
Abstract
A series of new pyrazolic heterocyclic compounds were prepared in good and excellent yields and characterized by proton and carbon nuclear magnetic resonance, infrared, and mass spectroscopy studies. These products were screened in vitro against three bacterial pathogens, namely Bacillus subtilis, Micrococcus luteus, and Escherichia coli and antifungal potential, against Fusarium oxysporum f.sp.albedinis. A considerable and excellent activity was recorded with respect to the two studied microorganisms. A good correlation was obtained between the experimental results and the theoretical predictions of bioavailability using Petra/Osiris/Molinspiration suite (Petra/Osiris/Molinspiration containing Lipinski’s rule-of-five). The quantitative structure activity relationship approach has been analyzed to support the Petra/Osiris/Molinspiration results and composite indexes of some quantum chemical parameters were constructed in order to characterize the inhibition performance of the tested molecules.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The incidence of bacterial and fungal infections has increased dramatically in recent years (Nilesh and Manish 2011). The decline of sensibility to antimicrobial agents in current use has also been increasing for a great variety of pathogens and the resistance to multiple drugs is more and more prevalent for several microorganisms (Fang et al. 2010). Therefore, the urgent need for discovery or optimization of antimicrobial agents active against these resistant strains is of paramount importance (Payne et al. 2007; Bayrak et al. 2009). The most widely used classes of antifungal drugs are known as the azoles, based on their common feature, an imidazole or triazole ring (Douglas and Jie 2007). In this context, pyrazole derivatives skeleton is a fertile source of biologically important molecules possessing a wide spectrum of biological and pharmacological activities such as anti-inflammatory (Tewari and Mishra 2001), anti-anxiety (Haufel and Breitmaier 1974; Wustrow et al. 1998), antipyretic (Wiley and Wiley 1964), antimicrobial (Pimerova and Voronina 2001), antiviral (Janus et al. 1999), antitumor (Park et al. 2005; Bouabdallah et al. 2006), anticonvulsant (Michon et al. 1995), antihistaminic (Yildirim et al. 2005), antidepressant (Bailey et al. 1985), insecticides (Chu and Cutler 1986), and fungicides (Manfredini et al. 2000). In view of these particulars, we report here the synthesis of some new derivatives of pyrazole. The constitution of all the products was characterized using 1H and 13C NMR, Infrared (IR) and mass spectrometry (MS) and their activities in vitro as antibacterial and antifungal agents were evaluated.
Results and discussion
Chemistry
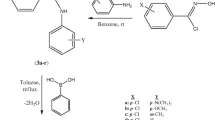
The synthetic route of target pyrazoles was illustrated in (Scheme 1). Compounds a and b were already reported by several old and recent works (Boussalah et al. 2009; Dvoretzky and Holmes 1950). All the pyrazolic compounds excepting 15, 16, and 18–20 were already described and published in the literature (Abrigach et al. 2014; Khoutoul et al. 2015; El Kodadi et al. 2004).

General synthetic pathway reaction
Biochemical evaluation of the synthetic compounds
The antifungal and antibacterial properties of compounds 1–14 and 17 have been previously reported by other work (El-youbi et al. 2015). In this part, we present the results obtained with the novel synthesized compounds 15, 16, and 18–20.
Antifungal activity
The pyrazolic derivatives 15, 16, and 18–20 were screened in vitro for their antifungal potential against Fusarium oxysporum f.sp.albedinis. The antifungal potential of each compound has been expressed in (Table 1) and the obtained results showed that the most of the compounds presented an inhibitory effect against using fungi. Especially, the compounds 15 and 20 which showed an excellent efficacy with an IC 50 (mM) = 0.086 and 0.168 respectively, may be due to the presence of the two phenyl moieties. The compound 18 showed a moderate potential with an IC50 (mM) = 0.284 probably due to the (–Br) group which is an important source of electronegativity. The two other tested pyrazoles, showed a poor antifungal activities.
Antibacterial activity
The examination of the data (Table 2) reveals that most of the tested compounds showed antibacterial activity against the three bacterial pathogens with varying efficacy from one bacterium to another and from one molecule to another. For each of the bacteria used in the test, we can classify the products tested by two different criteria, which are not correlated: the diameter of inhibition and the IC50.
Bacillus subtilis
Molecules (15—16—19—20) caused a diameter of inhibition greater than 18 mm. While molecule (18) had no effect (inhibition diameter = 0 mm) on this bacterium. However, the molecules 19 and 20 are the best inhibitors of this strain.
Micrococcus luteus
Molecule 19 caused a diameter of inhibition greater than 18 mm. While molecule 18 had no effect (inhibition diameter = 0 mm) on this bacterium.
