
Abstract
Background
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease. There is no cure currently. The discovery that mutations in the gene SOD1 are a cause of ALS marks a breakthrough in the search for effective treatments for ALS. SOD1 is an antioxidant that is highly expressed in motor neurons. Human SOD1 is prone to aberrant modifications. Familial ALS-linked SOD1 variants are particularly susceptible to aberrant modifications. Once modified, SOD1 undergoes conformational changes and becomes misfolded. This study aims to determine the effect of selective removal of misfolded SOD1 on the pathogenesis of ALS.
Methods
Based on the chaperone-mediated protein degradation pathway, we designed a fusion peptide named CT4 and tested its efficiency in knocking down intracellularly misfolded SOD1 and its efficacy in modifying the pathogenesis of ALS.
Results
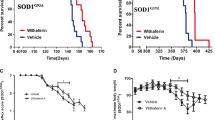
Expression of the plasmid carrying the CT4 sequence in human HEK cells resulted in robust removal of misfolded SOD1 induced by serum deprivation. Co-transfection of the CT4 and the G93A-hSOD1 plasmids at various ratios demonstrated a dose-dependent knockdown efficiency on G93A-hSOD1, which could be further increased when misfolding of SOD1 was enhanced by serum deprivation. Application of the full-length CT4 peptide to primary cultures of neurons expressing the G93A variant of human SOD1 revealed a time course of the degradation of misfolded SOD1; misfolded SOD1 started to decrease by 2 h after the application of CT4 and disappeared by 7 h. Intravenous administration of the CT4 peptide at 10 mg/kg to the G93A-hSOD1 reduced human SOD1 in spinal cord tissue by 68% in 24 h and 54% in 48 h in presymptomatic ALS mice. Intraperitoneal administration of the CT4 peptide starting from 60 days of age significantly delayed the onset of ALS and prolonged the lifespan of the G93A-hSOD1 mice.
Conclusions
The CT4 peptide directs the degradation of misfolded SOD1 in high efficiency and specificity. Selective removal of misfolded SOD1 significantly delays the onset of ALS, demonstrating that misfolded SOD1 is the toxic form of SOD1 that causes motor neuron death. The study proves that selective removal of misfolded SOD1 is a promising treatment for ALS.





Similar content being viewed by others


Availability of supporting data
All data published in the paper will be available upon request.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- CMA:
-
Chaperone-mediated autophagy
- CTM:
-
Chaperone-mediated autophagy targeting motif
- DBR:
-
Derlin-1 binding region
- Derlin-1:
-
Degradation in endoplasmic reticulum protein 1
- FALS:
-
Familial amyotrophic lateral sclerosis
- NSC:
-
Neural stem cell
- WT:
-
Wild-type
References
Gurney ME, Pu H, Chiu AY, Dal-Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264(5166):1772–1775
Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL (1995) An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14(6):1105–1116
Julien J-P (2001) Amyotrophic lateral sclerosis: unfold the toxicity of the misfolded. Cell 104:581–591
Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D et al (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 13(11):1396–1403
Guareschi S, Cova E, Cereda C, Ceroni M, Donetti E, Bosco DA, Trotti D, Pasinelli P (2012) An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc Natl Acad Sci USA 109(13):5074–5079
Ezzi SA, Urushitani M, Julien JP (2007) Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J Neurochem 102(1):170–178
Fan X, Jin WY, Lu J, Wang J, Wang YT (2014) Rapid and reversible knockdown of endogenous proteins by peptide-directed lysosomal degradation. Nat Neurosci 17(3):471–480
Dice JF, Terlecky SR, Chiang HL, Olson TS, Isenman LD, Short-Russell SR, Freundlieb S, Terlecky LJ (1990) A selective pathway for degradation of cytosolic proteins by lysosomes. Semin Cell Biol 1(6):449–455
Nishitoh H, Kadowaki H, Nagai A, Maruyama T, Yokota T, Fukutomi H, Noguchi T, Matsuzawa A, Takeda K, Ichijo H (2008) ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev 22(11):1451–1464
