
Abstract
Hypochaeris leontodontoides is a rupicolous species endemic to the Atlas Mountains (Morocco), where it occurs in scattered populations. This study aims to understand the biogeographic structure of a high mountain species in a rather small area of NW Africa. We used Amplified Fragment Length Polymorphism (AFLP) to investigate the population structure and phylogeography of H. leontodontoides in 19 populations sampled from the entire species distribution range. Multivariate analyses including PCoA, UPGMA analysis, and Bayesian clustering were applied to infer the influence of past biogeographic events. The AFLP differentiation among the populations was high (F ST = 0.508). A significant geographical pattern by mountain region was found. The different phylogeographical analyses revealed four main groups corresponding to four well-defined geographic regions: Middle Atlas, Eastern High Atlas, Central High Atlas, and Western High Atlas, and highlighted the Western High Atlas as the most divergent group. Our data also indicate two regions as refuges during the Pleistocene ice ages: the Middle Atlas and the northernmost area of the Western High Atlas.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Phylogeographical research has been conducted on many organisms worldwide. However, the geographical coverage of such studies is unevenly distributed, with some areas having numerous accounts, whilst others remain largely unexplored. A notable regional contrast exists between much of Europe vs North Africa, with the latter remaining poorly studied. This imbalance is unfortunate because North Africa harbours numerous major “hotspots” of plant biodiversity and endemism (Médail and Quézel 1997), and is also one of the regions of the world that is likely to suffer marked negative effects with the advance of global warming, in particular a significant increase in aridity (IPCC 2001). Due to its climatic history and strikingly montane topography, phylogeographical surveys typically detect a much greater allelic richness and evolutionary divergence of populations in this region compared with those further north (Fineschi et al. 2002; Hampe et al. 2003; Petit et al. 2003), and they may trace range dynamics dating back well into the Tertiary (Lumaret et al. 2002; Caujapé-Castells and Jansen 2003; Magri et al. 2007). For North African taxa, detailed range-wide population surveys of exclusively Moroccan plant species are limited (but see El Mousadik and Petit 1996; Terrab et al. 2006, 2009) and since the fossil record of this region is also fragmentary (Elenga et al. 2000; Magri and Parra 2002), the past vegetation dynamics in the area and their impacts on gene pools remain largely unknown.
The altiplano of the Atlas mountains of Morocco presents an extensive range that constitutes the biogeographical province known as Atlasica, which contains the highest mountains of North Africa, several of which exceed 4000 m, and which is subdivided into the Middle Atlas, High Atlas and Anti Atlas (Benabid and Fennane 1994; Deil and Galán de Mera 1996). The Atlas Mountains are situated at the Africa–Eurasia plate boundary, formed during the Mesozoic and Cenozoic by the uplift and deformations of Palaeozoic terrains (Hughes et al. 2011). The uppermost mountains comprise Palaeozoic granites and volcanic lavas such as basalt, andesite and rhyolite, whilst elsewhere the mountains are formed by uplifted and deformed Mesozoic carbonate rocks (Dresch 1941; Piqué 1994; Pouclet et al. 2007). The highest peaks are situated at the Western High Atlas and culminate in the Jbel Toubkal massif (4167 m).
In an ongoing study of the phylogeography and genetic structure of populations of species of the genus Hypochaeris L. (Tremetsberger et al. 2006, 2009; Ortiz et al. 2007, 2008; Terrab et al. 2009) we here focus on H. leontodontoides Ball, a rupicolous species that is endemic to the Atlasica region (NW Africa), where it occurs in isolated populations from 1800 to 3200 m. A molecular phylogenetic study using ITS and chloroplast (rps16 intron) markers has shown that H. leontodontoides is the basal species of the sect. Seriola Benth and Hooker (Tremetsberger et al. 2005), and molecular clock estimations indicate that sect. Seriola has been separated from sect. Hypochaeris since the Miocene, about 6.9 Mya ago. Within sect. Seriola, H. leontodontoides separated from H. laevigata (L.) Ces. and al. in the Pliocene, about 3.2 Mya ago (Tremetsberger et al. 2013).
We used Amplified Fragment Length Polymorphism (AFLP) to investigate the phylogeographic structure of H. leontodontoides in order to understand the processes that have shaped its current spatial genetic pattern. Additionally, the nuclear ribosomal internal transcribed spacer (nrITS) was analysed in six populations to potentially further assess genetic differences among populations. Few phylogeographical studies of rupicolous species are known for NW Africa (but see Vargas et al. 1999), contrary to the diverse studies in Europe, especially in the Alps and Pyrenees (e.g. López-Pujol et al. 2001; Jiménez et al. 2002; Segarra-Moragues et al. 2007; Segarra-Moragues and Catalán 2008; Kropf et al. 2012; Winkler et al. 2012, 2013; Christe et al. 2014). Within the context of our AFLP parameters, we aimed at evaluating the influence of geomorphological structures and past climate changes on population differentiation across the whole range of the species. The palaeoclimatologic information available for the studied region was used to infer the history of past changes leading to the present-day distribution of the species. Specifically, we addressed the following questions: (1) Does the current genetic population structure of H. leontodontoides show any geographical pattern? (2) Which region appears as the most probable ancestral area of the species, and what are the possible colonisation routes that gave rise to its current distribution? (3) Is the current genetic structure of the species a result of isolation by distance? (4) How did Pleistocene events shape the distribution of this species? (5) Did any areas serve as ice-age refugia for populations of this species?
Materials and methods
Species
Hypochaeris leontodontoides [section Seriola] was described by Ball (1873), as a perennial rupicolous plant growing on calcareous, porphyry or granite rocks between 1800 and 3650 m in the Middle and High Atlas mountain regions (Morocco). The plant is woody at the base, with monocephal scapes, and entire and toothed leaves that are glabrous to hairy. Plants with hairy leaves were described as var. villosa by Maire (1926), but this feature is very variable and does not warrant taxonomic recognition (Talavera, pers. obs). Galland and Favarger (1985) described H. leontodontoides var. glauca from the Rif Mountains (N Morocco), and Galán de Mera et al. (1999) described H. leontodontoides var. atlantica from the Middle Atlas but judging from their descriptions, both are synonyms of H. laevigata. The chromosome number of H. leontodontoides is 2n = 12 (Oberprieler and Vogt 2002). H. leontodontoides is an allogamous plant pollinated by small and medium-sized solitary bees (Talavera, pers. obs.), and greenhouse experiments (Ortiz et al. unpublished) have shown that it is a self-incompatible species.