E. coli
Molecule 16 caused a diameter of inhibition greater than 18 mm. While molecules 15 and 18 have no effect (inhibition diameter = 0 mm) on this bacterium.
From these preliminary antimicrobial screening results, it is interesting to note that a minor change in the molecular substitutes of the investigated compounds may have a pronounced effect on antimicrobial activity. It was clearly showed that the (–COOH) group and the phenyl moiety increase the antibacterial potential of our compounds.
Theoretical calculations of molecular properties
Computational methods have been used in several studies in order to comprehend differences between natural products and other sources of drug leads. Current drug discovery is vastly founded on screening of small molecules against macromolecular disease targets necessitating that molecular screening libraries comprising drug-like or lead-like compounds.
Semi empirical Hartree–Fock Austin model 1 (AM1) calculations
The geometric structures of molecules 1–20 have been optimized with the semi empirical Hartree–Fock AM1 method (Dewar et al. 1985), which has been parameterized to provide accurate geometries and physicochemical properties for organic molecules. Besides, the following chemical effects can also be quantified: bond dissociation energies, heats of formation, p-charge distribution, sigma charge distribution, resonance effect, inductive effect, delocalization energies, and polarizability effect. All the calculations were performed using the Gaussian 09 program and the calculated Mulliken atomic charges of the heteroatoms were used to model the bioactivity against bacterial and fungal. The result of the calculations of delocalized charge on heteroatoms for the molecules 1–20 under study is gathered in (Table 3) (Frisch et al. 2010).
The nitrogen atoms are negatively charged with the exception of N16 of the nitro group in molecule 3, which is positively charged because of the withdrawing character of the oxygen atoms of the nitro group O7 and O8. The sp2 hybridized nitrogen atom N3 of the pyrazole ring is slightly negatively charged and the maximum of negative charge for the nitrogen atoms is encountered for the atom N9 (in β position with respect to the nitrogen of the pyrazole ring) and lies between −0.234 for molecule 20 and −0.412 for molecule 13. The oxygen atoms bear negative charges while brome atoms are slightly positively charged. The maximum of negative charge for the oxygen atoms is encountered for the atom O7 (−0.379) of the nitro group in molecule 3.
Effort was made to integrate steric and indicator parameters which appeared as essential contributors from preceding pharmacologic analysis. The present results support the former observations that bulky substitution of the atom N9 such as: bromo-pyridine (compounds 4, 5, and 6), phenyl ring substituents (compounds 15 and 20) are conducive to the fungal activity. Meanwhile, antibacterial activity is enhanced by considering bulky substituents to the atom N9 like pyrimidine or pyrimidine with one or two (–OH) groups (Fig. 1).

Possible mechanisms for bacterial and fungal inactivation
The AM1 calculations established that all compounds 1–20 have a strong predilection for forming antibacterial and antifungal pharmacophore sites though their estimated partial p-charges respectively for nitrogen and oxygen atoms are of negative charges. Hence the negative charges of the oxygen and nitrogen atoms contribute positively in favor of an antibacterial activity and this is in good agreement with the mode of antibacterial action of the compounds bearing (Aδ−–Bδ−) involving coordination of the metal within the bacteria. It was assumed that alteration in charges between two heteroatoms of the same pharmacophore site (Aδ−–Bδ+) may help the inhibition of bacteria. It was also detected that the activity growths with growth in negative charge of the heteroatoms of the common pharmacophore fragment of the molecule. This is correlated with probable secondary electronic interaction with the positively charged side chains of the bacteria target(s).
On the basis of the above comments, it is proposed that compounds 1–20 display two combined antibacterial O and N pharmacophore sites in which the oxygen and nitrogen heteroatoms act as ligation centers and possibly accommodate themselves with two metals (M = Mg2+, and/or Mn2+) in such a way that a stable bimetallic complex [(L)M2(H2O)2] of the two pharmacophore sites for molecule 19 is formed therefore, triggering partial or complete bacterial inactivation. A different mechanism prevails for molecule 20, where nitrogen atoms behave as ligation centers and probably accommodate themselves with one metal (M = K+ or Na+) in such a way that a stable metallic complex of the pharmacophore site is formed therefore, prompting partial or complete bacterial inactivation.
The compounds 1–20 display one antifungal N, N-pharmacophore sites in which the nitrogen heteroatoms act as ligation centers and perhaps accommodate themselves with one metal (M = K+ or Na+) in such a way that a stable metallic complex of the pharmacophore site is formed (case of molecules 15 and 20 in Fig. 1).