Chen X, Zhang X, Li C, Guan T, Shang H, Cui L, Li XM, Kong J (2013) S-nitrosylated protein disulfide isomerase contributes to mutant SOD1 aggregates in amyotrophic lateral sclerosis. J Neurochem 124(1):45–58
Basu S, Campbell HM, Dittel BN, Ray A (2010) Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. 10(41):1546
Li W, Guan T, Zhang X, Wang Z, Wang M, Zhong W, Feng H, Xing M, Kong J (2015) The effect of layer-by-layer assembly coating on the proliferation and differentiation of neural stem cells. ACS Appl Mater Interfaces 7(5):3018–3029
Thomson CE, McCulloch M, Sorenson A, Barnett SC, Seed BV, Griffiths IR, McLaughlin M (2008) Myelinated, synapsing cultures of murine spinal cord—validation as an in vitro model of the central nervous system. Eur J Neurosci 28(8):1518–1535
Karch CM, Borchelt DR (2008) A limited role for disulfide cross-linking in the aggregation of mutant SOD1 linked to familial amyotrophic lateral sclerosis. J Biol Chem 283(20):13528–13537
Wang J, Slunt H, Gonzales V, Fromholt D, Coonfield M, Copeland NG, Jenkins NA, Borchelt DR (2003) Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet 12(21):2753–2764
Schwarze SRHA, Vocero-Akbani A, Dowdy SF (1999) In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285(5433):1569–1572
Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA (2004) A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 429(6994):841–847
Lilley BN, Ploegh HL (2004) A membrane protein required for dislocation of misfolded proteins from the ER. Nature 429(6994):834–840
Fujisawa T, Homma K, Yamaguchi N, Kadowaki H, Tsuburaya N, Naguro I, Matsuzawa A, Takeda K, Takahashi Y, Goto J et al (2012) A novel monoclonal antibody reveals a conformational alteration shared by amyotrophic lateral sclerosis-linked SOD1 mutants. Ann Neurol 72(5):739–749
Homma K, Fujisawa T, Tsuburaya N, Yamaguchi N, Kadowaki H, Takeda K, Nishitoh H, Matsuzawa A, Naguro I, Ichijo H (2013) SOD1 as a molecular switch for initiating the homeostatic ER stress response under zinc deficiency. Mol Cell 52(1):75–86
Chen X, Shang H, Qiu X, Fujiwara N, Cui L, Li XM, Gao TM, Kong J (2012) Oxidative modification of cysteine 111 promotes disulfide bond-independent aggregation of SOD1. Neurochem Res 37(4):835–845
Gaj T, Ojala DS, Ekman FK, Byrne LC, Limsirichai P, Schaffer DV (2017) In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci Adv 3(12):eaar3952
Lim CKW, Gapinske M, Brooks AK, Woods WS, Powell JE, Zeballos CM, Winter J, Perez-Pinera P, Gaj T (2020) Treatment of a mouse model of ALS by in vivo base editing. Mol Ther 28(4):1177–1189
Foust KD, Salazar DL, Likhite S, Ferraiuolo L, Ditsworth D, Ilieva H, Meyer K, Schmelzer L, Braun L, Cleveland DW et al (2013) Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol Ther 21(12):2148–2159
McCampbell A, Cole T, Wegener AJ, Tomassy GS, Setnicka A, Farley BJ, Schoch KM, Hoye ML, Shabsovich M, Sun L et al (2018) Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Investig 128(8):3558–3567
Miller T, Cudkowicz M, Shaw PJ, Andersen PM, Atassi N, Bucelli RC, Genge A, Glass J, Ladha S, Ludolph AL et al (2020) Phase 1–2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med 383(2):109–119
Mueller C, Berry JD, McKenna-Yasek DM, Gernoux G, Owegi MA, Pothier LM, Douthwright CL, Gelevski D, Luppino SD, Blackwood M et al (2020) SOD1 suppression with adeno-associated virus and microRNA in familial ALS. N Engl J Med 383(2):151–158
Ding H, Schwarz DS, Keene A, Affar EB, Fenton L, Xia X, Shi Y, Zamore PD, Xu Z (2003) Selective silencing by RNAi of a dominant allele that causes amyotrophic lateral sclerosis. Aging Cell 2(4):209–217
Raoul C, Abbas-Terki T, Bensadoun JC, Guillot S, Haase G, Szulc J, Henderson CE, Aebischer P (2005) Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med 11(4):423–428
Bravo-Hernandez M, Tadokoro T, Navarro MR, Platoshyn O, Kobayashi Y, Marsala S, Miyanohara A, Juhas S, Juhasova J, Skalnikova H et al (2020) Spinal subpial delivery of AAV9 enables widespread gene silencing and blocks motoneuron degeneration in ALS. Nat Med 26(1):118–130
Powell JE, Lim CKW, Krishnan R, McCallister TX, Saporito-Magrina C, Zeballos MA, McPheron GD, Gaj T (2022) Targeted gene silencing in the nervous system with CRISPR–Cas13. Sci Adv 8(3):eabk2485
