
Abstract
Endoplasmic reticulum (ER) stress initiates the unfolded protein response (UPR) and is decisive for tumor cell growth and tumor microenvironment (TME) maintenance. Tumor cells persistently undergo ER stress and could transmit it to the neighboring macrophages and surroundings. Tumor infiltrating macrophages can also adapt to the microenvironment variations to fulfill their highly energy-demanding and biological functions via ER stress. However, whether the different macrophage populations differentially sense ER stress and transmit ER stress to surrounding tumor cells has not yet been elucidated. Here, we aimed to investigate the role of transmissible ER stress, a novel regulator of intercellular communication in the TME. Murine bone marrow-derived macrophage (BMDM) can be polarized toward distinct functional endpoints termed classical (M1) and alternative (M2) activation, and their polarization status has been shown to be tightly correlated with their functional significance. We showed that tumor cells could receive the transmissible ER stress from two differentially polarized macrophage populations with different extent of ER stress activation. The proinflammatory M1-like macrophages respond to ER stress with less extent, however they could transmit more ER stress to tumor cells. Moreover, by analyzing the secreted components of two ER-stressed macrophage populations, we identified certain damage-associated molecular patterns (DAMPs), including S100A8 and S100A9, which are dominantly secreted by M1-like macrophages could lead to significant recipient tumor cells death in synergy with transferred ER stress.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Tumorigenesis is unceasingly occurred along with correlative chronic inflammatory responses, and the abundant tumor-associated inflammatory immune cells infiltration were observed in clinical tumor biopsies [1,2,3]. Numerous intrinsic and extrinsic factors, such as hypoxia, viral infection, nutrient deficiency, low pH and proto-oncogene activation, lead to aberrant endoplasmic reticulum (ER) stress activation in the tumor microenvironment (TME). To restore ER homeostasis, the unfolded protein response (UPR) increases ER chaperone content to elevate protein folding rate [4]. Transmissible ER stress has been shown to play a critical role in intercellular communication in the pathogenesis of various diseases, especially in malignant cancers [5,6,7,8,9]. The transmissible ER stress from tumor cells to neighboring non-tumor cells, such as macrophages and DCs, has been identified in the TME. It was also reported that ER stress could be transmitted from prostate cancer cells to homologous and heterologous cancer cells, promoting β-catenin-mediated Wnt signaling and enhancing tumorigenic effects in vivo [10]. Moreover, cancer cells under pharmacological or physiological ER stress can train the nearby macrophages to mirror the behaviors of cancer cells, and this transmissible ER stress is not sensed by TLR2 or IL-6R but TLR4 [5]. Meanwhile, limited evidence suggests that ER stress has the capacity to modulate therapeutic efficacy in cancer cells. However, whether ER stress could be transferred from non-tumor cells, like macrophages, to tumor cells has not yet been studied.
Macrophages are highly plastic and heterogeneous cell populations and account for up to almost 50% of cell components in some solid neoplasms [11, 12]. Accumulating studies have already proved that the clinical significance of the number and density of macrophages within TME is highly linked with poor patient prognosis and poor survival in some cancer types [13]. Moreover, the phenotypes of macrophages are suggested to be responsible for promoting activities of tumors, including tumor initiation, progression and metastasis [14]. Macrophages have continuous functional activity states with two extremely distinct polarized conditions, mostly named proinflammatory M1- or anti-inflammatory M2-phenotypes. In the invasive TME, macrophage colony-stimulating factor (M-CSF) was considered to have the effect to recruit macrophages in tumors by promoting migration and survival, by which mainly induces M2-like macrophages. M2-like macrophages secrete suppressive cytokines (such as IL4, IL10, TGFβ, CCL22, etc.) to suppress innate and adaptive anti-tumor immunity. On the other hand, the proinflammatory factors, such as IL6, IL12, TNF, granulocyte–macrophage colony-stimulating factor (GM-CSF) and other relative cytokines, could recruit the proinflammatory M1 macrophages to tumor local site [15, 16]. Even though numerous shreds of evidence suggested that macrophages in vivo do not exhibit hyper-polarized M1 or M2 phenotype and intermediate state of macrophages exist to perform M1 and M2 characteristics concurrently and mixed polarization phenotypes of macrophages have been described in several human malignant solid tumors [17], however, tumor-associated macrophages (TAMs) are still generally considered as M2-like macrophages. Notably, the previous study has reported that in non-small cell lung cancer, the density of M1-like macrophage is an independent factor of overall survival time [18], indicating that macrophage polarization status could be an indispensable factor when studying intratumoral cell communications.
In the current study, using well-documented GM-CSF cultured bone marrow-derived macrophages (GM-BMDM) and M-CSF cultured counterpart (M-BMDM) as the in vitro differentiated M1 and M2 macrophage models [19], we observed that ER stress transmission is a bidirectional process. ER stress can be transferred from tumor cells to macrophages and vice versa. Transcriptome analysis revealed that GM-BMDM has less potential to sense but could transfer more ER stress to tumor cells than M-BMDM. In further proteomics study, we screened the secreted proteins from ER-stressed macrophages and identified certain classic damage-associated molecular patterns (DAMPs) (S100A8 and S100A9) as the ER stress promoted transferable components dominantly from GM-BMDM, which could synergize with transferred ER stress to trigger significant tumor cell death. Both UPR and ER stress-induced activation of mitogen-activated protein kinase (MAPK) signals were involved and amplified by DAMPs and ER stress co-stimulation. Taken together, our findings suggested that M1 macrophage could prompt severe tumor cell death via transmissible ER stress and release of DAMPs, whereas M2 populations would facilitate tumor growth and metastasis under the same conditions. These findings suggested a novel understanding that different macrophage populations have distinct responses to ER stress and could remodel the TME via transmissible ER stress.
