-2-phenylthizole-5-carboxylate (2e)
The mixture of 2-bromo-3-oxoethylbutanoate (36.4 mmol) and benzothioamide (36.4 mmol) in 25 mL ethanol was refluxed for 8 h. After completion of reaction (TLC), half of the solvent was distilled-off and reaction mass cooled to room temperature. The product was filtered, washed with water and recrystallized from ethanol. Yield: 4.0 g, (80 %), m.p. 90–92 °C. The compounds 2f–h were synthesized by same method.
General procedure for synthesis of (4-Methyl-2-phenylthiazol-5-yl) methanol (3e)
To a cold solution of lithium aluminum hydride (20 mmol) in dry diethyl ether (25 mL), ethyl 4-methyl-2-phenylthiazole-5-carboxylate, 2e (10 mmol) in diethyl ether (25 mL) was added dropwise over a period of 30 min and the reaction mixture was further stirred for 1 h at 0 °C. After completion of the reaction (TLC), the reaction mixture was quenched by saturated solution of sodium sulphate. The reaction mixture was filtered and the aqueous layer was extracted with diethyl ether (2 × 30 mL), the combined organic layer was washed with water, brine and dried over sodium sulphate furnished (4-Methyl-2-phenylthiazol-5-yl) methanol.
General procedure for synthesis of 4-Methyl-2-phenylthiazole-5-carbaldehyde (4e)
To a solution of (4-methyl-2-phenylthiazole-5-yl) methanol, 3e (10 mmol) in DMSO (30 mL), IBX (11 mmol) was added and the reaction mixture was stirred at 0 to 20 °C. The progress of reaction was monitored on TLC. After completion of the reaction (20 min), reaction mixture was filtered and washed with DMSO (5 mL). Water (90 mL) was added in the filtrate and the filtrate was extracted with diethyl ether (3 × 30 mL). The organic layer was washed with water, brine and dried over sodium sulphate. The solvent was distilled to afford white solid 4e in 95% yield.
General procedure for synthesis of 1-((4-methyl-2-phenylthiazol-5-yl) methylene) thio-semicarbazide (5e)
The mixture of 4-methyl-2-phenylthiazole-5-carbaldehyde, 4e (2 mmol) and thiosemicarbazide (2.5 mmol) in ethanol (10 mL) was refluxed for 2 h. After completion of reaction (TLC) solvent was cooled to room temperature. The product was filtered, washed with water and recrystallized from ethanol.
1-((4-methyl-2-phenylthiazol-5-yl)methylene)thiosemicarbazide (5e)
Yield 66 %, m.p. 230 °C. IR (KBr): 3474, 3432, 3261, 3158, 3018, 2978, 1688, 1647, 1597, 1546, 1491, 1369, 1318, 1284, 1111, 841 cm−1; 1H NMR (DMSO-d 6 , 500 MHz) δ: 2.46 (s, 3H, Thiazole-CH3), 7.50–7.52 (m, 3H, H-3′, H-4′, H-5′), 7.60 (s, 1H, NH), 7.90–7.92 (m, 2H, H-2′, H-6′), 8.28 (s, 1H, NH), 8.35 (s, 2H, NH2), 11.45 (s, 1H, N = CH); 13C NMR (DMSO-d 6 ,125 MHz) δ: 15.5 (Thiazole-CH3), 128.0 (C-3′, -5′), 129.5 (C = N), 130.9 (C-4′), 132.5 (C-2′, -6′), 132.7 (C-1′), 135.4 (Thiazole C-5), 154.9 (Thiazole C-4), 166.5 (Thiazole C-2), 177.5 (C = S); LC–MS m/z: 277.1 (M + H)+.
1-((2-(4-chlorophenyl)-4-methylthiazol-5-yl)methylene)thiosemicarbazide (5f)
Yield 65 %, m.p. 240 °C (dec.); IR (KBr): 3465, 3428, 3260, 3166, 3010, 2976, 1687, 1646, 1598, 1546, 1502, 1368, 1320, 1284, 1124, 1001, 840, 801 cm−1; 1H NMR (DMSO-d 6 , 500 MHz) δ: 2.46 (s, 3H, Thiazole-CH3), 7.57 (d, J = 8 Hz, 2H, H-3′, H-5′), 8.60 (s, 1H, NH), 7.90 (d, J = 8 Hz, 2H, H-2′, H-6′), 8.28 (s, 1H, NH), 8.33 (s, 2H, NH2 ), 11.48 (s, 1H, N = CH); 13C NMR (DMSO-d 6 , 125 MHz) δ: 16.0 (Thiazole-CH3), 128.3 (C-2′, -6′), 128.8 (C = N), 129.9 (C-3′, -5′), 131.9 (C-4′), 135.7 (Thiazole C-5), 135.8 (C-1′), 155.3 (Thiazole C-4), 165.5 (Thiazole C-2), 178.0 (C = S); LC–MS m/z: 311.0 (M + H)+, m/z: 313.0 (M + H + 2)+.