Ivannikov MV, Van Remmen H (2015) Sod1 gene ablation in adult mice leads to physiological changes at the neuromuscular junction similar to changes that occur in old wild-type mice. Free Radic Biol Med 84:254–262
Urushitani M, Ezzi SA, Julien JP (2007) Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 104(7):2495–2500
Rakhit R, Robertson J, Vande Velde C, Horne P, Ruth DM, Griffin J, Cleveland DW, Cashman NR, Chakrabartty A (2007) An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat Med 13(6):754–759
Liu HN, Tjostheim S, Dasilva K, Taylor D, Zhao B, Rakhit R, Brown M, Chakrabartty A, McLaurin J, Robertson J (2012) Targeting of monomer/misfolded SOD1 as a therapeutic strategy for amyotrophic lateral sclerosis. J Neurosci 32(26):8791–8799
Maier M, Welt T, Wirth F, Montrasio F, Preisig D, McAfoose J, Vieira FG, Kulic L, Spani C, Stehle T et al (2018) A human-derived antibody targets misfolded SOD1 and ameliorates motor symptoms in mouse models of amyotrophic lateral sclerosis. Sci Transl Med 10(470):eaah3924
Hill MD, Goyal M, Menon BK, Nogueira RG, McTaggart RA, Demchuk AM, Poppe AY, Buck BH, Field TS, Dowlatshahi D et al (2020) Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): a multicentre, double-blind, randomised controlled trial. Lancet 395(10227):878–887
Chattopadhyay M, Valentine JS (2009) Aggregation of copper-zinc superoxide dismutase in familial and sporadic ALS. Antioxid Redox Signal 11(7):1603–1614
Chen X, Guan T, Li C, Shang H, Cui L, Li XM, Kong J (2012) SOD1 aggregation in astrocytes following ischemia/reperfusion injury: a role of NO-mediated S-nitrosylation of protein disulfide isomerase (PDI). J Neuroinflammation 9:237
Forman HJ, Fukuto JM, Torres M (2004) Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol 287(2):C246-256
Woo HA, Chae HZ, Hwang SC, Yang KS, Kang SW, Kim K, Rhee SG (2003) Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science 300(5619):653–656
Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ 3rd, Charrier V, Parsonage D (1999) Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry 38(47):15407–15416
Uchida K, Kawakishi S (1994) Identification of oxidized histidine generated at the active site of Cu, Zn-superoxide dismutase exposed to H2O2. Selective generation of 2-oxo-histidine at the histidine 118. J Biol Chem 269(4):2405–2410
Kurahashi T, Miyazaki A, Suwan S, Isobe M (2001) Extensive investigations on oxidized amino acid residues in H(2)O(2)-treated Cu, Zn-SOd protein with LC-ESI-Q-TOF-MS, MS/MS for the determination of the copper-binding site. J Am Chem Soc 123(38):9268–9278
Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281(5384):1851–1854
Furukawa Y, Fu R, Deng HX, Siddique T, O’Halloran TV (2006) Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc Natl Acad Sci USA 103(18):7148–7153
Tiwari A, Hayward LJ (2003) Familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase are susceptible to disulfide reduction. J Biol Chem 278(8):5984–5992
Tiwari A, Liba A, Sohn SH, Seetharaman SV, Bilsel O, Matthews CR, Hart PJ, Valentine JS, Hayward LJ (2009) Metal deficiency increases aberrant hydrophobicity of mutant superoxide dismutases that cause amyotrophic lateral sclerosis. J Biol Chem 284(40):27746–27758
Okado-Matsumoto A, Fridovich I (2002) Amyotrophic lateral sclerosis: a proposed mechanism. Proc Natl Acad Sci USA 99(13):9010–9014
Furukawa Y, O’Halloran TV (2006) Posttranslational modifications in Cu, Zn-superoxide dismutase and mutations associated with amyotrophic lateral sclerosis. Antioxid Redox Signal 8(5–6):847–867
Forsberg K, Andersen PM, Marklund SL, Brannstrom T (2011) Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol 121(5):623–634
Kabashi E, Valdmanis PN, Dion P, Rouleau GA (2007) Oxidized/misfolded superoxide dismutase-1: the cause of all amyotrophic lateral sclerosis? Ann Neurol 62(6):553–559
Bunton-Stasyshyn RK, Saccon RA, Fratta P, Fisher EM (2015) SOD1 function and its implications for amyotrophic lateral sclerosis pathology: new and renascent themes. Neuroscientist 21(5):519–529
Acknowledgements
The authors would like to acknowledge the assistance of Dr Lynda Kong in editing the manuscript.