Materials and methods
Reagents
Tunicamycin (Tm, ≥ 98%, 12,819), thapsigargin (Tg, ≥ 98%, 12,758) and cell lysis buffer (9803) were purchased from Cell Signaling Technology (CST). Dulbecco’s phosphate-buffered saline (PBS) was obtained from Biosharp. Phosphatase (4,906,837,001), protease (04,693,132,001) inhibitors cocktails, 2 × RNase inhibitor and FastStart Essential DNA Green Master (4,913,914,001) were from Roche. Recombinant mouse GM-CSF (415-ML, BJ2518091) and M-CSF (416-ML, ME4920031) were purchased from R&D. Murine recombinant S100A8 (50,228-M08B) and S100A9 (50,284-M07E) proteins were purchased from Sino Biological. PrimeScript™ RT reagent kit with gDNA eraser (RR047A) was acquired from Takara. BCA protein assay kit, Hoechst 33,342 (C0030) and PI (C0080) solutions were from Solarbio Life Sciences. Cell culture inserts (0.4 μm, PICM01250) were purchased from Millipore. Luminescent cell viability assay (G7570), 3D cell viability (G9681), Caspase-Glo 3/7 assay (G8091) and cell cytotoxicity assay (G8741) were from Promega.
Cell culture
Mouse Louis Lung Cancer cell lines (LLC) were purchased from the Cell Bank of the Chinese Academy of Sciences and tested without mycoplasma contamination. LLC cells were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco), 100 U/ml penicillin and 100 mg/ml streptavidin (Sangon Biotech). Cell lines were cultured at 37 °C in a carbon dioxide cell incubator with 5% CO2.
Animals and procedures
Male C57BL/6 mice (SPF degree, 6–12 weeks old) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. The animals were maintained in the specific-pathogen-free laboratory animal room with a 12-h light–dark cycle. All experimental protocols involved were approved by the Committee on the Ethics of Animal Experiments of the School of Life Sciences of Lanzhou University (EAF-2021026).
Murine primary BMDM isolation and culture
Mouse BMDMs were prepared by collecting the bone marrow cells from femurs as previously described [20]. The cell suspensions were passed through a 100 micron nylon cell strainer (BD Falcon), collected by centrifugation at 300 g for 10 min, and resuspended in DMEM containing 10% FBS, 100 U/ml penicillin, 100 mg/ml streptomycin, either with 50 ng/ml M-CSF or 20 ng/ml GM-CSF. Cells (5 × 106 for M-BMDMs or 10 × 106 for GM-BMDMs) were seeded in 100 mm dishes and cultured at 37 °C with 5% CO2 for 7 days. Non-adherent cells were removed and the adherent BMDMs were treated as indicated in individual experiment. The BMDMs cultured in upper chambers of the transwell plates (0.2 × 106 for M-BMDMs or 0.4 × 106 for GM-BMDMs) were seeded in 24-well plates and cultured at 37 °C with 5% CO2 for 7 days.
Immunoblotting and antibodies
Cell lysates were centrifuged at 12,000g for 10 min, and the supernatants were collected. The protein concentrations were measured by the BCA protein assay kit (Solarbio, PC0020). The proteins were fractionated by SDS-PAGE and transferred to the 0.2 μm polyvinylidene difluoride membrane (Millipore). After blocking with 5% nonfat milk in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.1% Tween 20) for 1 h at room temperature, the membranes were incubated with the specified primary antibodies overnight at 4 °C. After being washed by TBST, the membranes were incubated with the secondary antibody in 5% nonfat milk for 2 h at room temperature. Then, the binds were detected with ECL Detection Reagent (Millipore, WBKLS0500) and photographed by Tanon 5200 imaging system.
The primary antibodies used were: Purified mouse monoclonal antibody to Grp78 (610,978, 1:1000, BD Biosciences, RRID:AB_398291), mouse monoclonal antibody to CHOP (2895, 1:1,000, CST, RRID:AB_2089254), rabbit monoclonal antibodies to Xbp1 (ab220783, 1:1,000, Abcam), rabbit monoclonal to caspase-1 (ab179515, 1:1,000, Abcam, RRID:AB_2884954), mouse monoclonal antibody to caspase-9 (9508, 1:1,000, CST, RRID:AB_2068620), rabbit monoclonal antibody to GAPDH (5174, 1:1,000, CST, RRID:AB_10622025), rabbit polyclonal antibody to LC3 (4108, 1:1,000, CST, RRID:AB_2137703), rabbit monoclonal antibody to Beclin-1 (3495, 1:1,000, CST, RRID:AB_1903911), rabbit monoclonal antibody to p-ULK1 (5869, 1:1,000, CST, RRID:AB_10707365), rabbit polyclonal antibody to cleaved-PARP (9544, 1:1,000, CST, RRID:AB_2160724), rabbit monoclonal antibody to p-JNK (4668, 1:1,000, CST, RRID:AB_823588), rabbit polyclonal antibody to JNK (9252, 1:1,000, CST, RRID:AB_2250373), rabbit monoclonal antibody to p-ERK (4370, 1:1,000, CST, RRID:AB_2315112), rabbit monoclonal antibody to ERK (4695, 1:1,000, CST, RRID:AB_390779), rabbit monoclonal antibody to p-p38 (4511, 1:1,000, CST, RRID:AB_2139682), rabbit monoclonal antibody to p38 (8690, 1:1,000, CST, RRID:AB_10999090), rabbit monoclonal antibody to S100A9 (73,425, 1:1,000, CST, RRID:AB_2799839). Secondary antibodies were horseradish peroxide-conjugated anti-mouse IgG (abs20001, RRID: AB_2716555) or anti-rabbit IgG (abs20002, RRID: AB_2716554) obtained from Absin.
Quantitative real-time PCR
Total RNA was isolated with Trizol reagent (Takara, 9109) following the manufacturer’s instructions. The concentration, integrity, and purity of RNAs were quantified on a Nanodrop (ND-ONE) spectrophotometer (Thermo Scientific). RNAs reverse transcribed by PrimeScript™ RT reagent kit following the manufacturer’s protocol. Real-time PCR was performed with an ABI Stepone Plus system to quantify the mRNA expression levels using FastStart Essential DNA Green Master. The target genes were normalized to that of β-actin [20], and relative expression was determined using the ΔCt relative Quantification method. The primers used were listed in Table S1.
Conditioned media preparation
The ER-stressed conditioned media (CM) were generated as follows. GM-BMDMs, M-BMDMs or LLC cells were treated with 1 μg/ml Tm or 300 nM Tg for the indicated times. After stimulation, cells were washed 3 × with Dulbecco’s PBS and were added with fresh serum-containing media. After 16 h’ incubation, media were collected and filtered through a 0.22 μm syringe filter (Millipore) to obtain CM.