1-((2-(4-fluorophenyl)-4-methylthiazol-5-yl)methylene)thiosemicarbazide (5g)
Yield 75 %, m.p. 230 °C (dec.);IR (KBr): 3468, 3434, 3265, 3160, 3012, 2975, 1688, 1649, 1600, 1549, 1500, 1368, 1320,1284, 1124, 1001, 839, 800 cm−1: 1H NMR (500 MHz, DMSO-d 6 ) δ: 2.43, (s, 3H, Thiazole-CH3), 7.40 (t, J = 7.6 Hz, 2H, H-3′, H-5′), 7.60 (s, 1H, NH), 7.94–7.96 (m, 2H, H-2′, H-6′), 8.28 (s, 1H, NH), 8.35 (s, 1H, NH), 11.46 (s, 1H, N = CH); 13C NMR (125 MHz, DMSO-d 6 ) δ: 16.6 (Thiazole-CH3), 115.8 (C-3′, -5′), 129.3 (C = N), 129.6 (C-2′, -6′), 130.5 (C-1′), 135.4 (Thiazole C-5), 155.8 (Thiazole C-4), 162.2 (C-4′), 166.9 (Thiazole C-2), 178.5 (C = S); LC–MS m/z: 295.1 (M + H)+.
1-((4-methyl-2-p-tolylthiazol-5-yl)methylene)thiosemicarbazide (5h)
Yield 72 %, m.p. 240 °C (dec.); IR (KBr): 3466, 3430, 3261, 3163, 3009, 2974, 1687, 1645, 1597, 1545, 1500, 1368, 1320,1284, 1124, 1001, 841, 801 cm−1 ; 1H NMR (500 MHz, DMSO-d 6 ) δ: 2.45, (s, 3H, Thiazole-CH3), 2.56 (s, 3H, Ar-CH3), 7.35 (d, J = 6.8 Hz, 2H, H-3′, H-5′), 7.60 (s, 1H, NH), 7.84 (d, J = 6.8 Hz, 2H, H-2′, H-6′), 8.25 (s, 1H, NH), 8.35 (s, 1H, NH), 11.48 (s, 1H, N = CH); 13C NMR (125 MHz, DMSO-d 6 ) δ: 16.0 (Thiazole-CH3), 21.5 (Ar–CH3), 126.8 (C-2′, -6′), 127.5 (C = N), 130.5 (C-3′, -5′), 130.6 (C-4′), 135.5 (Thiazole C-5), 141.2 (C-1′), 155.1 (Thiazole C-4), 167.1 (Thiazole C-2), 178.5 (C = S); LC–MS, m/z: 291.07 (M + H)+.
General procedure for synthesis of 1-(4-methyl-2-phenylthiazol-5-yl) ethanone (4i)
The mixture of thiobenzamide (6.62 mmol) and 3-bromopentane-2,4-dione, (6.62 mmol) was refluxed in ethanol. After completion of reaction (TLC), solvent was removed under reduced pressure and the residue was dissolved in ethyl acetate. Organic layer was washed with sodium bicarbonate and then water. Organic layer was dried over sodium sulphate and distilled under vacuum. The product obtained was purified by column chromatography using hexane:ethyl acetate (9:1) as eluent.
General procedure for synthesis of 1-(1-(4-methyl-2-phenylthiazol-5-yl) ethylidene) thio-semicarbazide (5i)
The mixture of 1-(4-methyl-2-phenylthiazol-5-yl) ethanone, 4i (2 mmol) and thiosemicarbazide (2.5 mmol) in ethanol (10 mL) was refluxed for 2 h. After completion of reaction (TLC) solvent was cooled to room temperature. The product was filtered, washed with water and recrystallized from ethanol.
1-(1-(4-methyl-2-phenylthiazol-5-yl)ethylidene)thiosemicarbazide (5i)
Yield 66 %, m.p. 230 °C (dec.); IR (KBr): 3470, 3425, 3255, 3145, 3018, 2972, 1679, 1640, 1595, 1546, 1491,1311, 1279, 1111, 840 cm−1; 1H NMR (DMSO-d 6 , 500 MHz) δ: 2.38 (s, 3H, Thiazole-CH3), 2.60 (s, 3H, N = CCH3), 7.38–7.42 (m, 4H, H-3′, H-4′, H-5′, NH), 7.80–7.82 (m, 2H, H-2′, H-6′), 8.45 (s, 1H, NH), 10.51 (s, 1H, NH).13C NMR (DMSO-d 6 ,125 MHz) δ: 18.4 (Thiazole-CH3), 31.1 (N = C–CH3), 128.3 (C-3′, -5′), 129.5 (C-4′), 129.6 (C-2′, -6′), 130.5 (Thiazole C-5), 132.2 (C = N), 135.8 (C-1′), 157.9 (Thiazole C-4), 167.5 (Thiazole C-2), 180.5 (C = S); LC–MS m/z: 291.08 (M + H).+
1-(1-(2-(4-chlorophenyl)-4-methylthiazol-5-yl)ethylidene)thiosemicarbazide (5j)
Yield 65 %, m.p. 240 °C (dec.); IR (KBr): 3468, 3430, 3266, 3167, 3012,2975, 1689, 1644, 1596, 1544, 1498, 1365, 1321,1284, 1124, 1001, 838, 800 cm−1; 1H NMR (500 MHz, DMSO-d 6 ) δ: 2.35 (s, 3H, Thiazole-CH3 ), 2.62 (s, 3H, N = CCH3 ), 7.41 (s, 1H, NH), 7.50 (d, J = 7.6 Hz, 2H, H-3′, H-5′), 7.85 (d, J = 7.6 Hz, 2H, H-2′, H-6′), 8.40 (s, 1H, NH), 10.53 (s, 1H, NH); 13C NMR (DMSO-d 6 , 125 MHz) δ: 20.0 (Thiazole-CH3), 31.5 (N = C–CH3), 126.0 (C-2′, -6′), 129.5 (C-3′, -5′), 130.3 (Thiazole C-5), 132.0 (C = N), 132.8 (C-4′), 143.9 (C-1′), 152.3 (Thiazole C-4), 165.2 (Thiazole C-2), 178.6 (C = S); LC–MS m/z: 325.03 (M + H)+, m/z: 327.2 (M + H + 2).+
1-(1-(2-(4-fluorophenyl)-4-methylthiazol-5-yl)ethylidene)thiosemicarbazide (5k)