Osiris calculations
The Osiris property explorer is one of the main computational programs available online. With this program, we can draw chemical structures and calculates different drug-relevant properties and therefore the prediction results are valued and coded. Properties with high risks of undesired effects like mutagenicity or a poor intestinal absorption are symbolized (−), whereas a (+) symbol indicates drug-conform behavior (Table 4). Scanning literatures shows that it is possible, using a combined electronic/structure docking procedure, to predict activity and/or inhibition with increasing success in two targets such as bacteria and HIV (Ben Hadda et al. 2013; Al Houari et al. 2008). The very well behaved mutagenicity of various synthetic molecules can be used to enumerate the role played by a number of organic groups in promoting or interfering with the way a drug can associate with DNA.
Molinspiration calculations
In Molinspiration program, the methodology used for the calculation of cLogP (octanol/water partition coefficient) is very robust and is able to process practically all organic and most organometallic molecules. The method is a sum of fragment-based contributions and correction factors. Molecular polar surface area (TPSA) is calculated on the methodology published by (Ertl et al. 2003) as a sum of fragment contributions. O- and N-centered polar fragments are considered. TPSA has been shown to be a very good descriptor characterizing drug absorption, including intestinal absorption, bioavailability, Caco-2 permeability and blood–brain barrier penetration. Prediction results of compounds 1–20 molecular properties (TPSA, GPCR ligand, and ICM) are valued (Table 5).
3D-QSAR
Data set and statistical analysis
Quantitative structure activity relationship (QSAR) has been derived for several sets of biological inhibitors (Goodarzi et al. 2010; Pourbasheer et al. 2010; Papa et al. 2010; Luilo and Cabaniss 2010) as attempts to find consistent relationship between the variations in the values of molecular properties and the inhibitor activity for a series of compounds. We have performed 3D-QSAR of antimicrobial and antifungal activities for pyrazole derivatives reported as IC50 (mM). The alignment independent descriptors viz. WHIM, Eva etc. available through e-Dragon (Tetko et al. 2005) were checked to develop robust QSAR model. In our statistical analysis, Matlab (Solomon and Breckon 2011) and Microsoft excel were used for various statistical calculations.
Therefore, in the present study, mathematical models were fitted to the experimental values of the fungal and microbial inhibition efficiencies of the 20 compounds, respectively (Table 6). The goals were, on the one hand, to find suitable equations in predicting IC50 from the concentrations of the inhibitors and their quantum chemical parameters, and on the other hand, to provide theoretical elucidations for the effects of the different variables studied. The model we have investigated is an empirical linear model expressed as:
IC 50 is the calculated dependent variable, β i the regression coefficient or constant obtained by regression analysis, X i the predictor variable or independent variable, ε the error.Where R 2 is the coefficient of determination, and SSE is the sum of squared errors defined as:
The experimental results of antifungal and antibacterial activities of compounds 1–20 gathered in Table 6 were fitted to the empirical model of Eq. (1) by forward multiple linear-regression, using the software package Matlab. For each form of activity, the size of the selected subset of independent variables was derived from an initial set of 1345 variables.
Multiple linear regression equations
The purpose of the statistical method used here (multiple linear regression) is to establish a quantitative relationship between a group of predictor variables and a response IC50 in the different studied cases (Antifungal, Bacillus subtilis, Micrococcus luteus and Escherichia coli). This relationship is useful for understanding which predictors have the greatest effect on the inhibition efficiency of a given compound. The variant used here after is based on least-squares fit, i.e., by minimizing the squares of the deviations of the data from the model. This Model can then be used to predict future values of IC50 when only reduced numbers of predictors are currently known.
In order to validate the model, the maximum of the absolute values of the deviation of the data from the model is given by:
where IC50exp is the vector of the experimental entity while IC50cal denote the vector of the corresponding calculated entity witch is given by the multilinear regression method. Besides, \({\rm{IC}}_{50{\rm{exp}}}^i\) denote the ith component of the IC50exp vector while \({\rm{IC}}_{{\rm{50cal}}}^i\) is the ith component of the IC50cal vector.