Funding
The study was supported by Grants from ALS Society of Canada, Brain Canada and By-Health China.
Author information
Authors and Affiliations
Contributions
TG coordinated the project and drafted the manuscript. TZ performed most of the in vivo experiments. XZ conducted plasmid construction and in vitro validation. YG, CY, YC aided in animal experiments. JL, JVZ helped cell-based experiments. HM, YTW made a conceptual contribution and reviewed the manuscript. JK initiated and supervised the project. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval and consent to participate
All our work was reviewed and approved by the animal use subcommittee at the University of Manitoba.
Consent for publication
The authors consent to publishing this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
18_2023_4956_MOESM1_ESM.tiff
Supplementary file1 Supplementary Figure 1. Knockdown of misfolded SOD1 by CT4 in glial cells. (A) G93A-hSOD1 expressing astrocytes (GFAP+), oligodendrocyte precursor cells (OPCs, PDGFRα+), and mature oligodendrocytes (MBP+) were treated with 5 µM CT4 for 2 hours. Cells treated with mCT4 were used as controls. Scale bar = 20 μm. G93A-hSOD1 fluorescence intensities were measured in each type of cell. n = 6 from 3 separate cultures. (B) Knockdown of misfolded SOD1 in G93A-hSOD1 mice by intravenous injection of CT4 or mCT4 at 10 mg/kg daily for 3 days. Immunostaining images with anti-misfolded SOD1 (B8H10) antibody and glial markers in the lumbar spinal cord are shown. Scale bar: 100 μm (for images in lower magnification) and 20 μm (higher magnification). G93A-hSOD1 fluorescence intensities (mean ± SD) were measured in each cell type and analyzed with two-way ANOVA followed by Sidak’s multiple comparisons test. **P<0.01 (TIFF 10628 KB)
18_2023_4956_MOESM2_ESM.tiff
Supplementary file2 Supplementary Figure 2. Short-term knockdown of misfolded SOD1 by CT4 in vivo. (A) Western blotting of misfolded SOD1 with the A5C3 antibody in mice expressing WT-hSOD1 and G93A-hSOD1 at the age of 150 days. (B) The G93A-hSOD1 mice received intravenous injections of either mCT4 or CT4 peptide at 10 mg/kg body weight daily for three days at the age of 100 days. Spinal cords were harvested 24 hours after the last injection. Equal volumes of lysates with a total of 10 µg protein were applied to 12% TGX Stain-Free polyacrylamide gels. Total protein generated by stain-free visualization was used to ensure equivalent loading. A two-tailed t-test was performed to establish significant differences with the control group. *P < 0.05; **P < 0.01. Shown are mean ± SD. n = 4 in mCT4 group and 3 in CT4 group (TIFF 4100 KB)
18_2023_4956_MOESM3_ESM.tiff
Supplementary file3 Supplementary Figure 3. No toxicity was observed in wild-type mice exposed to CT4. Beginning at 60 days of age, wild-type mice received an IP injection of CT4 peptide at a dose of 10 mg/kg q.o.d until 173 days of age. Sections of mouse brain, heart, spinal cord, kidney, liver, muscle, lung, and spleen were stained with H&E. Scale bar represents 100 μm. (TIFF 14391 KB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Guan, T., Zhou, T., Zhang, X. et al. Selective removal of misfolded SOD1 delays disease onset in a mouse model of amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 80, 304 (2023). https://doi.org/10.1007/s00018-023-04956-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-023-04956-9