Yield 70 %, m.p. 203 °C (dec.); IR (KBr): 3462, 3433, 3265, 3015, 2976, 1685, 1642, 1599, 1540, 1496, 1365, 1321,1284, 998, 841, 802 cm−1; 1H NMR (500 MHz, DMSO-d 6 ) δ: 2.34 (s, 3H, Thiazole-CH3), 2.58, (s, 3H, N = CCH3), 7.32 (t, J = 7.6 Hz, 2H, H-3′, H-5′), 7.42 (s, 1H, NH), 8.84–8.86 (m, 2H, H-2′, H-6′), 8.36 (s, 1H, NH), 10.56 (s, 1H, NH); 13C NMR (125 MHz, DMSO-d 6 ) δ: 20.0 (Thiazole-CH3), 31.4 (N = C–CH3), 116.8 (C-3′, -5′), 130.0 (C-2′, -6′), 133.0 (C = N), 135.8 (Thiazole C-5), 142.6 (C-1′), 154.6 (Thiazole C-4), 163.0 (C-4′), 167.2 (Thiazole C-2), 178.7 (C = S); LC–MS m/z: 309.06 (M + H).+
1-(1-(4-methyl-2-p-tolylthiazol-5-yl)ethylidene)thiosemicarbazide (5l)
Yield 75 %, m.p. 230 °C (dec.); IR (KBr): 3460, 3430, 3260, 3011, 2975, 1686, 1640, 1600, 1544, 1499, 1366, 1322, 1285, 1000, 841, 800 cm−1; 1H NMR (500 MHz, DMSO- d 6 ) δ: 2.15 (s, 3H, Ar-CH3), 2.40 (s, 3H, Thiazole-CH3), 2.66 (s, 3H, N = CCH3), 7.42 (s, 1H, NH), 7.62 (d, J = 6.8 Hz, 2H, H-3′, H-5′), 8.05 (d, J = 6.8 Hz, 2H, H-2′, H-6′), 8.35 (s, 1H, NH), 10.58 (s, 1H, NH); 13C NMR (125 MHz, DMSO-d 6 ) δ: 17.7 (Thiazole-CH3), 21.4 (Ar–CH3), 31.2 (N = C–CH3), 126.4 (C-2′, -6′), 128.5 (C-3′, -5′), 129.9 (C-4′), 130.3 (C = N), 136.0 (Thiazole C-5), 142.2 (C-1′), 154.0 (Thiazole C-4), 167.4 (Thiazole C-2), 178.3 (C = S); LC–MS m/z: 305.1 (M + H).+
Pharmacology
Antitubercular activity
Standard cultures of M. tuberculosis H37Ra (ATCC 25177) (MTB) and M. bovis Bacille Calmette Guerin (BCG) (ATCC 35743) were procured from the American Type Culture Collection (Manassas, VA). MTB and M. bovis BCG were grown in a defined Mycobacterium phlei medium and in 50 mM sodium nitrate-containing Dubos medium (Difco, Detroit, MI), respectively. These were maintained as glycerol stocks at −70 °C. Prior to inoculation for experiments, 50 µL of glycerol stock was pre-inoculated in the corresponding medium to obtain metabolically active mycobacteria. For each experiment, cultures were grown to log phase [optical density at 595 nm (O.D.595) = 1] under aerobic conditions at 37 °C and 150 r.p.m. As mycobacteria grow in visibly aggregated clumps in the culture medium, cultures were sonicated for 2 min using a water-bath sonicator (Ultrasonic, Freeport, IL) to obtain viable dispersed cells. This step was introduced to reproducibly inoculate mycobacterial bacilli in fresh medium for carrying out the experiments.
Primary screening
Activity against dormant stage MTB was determined through the XTT reduction menadione assay (XRMA), reading absorbance at 470 nm as per the protocol described by Singh et al. A compound solution (2.5 μL) was added in a total volume of 250 μL of M. pheli medium consisting of the MTB and BCG; sealed with plate sealers and allowed to incubate for 12 days at 37 °C. The XRMA was then carried out to estimate viable cells present in different wells of the assay plate. To all wells, 200 μM XTT was added and incubated at 37 °C for another 20 min. It was followed by the addition of 60 μM of menadione and incubated at 37 °C for 40 min. The optical density was measured using a microplate reader (SpectraMaxPlus 384 plate reader, Molecular Devices Inc.) at 470 nm filter against a blank prepared from a well free of cells. Absorbance obtained from the cells treated with 1% DMSO alone was considered as 100% cell growth. The nitrate reductase (NR) assay was performed to estimate inhibition of M. bovis BCG by the compounds. In the NR assay, 80 μL of culture from an incubated 96-well-plate was taken into another 96-well-plate, then 80 μL of 1% sulfanilic acid in 20% of conc. HCl was added, incubated for 10 min. at room temperature and then 80 μL of 0.1% N-(1-naphthyl)ethylenediaminedihydrochloride solution in distilled water was added. Finally, absorbance for the NR assay was measured at 540 nm.