Our computations of the least-squares fit model using the multilinear regression equation for Fungal, B. subtilis, M. luteus, and E. coli give the following equations (2, 3, 4, and 5, respectively):
\(IC_{50}^{{\rm{Antifungal}}}\) = 103 (8.1 SMTIV—31.9 GMTI—8.0 GMTIV + 27.7 HyDp + 3.0 ww—2.8 Wap + 63.1 Whetp + 101.0 SRW08—11.9 SRW09—16.6 SRW10 + 2.4 IDMT—158.9 TIC2—9.1 W3D—159.6 L/Bw—3.5 QYYm + 4.8 QXXe—12.1 QZZe—58.8 Mor01m)
\(IC_{50}^{Bacillus\,\,subtilis}\) = 103 (19.4 GMTI—21.4 GMTIV + 83.7 HyDp + 12.0 ww—82.1 Wap—32.8 SRW08 + 18.5 SRW09 + 19.7 SRW10 + 27.8 IDMT—71.3 W3D + 7.7 QYYm + 61.8 QZZe −382.2 Mor01m)
\(IC_{50}^{Micrococcus\,luteus}\) = 103 (1017.5 SMTIV + 14.5 GMTI—290.7 GMTIV—545.2 HyDp—39.2 ww + 164.3 Wap—157.4 Whetp—1747.3 T(N..N)—392.9 SRW08 + 50.2 SRW09 + 17.0 SRW10 −183.8 IDMT + 193.1 W3D + 246.4 QYYm—974.2 QXXe—589.0 QZZe)
\({\rm{IC}}_{50}^{Escherichia\,\,coli}\) = 103 (109.6 SMTIV—146.6 GMTI + 7.8 GMTIV—839.5 HyDp—22.5 ww + 164.1 Wap—258.6 Whetp + 696.4 SRW08 + 92.3 SRW09—204.0 SRW10 + 444.4 IDMT + 56.1 W3D—94.1 QZZm + 1046.6 QYYe—568.5 QZZe).
These equations produce close estimate of IC50cal and are therefore useful in predicting the inhibition efficiency. In other words, these equations traduce the correlation between experimental inhibition efficiency IC50exp and calculated inhibition efficiency IC50cal obtained for fungal, B. subtilis, M. luteus, and E. coli QSAR models, respectively from Eq. (1).
Noteworthy, the calculation of R2 and \(R_{{\rm{adj}}}^2\) for the four models give the value of 1 while the values of SSE are: 9.3064 × 10−26, 1.4929 × 10−25, 1.9288 × 10−24, and 4.9425 × 10−24 for fungal, B. subtilis, M. luteus, and E. coli QSAR models, respectively.
From Table 7 gathering the calculated deviation between IC50cal and IC50exp for the different QSAR models under study, it can be seen that the estimates are very close to the experimental values. An inspection of the residuals shows that the maximum of the absolute value of the deviation of the data calculated for fungal, B. subtilis, M. luteus, and E. coli QSAR models (ErrMax) is 2.3415 × 10−13 (10−3 M), 1.4744 × 10−13 (10−3 M), 1.1546 × 10−12 (10−3 M) and 7.7094 × 10−13 (10−3 M), respectively.
The multiple-linear regression analyses fitted the theoretical data very well and the calculated inhibition efficiencies of fungal, B. subtilis, M. luteus, and E. coli, were found to be close to their experimental inhibition efficiencies. The results obtained in this study showed that undeniably, the QSAR approach is adequately appropriate to forecast the inhibitor efficiency using the theoretical approach.
Experimental
General
Melting points were determined on a Kofler bench melting point apparatus and are uncorrected. The 1H NMR spectra and 13C NMR spectra were recorded on a Bruker 300 (operating at 300.13 MHz for 1H, 75.47 MHz for 13C) spectrometer. Chemical shifts are reported in parts per million (ppm) downfield from an internal trimethylsilane reference. The band positions on IR spectra are reported in reciprocal centimeters (cm−1) on a Shimadzu IR spectrophotometer using the KBr disk technique. Mass spectra (MS) were obtained by using electrospray ionization (ESI) technique.
General procedure for the synthesis of ligands
The target pyrazoly compounds (Scheme 2), were prepared in one step by condensation of one equivalent of (3,5-dimethyl-1H-pyrazol-1-yl) methanol ((1H-pyrazol-1-yl) methanol) with one equivalent of an appropriate amine in 20 mL of acetonitrile (CH3CN) as solvent. All reactions were carried out at reflux for 4 h. The liquid residue is dried over MgSO4, filtered and concentrated in vacuum.

Chemical structures of studied compounds
N-((3,5-dimethyl-1H-pyrazol-1-yl)methyl)-N-phenylbenzenamine (15)
White solid; yield: 61.92%. m.p. 78–80 °C. IR (KBr, ν (cm−1)): 3203–2870 (CH); 1591 (C=N); 1555 (C=C); 1496 (C–N aromatic); 1220 (C–N aliphatic). 1H NMR (300 MHz, CDCl3) δ ppm: 7.33–6.90 (m, 10H, two phenyl); 5.81 (s, 2H, N–CH 2 –N); 5.72 (s, 1H, CH (4) of pyrazole) 2.24 (s, 3H, CH3 (5) of pyrazole); 1.76 (s, 3H, CH3 (3) of pyrazole). 13C NMR (75 MHz, CDCl3) δppm: 147.73 (N–C phenyl); 129 (N–C–C phenyl); 122 (N–C–C–C phenyl); 105.91 (CH (4) of pyrazole); 65.20(N–CH 2 –N); 13.63 (CH3 (3) of pyrazole); 10.95 (CH3 (5) of pyrazole). MS [M+] (m/z): calculated 277.16, found 317 (M+ + K + 1).