Plant material and DNA extraction
We obtained plant material from 19 populations of H. leontodontoides (Table 1; Fig. 1a) from four mountain regions: Middle Atlas (five populations), Eastern High Atlas (five populations), Central High Atlas (three populations) and Western High Atlas (six populations). These areas effectively represent the entire distribution range of this species. For each locality we collected fresh leaf samples from 9–19 individuals (mean = 14), with a total of 265 individuals. Samples were dried and stored in silica gel until DNA extraction. Vouchers of all populations sampled were deposited in the Herbarium of the University of Seville (SEV, Spain).

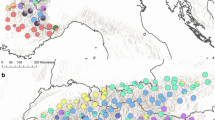
a Localities of the 19 sampled populations of H. leontodontoides (for details of numbered circles see Table 1). Grey area indicates the Atlas Mountain range, in Morocco. Elevations shown at 1000 and 3000 m a. s. l. (Map modified from Deil and Galán de Mera 1996). The bars indicate the mean number ±SE of the private fragments (F priv; min–max:4–10), the rare fragments index (DW; min–max:34–58) and the genetic diversity (H D; min–max: 0.065–0.094). Colour coding of populations indicates results of Bayesian clustering at K = 4. b Population structure at individual level for Bayesian Analysis conducted with STRUCTURE with optimal value, K = 2, and suboptimal values K = 3 and K = 4
Total genomic DNA was extracted from dry leaf using the unmodified Invisorb® Spin Plant Mini Kit protocol. Quality and quantity of extracted DNA were determined electrophoretically after SYBR green staining using a ladder with known amounts of DNA as standards (HyperLadder™, Bioline) and by using a NanoDrop UV–Vis spectrophotometer (Thermo®).
nrDNA ITS analyses
One randomly chosen individual per population from six populations (pops. 1, 5, 8, 14, 15 and 17) of H. leontodontoides was used in the nrDNA ITS analyses (Genbank No. KP226625 through KP226630; see Table 1). In the ITS analysis we also included the only sequence of H. leontodontoides available in GenBank (AJ627266.1), and two individuals of H. achyrophorus L. (sect. Seriola) were used as outgroup (GenBank No. KP226631 and KP226632). ITS of 18S–5.8S–28S nuclear ribosomal DNA was amplified using primers ITS4 and ITS5 published by White et al. (1990). The Polymerase Chain Reaction (PCR) was optimised with the following conditions for each sample: 25 μl reaction contained 12.6 µl H2O, 2.5 μl of buffer 10×, 1.6 μl MgCl 50 mM, 2 µl dNTPs 10 mM, 1 µl BSA 1×, 1 µl DMSO, 1 µl of each primer (forward and reverse), 0.3 μl of Taq DNA polymerase (Bioline) 5 and 2 μl template DNA (approx. 30–100 ng/µl). Amplification was performed in a thermal cycler (Applied Biosystems Thermal Cycler) with the following reaction conditions: 94 °C/5 min (one cycle); 94 °C/1 min, 49 °C/30 s, 72/1 min (30 cycles) and 72 °C/15 min (1 cycle); 4 °C at the end. The amplified fragments were checked with 1.5 % agarose gel and purified using Exosap-IT enzyme (Bioline). The purified fragments were sequenced on an ABI 3730xl sequencer in collaboration with STAB vida laboratory. The sequences were analysed using Geneious software to generate consensus sequences for each sample (Drummond et al. 2011). The alignment of the complete sequences of ITS1, 5.8S and ITS2 were analysed using the MEGA version 6 (Tamura et al. 2013) and Clustal IW software (Thompson et al. 2002). To select the model of nucleotide evolution that better suited to the data we used Modeltest 3.06 (Posada and Crandall 1998), according to the Akaike criterion. The Model used was the Kimura 2 parameter (K2). A heuristic search was carried out under the maximum likelihood (ML) criterion with 100 random addition sequence replicates and other settings kept as default with PAUP* ver. 4.0b10 (Swofford 2003). Bootstrap support (BP) for each node was estimated with 500 bootstrap replicates with PAUP* ver. 4.0b10 (Swofford 2003).
AFLP analyses
The AFLP procedure followed established protocols (Vos et al. 1995). An initial screening of selective primers using 36 primer combinations with three selective nucleotides was performed on eight individuals of four populations (2 individuals per population). The six primer combinations selected for the selective PCR were EcoRI (Fam)-ATC/MseI-CTC, EcoRI (Fam)-ATC/MseI-CTGA, EcoRI (Vic)-ACG/MseI CAT, EcoRI (Vic)-AGG/MseI CTC, EcoRI (Ned)-AGC/MseI-CAG, and EcoRI (Ned)-ACC/MseI-CTA. We analysed the 265 collected samples and, additionally, 25 individuals were replicated in order to exclude non-reproducible bands and to calculate the error rate according to Bonin et al. (2004). The fluorescence labelled selective amplification products were separated by capillary gel electrophoresis at the ‘‘Genomic Unit’’ (Universidad Complutense, Madrid, Spain), on an automated sequencer (3730 DNA Analyser, PE Applied Biosystems, Foster City, CA, USA) with an internal size standard (GeneScan 500 LIZ, Applied Biosystems). Raw data were exported to GeneMarker 1.8 (SoftGenetics, PA, USA) for automatic scoring of fragments, after normalisation of the profiles. The peaks were considered present when they were stronger than a scoring fluorescence intensity threshold set at 50 relative fluorescent units. The minimum percent of allele peaks to the highest peak in the lane was set at 1 %, and the local region percent, that defines the peak detection threshold based upon the percentage of the highest peak in one locus, was set at 1 %. Amplified fragments from 100 to 500 base pairs were scored. The results of the scoring were exported as a presence/absence matrix.