The % Inhibition in the presence of test material is calculated by using formula, % inhibition = (Average of Control−Average of Compound)/(Average of Control−Average of Blank) × 100). Where, Control is culture medium with cells and DMSO and blank is culture medium without cells. For all samples, each compound concentration was tested in triplicates in a single experiment and the quantitative value was expressed as the mean ± standard deviation (S.D.).
Antibacterial activity
All bacterial cultures were first grown in Luria Burtony media at 37 °C at 180 r.p.m. Once the culture reaches one O.D., it is used for anti-bacterial assay. Bacterial strains E. coli (NCIM 2576), P. flurescence (NCIM 2059) as Gram-negative and S. aureus (NCIM 2602), B. subtilis (NCIM 2162) as Gram-positive were obtained from NCIM (NCL, Pune) and were grown in Luria Burtony medium from Hi Media, India. The assay was performed in 96-well-plates after 8 and 12 h for Gram negative and Gram positive bacteria, respectively (Ciapetti et al. 1993). 0.1% of one O.D. culture at 620 nm was used for screening inoculated culture was added into each well of 96-well-plate containing the compounds to be tested. Optical density for each plate was measured at 620 nm after 8 h for Gram-negative bacteria and after 12 h for Gram-positive bacteria.
Antifungal activity
The in vitro antifungal activity of all the synthesized compounds was done by the disk diffusion method (Alley et al. 1988; Singh et al. 2015) against the C. albicans (NCIM 3100) obtained from National Chemical Laboratory Pune, India. All cultures were maintained at 4 °C over nutrient agar slants throughout the experiment. The cultures incubated overnight at 37 °C in nutrient broth before using for antifungal activity. Five hundred microliters of overnight old fungal suspension were spread over the nutrient agar plates using a sterile cotton swab in order to get a uniform microbial growth. The synthesized compounds were dissolved in DMSO. Under aseptic conditions, empty sterilized disks (Whatman no. 5, 6 mm diameter) were impregnated with concentration 100 µg/disk of respective synthesized compounds and placed on the agar surface. Paper disk moistened with aqueous DMSO was placed on seeded petriplates as a vehicle control. A standard disk containing Fluconazole (25 µg/disk) was used as positive control. The plates were left for 30 min. at room temperature to allow the diffusion of synthesized compounds and then incubated at 37 °C for 24 h. The antifungal activity was evaluated by measuring the zone of inhibition against the test microorganism. All experiments were carried in triplicates.
Cytotoxic activity
Cell lines were maintained under standard cell culture conditions at 37 °C and 5% CO2 in a humidified environment. The cytotoxic effect of synthesized compounds was checked on cancer cell lines using the concentrations 30, 10, 3 μg/mL to determine the growth inhibition. In short, log-phase cells were harvested using trypsin (0.05% trypsin and 0.02% ethylene diamine tetra-acetic acid in PBS) from tissue culture flasks and the suspension was diluted with appropriate culture medium to obtain a cell density of 105 cells/mL was determined by haemocytometry. An aliquot of 100 µL of each suspension was seeded in 96-well cell culture plates and was incubated at 37 °C in an atmosphere of 5% CO2 and 95% relative humidity in a CO2 incubator. After 24 h, synthesized compounds at varying concentrations of 3, 10, and 30 µg/mL were added to the wells containing cells. Rifampicin was used as positive control. Suitable controls with an equivalent concentration of dimethyl sulphoxide (DMSO) (Sigma-Aldrich) were also included. The plates were further incubated for 48 h. At the end of the incubation period, the solution containing the unattached cells was discarded and the wells were washed three times with 1 mL of PBS followed by addition of 10 µL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (5 mg/mL in PBS) to adherent cells in growth medium. After 4 h at 37 °C for MTT cleavage, the formazan product was solubilised by addition of 100 µL of 0.04 N HCl in isopropanol. Absorbance was measured on a SpectraMax® PLUS 384 plate reader (Molecular Devices, Sunnyvale, CA) at a wavelength of 570 nm. Percentage cytotoxicity was calculated using the formula: % cytotoxicity = (average of control−average of compound)/(average of control−average of blank) × 100). Each concentration was tested in triplicate in a single experiment and the quantitative value was expressed as the mean ± S.D.
Results and discussion
Chemistry
A general route for the synthesis of title compounds 5a–l is depicted in Scheme 1.

Synthesis of Target Compounds 5a–l
All the compounds, 5a–l were characterized by spectroscopic analysis and were studied for their anti-tuberculosis and antimicrobial activity. Their toxicity was also studied against two human cancer cell lines.
Anti-tubercular activity
The antitubercular activity for each synthesized compound was determined by measuring inhibition of growth against M. tuberculosis H37Ra (MTB, ATCC 25177) and M. bovis (BCG, ATCC 35743) in liquid medium. In a preliminary screening, the antimycobacterial activity of these compounds was assessed at concentrations of 30, 10, 3 μg/mL using first-line antitubercular drug Rifampicin as reference standard. In vitro activity studies against MTB and M. bovis BCG were performed using the XRMA (Singh et al. 2011; Khan et al. 2008) and NR assays (Khan et al. 2008; Sarkar and Sarkar 2012), respectively. The results of antitubercular activity are summarized in Table 1.