Co-cultured 3D system
3D cell culture was performed as managed previously [21, 22]. Briefly, 0.8 × 104 cells/well were seeded into 24-well plates. GM-BMDMs and M-BMDMs cultured in cell inserts were treated with Tm (1 μg/ml) for 0 or 6 h. Then, the inserts were washed with PBS for 3 times and incubated with 3D cultured LLC for additional 40 h.
The high content imaging analysis system and fluorescence microscope
LLC (0.5 × 104 cells in 1 ml) were seeded into the 24-well plates (Corning). PI (a membrane-impermeant nuclear dye, red) and Hoechst 33,342 (a cell-permeable nuclear dye, blue) were added shortly before automated image acquisition in an atmosphere-controlled chamber (37 °C, 5% CO2) using High-quality cell imaging instrument (HCI, Operetta, PerkinElmer, America) or the ZEISS Ver.A1 microscope (Carl Zeiss, Heidenheim, Germany). The living cells were visualized by Hoechst 33,342 and the dead cells were positive for PI. The PI+ or Hoechest+ cell numbers were quantified using Image J software.
Elisa
The concentrations of S100A8, S100A9 and S100A8/A9 in the supernatants of BMDMs post the indicated treatment were measured using commercial ELISA kits (Cloud Clone) following the manufacture’ instructions.
RNA-sequencing
Total RNA was isolated from cells using the Trizol reagent (Takara). The 3 µg of RNA were used as input material for the RNA sample preparations to generate sequencing libraries. Products were purified (AMPure XP system) and the library was then sequenced on NovaSeq 6000 platform (Illumina). The high qualities sequences (clean data) were filtered using Cutadapt (v1.15) and mapped to the reference genome using HISAT2 v2.0.5 [23]. The gene read counts were acquired via htseq-count [24]. Differentially expressed genes (DEGs) were analyzed using DESeq [25] and identified the threshold of P value < 0.05 and fold change (FC) > 1.5 as significantly differential expression. Hierarchical cluster analysis of DEGs was performed to explore genes expression patterns. The Gene Ontology (GO) Annotation analysis [26] and IPA analysis [27] of DEGs were, respectively, performed.
Protein mass spectrometry
CMs were centrifugated at 12,000g at 4 °C for 10 min in an ultrafiltration device to remove the putative cell components. The protein concentration was determined with the BCA kit and the tryptic peptides were fractionated by high pH reverse-phase HPLC. The resulting MS/MS data were processed using the Maxquant search engine (v.1.5.2.8). Tandem mass spectra were searched against the UniProt database concatenated with reverse decoy database [28]. Proteins containing secreted keywords in UniProt database are filtered as secreted proteins and shown in Table S4. Fold change > 1.2 was set as the threshold for significantly differential expression.
Mass spectrometry analysis
Tunicamycin in culture medium was quantified by mass spectrometry analysis. Chromatographic separation was performed on an Agilent 1290 Infinity II ultrahigh-performance liquid chromatography system, using a poroshell 120 EC C18 column (Agilent Technologies, California, USA). The injection volume was 10 μl. The separation was carried out with a mobile phase consisting of 10% solvent A (water) and 90% solvent B (methanol) at a flow rate of 0.3 ml/min.
Mass spectrometry analyses were performed on a 6460 Triple Quad mass spectrometer (Agilent Technologies, California, USA) with electrospray ionization (ESI) source in positive mode. Quantitation was based on peak area generated by scanning for product ions (m/z 755.3) from the protonated molecular ion parent (m/z 867.4).
Statistical analysis
The experimental data are presented as Mean ± SEM of triplicate experiments. P value s < 0.05 were indicated statistically significant in the figure legends as indicated. Statistical analysis was performed using one-way or two-way ANOVA and Tukey’s multiple comparisons using GraphPad Prism version 8.0 software.
Results
Tumor cells sense transferable ER stress from two different polarized macrophage populations
Recent data from single cell RNA-sequencing analysis of patient tumor samples showed that ER stress related genes are highly expressed not only in malignant cells but also in intratumoral macrophages, for example in lung and liver tumors (Fig. S1a and S1b) (https://omnibrowser.abiosciences.com) [29, 30]. A previous study reported that tumor cells undergoing ER stress could promote ER stress signals in TAMs, and this transmissible ER stress was sensed by TLR4 [5]. Moreover, the infiltrating macrophages are also in the stress TME and could have high protein folding demand, which could also initiate ER stress [31]. Therefore, we wondered whether the transferable ER stress is bidirectional? Do differentially polarized macrophages have similar behavior on initiating and transferring ER stress? To investigate these questions, Tunicamycin (Tm, 1 μg/ml) was used to block protein glycosylation and trigger the ER stress [32] in donor GM-BMDMs (M1-like macrophages) and M-BMDMs (M2-like macrophages) [19]. Upon Tm treatment, the generated ER-stressed conditioned media (CM) were further applied to the recipient mouse Louis Lung Cancer cell (LLC) (Fig. 1a). After 24-h incubation with CMs from ER-stressed M-BMDMs or GM-BMDMs, the significantly increased abundance of critical ER stress master regulator glucose-regulated protein-78 (Grp78) was observed, in parallel with the induction of other ER stress signature molecules, such as spliced X-box binding protein 1 (sXbp1) and C/EBP homologous protein (CHOP) in LLC cells (Fig. 1b–d). The effects were further confirmed by immunoblotting assays, demonstrating enhanced activation and generation of UPR key sensors upon CMs treatment (Fig. 1e–h). Of note, LLC treated with ER-stressed GM-BMDM-CMs showed a more potent ER stress response at every Tm treatment time point, indicating that GM-BMDMs could transmit more ER stress to LLC cells than M-BMDMs. Interestingly, notably upregulated expressions of inflammatory cytokines IL6 and IL1α, and chemokine CXCL1 were observed in LLC cells treated with CMs from ER-stressed GM-BMDMs (Fig. 1i–k), suggesting that different transmissible ER stress could induce distinct responses in recipient tumor cells. To better mimic the TME and evaluate the ER stress transmitted by components in CMs more accurately, we stressed the macrophages in a cell insert and then evaluated the transmissible ER stress in transwell chambers with the established LLC spheres embedded in a 3D Matrigel matrix (Fig. S1c). Consistent with the observation in Fig. 1, UPR mediators were elevated in LLC cells after receiving transferred ER stress from macrophages (Fig. S2d–f). Both proinflammatory cytokine IL6 (Fig. S2g) and chemokine CXCL1 (Fig. S2h) were predominantly expressed in spheres co-cultured with ER-stressed GM-BMDMs. In addition, another ER stress inducer thapsigargin (Tg) who inhibits calcium ion uptake [5] was used to drive ER stress and similar results were observed (Fig. S2i–n). To rule out the possibility that remaining Tm carried over in CMs activated UPR in recipient tumor cells, mass spectroscopy was used to analyze the abundance of Tm in ER-stressed CMs. The result showed that there is no detectable Tm existed in CMs (Fig. S3a). Besides, the minimum dose of Tm can be detected is 0.25 μg/ml while Tm at this dose could not successfully prompt stress responses in both macrophages (Fig. S3b–f) and tumor cells (Fig. S3g–i), indicating that the transmission of ER stress from macrophage populations to tumor cells is not because of the carryover Tm in the CMs. Hence, the results suggested that macrophages who underwent ER stress could transfer UPR to the nearby tumor cells, with the more potent induction of ER stress from GM-BMDMs-derived CMs.