The presence/absence matrix that originated with the six primer combinations was imported into PAUP* (v. 4.0b10; Sinauer Associates). Within-population genetic diversity was assessed for each population using the total number of AFLP fragments present (Frag tot), the percentage of polymorphic fragments (Frag poly), the number of private fragments (Frag priv), and the average gene diversity (H D). Additionally, the frequency down-weighed marker value (DW), also called Rarity Index (Schönswetter and Tribsch 2005), was calculated using the function DW of the AFLPdat sourcecode in R (Ehrich 2006). This function calculate the rarity index per population (rarity 2) by making a table with the presence of markers by population; then each marker is divided by the total number of occurrences of this marker in the dataset.
We assessed patterns and levels of population genetic differentiation using analyses of molecular variance (AMOVA), conducted with ARLEQUIN v. 3.01, (Excoffier et al. 2005) that we undertook with four different groupings of the H. leontodontoides populations. The first grouping (a) has two hierarchical levels and describes differentiation among all populations of the species. Groupings b, c and d each have three hierarchical levels that additionally describe differentiation among geographical regions: between Middle Atlas populations versus High Atlas populations (grouping b); between Western High Atlas populations versus the remaining populations (grouping c); and between Middle Atlas versus Eastern High Atlas versus Central High Atlas versus Western High Atlas populations (grouping d). In this way, we aimed to test which potential geographical barrier (represented by groupings b, c and d), if any, had the largest effect on genetic differentiation in H. leontodontoides. We also calculated the AMOVA-derived fixation index F ST (ARLEQUIN v. 3.01), which describes the reduction in heterozygosity within populations relative to the total population (Wright 1951) and is an indirect approach to estimate gene flow.
We used principal co-ordinate analysis (PCoA) among individuals of the different populations to investigate genetic distances and relationships between populations. The PCoA was calculated and plotted in R software. The matrix of Jaccard similarities among individuals was calculated with the function “vegdist” of the vegan package (Oksanen et al. 2013), and the PCoA was calculated using the resulting matrix and the function “pco” of the ecodist package (Goslee and Urban 2007).
To represent overall genetic relationships among all the analysed individuals of H. leontodontoides, we constructed a dendrogram applying the UPGMA algorithm based on Nei and Li (1979) genetic distances. Support for each node was tested by 100,000 bootstrap replicates. The NJ method was also applied to the distance matrix and resulted in a similar dendrogram (data not shown). The UPGMA tree was rooted with H. glabra L. and H. achyrophorus.
To test for isolation by distance (IBD), we compared the 19 populations pairwise F ST values with their geographical distance (in kilometres) using Mantel test based on Spearman correlations (on 9,999 random permutations). Also, Mantel tests were performed within each of the four Atlas Mountain regions (Middle Atlas, Eastern High Atlas, Central High Atlas and Western High Atlas), with Bonferroni correction. The matrix of geographic distances between population pairs were calculated using the spDistsN1 function of the “sp” package in R, with the Great Circle distance (WGS84 ellipsoid) method. Mantel test was performed using the “mantel” function of the “vegan” package in R (Oksanen et al. 2013).
The overall population genetic structure was explored using model-based Bayesian assignment running STRUCTURE 2.3.4 (Pritchard et al. 2000; Pritchard 2010). Clustering of individuals was conducted without using the geographic origin of the samples as a prior informative. Analyses were based on an admixture ancestry model with correlated allele frequencies, for a range of K genetic clusters from one to 11, with 12 replicates for each K. The analyses were performed with a burn-in period of 150,000 and a run length of the Monte Carlo Markov Chain (MCMC) of 106 iterations, respectively. The most likely number of genetic clusters (K) was determined according to the ∆K and Mean LnP(K) (Evanno et al. 2005; Pritchard 2010), using STRUCTURE Harvester (Earl and vonHoldt 2012). Afterwards, CLUMPP 1.1.2 (Jakobson and Rosenberg 2007) was used to align the 12 runs of the most representative K values, the average pairwise similarity (H) for the 12 replicates, and the average probability of belonging to each cluster (Q). For K = 2, the Full search method with 1000 replicated was used. For K = 3 and K = 4, we used the Large K-Greedy algorithm, with 30,000 random input orders.
Results
ITS data
PCR-amplified ITS fragments showed a single band when examined on agarose gels. The complete sequence comprised ITS1, 5.8 rDNA gene and ITS2. The aligned ITS1 was 251 nucleotides (nt) long. The 5.8S rDNA gene was 163 nt long, and had no gaps. The aligned ITS2 was 220 nt long. There was no change in the sequences of ITS1 and ITS2 among individuals of the different populations. The six accessions of H. leontodontoides had almost identical ITS sequences.
AFLP data
The six AFLP primer combinations generated 873 unambiguously scorable DNA fragments (EcoRI-AGC/MseI-CAG: 138; EcoRI-ACG/MseI-CAT: 131; EcoRI- ACC/MseI-CTA: 177; EcoRI- ATC/MseI-CTC: 128; EcoRI- AGG/MseI-CTC: 155; EcoRI -ATC/MseI-CTGA: 144) of which 848 were polymorphic. All 265 analysed individuals had unique AFLP profiles. The error rate, based on phenotypic comparisons among the 25 replicated individuals, amounted to 2 %.
Genetic diversity
The Middle Atlas region included three populations with moderate high gene diversity (H D), namely pop. 3 (H D=0.126), pop. 2 (H D=0.119), and pop. 4 (H D=0.105) with a mean value for the five populations of H D = 0.094 (Table 1). In contrast, all High Atlas populations had low or very low gene diversity (H D range between 0.050 and 0.082) and mean values of H D between 0.065 and 0.075 with the exception of the pop. 15 from the Western High Atlas, with H D = 0.105. The highest number of private fragments was found in populations of the Middle Atlas (between 10 and 24 Priv frag in pops. 2–4), and also in the pop. 15 from the Western High Atlas (Priv frag = 22). In addition, the highest values of rarity index (DW) were found in the same populations (Table 1).