Analysis of the anti-tubercular activity results provides some lead molecules with good anti-tubercular activity. The results revealed that, in case of thiosemicarbazide derivatives of 2-arylthiazole-4-carbaldehyde the compounds 5a (R = H), 5b (R = 4-Cl), 5c (R = 4-F) and 5d (R = 4-CH3) showed good activity against M. bovis at 30 µg/mL concentration whereas in compounds 5a (R = H) and 5b (R = 4-Cl) anti-tubercular activity is retained after the concentration of compounds was decreased by 10 times (3 µg/mL). All the compounds showed moderate activity against M. tuberculosis H37Ra. From the preliminary structure activity relationship (SAR) study it is noteworthy to mention that thiosemicarbazide of unsubstituted 2-phenylthiazole-4-carbaldehyde, 5a showed excellent activity against M. bovis, whereas replacement of hydrogen atom of phenyl ring by substituent groups like 4-Cl, 4-F, or 4-CH3 significantly decrease the anti-tubercular activity.
Similarly, thiosemicarbazide derivatives of 4-methyl-2-arylthiazole-5-carbaldehyde 5e (R = H), 5f (R = 4-Cl), 5g (R = 4-F) and 5h (R = 4-CH3) showed moderate activity against M. tuberculosis H37Ra at 30 µg/mL concentration. Compounds 5e (R = H) and 5g (R = 4-F) showed moderate anti-tubercular activity against M. bovis BCG, whereas compounds 5f (R = 4-Cl) and 5h (R = 4-CH3) reported good activity and it retained at 3 µg/mL concentration. From the SAR study, it is noteworthy to mention that as phenyl ring of 4-methyl-2-arylthiazole-5-carbaldehyde is substituted by 4-Cl or 4-CH3 group, thiosemicarbazide derivative showed good anti-tubercular activity against M. bovis.
Also, in case of thiosemicarbazide derivatives of 1-(4-methyl-2-arylthiazol-5-yl) ethanone, compound 5j (R = 4-Cl) presented good anti-tubercular activity against both tested strains, whereas compounds 5i (R = H) and 5k (R = 4-F) showed good activity against M. bovis at 30 µg/mL concentration. The compounds 5i, 5k, and 5l showed moderate activity against M. tuberculosis H37Ra. From the SAR study, it is noteworthy to mention that thiosemicarbazide derivative of unsubstituted 2-phenylthiazole-4-carbaldehyde showed excellent activity against M. bovis. When phenyl ring is substituted by 4-Cl, activity decreases for M. bovis but it shows good activity against M. tuberculosis H37Ra.
Antimicrobial activity
The antibacterial activity (Singh et al. 2011; Khan et al. 2008) of synthesized compounds was against the standard Gram-negative bacteria, E.coli (NCIM 2576), P. flurescence (NCIM 2059) and Gram-positive bacteria, S. aureus (NCIM 2602), B. subtilis (NCIM 2162); while the antifungal activity Khan and Sarkar (2008); Khan et al. 2008; Sarkar and Sarkar 2012; Mosmann 1983) was against C. albicans (NCIM 3100). Ampicillin served as positive control for antibacterial activity. The in vitro preliminary screening values (% inhibition) and MIC90 against microorganisms tested are summarized in Tables 2 and 3, respectively. Fluconazole served as positive control for antifungal activity and in vitro preliminary screening values (zone of inhibition) are summarized in Table 4.
Most of the synthesized compounds exhibited moderate to good antibacterial activity. The compounds 5a, 5b, and 5c showed moderate activity against all tested strains. Compound 5d (R = 4-CH3) showed good antibacterial activity against all tested strains. It is also noticed that the activity retained at 3 µg/mL concentration. Thus, it is concluded that in case of thiosemicarbazide derivatives of 2-arylthiazole-4-carbaldehyde, compounds with R = 4-CH3 group show good antibacterial activity whereas compounds with unsubstituted phenyl ring and substitution with 4-Cl or 4-F phenyl group showed moderate activity.
Similarly, in case of thiosemicarbazide derivatives of 4-methyl-2-arylthiazole-5-carbaldehyde, the compounds 5f (R = 4-Cl) and 5h (R = 4-CH3) showed good antibacterial activity at 30 µg/mL concentration against all tested strains. Compounds 5e (R = 4-H) and 5g (R = 4-F) showed moderate antibacterial activity against all tested strains. Thus, it is concluded that compounds with R = 4-Cl or 4-CH3 group showed good antibacterial activity.
Among the thiosemicarbazide derivatives of 1-(4-methyl-2-arylthiazol-5-yl) ethanone, the compounds 5i (R = H) and 5k (R = 4-F) reported good activity at 30 µg/mL concentration, against all the four species. The compounds 5j (R = 4-Cl) and 5 l (R = 4-CH3) were found less active against all tested strains. In general, compound with phenyl group or 4-fluorophenyl at 2-position of thiazole moiety showed enhanced activity.
Careful analysis of the MICs in Table 3 provides compound 5d, 5h, 5i, and 5k as lead molecules with good antibacterial activity.