N-((3,5-dimethyl-1H-pyrazol-1-yl)methyl)-1-phenylethanamine (16)
White solid; yield: 69.35%. m.p. 68 °C. IR (KBr, ν (cm−1)): 3444 (NH); 3290–2843 (CH); 1604 (C=N); 1545 (C=C); 1454 (C–N aromatic); 1269 (C–N aliphatic). 1H NMR (300 MHz, CDCl3) δ ppm: 7.31–7.21(m, 5H, phenyl); 5.72 (s, 1H,CH(4) of pyrazole); 4.72 (s, 2H, N–CH 2 –N); 4.56(s, 1H, NH);3.60 (q, 1H, CH aliphatic); 2.22 (s, 3H, CH3 (5) of pyrazole); 1.89 (s, 3H, CH3 (3) of pyrazole). 13C NMR (75 MHz, CDCl3) δ ppm: 104.74 (CH (4) of pyrazole); 59.65(N–CH 2 –N); 53.63 (CH aliphatic); 13.63 (CH3 (3) of pyrazole); 10.95 (CH3 (5) of pyrazole). MS [M+] (m/z): calculated 229.16, found 230 (M+ + 1).
N-((1H-pyrazol-1-yl)methyl)-5-bromopyridin-2-amine (18)
Solid; Yield: 33.40%; m.p. 168 °C. IR (KBr, ν (cm−1)): 3422(NH); 3291–2959 (CH); 1581(C=N); 1498(C=C); 1365(C–N aromatic); 1280 (C–N aliphatic); 626 (C–Br). 1H NMR (300 MHz, CDCl3) δ ppm: 8.16 (d, 1H, CH(6) of pyridine, J H–H = 3 Hz); 8.13 (d, 1H, CH(4) of pyridine, J H–H = 3 Hz); 7.77 (d, 1H, CH(3) of pyrazole, J H–H = 3 Hz); 7.73 (d, 1H, CH(5) of pyrazole, J H–H = 3 Hz); 7.52 (d, 1H, CH(3) of pyrazole, J H–H = 3 Hz); 6.21 (t, 1H, CH(4) of pyrazole, J H–H = 3 Hz); 5.78 (d, 2H, N–CH 2 –N, J H–H = 6 Hz); 5.08 (t, 1H, NH, J H–H = 6 Hz). 13C NMR (75 MHz, CDCl3) δ ppm: 147(CH(4) of pyridine); 139 (CH(3) of pyrazole); 130.23 (CH(5) of pyrazole); 105.45 (CH(4) of pyrazole); 56.73 (N–CH 2 –N). MS [M+] (m/z): calculated 252, found 252.
4-((1H-pyrazol-1-yl)methylamino)benzoic acid (19)
White solid; yield: 91.29%; m.p. 158–162 °C. IR (KBr, ν (cm−1)): 3296 (NH); 3118–2524 (OH); 1683 (C=O); 1606 (C=N); 1546 (C=C); 1431 (C–N aromatic); 1267 (C–N aliphatic). 1H NMR (300 MHz, DMSO) δ ppm: 12.18 (s, 1H, OH); 7.79 (d, 2H, CH (2) of benzene, J H–H = 3 Hz); 7.66 (d, 1H, CH(3) of pyrazole, J H–H = 3 Hz); 7.43 (d, 1H, CH(5) of pyrazole, J H–H = 3 Hz); 6.82 (d, 2H, CH(3) of benzene, J H–H = 9 Hz); 6.21 (t, 1H, CH(4) of pyrazole, J H–H = 9 Hz); 5.49 (d, 2H, N–CH 2 –N, J H–H = 6 Hz); 3.39 (s, 1H, NH). 13C NMR (75 MHz, DMSO) δ ppm: 167.78 (–C=O); 150.99 (C (4)–NH); 139.15 (C(3) of pyrazole); 131.46 (CH(2) of benzene); 129.57 (C(5) of pyrazole); 119.58 (C(1) of benzene); 112.52 (CH(3) of benzene); 105.88 (CH(4) of pyrazole); 58.81 (N–CH 2 –N). MS [M+] (m/z): calculated 217.09, found 243 (M+ + Na + 3).