Tumor cells sense transferable ER stress from two different polarized macrophage populations. a Schematic diagram of experimental workflow. GM-BMDMs and M-BMDMs were treated with Tm (1 μg/ml) for 0, 4, 6, 8 and 12 h. After washing 3 × with Dulbecco’s PBS, fresh serum-containing media were added. 16 h later, conditioned media (CM_GM and CM_M) were collected and used to culture LLC cells for 24 h. LLC cells were harvested and processed for the indicated measurement. b–h LLC cells were treated as in a and total RNAs were prepared and used to determine mRNA levels of UPR activation markers CHOP (b), Grp78 (c) and sXbp1 (d), and the whole cell lysates were processed for Immunoblot analysis and probed with the indicated primary antibodies including Grp78, sXbp1, CHOP (e). Molecular weights in kDa are indicated to the right. Data are representative of triplicate experiments and the quantitative analysis of Western blot data are shown in f–h. i–k LLC cells were treated as in a and total RNAs were analyzed by qRT-PCR for inflammatory mediators IL6 (i), IL1α (j) and CXCL1 (k). Data are presented as Mean ± SEM of triplicate experiments and the differences were evaluated by two-way ANOVA and multiple comparisons test. P < 0.05 is indicated by * for the comparison of Tm-treated groups vs untreated ones in CM_GM culture, Ψ for comparison of Tm-treated groups vs untreated ones in CM_M culture, and # for comparison CM_GM culture vs CM_M culture with the same Tm treatment time. ns, no significance. CM, conditioned media
Transferred ER stress induces LLC tumor cells death
One outstanding phenotypic switch of recipient cells we observed was the development of cell death upon transmitted ER stress (Fig. 2a–c). As shown, LLC cells dramatically died after CM incubation, and the longer ER stress received by donor cells the severe death the recipient cells experienced. Consistently, more LLC cells survived in M-BMDM-CMs than GM-BMDM-CMs both in 2D (Fig. S3a) and transwell 3D system (Fig. S3b-d). These results showed that the induction of cell death in recipient LLC tumor cells differed due to transmitted ER stress donor cells, with GM-BMDM-donor cells giving more dramatic cell death in LLC. Since both autophagy and apoptosis could be activated by ER stress [33], we further examined the typical apoptosis and autophagy markers in CM-treated LLC cells. The apoptotic signals, including cleaved-PARP, caspase-1 and caspase-9 were more abundant in LLCs treated with CMs from GM-BMDMs (Figs. 2d, e and S3e–g). Interestingly, the autophagy-related proteins were also higher in LLC tumor cells receiving transferable ER stress from GM-BMDMs (Figs. 2f, g and S3h, i). These results suggested that LLC cells initiated both autophagy and apoptosis upon this transferred ER stress in LLC cells, while autophagy initially activated to aid cell survival, prolonged stress might also induce autophagic and apoptotic cell death at later stage [33].

Transferred ER stress induces LLC tumor cell death. a–c LLC cells were treated as in Fig. 1a for 36 h. Cells were co-stained with PI (red) and Hoechst 33,342 (blue) for 30 min and photographed by a High-quality cell imaging instrument (HCI). Representative images were shown (a) and percentages of Hoechst+ cells (b) and PI+ cells (c) were calculated. Scale bars, 100 μm. Data are presented as Mean ± SEM of triplicate experiments and the differences were evaluated by two-way ANOVA and multiple comparisons test. P < 0.05 is indicated by * for the comparison of Tm-treated groups vs untreated ones in CM_GM culture, Ψ for comparison of Tm-treated groups vs untreated ones in CM_M culture, and by # for comparison CM_GM culture vs CM_M culture with the same TM treatment time. ns, no significance. d–g LLC tumor cells treated as in a for 36 h. Whole cell lysates were processed for Western Blot analysis and probed with the indicated primary antibodies including caspase-1, caspase-9, cleaved-PARP, p-ULK1, Beclin-1 and LC3 in d and p-JNK, p-ERK, p-p38, JNK, ERK and p38 in f. Molecular weights in kDa are indicated to the right. Data are representative of triplicate experiments and the quantitative analysis of Western blot data are in e and g. Data are presented as Mean ± SEM and the differences were evaluated by two-way ANOVA and multiple comparisons test. P < 0.05 is indicated by * for the comparison of Tm-treated groups vs untreated ones in CM_GM culture, Ψ for comparison of Tm-treated groups vs untreated ones in CM_M culture, and # for comparison CM_GM culture vs CM_M culture with the same Tm treatment time. CM, conditioned media
Both autophagy and apoptosis could be modulated by cascade signaling pathways including MAPK signaling pathways [34]. LLC recipient cells treated with ER-stressed CMs from GM-BMDMs had much higher levels of phosphorylated JNK (p-JNK) throughout all Tm treatment times (Fig. 2f, g). While extracellular signal-regulated kinase (p-ERK) was transiently induced in M-BMDMs stressed LLCs with the peak at 4 h of Tm treatment. In contrast, the level of p-ERK was gradually enhanced in GM-BMDMs stressed LLCs (Fig. 2f, g). The phosphorylated p38 (p-p38) abundance was highest in both populations at 4 h of Tm treatment with a more sustained level in GM-BMDMs stressed LLCs (Figs. 2f and S3j). These findings demonstrated that MAPK signaling pathways could be the potential targets of this transmissible ER stress and different macrophage populations activate these pathways with different patterns.