The result of antifungal activity (Table 4) revealed that most of the synthesized compounds showed moderate activity against C. albicans.
Cytotoxic activity
The synthesized compounds 5a–l were further assayed for their cytotoxic activity against the two different human cancer cell lines, cervix adenocarcinoma HeLa and colorectal carcinoma human colon carcinoma (HCT) 116 using MTT assay (Mosmann 1983; Ciapetti et al. 1993) with 48 h exposure time of the tested compounds. Rifampicin was used as positive control. The observed results are summarized in Table 5. The cytotoxic effect of these compounds was checked on cancer cell lines using the concentration range between 30, 10 and 3 μg/mL to determine the growth inhibition. The results indicated that, in MTT cytotoxicity studies most active compounds are leads as antimicrobials owing to no significant cell toxicity against HeLa and HCT 116 cell lines at the maximum concentration evaluated.
Conclusions
In conclusion, three series of thiosemicarbazide derivatives of 2-arylthiazole-4-carbaldehyde, 4-methyl-2-arylthiazole-5-carbaldehyde and 1-(4-methyl-2-arylthiazol-5-yl) ethanone were synthesized. The antimycobacterial activity studies were undertaken to evaluate the effects of substituents/group on the antitubercular and antimicrobial activities. From the SAR, in case of thiosemicarbazide derivatives of 2-arylthiazole-4-carbaldehyde, compounds with un-substituted phenyl ring showed excellent antitubercular activity whereas, substituents on phenyl ring significantly decrease the antitubercular activity. However, 4-CH3 substituted phenyl ring at 2-position of thiazole moiety is required for antibacterial activity. In case of thiosemicarbazide derivatives of 4-methyl-2-arylthiazole-5-carbaldehyde, 4-Cl and 4-CH3 substituted phenyl ring at 2-position of thiazole moiety is required for anti-tubercular, as well as antibacterial activities. However in case of thiosemicarbazide derivatives of 1-(4-methyl-2-arylthiazol-5-yl) ethanone, it is concluded that, compound with phenyl group or 4-fluorophenyl at 2-position of thiazole moiety showed enhanced activity. Most of the thiosemicarbazide derivatives showed moderate to good antimycobacterial activity.
References
Abhale YK, Deshmukh KK, Sasane AV, Chavan AP, Mhaske PC (2016) Fused heterocycles: synthesis and antitubercular activity of novel 6-substituted-2-(4-methyl-2-substituted phenylthiazol-5-yl)H imidazo [1,2-a]pyridine. J Heterocyclic Chem 53:229–233
Abhale YK, Sasane AV, Chavan AP, Deshmukh KK, Kotapalli SS, Ummanni R, Sayyad SF, Mhaske PC (2015) Synthesis and biological screening of 2′-aryl/benzyl-2-aryl-4-methyl-4′,5-bithiazolyls as possible anti-tubercular and antimicrobial agents. Eur J Med Chem 94:340–347
Abhale YK, Sasane AV, Chavan AP, Shekh SH, Deshmukh KK, Bhansali S, Nawale L, Sarkar D, Mhaske PC (2017) Synthesis and antimycobacterial screening of new thiazolyl-oxazole derivatives. Eur J Med Chem 132:333–340
Achkar JM, Fries BC (2010) Candida infections of the genitourinary tract. Clin Microbiol Rev 23(2):253–273
Alagarsamy V, Arun kumar B, Parthiban P, V Sheorey R, Raja Solomon V (2012) Synthesis and Antibacterial Activity of Some Novel 1-(4-oxo-3-(3- methoxyphenyl)-3H-quinazolin-2-yl)-4-(substituted) thiosemicarbazides. Anti-Infect Agents 10(2):105–110
Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR (1988) Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res 48(3):589–601
Arora S, Agarwal S, Singhal S (2014) Anticancer activities of thiosemicarbazides / thiosemi-carbazones: a review. Int J Pharm Pharm Sci 6:34–41
Cannon RD, Lamping E, Holmes AR, Niimi K, Baret PV, Keniya MV, Tanabe K, Niimi M, Goffeau A, Monk BC (2009) Efflux-mediated antifungal drug resistance. Clin Microbiol Rev 22(2):291–321
Casas JS, Garcia-Tasende MS, Sordo J (2000) Main Group metal complexes of semicarbazones and thiosemicarbazones. a structural review. Coord Chem Rev 209:197–261
Chornous VA, Grozav AN, Todoriko LD, Vovk MV (2014) Synthesis and biological activity of 4-chloro-1H-imidazole-5-carbaldehyde thiosemicarbazones. Pharma Chem J 47(10):524–526
Ciapetti G, Cenni E, Pratelli L, Pizzoferrato A (1993) In vitro evaluation of cell/ biomaterial interaction by MTT assay. Biomaterials 14(5):359–364
Gaikwad ND, Patil SV, Bobade VD (2012) Synthesis and biological evaluation of some novel thiazole substituted benzotriazole derivatives. Bioorg Med Chem Lett 22:3449–3454