Phenetic phylogeographical analyses
In the PCoA conducted at the level of individuals (Fig. 2), the first two axes explained 65.5 and 21.7 % of the total variation, respectively, and separated perfectly the Western High Atlas populations (pops. 14–19) from the rest of the data set. Middle Atlas populations formed another group, albeit individuals were more scattered, while populations from Eastern (pops. 6–10) and Central (pops. 11–13) High Atlas were situated in a relatively intermediate position between the Middle Atlas and Western High Atlas populations. The PCoA conducted at the level of populations (Fig. S1) showed similar results, and highlighted the genetic divergence of the population 15 from the other Western High Atlas populations, and of populations 2 and 3 from the other Middle Atlas ones. Otherwise, the Eastern and Central High Atlas population groups appeared more closely related.

Principal coordinates analysis (2D-PCoA) based on Nei and Li distances at the individual level
The UPGMA tree (Fig. 3) rooted with H. glabra and H. achyrophorus was highly congruent with the results of the PCoA. All H. leontodontoides populations/samples clustered together with high bootstrap support (BS). The Western High Atlas populations (pops. 14–19) clustered with (100 % BS) and formed the sister group to the remaining populations (75 % BS). The latter comprised three groups: (a) Middle Atlas populations (pops. 1–5; 92 % BS); (b) Eastern High Atlas populations (pops. 6–10; 77 % BS); and (c) Central High Atlas populations (pops. 11–13; 64 % BS).

UPGMA analysis of AFLP phenotypes of H. leontodontoides, based on Nei and Li’s genetic distance. Bootstrap values higher than 50 % are indicated at each node (based on 10,000 replicates). O.gr. Outgroup
A non-hierarchical AMOVA (Table 2) revealed that half of the overall genetic variation was explained by the within-population component (49.17 %; Φ ST = 0.508). A nested AMOVA (i.e. grouping Middle Atlas vs. High Atlas) attributed 57.2 % of the global variation to differences between the different regions, and 33.99 % to within-region population differentiation (both at p < 0.001). These variation components were increased when we considered the grouping Western High Atlas vs remaining populations (Φ ST = 0.595), and the among-region component increased even more when we considered the grouping: Middle Atlas vs Eastern High Atlas vs Central High Atlas vs Western High Atlas (35.78 %).
The values of the pairwise fixation index (F ST) between populations (Table S1) indicated, mostly, high values. Populations 18 and 19 (F ST = 0.119), 9 and 10 (F ST = 0.149), 11 and 13 (F ST = 0.154), 6 and 8 (F ST = 0.163), 8 and 9 (F ST = 0.167), and 2 and 3 (F ST = 0.175) were the most genetically similar populations.
The Mantel test, which compared the geographical distances between populations with the respective pairwise F ST values, indicated a very strong positive correlation for these parameters (r = 0.846, p = 0.0001. By contrast, the Mantel test indicated no correlation within the four mountain regions (p > 0.05).
Bayesian analyses
Bayesian clustering with STRUCTURE was consistent with the results of the ordination and UPGMA analyses. Bayesian clustering showed that the greatest informative representation of overall genetic structure was achieved with K = 2 (∆K = 306, and Mean LnP(K) = −64402; Fig. S2). For the 12 replicates of K = 2, the average pairwise similarity (H′) was 0.998 for the matrices of populations. The first cluster was very homogeneous and consisted exclusively of Middle Atlas, Eastern and Central High Atlas individuals while the second cluster comprised only individuals of the Western High Atlas populations (Fig. 1b). Bayesian mean F ST values corresponding to the divergence of each inferred K cluster from the hypothetical ancestral population were 0.39 for the first cluster (Middle Atlas, Eastern and Central High Atlas group) and 0.59 for the second cluster (Western High Atlas group), indicating that populations from the first cluster were less diverged from the hypothetical ancestral population (Viruel et al. 2012).
A second maximum ΔK = 95 value, with a Mean LnP(K) = −57203, was obtained for K = 3 (Fig. S2), which separated the populations of Middle Atlas (pops. 1–5) from those of Eastern and Central High Atlas (pops. 6–13), and Western High Atlas (pops. 14-19) in three distinct clusters (Fig. 1b). The average pairwise similarity (H’) in this case was 0.925 for the matrices of populations. Bayesian mean F ST values oscillated between 0.678 (Western High Atlas populations) and 0.430 (Middle Atlas populations). The lowest Bayesian mean F ST value of this latter cluster points out this group as the least diverged from the hypothetical ancestral population. A further maximum ΔK = 55 value, with a Mean LnP(K) = -54539; was obtained for K = 4 (Fig. S2) which separated the 19 analysed populations in four distinct clusters: Middle Atlas, Eastern High Atlas, Central High Atlas, and Western High Atlas (Fig. 1b). The average pairwise similarity (H′) was 0.779 for the matrices of populations. Bayesian mean F ST values oscillated between 0.649 (Western High Atlas) and 0.384 (Middle Atlas), indicating once again that the Middle Atlas population group is the least diverged from the hypothetical ancestral population. The populations 1, 4 and 5 from Middle Atlas appeared as a mixed population in K = 3 and even more in K = 4 clustering.
Discussion
Strong genetic differentiation among Mountain regions
Our AFLP data showed a highly geographical structuring of the populations of H. leontodontoides along the Atlas Mountain range. Two main outcomes deserve to be highlighted. The first one is the marked group differentiation between the Western High Atlas populations from the remainder, which were supported by the UPGMA dendogram and PCoA analyses and also the Bayesian analysis at the optimal peak, K = 2. It is possible that the basins that delimit Western High Atlas region, that is, the Oued Tensift basin to the north, and the Oued Tassaout basin to the east, together with the Ourika river valley, act as insurmountable barriers that limit gene flow between the Western High Atlas populations and the other populations. Similarly, the Aosta valley (western Alps) and River Adige valley (eastern Alps) have been identified as efficient barriers against gene flow for many alpine herbaceous plants, such as Androsace obtusifolia, Phyteuma betonicifolium, Ranunculus glacialis, etc. (Schönswetter et al. 2005; Thiel-Egenter et al. 2011). Moreover, it is interesting to note that the populations from the Western High Atlas are at higher altitudes compared with the remainder populations (see Table 1), a fact that also could limit gene flow between the populations in this area and those of other areas.