As previously reported [5], ER stress could transfer from tumor cells to macrophages. We next tested if tumor cells influenced the survival of different macrophage populations equally. Interestingly, the transmission of ER stress from tumor cells led to the induction of cell death in macrophages, and GM-BMDMs died more after receiving transferable ER stress (Fig. S3k–n). These data suggested that the bidirectional transmissible ER stress can drive recipient cell death and play a significant role in the hostile condition between tumor and GM-like M1 macrophage.
LLC cells exhibit different gene expression profiles in response to transferred ER stress from two polarized macrophage populations
To better characterize the influence of transmissible ER stress on the recipient cells, the high-throughput RNA-seq [35] was performed with LLC cells cultured in various CMs. We compared normally cultured LLC cells (NT) with LLC cultured in CMs from unstressed M-BMDMs (CM_M0) or GM-BMDMs (CM_G0) as well as with LLC cultured in CMs from 12-h ER-stressed M-BMDMs (CM_M12) or GM-BMDMs (CM_G12). The heatmap of differentially expressed genes (DEGs) showed that treatment with CMs could alter gene expression in LLC cells (Fig. 3a). There were 101 (CM_G12 vs CM_G0) and 144 (CM_M12 vs CM_M0) upregulated DEGs in LLC treated with stressed CMs, respectively (Fig. 3b), in which multiple ER stress-inducible genes were among the top upregulated genes in both CM_G12 and CM_M12 treated LLC cells (Fig. 3c), such as Avil, Car6, Lcn2, Sspo, Ppp1r15a and Hspa5, demonstrating that the transmission of ER stress from two macrophage populations to recipient tumor cells took place. Interestingly, some inflammation-related genes including Ptgs2, Sh2d6 and ATf3 were also included in the top changed DEGs.

LLC cells exhibit different gene expression profiles in response to transferred ER stress from two polarized BMDMs. a The hierarchical cluster of DEGs (fold change > 1.5 and P value < 0.05) of pairwise comparison between five groups. LLC cells were treated as in Fig. 1a. 24 h later, the total RNA of LLC cells was extracted and performed for RNA-seq analysis. NT represented LLC cells without CMs treatment. CM_G0 and CM_M0 for LLC cells treated with CM_GM or CM_M without Tm incubation, respectively, CM_G12 and CM_M12 for LLC cells treated with CM_GM or CM_M with Tm incubation for 12 h. The upregulated DEGs were indicated in red and down-regulated in green. b Bar chart of DEGs between each comparison in a. c The Volcano Plots of DEGs among LLC cultivated in ER-stressed conditioned media, CM_G12 vs CM_G0 (left) and CM_M12 vs CM_M0 (right). Red dots showed the upregulated DEGs in CM_G12 or CM_M12 and blue dots for the down-regulated DEGs. d GO enrichment analysis of the shared upregulated DEGs in CM_G12 vs CM_G0 and CM_M12 vs CM_M0. The bar charts represent the enrichment scores of relevant pathways. The dotted lines indicate the gene numbers enriched to related pathways. GO enrichment analysis of the upregulated DEGs (e) and down-regulated DEGs (f) in the comparison of CM_M12 vs CM_G12 group. The blue bar charts represent enrichment scores of the relevant pathways. The orange dotted lines indicate the gene numbers enriched to related pathways. g The relative distribution of apoptotic-associated genes (above) and autophagy-related genes (below). The gene lists were seen in Table S2. h The upregulated networks of the term “inflammatory response” by IPA analysis. Orange indicates upregulated disease/function in GM_G12 group compared with CM_M12. Green indicates down-regulated DEGs while red indicates upregulated DEGs in CM_G12 vs CM_M12 group. DEG, differentially expressed genes; FC, fold change
Consistently, the GO enrichment analysis indicated that the pathways of responses to unfolded protein, cytokine activity, as well as protein folding were significantly enriched in the functions of upregulated DEGs both for CM_G12 vs CM_G0 and CM_M12 vs CM_M0 (Fig. 3d). In addition, the upregulated DEGs of the CM_M12 group compared with the CM_G12 group (CM_M12 vs CM_G12 up) were specifically enriched in the functions beneficial to blood vessel development and cell migration (Fig. 3e), whereas the down-regulated DEGs (CM_M12 vs CM_G12 down) were enriched in pathways of cell killing and inflammatory responses (Fig. 3f), such as the response to IFNγ and IL1. Moreover, the apoptosis- and autophagy-associated genes were highly expressed in LLCs receiving GM-BMDMs stressed CMs (Fig. 3g; Table S2). The IPA analysis of significant DEGs showed that tumor cells treated with CMs from stressed M1-like macrophages (CM_G12) possessed more ability to recruit and activate immune cells, which may, in turn, be a disadvantage to tumor growth (Fig. 3h). Collectively, these results suggested that tumor cells received transmissible ER stress from M2-like macrophages have the potential to facilitate tumor survival, while transmissible ER stress from M1-like macrophages could lead to more acute cell death and inflammation.
Differential ER stress responses in two polarized macrophage populations
The different responses of recipient LLC cells receiving transmissible ER stress from two donor macrophage populations suggested that these two types of macrophages might have different responses to ER stress. To assess this possibility, both GM-BMDMs and M-BMDMs were treated with Tm (1 μg/ml) for indicated times. The expression of ER stress genes (such as CHOP, Grp78 and sXbp1) were upregulated after 4–12-h Tm treatment (Figs. 4a–e and S4a–c) and M-BMDMs sense more ER stress than GM-BMDMs. Interestingly, the expression of proinflammatory mediators, including IL1β (Fig. 4f) and IL6 (Fig. 4g), were enhanced in Tm-treated macrophages more dominantly in GM-BMDMs. Under the same treatment condition, anti-inflammatory cytokine IL10 expressed dominantly in M-BMDMs, but its expression decreased upon stress (Fig. 4h). However, proinflammatory chemokine CXCL1 was induced early in stressed GM-BMDMs, but more abundant later in stressed M-BMDMs, indicating the diverse gene expression regulations in stressed macrophages (Fig. 4i).