Haraguchi SK, Silva AA, Vidotti GJ, V dos Santos P, Garcia FP, Pedroso RB (2011) Antitrypanosomal activity of novel benzaldehyde-thiosemicarbazone derivatives from kaurenoic acid. Molecules 16(2):1166–1180
Inamdar GS, Pandya AN, Thakar HM, Sudarsanam V, Kachler S, Sabbadin D, Moro S, Klotz KN, Vasu KK (2013) New insight into adenosine receptors selectivity derived from a novel series of [5-substituted-4-phenyl-1,3-thiazol-2-yl] benzamides and furamides. Eur J Med Chem 63:924–934
Johnson M, Antonio T, Reith ME, Dutta AK (2012) Structure-activity relationship study of N6-(2-(4-(1H-Indol-5-yl)piperazin-1-yl)ethyl)-N6-propyl-4,5,6,7-tetrahydrobenzo[d]thiazole-2,6-diamine analogues: development of highly selective D3 dopamine receptor agonists along with a highly potent D2/D3 agonist and their pharmacological characterization. J Med Chem 55(12):5826–5840
Kathiravan MK, Salake AB, Chothe AS, Dudhe PB, Watode RP, Mukta MS, Gadhwe S (2012) The biology and chemistry of antifungal agents: a review. Bioorg Med Chem 20(19):5678–5698
Khan A, Sarkar D (2008) A simple whole cell based high throughput screening protocol using Mycobacterium bovis BCG for inhibitors against dormant and active tubercle bacilli. J Microbiol Methods 73:62–68
Khan A, Sarkar S, Sarkar D (2008) Bactericidal activity of 2-nitroimidazole against the active replicating stage of Mycobacterium bovis BCG and Mycobacterium tuberculosis with intracellular efficacy in THP-1 macrophages. Int J Antimicrob Agents 32(1):40–45
Kucukguzel G, Kocatepe A, De Clercq E, Şahin F, Gulluce M (2006) Synthesis and biological activity of 4-thiazolidinones, thiosemicarbazides derived from diflunisal hydrazide. Eur J Med Chem 41:353–359
Kumari S, Sharma NK, KohliS (2012) Synthesis, characterization and antimicrobial studies of copper (II) complexes of semicarbazone and thiosemicarbazone of m-Hydroxy benzaldehydeand p-hydroxy benzaldehyde. Orient J Chem 28:969–974
Kunin CM, Ellis WY (2000) Antimicrobial activities of mefloquine and a series of related compounds. Antimicrob Agents Chemother 44(4):848–852
Lee YS, Kim H, Kim YH, Roh EJ, Han H, Shin KJ (2012) Synthesis and structure-activity relationships of tri-substituted thiazoles as RAGE antagonists for the treatment of Alzheimer’s disease. Bioorg Med Chem Lett 22(24):7555–7561
Leite ACL, de Lima RS, de M Moreira DR, de O Cardoso MV, de Brito Ana CG, dos Santos LMF (2006) Synthesis, docking, and in vitro activity of thiosemicarbazones, aminoacyl-thiosemicarbazides and acyl-thiazolidones against Trypanosoma cruzi. Bioorg Med Chem 14:3749–3757
Malik S, Bahare RS, Khan SA (2013) Design, synthesis and anticonvulsant evaluation of N-(benzo[d]thiazol-2-ylcarbamoyl)-2-methyl-4-oxoquinazoline-3(4H)-carbothioamide derivatives: a hybrid pharmacophore approach. Eur J Med Chem 67:1–13
Malki A, Elbayaa RY, Ashour HM, Loffredo CA, Youssef AM (2015) Novel thiosemicarbazides induced apoptosis in human MCF-7 breast cancer cells via JNK signaling. J Enzyme Inhib Med Chem 30(5):786–795
Moglea PP, Meshramb RJ, Hesea SV, Kamblea RD, Kambleb SS, Gaccheb RN, Dawanea BS (2016) Synthesis and molecular docking studies of new series of bipyrazol-ylthiazol-ylidene-hydrazinecarbothioamide derivatives as potential antitubercular agents. MedChemCom 7:1405-1420
Mavrova AT, Wesselinova D, Tsenov JA, Lubenov LA (2014) Synthesis and antiproliferative activity of some new thieno[2,3-d]pyrimidin-4(3H)-ones containing 1,2,4-triazole and 1,3,4-thiadiazole moiety. Eur J Med Chem 86:676–683
Mishra CB, Kumari S, Tiwari M (2015) Thiazole: a promising heterocycle for the development of potent CNS active agents. Eur J Med Chem 92:1–34
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65(1-2):55–63
Navale A, Pawar S, Deodhar M, Kale A (2013) Synthesis of substituted benzo[d]thiazol-2-ylcarbamates as potential anticonvulsants. Med Chem Res 22(9):4316–4321
Patel SR, Gangwal R, Sangamwar AT, Jain R (2014) Synthesis, biological evaluation and 3D-QSAR study of hydrazide, semicarbazide and thiosemicarbazide derivatives of 4-(adamantan-1- yl)quinoline as anti-tuberculosis agents. Eur J Med Chem 85:255–267
Perkoviisć I, Butula I, Kralj M, Martin-Kleiner I, Balzarini J, Hadjipavlou-Litina D, Katsori AM, Zorc B (2012) Novel NSAID 1-acyl-4-cycloalkyl/arylsemicarbazides and 1-acyl-5-benzyloxy/hydroxyl carbamoylcarbazides as potential anticancer agents and antioxidants. Eur J Med Chem 51:227–238