The second outcome from our study is the appreciable differentiation found between the three remaining geographical regions: Middle Atlas, Eastern High Atlas and Central High Atlas, with the two latter groups being more genetically related (see Bayesian analyses at K = 3). Again, geomorphological barriers such as high montane topography or river valleys might explain this genetic structure. Similar patterns of genetic differentiation have been observed in other organisms (Brown et al. 2002; Fritz et al. 2006; Sousa et al. 2011). Likewise, a differential genetic structure among different mountain regions from the Atlas mountain range (Western High Atlas vs Central High Atlas and South Middle Atlas) was observed in Arabidopsis thaliana (Brennan et al. 2014).
Despite the genetic differentiation among regions found with AFLP markers, almost identical ITS sequences were found for individuals from the different regions, which suggest that H. leontodontoides is a well supported taxa.
Current gene flow patterns
Our AFLP data reveals a sequence of genetically distinct populations of H. leontodontoides occurring across the Atlas Mountain range, which is exemplified a high genetic differentiation (F ST = 0.508). Although significant correlation between populations pairwise F ST values with their geographical distance (Mantel test) could suggest isolation by distance (IBD), this result should be considered with caution. Indeed, geographically distant populations did show low F ST values, (e.g. Eastern High Atlas vs Central High Atlas populations), while geographically closer populations showed higher F ST values (e.g. Eastern High Atlas vs Middle Atlas populations; see Table 1S and Fig. 1a). Meirmans (2012) argued that IBD tests can lead to a large number of false positives, and suggested that a partial Mantel test can be used to test whether geography contributes to apparent clustering. In our scenario, the Mantel tests performed within each of the four mountain regions (Middle Atlas, Eastern, Central and Western High Atlas) were not significant, which indicate that at regional scale, gene flow between populations was not correlated with geographical distance. Indeed, Mantel tests do not distinguish between patterns resulting from clustering and those resulting from isolation by distance (Meirmans et al. 2011). Our results could be due to two different factors: (1) the scattered distribution populations of H. leontodontoides, occurring only rocky outcrops on north slopes, and (2) geographical barriers due to the abrupt topography of this region.
The isolation pattern found among populations of H. leontodontoides is also found in several European montane plant species of the Alps, Pyrenees, Apennines, Betic, Balkan and Carpathian systems (Zhang et al. 2001; Despres et al. 2002; Schönswetter et al. 2004; Schönswetter and Tribsch 2005; Kropf et al. 2006). Also, the congeneric H. angustifolia, another species from the Altas Mountain range, showed a similar sequence of isolated populations across the high mountain altiplano (Terrab et al. 2009). However, H. angustifolia inhabits high montane marshlands of the Atlas and it seems that its current population structure pattern most likely arose by habitat fragmentation following volcanic activity during the Quaternary of a previously much more extensive population system (i.e. a vicariance origin).
Diversification centre and colonization throughout the Atlas Mountain range
Morocco has been inferred as the ancestral area of Section Hypochaeris, which is sister to section Seriola that include H. leontodontoides. The Sebou valley seems to have played an important role in diversification within sect. Hypochaeris. Three species of that section (H. glabra, H. radicata and H. salzmanniana) evolved in the north-western foothills of the Middle Atlas, and another, H. arachnoidea, in the Atlas Mountain range (Ortiz et al. 2009).
Since all species from section Seriola (H. leontodontoides and their closest relatives H. laevigata, H. achyrophorus; Tremetsberger et al. 2005) inhabits the Middle Atlas, this region seems to be the diversification centre of section Seriola. Our molecular results also imply a possible origin of the species in the Middle Atlas. This view is supported by the Bayesian F ST values, and the highest number of private fragments and rare fragments index (DW), as well as the highest genetic diversity index, all found within this region (see Table 1; Fig. 1a). Although we cannot trace in detail the routes of the colonisation by H. leontodontoides, the most likely scenario, based on the Bayesian Analysis, is that populations subsequently dispersed southwards from the Middle Atlas, to the Eastern, Central and Western High Atlas. Moreover, the genetic structure of pop. 15 could suggest another colonisation event by long distance dispersal, from the Middle Atlas to the Western High Atlas (see Fig. 1a, b).
Floristic consequences of Quaternary climate fluctuations in the Atlas Mountain
Pliocene–Pleistocene ice ages are likely to have modelled the distribution and genetic structure of populations of H. leontodontoides. In fact, there is evidence that most of the Atlas Mountain range was affected at least by the last glacial maximum (Peulvast et al. 2000; Hughes et al. 2011). The largest glaciers in the whole Atlas mountain range were found in Toubkal massif (Western High Atlas), where valley glaciers emanated from a central ice field which formed between the two highest summits, Toubkal (4167 m a.s.l.) and Ouanuokrim (4067 m a.s.l.) (Hughes et al. 2011; see Fig. 1a). This glaciated area is coincident with the location of the pops. 16 and 17. In fact, these populations exhibit a depauperate genetic structure with low values of gene diversity, private fragment and rare fragment index. The westernmost populations (pops. 18 and 19) of the Western high Atlas, also showed similar impoverished gene diversity (see Table 1), which suggest that this area was also colonized more recently.
Overall, our data suggest that two regions within the Atlas Mountain range may have acted as refuges during the Pleistocene ice ages: The Middle Atlas and, to a lesser extent, the northernmost part of the Western High Atlas. Effectively, the highest values of private and rare fragments, and population gene diversity, were found in most populations of the Middle Atlas (pops. 2-4), and also in the northernmost population (pop. 15) of the Western High Atlas (Table 1). Other authors have proposed the occurrence of glacial refuges in the Middle and High Atlas regions (Médail and Diadema 2009; Hughes et al. 2011; Brennan et al. 2014). Populations of H. leontodontoides in the Middle Atlas region, currently located at 1900–2200 m a.s.l., should have been weakly affected by the ice ages, since the glacial snowline, at least of the last glacial period, were situated much higher at 2800 m (Hughes et al. 2011). The High Atlas region, however, showed a more severe impact of the ice ages, especially the Western High Atlas, a region that was extensively covered by ice. Despite this, it seems that in the northernmost part of this region, H. leontodontoides may have found a suitable area that permitted its survival, possibly by changing elevation during the coldest periods. Elevational shifts could also assure glacial survival in other regions; however, genetic structuring and diversity patterns found make a colonisation history for the populations from the Eastern and Central High Atlas more likely.