N-((1H-pyrazol-1-yl)methyl)-N-phenylbenzenamine (20)
White solid; yield: 68.53%. m.p. 74–78 °C .IR (KBr, ν (cm−1)): 3104–2837 (CH); 1593 (C=N); 1492 (C=C); 1357 (C–N aromatic); 1242 (C–N aliphatic). 1H NMR (300 MHz, CDCl3) δ ppm: 5.76 (d, 1H, CH(3) of pyrazole, J H–H = 3 Hz); 5.38(d, 1H, CH(5) of pyrazole, J H–H = 3 Hz) 7.33–7.02 (m, 10H, two phenyl); 6.22 (t, 1H,CH(4) of pyrazole, JH–H = 3 Hz) ;6.01 (s, 2H, N–CH 2 –N). 13C NMR (75 MHz, CDCl3) δppm: 146.59 (N–C phenyl); 129.58 (N–C–C phenyl); 12,290 (N–C–C–C phenyl); 105.96 (CH(4) of pyrazole); 68.08 (N–CH 2 –N). MS [M+] (m/z): calculated 249.13, found 249.
Screening for antifungal activity
The antifungal activity was determined by the agar diffusion technique as described in the literature. The Fusarium oxysporum f.sp.albedinis was isolated and prepared in PDA (potato dextrose agar) medium (Boussalah et al. 2013). The agar media were incubated (at 28 °C/7 days) with the micro-organisms and a solution of the tested compound in DMSO at different concentrations (40, 80, 160, 320, and 640 μg mL−1). The inhibition percentage of a molecule is equal to the mycelium diameter of the culture in the presence of a dose of the tested compound over the mycelium diameter of the reference culture multiplied by 100. The half-maximal inhibitory concentration (IC50) was determined by the linear regression equation between the natural logarithm of the concentrations and the growth inhibition percentages.
Screening for antibacterial activity
The antibiotic effect of the pyrazolic agents was tested on three bacterial strains (Table 8) in accordance with the requirements of the National Committee for Clinical Laboratory Standards (NCCLS) recommended by the World Health Organization (WHO) and the French norm NF—U—47-107 (Wallace 2009; Olajuyigbe et al. 2014).
A Petri plate containing Muller–Hinton agar was seeded by 5 mL of bacterial test inoculum at 106 CFU mL−1. The excess of bacterial solution was removed and the WATTMAN paper disks (6 mm diameter) which had previously been soaked in the different tested compounds were placed on the inoculated agar plates. After 24 h of incubation, the antibacterial activity was assessed by measuring the diameter of the growth-inhibition zone in millimeters. The measurements of inhibition zones were carried out three times for each drug including Gentamicin (1 mg mL−1) as a positive control and distilled water as a negative control. The half-maximal inhibitory concentration (IC50) was calculated using the same bacterial strains mentioned above with decreasing concentration of the tested drugs. The Optical density was measured of each culture at 625 nm after 6 h of incubation.
Conclusion
In conclusion, a series of pyrazole derivatives were prepared by simple condensation between a pyrazole moiety and primary amines. All new compounds were characterized by MS, IR, 1H, and 13C NMR. The in vitro antimicrobial activities of these pyrazole derivatives were determined against three bacterial strains (B. subtilus, M. luteus, and E. coli) and one fungal strain (Fusarium oxysporum f.sp.albedinis). The preliminary results of these compounds against the studied micro-organisms showed that some compounds exhibited significant antibacterial (19 and 20) and antifungal activities (15 and 20). However, the other compounds gave moderate to poor potency. In these series compounds, it was clearly showed that the (–COOH), (–Br) groups and the phenyl moiety are more helpful for increasing antibacterial and antifungal efficacy respectably, in comparison with other substitutes. The present study throws light on the identification of these new structural classes as antibacterial and antifungal. Further developments on this subject are currently in progress at our laboratory to determine and understand the mechanism of this remarkable process. The QSAR approach has been employed in this study and a good relationship was found between our experimental and calculated results in this work. The calculated inhibition efficiency was found to be very close to the experimental inhibition with R2 and \(R_{{\rm{adj}}}^2\) equal to 1 while the values of SSE are: 9.3064 × 10−26, 1.4929 × 10−25, 1.9288 × 10−24, and 4.9425 × 10−24 for fungal, B. subtilis, M. luteus and E. coli QSAR models, respectively.