Differential ER stress responses in two polarized macrophage populations. a Schematic diagram of experimental workflow. b–i GM-BMDMs and M-BMDMs were treated with Tm (1 μg/ml) for 0, 4, 6, 8 and 12 h. Total RNA was prepared and used to determine mRNA levels of UPR sensors CHOP (b), Grp78 (c) and sXbp1 (d) and inflammatory mediators IL1β (f), IL6 (g), IL10 (h) and CXCL1 (i) by RT-PCR. Whole cell lysates were processed for Western Blot analysis and probed with the indicated primary antibodies including Grp78, CHOP, sXbp1 (e). Molecular weights in kDa are indicated to the right. All data were represented as mean ± SEM of triplicate experiments and the differences were evaluated by two-way ANOVA and multiple comparisons. P < 0.05 is indicated by * for comparison of vehicle—(0 h) and Tm-treated GM-BMDMs, Ψ for comparison of vehicle—(0 h) and Tm-treated M-BMDMs, and # for comparison of GM-BMDMs and M-BMDMs with the same Tm treatment time. ns, no significance. j GM-BMDMs and M-BMDMs were treated with Tm (1 μg/ml) for 0 and 4 h. Total RNAs were processed for RNA-seq analysis. The hierarchically clustered heatmap of DEGs (fold change > 1.5 and P value < 0.05) of pairwise comparison of vehicle- and Tm-treated groups for GM-BMDMs (G0 and GTm4h) and M-BMDMs (M0 and MTm4h). The upregulated DEGs were indicated in red and down-regulated in green. GO enrichment analysis of the upregulated (k) and down-regulated (l) DEGs in MTm4h compared with GTm4h. The blue bar charts represent enrichment scores of the relevant pathways. The orange dotted lines indicate the gene numbers enriched to related pathways
To better elucidate ER stress-induced responses in different macrophage populations, we also performed RNA-seq studies in Tm-challenged GM-BMDMs and M-BMDMs. The unbiased hierarchically clustered heatmap of DEGs indicated that stress induces a subset of genes expression in both cell types, however, the differentiation factors appeared to be the dominant determinator of gene expression profiles (Fig. 4j). Under normal physiological conditions, GM-BMDMs (G0 vs M0 up) promoted proinflammatory responses, such as IL1, IFNγ and TNF responses and provided hostile surroundings for tumor cells (Fig. S4d). Meanwhile, M-BMDMs exhibited functions (G0 vs M0 down) including tissue remodeling and angiogenesis (Fig. S4e), indicating M-BMDMs could help the tumor to remold TME and hold the tumorigenic abilities. Furthermore, ER stress-induced genes in M-BMDMs (MTm4h vs GTm4h up) were enriched in angiogenesis and cell–cell adhesion (Fig. 4k). On the other hand, multiple inflammatory response-related cell death and apoptosis signaling pathways were enriched in stressed GM-BMDMs (MTm4h vs GTm4h down) (Fig. 4l). Consistently, apoptosis-, autophagy- and necroptosis-related genes were significantly heightened in ER-stressed GM-BMDMs (Fig. S4f–h; Table S3). Altogether, the results suggested that macrophages with different polarization characteristics could respond to ER stress differentially. Notably, M-BMDMs were more susceptible to ER stress, but GM-BMDMs facilitated the induction of proinflammatory signals while M-BMDMs facilitate tumor growth.
GM-BMDM-derived CMs contain more proinflammatory mediators and enrich cell death functions
As the CM from stressed GM-BMDMs and M-BMDMs have distinct effects on LLC cell survival, the next question we asked is which component(s) in the CM exert this function. BMDMs were treated with Tm for 6 h when there is no measurable cell death (Fig. S5a) and the supernatants were collected and analyzed by mass spectrometry [36]. The differential secreted proteins were identified as in Fig. 5a and Table S4. There were 157 upregulated secreted proteins released from stressed GM-BMDMs (CM_G6 vs CM_GM_C) and 59 proteins in stressed M-BMDMs (CM_M6 vs CM_M_C) (Fig. 5b). Functions of inflammatory response, ERK cascade, protein secretion, extracellular exosome and vesicle transport were found in ER stress-induced changes of secreted proteins in both stressed macrophage populations (CM_G6 vs CM_GM_C up and CM_M6 vs CM_M_C up), indicating ER stress was capable of prompting molecules release in both cells (Fig. 5c). Not surprisingly, the functions of secretory proteins from M-BMDMs (CM_GM_C vs CM_M_C down) were beneficial for remodeling (Fig. S5b) while those from GM-BMDMs (CM_GM_C vs CM_M_C up) were inflammation-related (Fig. S5c). Furthermore, the upregulated secreted proteins from ER-stressed GM-BMDMs (CM_G6 vs CM_M6) were enriched in apoptotic signaling process and secretory regulation (Fig. 5d). In contrast, the upregulated proteins in Tm-challenged M-BMDMs (CM_M6 vs CM_G6) were strongly related to proliferation and regeneration (Fig. 5e), suggesting the different responses to UPR between two characteristic macrophage populations. Consistent with the shown observations (Fig. 4f–1), in the CMs from stressed GM- and M-BMDMs, the levels of proinflammatory cytokines, such as IL2, IL6, IL1β, IL12p70, TNFα and IFNγ were much higher in CM_G6, whereas the anti-inflammatory cytokine IL10 was higher in CM_M6 (Fig. 5f). In-depth analysis of these differentially secreted proteins (CM_G6 vs CM_M6 up) revealed that cell death-associated signaling pathways were predicted to be upregulated in CMs from ER-stressed GM-BMDMs, and correspondingly the cell survival was predicted to be upregulated (Fig. 5g) in ER-stressed M-BMDM-CMs, which may partially explain the phenomenon observed in Fig. 2. These results demonstrated GM-BMDMs under ER stress released much more molecules heightened with cell death signals.