Conclusions
The present study shows a highly structured phylogeographic pattern in H. leontodontoides, with notable population differentiation within the Atlas Mountain range, with the population group from the Western High Atlas being most differentiated. Furthermore, a substructuring of the populations from the Middle Atlas, the Eastern High Atlas and the Central High Atlas was also detected. The high genetic differentiation among those groups of populations possibly reflects the impact of geomorphological features in the context of Quaternary climatic fluctuations as dispersal barriers.
The most likely scenario of the colonisation history of H. leontodontoides is that populations dispersed southwards from the Middle Atlas, to the Eastern, Central and Western High Atlas. Two regions could be suggested as refuges during the Pleistocene ice ages: the Middle Atlas and, more strikingly given its glacial history, the northernmost area of the Western High Atlas. Similar patterns of genetic structure of populations have been found in other Atlas Mountain species such as H. angustifolia (Terrab et al. 2009), or Arabidopsis thaliana (Brennan et al. 2014), although the hypotheses explaining these patterns are in detail not necessarily the same (Terrab et al. 2009).
References
Ball J (1873) Description of some new species, subspecies and varieties of plants collected in Morocco by J. D. Hooker, G. Maw and J. Ball. J Bot 11:371–372
Benabid A, Fennane M (1994) Connaissances sur la végétation du Maroc. Phytogéographie, phytosociologie et séries de végétation. Lazaroa 14:21–97
Bonin A, Bellemain E, Bronken Eidesen P, Pompanon F, Brochmann C, Taberlet P (2004) How to track and assess genotyping errors in population genetic studies. Mol Ecol 13:3261–3273
Brennan AC, Méndez-Vigo B, Haddioui A, Martínez-Zapater JM, Picó FX, Alonso-Blanco C (2014) The genetic structure of Arabidopsis thaliana in the south-western Mediterranean range reveals a shared history between North Africa and southern Europe. BMC Plant Biol 14:17
Brown RP, Suárez NM, Pestano J (2002) The Atlas mountains as a biogeographical divide in North-West Africa: evidence from mtDNA evolution in the Agamid lizard Agama impalearis. Mol Phylogenet Evol 24:324–332
Caujapé-Castells J, Jansen RK (2003) The influence of the Miocene Mediterranean desiccation on the geographical expansion and genetic variation of Androcymbium gramineum (Cav.) McBride (Colchicaceae). Mol Ecol 12:1515–1525
Christe C, Caetano S, Aeschimann D, Kropf M, Diadema K, Naciri Y (2014) The intraspecific genetic variability of siliceous and calcareous Gentiana species is shaped by contrasting demographic and re-colonization processes. Mol Phylogenet Evol 70:323–336
Deil V, Galán de Mera A (1996) Contribution à la connaissance de la phytosociologique et biogéographie des groupement rupicoles calcaires du Maroc. Bull Inst Sci Rabat 20:87–111
Despres L, Loriot S, Gaudeul M (2002) Geographic pattern of genetic variation in the European globeflower Trollius europaeus L. (Ranunculaceae) inferred from amplified fragment length polymorphism markers. Mol Ecol 11:2337–2347
Dresch J (1941) Recherches sur l’evolution du relief dans le massif central du Grand Atlas: le Haouz et le Sous Tours. Publications de l’lnstitut des Hautes-Etudes Marocaines, Rabat, pp 429–434
Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A, Duran C, Field M, Heled J, Kearse M, Markowitz S (2011) Geneious v5.4. Biomatter Ltd, Auckland
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Ehrich D (2006) AFLPdat: a collection of R functions for convenient handling of AFLPdata. Mol Ecol Notes 6:603–604
El Mousadik A, Petit RJ (1996) Chloroplast DNA phylogeography of the Argan tree in Morocco. Mol Ecol 5:547–555
Elenga H, Peyron O, Bonnefille R, Jolly D, Cheddadi R, Guiot J, Andrieu V, Bottema S, Buchet G, de Beaulieu JL, Hamilton AC, Maley J, Marchant R, Perez-Obiol R, Reille M, Riollet G, Scott L, Straka H, Taylor D, Van Campo E, Vincens A, Laarif F, Jonson H (2000) Pollen-based biome reconstruction for southern Europe and Africa 18,000 year BP. J Biogeogr 27:621–634
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Fineschi S, Anzidei M, Cafasso D, Cozzolino S, Garfi G, Pastorelli R, Salvini D, Taurchini D, Vendramin GG (2002) Molecular markers reveal a strong genetic differentiation between two European relic tree species: Zelkova abelicea (Lam.) Boissier and Z. sicula Di Pasquale, Garfî and Quézel (Ulmaceae). Conserv Genet 3:145–153