References
Abrigach F, Khoutoul M, Benchat N, Radi S, Draoui N, Feron O, Riant O, Touzani R (2014) Library of synthetic compounds based on pyrazole unit: design and screening against breast and colorectal cancer. Lett Drug Des Discov 11:1010–1016
Al Houari G, Kerbal A, Bennani B, Baba MF, Daoudi M, Ben Hadda T (2008) Drug design of new antitubercular agents: 1,3-dipolar cycloaddition reaction of arylnitriloxides and 3-para-methoxybenzylidene-isochroman-4-ones. Arkivoc 12:42–50
Bailey DM, Hansen PE, Hlavac AG, Baizman ER, Pearl J, Defelice AF, Feigenson ME (1985) 3,4-Diphenyl-1H-pyrazole-1-propanamine antidepressants. J Med Chem 28:256–260
Bayrak H, Demirbas A, Karaoglu SA, Demirbas N (2009) Synthesis of some new 1,2,4-triazoles, their Mannich and Schiff bases and evaluation of their antimicrobial activities. Eur J Med Chem 44:1057–1066
Ben Hadda T, Fathi J, Chafchaouni I, Masand V, Charrouf Z, Chohan ZH, Jawarkar R, Fergoug T, Warad I (2013) Computational POM and 3D-QSAR evaluation of experimental in vitro HIV-1-Integrase inhibition of amide-containing diketoacids. Med Chem Res 22:1456–1464
Bouabdallah I, M’barek LA, Zyad A, Ramadan A, Zidane I, Melhaoui A (2006) Anticancer effect of three pyrazole derivatives. Nat Prod Res 20:1024–1030
Boussalah N, Touzani R, Bouabdallah I, El Kadiri S, Ghalem S (2009) Synthesis, structure and catalytic properties of tripodal amino-acid derivatized pyrazole-based ligands. J Mol Catal A Chem 306:113–117
Boussalah N, Touzani R, Souna F, Himri I, Bouakka M, Hakkou A, Ghalem S, El Kadiri S (2013) Antifungal activities of amino acid ester functionalpyrazolyl compounds against Fusarium oxysporum f.sp.albedinis and Saccharomyces cerevisiae yeast. J Saudi Chem Soc 17:17–21
Chu CK, Cutler J (1986) Chemistry and antiviral activities of acyclonucleosides. J Heterocycl Chem 23:289–319
Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP (1985) Development and use of quantum mechanical molecular models. AM1: a new general purpose quantum mechanical molecular model. J Am Chem Soc 107:3902–3909
Douglas SJ, Jie JL (2007) The Art of drug synthesis. Wiley, Hoboken, NJ
Dvoretzky R, Holmes G (1950) Formaldehyde condensation in the pyrazole series. J Org Chem 15:1285–1288
El Kodadi M, Malek F, Ramdani A (2004) 1-(4-{[(3,5-dimethyl-1H-pyrazol-1-yl)methyl] amino} phenyl) ethanone. Molbank 1:M369
El-youbi M, Benabbes R, Lahmass I, Abrigach F, Khoutoul M, Benchat N, Bouakka M, Touzani R, Saalaoui E (2015) Antibacterial and antifungal activities of new pyrazolic compounds. Moroccan J Biol 12:9–13
Ertl P, Rohde B, Selzer P (2003) Fast calculation of molecular Polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem 43:3714–3717
Fang B, Cheng HZ, Xian CR (2010) Synthesis and biological activities of novel amine-derived bis-azoles as potential antibacterial and antifungal agents. Eur J Med Chem 45:4388–4398
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken B, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski KG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2010) Gaussian 09, Revision D.01. Gaussian, Inc., Wallingford CT
Goodarzi M, Freitas MP, Ghasemi N (2010) QSAR studies of bioactivities of 1-(azacyclyl)-3-arylsulfonyl-1H-pyrrolo[2,3-b]pyridines as 5-HT6 receptor ligands using physicochemical descriptors and MLR and ANN-modeling. Eur J Med Chem 45:3911–3915
Haufel J, Breitmaier E (1974) Synthesis of pyrazolo heteroaromatic compounds by means of 5-amino-3-methyl-l-phenylpyrazole-4-carbaldehyde. Angew Chem 13:604–604
Janus SL, Magdif AZ, Erik PB, Claus N (1999) Synthesis of triazenopyrazole derivatives as potential inhibitors of HIV-1. Monatsh Chem 130:1167–1174
Khoutoul M, Abrigach F, Zarrouk A, Benchat N, Lamsayah M, Touzani R (2015) New nitrogen donor pyrazolic ligands for an excellent liquid–liquid extraction of Fe2+ metal ion in aqueous solution with theoretical study. Res Chem Intermed 41:3319–3334