GM-BMDM-derived conditioned medium contains more proinflammatory mediators and enriches in cell death functions. a GM-BMDMs and M-BMDMs were washed 3 × with Dulbecco’s PBS and fresh serum-free media or media containing Tm (1 μg/ml) were added. 6 h later, CMs of GM-BMDMs (CM_GM_C and CM_G6) and M-BMDMs (CM_M_C and CM_M6) were harvested and processed for proteomic analysis. The heatmap of secreted proteins filtered using UniProt database was shown (Fold change > 1.2). b Venn diagram of the upregulated secreted proteins among the indicated comparison groups. c GO enrichment analysis of the shared upregulated secreted molecular pathways in CM_G6 vs CM_GM_C and CM_M6 vs CM_M_C. The bar charts represent the enrichment scores of the relevant enriched pathways. The dotted lines indicate the gene numbers enriched to related pathways. Pathway analysis of the upregulated secreted proteins of CM_G6 compared with CM_M6 (d) and CM_M6 compared with CM_G6 (e). f GM-BMDMs and M-BMDMs were treated with Tm (1 μg/ml) for 6 h. After washing 3 × with Dulbecco’s PBS, fresh serum-containing media were added. 16 h later, CMs for GM-BMDMs (CM_G6) and M-BMDMs (CM_M6) were collected and 10 inflammatory cytokines/chemokines were analyzed. g The predicted disease/function networks of the differently expressed secreted proteins of CM_G6 compared with CM_M6 were evaluated by IPA analysis. Orange indicates upregulated disease/function in the CM_G6 group compared with CM_M6 while blue indicated down-regulated ones
GM-BMDMs store and release more DAMPs and induce tumor cell death in synergy with ER stress
To verify which kind of molecules in the CMs might contribute significantly towards death of the recipient cell, the IPA upstream analysis was applied to the DEGs of LLC recipient cells under the transmissible ER stress from ER-stressed GM-BMDMs and M-BMDMs (CM_G12 vs CM_M12). The result revealed that the S100 family could be the causal factor resulting in subsets of gene expression changes in recipient cells, which are connected with the functions of cell death and inflammatory responses (Fig. 6a). We noticed that some of them are DAMPs, which could be released upon ER stress and trigger cell death and immune responses [37]. Therefore, we hypothesized that GM-BMDMs might release more DAMPs into the CMs under stress conditions than M-BMDMs. By looking at the known DAMPs detected in the transcriptome and proteomics in GM- and M-BMDMs, S100A8 and S100A9 with relatively high expression levels were found in GM-BMDMs and enriched in stressed GM-BMDMs (Fig. S5d). We further verified these findings by RT-PCR, Western blot and ELISA. As shown in Figs. 6b, c and S5e, f, GM-BMDMs showed higher levels of S100A8 and S100A9 mRNA transcripts and S100A9 protein. Moreover, the levels of S100A8, S100A9 and the dimer of S100A8/A9 were higher in CMs derived from GM-BMDMs (Fig. 6d–f). The results indicated that GM-BMDMs could store more DAMPs in normal physiological conditions and release more DAMPs under ER stress.

GM-BMDMs store and release more DAMPs. a GM-BMDMs and M-BMDMs were treated with Tm (1 μg/ml) for 12 h. After washing 3 × with Dulbecco’s PBS, fresh serum-containing media were added. 16 h later, conditioned media were collected and used to culture LLC cells for 24 h. The total RNA of LLC cells was extracted and performed for RNA-seq analysis. The direct and indirect connection of S100 with the upregulated DEGs of CM_G12 compared with CM_M12 by IPA analysis. CM_G12 and CM_M12 represent LLC cells treated with CM_GM or CM_M derived from GM-BMDMs and M-BMDMs incubated with Tm for 12 h. The orange box indicates the upstream regulatory gene family, red indicates the certain upstream regulatory genes, green indicates the detectable upregulated genes in sequencing, and blue shows the relevant biological functions displayed by detectable genes in green box. b–c GM-BMDMs and M-BMDMs were treated with Tm (1 μg/ml) for 0, 4, 6, 8 and 12 h. The S100A9 (b) and S100A8 (c) mRNA levels were determined using RT-PCR. d–f GM-BMDMs and M-BMDMs were treated with Tm (1 μg/ml) for the indicated time. After washing 3 × with Dulbecco’s PBS, fresh serum-containing media were added. 16 h later, CMs from GM-BMDMs (CM_GM) and M-BMDMs (CM_M) were collected and S100s were analyzed including S100A9 (d), S100A8 (e) and S100A8/A9 (f). Data were represented as mean ± SEM of triplicate experiments and the differences were evaluated by two-way ANOVA and multiple comparisons. P < 0.05 is indicated by * for comparison of vehicle—(0 h) and Tm-treated GM-BMDMs and by # for comparison of GM-BMDMs and M-BMDMs with the same Tm treatment time
ER stress is widely known to induce the efficient emission of DAMPs and serve as an important mechanism accentuating immunogenicity, especially decisive for immunogenic cell death (ICD) [38]. Hence, we postulated that these two proteins may affect cell survival along with ER stress in the process of ER stress transmission. To test this hypothesis, recombinant S100A8/A9 proteins were used to treat LLC cells with or without ER stress. As previously reported, both DAMPs and ER stress could induce certain cell death, however, surprisingly, the combination of DAMPs and stress could dramatically enhance tumor cell death (Fig. 7a, b).

DAMP signaling acts in synergy with ER stress signaling to induce tumor cell death. a, b LLC cells were treated with Tm (0.5 μg/ml), S100A8 (5 μg/ml) and S100A9 (5 μg/ml) individually or in combination for 6 h. Cells were co-stained with PI (red) and Hoechst 33,342 (blue) for 30 min and photographed by microscope. Representative images were shown (a) and the percentage of PI+ cells (b) was calculated. Scale bars, 100 μm. c Schematic diagram of experimental workflow. LLC cells were pretreated with DMSO or Tm (0.5 μg/ml) for 6 h and S100A9 (5 μg/ml) was added for the indicated times. d LLC cells were treated as in c. Whole-cell lysates were processed for Western Blot analysis and probed with the indicated primary antibodies including Grp78, CHOP, sXbp1, p-ERK, p-JNK, and p-p38, ERK, JNK and p38. Molecular weights in kDa are indicated to the right. Data are representative of triplicate experiments and the quantitative analysis of Western blot data is shown in right panel. Data were represented as mean ± SEM and evaluated by one-way ANOVA, *P < 0.05 is for comparison of vehicle (0 h) with other treatments
Mitogen-activated protein kinase (MAPK) signaling is predominantly activated upon DAMP stimulation [39]. S100A8 and S100A9 could induce the activation of NF-κB via increasing the phosphorylation of p38 and (or) p-ERK MAP kinases and play a pivotal role in the regulation of inflammatory responses [34]. To investigate how stress synergizes with DAMP signaling in inducing tumor cells death, LLC cells were pretreated with or without Tm to trigger the activation of UPR signals followed by S100A9 treatment as shown in Fig. 7c. Interestingly, pre-activated UPR signaling could further enlarge the S100A9-induced MAPK signaling pathways, especially the levels of p-JNK and p-p38. Meanwhile, S100A9 treatment following the activation of stress could significantly enhance the expression of ER stress markers, such as Grp78, CHOP and sXbp1 even with short time treatment (Fig. 7d and S6 a–d). The results suggested that S100A9 and S100A8 could severely induce cell death in synergy with ER stress, which may at least partially result from synergetic stress and DAMP-induced signaling (Fig. 8).