Fritz U, Barata M, Busack SD, Fritzsch G, Astilho R (2006) Impact of mountain chains, sea straits and peripheral populations on genetic and taxonomic structure of a freshwater turtle, Mauremys leprosa (Reptilia, Testudines, Geoemydidae). Zool Scr 35:97–108
Galán de Mera A, de Castro E, Vicente Orellana JA (1999) Hypochaeris alliatae group (Asteraceae) in the western Mediterranean Region. Nord J Bot 19:587–595
Galland N, Favarger C (1985) Notes de taxonomie et de nomenclature sur la flore orophile du Maroc. Candollea 40:231–235
Goslee SC, Urban DL (2007) The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw 22:1–19
Hampe A, Arroyo J, Jordano P, Petit RJ (2003) Rangewide phylogeography of a bird-dispersed Eurasian shrub: contrasting Mediterranean and temperate glacial refugia. Mol Ecol 12:3415–3426
Hughes PD, Gibbard PL, Woodward JC (2011) Quaternary glaciation in the Atlas Mountains of North Africa. In: Ehlers J, Gibbard PL, Hughes PD (eds) Quaternary glaciations—Extent and Chronology. A closer look, 15th edn. Developments in Quaternary Science, Amsterdam, pp 1065–1074
IPCC (2001) Climate Change 2001: The Scientific Basis. Cambridge University Press, Cambridge
Jakobson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program with label switching and multimodality in analysis of population structure. Bioinformatics 3:1801–1806
Jiménez JF, Sánchez-Gómez P, Güemes J, Werner O, Rosselló JA (2002) Genetic variability in a narrow endemic snapdragon (Antirrhinum subbaeticum, Scrophulariaceae) using RAPD markers. Heredity 89:387–393
Kropf M, Comes HP, Kadereit JW (2006) Long-distance dispersal vs vicariance: the origin and genetic diversity of alpine plants in the Spanish Sierra Nevada. New Phytol 172:169–184
Kropf M, Comes HP, Kadereit JW (2012) Past, present and future of mountain species of the French Massif Central–the case of Soldanella alpina L. subsp. alpina (Primulaceae) and a review of other plant and animal studies. J Biogeogr 39:799–812
López-Pujol J, Bosch M, Simon J, Blanché C (2001) Allozyme diversity of the two endemic Petrocoptis: P. montsicciana and its close related P. pardoi (Caryophyllaceae). Can J Bot 79:1379–1389
Lumaret R, Mir C, Michaud H, Raynal V (2002) Phylogeographical variation of chloroplast DNA in holm oak (Quercus ilex L.). Mol Ecol 11:2327–2336
Magri D, Parra I (2002) Late quaternary western Mediterranean pollen records and African winds. Earth Planet Sci Lett 200:401–408
Magri D, Fineschi S, Bellarosa R, Buonamici A, Sebastiani F, Schirone B, Simeone MC, Vendramin GG (2007) The distribution of Quercus suber chloroplast haplotypes matches the palaeogeographical history of the western Mediterranean. Mol Ecol 16:5259–5266
Maire R (1926) Contribution à l’étude de la flore de l’Afrique du Nord. Mém Soc Sci Nat Maroc 15:1–58
Médail F, Diadema K (2009) Glacial refugia influence plant diversity patterns in the Mediterranean Basin. J Biogeogr 36:1333–1345
Médail F, Quézel P (1997) Hot-spots analysis for conservation of plant biodiversity in the Mediterranean Basin. Ann Mo Bot Gard 84:112–127
Meirmans PG (2012) The trouble with isolation by distance. Mol Ecol 21:2839–2846
Meirmans PG, Goudet J, Gaggiotti OE (2011) Ecology and life history affect different aspects of the population structure of 27 high-alpine plants. Mol Ecol 20:3144–3155
Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA 76:5269–5273
Oberprieler Ch, Vogt R (2002) Hypochaeris arachnoidea Poir., a hitherto neglected species in NW Africa. Willdenowia 32:231–236
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H (2013) vegan: community ecology package. R package version 2.0–10
Ortiz MÁ, Tremetsberger K, Talavera S, Stuessy TF, García-Castaño JL (2007) Population structure of Hypochaeris salzmanniana DC. (Asteraceae), an endemic species to the Atlantic coast on both sides of the Strait of Gibraltar, in relation to Quaternary sea level changes. Mol Ecol 16:541–552
Ortiz MÁ, Tremetsberger K, Terrab A, Stuessy TF, García-Castaño JL, Urtubey E, Baeza CM, Ruas CF, Gibbs PE, Talavera S (2008) Phylogeography of the invasive weed Hypochaeris radicata (Asteraceae): from Moroccan origin to world-wide introduced populations. Mol Ecol 17:3654–3667
Petit RJ, Aguinagalde I, de Beaulieu JL, Bittkau C, Brewer S, Cheddadi R, Ennos R, Fineschi S, Grivet D, Lascoux M, Mohanty A, Müller-Starck JM, Demesure-Musch B, Palmé A, Martín JP, Rendell S, Vendramin GG (2003) Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300:1563–1565
Peulvast J-P, Barrier E, Ragala R, Doubre C, Vanney N, Vergne N, Bergerat F, Brunet M-F, Cadet J-P (2000) Last Glacial Maximum (20–18 ka BP), Programme PERI-THETYS, Map 24. (Eds: Dercourt et al.)