Luilo GB, Cabaniss SE (2010) Quantitative structure−property relationship for predicting chlorine demand by organic molecules. Environ Sci Technol 44:2503–2508
Manfredini S, Baraldi PG, Bazzanini R, Durini E, Vertuani S, Pani A, Marceddu T, Demontis F, Vargiu L, La Colla P (2000) Pyrazole related nucleosides 5. Synthesis and biological activity of 2′-deoxy-2′,3′-dideoxy- and acyclo-analogues of 4-iodo-1-beta-d-ribofuranosyl-3-carboxymethyl pyrazole. Nucleosides Nucleotides Nucleic Acids 19:705–722
Michon V, DuPenhoat CH, Tombret F, Gillardin JM, Lepagez F, Berthon L (1995) Preparation, structural analysis and anticonvulsant activity of 3- and 5-aminopyrazole N-benzoyl derivatives. Eur J Med Chem 30:147–155
Nilesh JT, Manish PP (2011) Synthesis, characterization, and antimicrobial evaluation of carbostyril derivatives of 1H-pyrazole. Saudi Pharm J 19:75–83
Olajuyigbe OO, Oyedeji O, Adedayo O (2014) Evaluation of the vitro interaction of amoxicillin and cotrimoxazole antibiotics against resistant bacterial strains. J Appl Pharm Sci 4:94–100
Papa E, Kovarich S, Gramatica P (2010) QSAR modeling and prediction of the endocrine-disrupting potencies of brominated flame retardants. Chem Res Toxicol 23:946–954
Park HJ, Lee K, Park S, Ahn B, Lee JC, Cho HY, Lee KI (2005) Identification of antitumor activity of pyrazole oxime ethers. Bioorg Med Chem Lett 15:3307–3312
Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL (2007) Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 6:29–40
Pimerova EV, Voronina EV (2001) Antimicrobial activity of pyrazoles and pyridazines obtained by interaction of 4-aryl-3-arylhydrazono-2,4-dioxobutanoic acids and their esters with hydrazines. Pharm Chem J 35:602–604
Pourbasheer E, Riahi S, Ganjali MR, Norouzi P (2010) Quantitative structure–activity relationship (QSAR) study of interleukin-1 receptor associated kinase 4 (IRAK-4) inhibitor activities by the genetic algorithm and multiple linear regression (GA-MLR) method. J Enzyme Inhib Med Chem 25:844–853
Solomon C, Breckon T (2011) Fundamentals of digital image processing: A practical approach with examples in Matlab. John Wiley & Sons, Ltd., Publication.
Tetko IV, Gasteiger J, Todeschini R, Mauri A, Livingstone D, Ertl P, Palyulin VA, Radchenko EV, Zefirov NS, Makarenko AS, Tanchuk VY, Prokopenko VV (2005) Virtual computational chemistry laboratory - design and description. J Comput Aid Mol Des 19:453–463
Tewari AK, Mishra A (2001) Synthesis and anti-inflammatory activities of N4,N5-disubstituted-3-methyl-1H-pyrazolo[3,4-c]pyridazines. Bioorg Med Chem 9:715–718
Wallace D (2009) Rapid culture methods for the microbiological examination of foods. Food Sci Technol 23:44–45
Wiley RH, Wiley P (1964) Chemistry of heterocyclic compounds: pyrazolones, pyrazolidones, and derivatives. Wiley, New York, Vol 20
Wustrow DJ, Capiris T, Rubin R, Knobelsdorf JA, Akunne H, Davis MD, MacKenzie R, Pugsley TA, Zoski KT, Heffner TG, Wise LD (1998) Pyrazolo[1,5a]pyrimidine CRF-1 receptor antagonists. Bioorg Med Chem Lett 8:2067–2070
Yildirim I, Ozdemir N, Akçamur Y, Dinçer M, Andaç O (2005) 4-Benzoyl-1,5-diphenyl-1H-pyrazole-3-carboxylic acid methanol solvate. Acta Cryst E61:256–258
Acknowledgements
The authors want to thank professor Olivier Riant from UNIVERSITE CATHOLIQUE DE LOUVAIN, Institut de la Matière Condensée et des Nanosciences (IMCN), Pôle Molécules, Solides et Réactivité (MOST), the CUD of Belgium and the CNRST of Morocco for its generous support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors confirm that this article content has no conflict of interest.
Rights and permissions
About this article

Cite this article
Abrigach, F., Karzazi, Y., Benabbes, R. et al. Synthesis, biological screening, POM, and 3D-QSAR analyses of some novel pyrazolic compounds. Med Chem Res 26, 1784–1795 (2017). https://doi.org/10.1007/s00044-017-1888-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-017-1888-8