Pitucha M, Wos´ M, Miazga-Karska M, Klimek K, Mirosław B, Pachuta-Stec A, Gładysz A, Ginalska G (2016) Synthesis, antibacterial and antiproliferative potential of somenew 1-pyridinecarbonyl-4-substituted thiosemicarbazidederivatives. Med Chem Res 25:1666–1677
Plech T, Wujec M, Siwek A, Kosikowska U, Malm A (2011) Synthesis and antimicrobial activity of thiosemicarbazides, s-triazoles and their Mannich bases bearing 3-chlorophenyl moiety. Eur J Med Chem 46(1):241–248
Rane RA, Naphade SS, Bangalore PK, Palkar MB, Shaikh MS, Karpoormath R (2014) Synthesis of novel 4-nitropyrrole-basedsemicarbazide and thiosemicarbazide hybrids with antimicrobial and anti-tubercular activity. Bioorg Med Chem Lett 24:3079–3083
Sarkar S, Sarkar D (2012) Potential Use of Nitrate Reductase as a Biomarker for the Identification of Active and Dormant Inhibitors of Mycobacterium tuberculosis in a THP1 Infection Model. J Biomol Screen 17(7):966–973
Salgın-Goksen U, Gokhan-Kelekci N, Goktas O, Koysal Y, Kılıc E, Isık S, Aktay G, Ozalp M (2007) 1-Acylthiosemicarbazides,1,2,4-triazole-5(4H)-thiones, 1,3,4-thiadiazoles and hydrazones containing 5-methyl-2-benzoxazolinones: synthesis, analgesic, anti-inflammatory and antimicrobial activities. Bioorg Med Chem 15:5738–5751
Sheikhy M, Jalilian AR, Novinrooz A, Motamedi-Sedeh F (2012) Synthesis and in vitro antibacterial evaluation of some thiosemicarbazides and thiosemicarbazones. J Biomed Sci Eng 5:39–42
Siddiqui N, Ahsan W (2010) Triazole incorporated thiazoles as a new class of anticonvulsants: design, synthesis and in vivo screening. Eur J Med Chem 45(4):1536–1543
Singh R, Nawale LU, Arkile M, Shedbalkar UU, Wadhwani SA, Sarkar D, Chopade BA (2015) Chemical and biological metal nanoparticles as antimycobacterial agents: a comparative study. Int J Antimicrob Agents 46(2):183–188
Singh U, Akhtar S, Mishra A, Sarkar D (2011) A novel screening method based on menadione mediated rapid reduction of tetrazolium salt for testing of anti-mycobacterial agents. J Microbiol Methods 84:202–207
Sriram D, Yogeeswari P, Thirumurugan R, Pavana RK (2006) Discovery of new antitubercular oxazolyl thiosemicarbazones. J Med Chem 49:3448–3450
Tan OU, Ozadali K, Yogeeswari P, Sriram D, Balkan A (2012) Synthesis and antimycobacterial activities of some new N-acylhydrazoneand thiosemicarbazide derivatives of 6-methyl-4,5-dihydropyridazin-3(2H)-one. Med Chem Res 21:2388–2394
Tomasic T, Katsamakas S, Hodnik Z, Ilas J, Brvar M, Solmajer T, Montalvao S, Tammela P, Banjanac M, Ergovic G, Anderluh M, Masic LP, Kikelj D (2015) Discovery of 4,5,6,7-Tetrahydrobenzo[1,2-d]thiazoles as Novel DNA Gyrase Inhibitors Targeting the ATP-Binding Site. J Med Chem 58:5501–5521
Umadevi P, Deepti K, Srinath I, Vijayalakshmi G, Tarakaramji M (2012) Synthesis and in vitro antibacterial activity of some urea, thiourea and thiosemicarbazide derivatives. Int J Pharm Pharm Sci 4(3):379–383
Urbanek RA, Xiong H, Wu Y, Blackwell W, Steelman G, Rosamond J, Wesolowski SS, Campbell JB, Zhang M, Brockel B, Widzowski DV (2013) Synthesis and SAR of aminothiazole fused benzazepines as selective dopamine D2 partial agonists. Bioorg Med Chem Lett 23(2):543–547
Yusufzai SK, Osman H, Khan MS, Mohamad S, Sulaiman O, Parumasivam T, Gansau JA, Johansah N, Noviany (2017) Design, characterization, in vitro antibacterial, antitubercular evaluation and structure–activity relationships of new hydrazinyl thiazolyl coumarin derivatives. Med Chem Res. doi:10.1007/s00044-017-1820-2
Zhang HJ, Qian Y, Zhu DD, Yang XG, Zhu HL (2011) Synthesis, molecular modeling and biological evaluation of chalcone thiosemicarbazide derivatives as novel anticancer agents. Eur J Med Chem 46:4702–4708
Acknowledgements
Y.K.A. expresses her gratefulness to CSIR [File No. 08/590(0001)/2012-EMR-I] for the financial support. P.C.M. would like to thank University Grant Commission (UGC No. 42-355/2013(SR), New Delhi, India for the financial assistance and CSIR-NCL, Pune for supporting biological activity. Central Analysis facility, Savitribai Phule Pune University, Pune is also acknowledged for spectral analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Abhale, Y.K., Shinde, A., Deshmukh, K.K. et al. Synthesis, antitubercular and antimicrobial potential of some new thiazole substituted thiosemicarbazide derivatives. Med Chem Res 26, 2557–2567 (2017). https://doi.org/10.1007/s00044-017-1955-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-017-1955-1