Piqué A (1994) Géologie du Maroc. Imprimerie El Maarif Al Jadida, Rabat
Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818
Pouclet A, Ouazzani H, Fekkak A (2007) The Cambrian volcano-sedimentary formations of the westernmost High Atlas (Morocco): their place in the geodynamic evolution of the West African Palaeo-Gondwana northern margin. Geol Soc Lond Spec Publ 297:303–327
Pritchard JK (2010) Documentation for STRUCTURE software: version 2.3. Available at:http://pritchardlab.stanford.edu/structure_software/release_versions/v2.3.4/html/structure.htm
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Schönswetter P, Tribsch A (2005) Vicariance and dispersal in the alpine perennial Bupleurum stellatum L. (Apiaceae). Taxon 54:725–732
Schönswetter P, Tribsch A, Niklfeld H (2004) Amplified fragment length polymorphism (AFLP) suggests old and recent immigration into the Alps by the arctic-alpine annual Comastoma tenellum (Gentianaceae). J Biogeogr 31:1673–1681
Schönswetter P, Stehlik I, Holderegger R, Tribsch A (2005) Molecular evidence for glacial refugia of mountain plants in the European Alps. Mol Ecol 14:3547–3555
Segarra-Moragues JG, Catalán P (2008) Glacial survival, phylogeography, and a comparison of microsatellite evolution models for explaining population structure in two species of dwarf yams (Borderea, Dioscoreaceae) endemic to the central Pyrenees. Plant Ecol Divers 1:229–243
Segarra-Moragues JG, Palop-Esteban M, González-Candelas F, Catalán P (2007) Nunatak survival vs. tabula rasa in the Central Pyrenees: a study on the endemic plant species Borderea pyrenaica (Dioscoreaceae). J Biogeogr 34:1893–1906
Sousa P, Froufe E, Harris DJ, Alves PC, van der Meijden A (2011) Genetic diversity of Maghrebian Hottentotta (Scorpiones: Buthidae) scorpions based on CO1: new insights on the genus phylogeny and distribution. Afr Invertebr 52:135–143
Swofford DL (2003) PAUP*: Phylogenetic analysis using parsimony, (* and other methods), Version 4.0b10. Sinauer, Sunderland
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Terrab A, Paun O, Talavera S, Tremetsberger K, Arista M, Stuessy TF (2006) Genetic diversity and population structure in natural populations of Moroccan Atlas cedar (Cedrus atlantica; Pinaceae) determined with cpSSR markers. Am J Bot 93:1114–1121
Terrab A, Ortiz MA, Talavera M, Ariza MJ, Moriana MC, García-Castaño JL, Tremetsberger K, Stuessy TF, Baeza CM, Urtubey E, Ruas CF, Casimiro-Soriguer R, Balao F, Gibbs PE, Talavera S (2009) AFLP and breeding system studies indicate vicariance origin for scattered populations and enigmatic low fecundity in the Moroccan endemic Hypochaeris angustifolia (Asteraceae), sister taxon to all of the South American Hypochaeris species. Mol Phylogenet Evol 53:13–22
Thiel-Egenter C, Alvarez N, Holderegger R, Tribsch A, Englisch T, Wohlgemuth T, Colli L, Gaudeul M, Gielly L, Jogan N, Linder HP, Negrini R, Niklfeld H, Pellecchia M, Rioux D, Schönswetter P, Taberlet P, van Loo M, Winkler M, Consortium IBD, Gugerli F (2011) Break zones in the distributions of alleles and species in alpine plants. J Biogeogr 38:772–782
Thompson JD, Gibson T, Higgins DG (2002) Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics 2:3
Tremetsberger K, Weiss-Schneeweiss H, Stuessy T, Samuel R, Kadlec G, Ortiz MÁ, Talavera S (2005) Nuclear ribosomal DNA and karyotypes indicate a NW African origin of South American Hypochaeris (Asteraceae, Cichorieae). Mol Phylogenet Evol 35:102–116
Tremetsberger K, Stuessy TF, Kadlec G, Urtubey E, Baeza CM, Beck SG, Valdebenito HA, Ruas CF, Matzenbacher NI (2006) AFLP phylogeny of South American species of Hypochaeris (Asteraceae, Lactuceae). Syst Bot 31:610–626
Tremetsberger K, Urtubey E, Terrab A, Baeza CM, Ortiz MA, Talavera M, König C, Temsch EM, Kohl G, Talavera S, Stuessy TF (2009) Pleistocene refugia and polytopic replacement of diploids by tetraploids in the Patagonian and Subantarctic plant Hypochaeris incana (Asteraceae, Cichorieae). Mol Ecol 18:3668–3682
Tremetsberger K, Gemeinholzer B, Zetzsche H, Blackmore S, Kilian N, Talavera S (2013) Divergence time estimation in Cichorieae (Asteraceae) using a fossil-calibrated relaxed molecular clock. Org Divers Evol 13:1–13
Vargas P, Morton CM, Jury SL (1999) Biogeographic patterns in Mediterranean and Macaronesian species of Saxifraga (Saxifragaceae) inferred from phylogenetic analyses of ITS sequences. Am J Bot 86:724–734
Viruel J, Catalán P, Segarra-Moragues JG (2012) Disrupted phylogeographical microsatellite and chloroplast DNA patterns indicate a vicariance rather than long-distance dispersal origin for the disjunct distribution of the Chilean endemic Dioscorea biloba (Dioscoreaceae) around the Atacama Desert. J Biogeogr 39:1073–1085
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M et al (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
White TJ, Bruns TD, Lee SB, Taylor JW (1990) Amplification and direct sequencing of fungal ribosomal RNA Genes for phylogenetics. In: Inns MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic Press, San Diego, pp 315–322
Winkler M, Tribsch A, Schneeweiss GM, Brodbeck S, Gugerli F, Holderegger R, Abbott RJ, Schönswetter P (2012) Tales of the unexpected: phylogeography of the arctic–alpine model plant Saxifraga oppositifolia (Saxifragaceae) revisited. Mol Ecol 21:4618–4630
Winkler M, Tribsch A, Schneeweiss GM, Brodbeck S, Gugerli F, Holderegger R, Schönswetter P (2013) Strong nuclear differentiation contrasts with widespread sharing of plastid DNA haplotypes across taxa in European purple saxifrages (Saxifraga section Porphyrion subsection Oppositifoliae). Bot J Linn Soc 173:622–636
Wright S (1951) The genetical structure of populations. Ann Eugen 15:323–354
Zhang LB, Comes HP, Kadereit JW (2001) Phylogeny and quaternary history of the European montane/alpine endemic Soldanella (Primulaceae) based on ITS and AFLP variation. Am J Bot 88:2331–2345
Acknowledgments
This work was funded by the Spanish Ministerio de Ciencia e Innovación and FEDER projects (CGL2012-32914). We acknowledge Peter Gibbs for helpful comments on the manuscript and language review, Juan Viruel for advice in statistical analysis and Luis Aguilar for help in the field and in the plant collections. We are grateful to two anonymous reviewers helped to improve previous versions of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
This research focuses on genetic structure of Hypochaeris leontodontoides (Asteraceae), M. A. Ortiz and R. Berjano contributed to data analysis and manuscript writing. F. J. Jiménez-López helped with field sampling molecular data analysis. A. Terrab and S. Talavera substantially contributed to the conception, manuscript writing and design of this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jiménez-López, F.J., Ortiz, M.A., Berjano, R. et al. High population genetic substructure in Hypochaeris leontodontoides (Asteraceae), an endemic rupicolous species of the Atlas Mountains in NW Africa. Alp Botany 126, 73–85 (2016). https://doi.org/10.1007/s00035-016-0163-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00035-016-0163-9