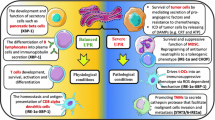
Schematic illustration of bidirectional ER stress transmission among macrophages and tumor cells in the tumor microenvironment. In the TME, ER stress could be sensed by both tumor cells and two types of macrophages. M1-like macrophages undergo less intracellular ER stress but could transfer more ER stress to the surrounding tumor cells in company with substantial release of proinflammatory cytokines, chemokines (IL6, IL1β, IL2, TNFα, etc.) and DAMPs (S100A8 and S100A9). On the contrary, M2-like macrophages sense more but transfer less ER stress to surrounding tumor cells with the release of anti-inflammatory molecules such as IL10, TGFβ and VEGF and less DAMPs (S100A8 and S100A9). The DAMPs-induced MAPK signaling could synergy with transferred ER stress signaling, consequently tumor cells are more likely to be killed by M1 macrophages but survive better with M2 macrophages. In turn, ER stress initiated by tumor cells within TME could vice versa be transferred to the nearby M1 and M2 macrophages, resulting in the more cell death in M1 macrophages but not much in M2 macrophages. Eventually, tumor cells and anti-inflammatory M2 macrophages (e.g., TAMs) survive under ER stress within TME to provoke tumor progression
Discussion
Persistent ER stress, an essential feature of cancer, is driven by multiple metabolic and oncogenic abnormalities in the tumor microenvironment (TME) that disrupt protein-folding homeostasis in malignant cells and infiltrating immune cells. The active ER stress response enables malignant cells to adapt to carcinogenesis and environmental challenges and coordinates with various immunomodulatory mechanisms to promote malignant progression [40]. In addition to directly mediating the activities of cancer cells, it has been reported that ER stress is also involved in intercellular communication. ER stress can be transferred from tumor cells to the surrounding bone marrow-derived myeloid cells, macrophages, and dendritic cells [5, 10]. And the receiver macrophages can sense and potentiate the transferred ER stress in a TLR4-dependent manner [5]. Moreover, cancer cells could regulate the macrophage transforming into the anti-inflammatory phenotype during the ER stress transmission, the anti-tumor effects of immune cells in the tumor were repressed and therefore the tumor growth and development can be promoted [41]. Along with the production of tumorigenic cytokines and immunosuppressive enzyme arginase, the transmission of ER stress could also suppress cross-priming in dendritic cells and further block the T-cell proliferation [42]. Furthermore, transmissible ER stress was shown to induce the activation of Wnt signaling in recipient human prostate cancer cells and enhance the resistance to common chemotherapies [10].
In the previous reports [5, 10, 41], the source of transferable ER stress has been focused on the tumor intrinsic ER stress. As the non-cancer cells in TME should also sense the hypoxia, nutrient deprivation, lactic acidosis resulted from oxygen depletion, these cells could also initiate intrinsic ER stress. Macrophages, an abundant tumor infiltration immune cell populations, could experience the high demand of protein folding or secretion during chemoattraction and response to a variety of intratumoral stimuli, therefore, it is likely that the ER stress could be initiated from macrophage as well. In the current study, our results suggested that the transferable ER stress indeed is bidirectional. It could also be transferred from stressed macrophages to nearby tumor cells. This bidirectional transmissible ER stress might be a fundamental mechanism of intercellular communication when tissue adapted to various stresses, especially in the TME.
Macrophage polarization present in the TME has complex functions in shaping the TME and regulating immunity. For example, the ratio of M1 to M2-like macrophages is closely correlated with the survival rate in ovarian cancer patients, and the overall M1/M2 TAM ratio decreased as the cancer stage increased [43]. Similar to this, a better overall survival was observed in pediatric classical Hodgkin Lymphoma patients with a predominant M1 polarization [44]. Hypoxia, lactate production and other cytokines can shift macrophage polarization from an anti-tumor M1-like phenotype towards a pro-tumor M2-like based on the environmental changes in the TME [45]. Several preclinical studies also manifested some strategies switch M2-like to M1-like phenotypes in order to inhibit tumor growth and progression [46, 47]. Approximately 30 compounds can shift M1-like phenotypes toward the M2-like macrophages and 20 compounds switch M2-like TAMs to the M1 phenotype through high-throughput screening were also identified in the reported study [48]. M-CSF, a M2 polarization factor, is overexpressed in many malignancies [49,50,51]. Treatment of tumor-bearing mice with anti-M-CSFR monoclonal blocking antibody resulted in a reduction of mature TAMs, and the block of M-CSFR is one of the oncotherapy strategies clinically [52]. On the other hand, GM-CSF has been used in cancer immunotherapy as an adjuvant by activation of both innate and adaptive immunity [53]. In our study, we use GM-CSF- and M-CSF-derived BMDMs as the model cells and found that though GM-CSF primed M1-like macrophages respond to ER stress weaker than M-CSF primed M2-like ones but they could transfer stronger ER stress signals to surrounding tumor cells. The elevated ER stress in recipient tumor cells initiates the cell death signaling leading to notable tumor killing. More interestingly, this M1-like macrophage is also disliked by the tumor cells. Once the tumor cells facing the M1–like macrophage, it also prefers to trigger this kind of M1 macrophage to die. Contrast to that, M2-like macrophage survived better in the stressed TME even though they sensed more stress. The intercellular signals M2-like macrophage sent to tumor cells are enriched in supporting tumor growth. Therefore, intratumoral macrophage polarization status not only influence the condition of tumor proliferation, but also is decisive for the survival of macrophages themselves (Fig. 8). That is consistent with the observations that most TAM are M2 like. As eliminating the tumor-associated immune monocytes in tumor tissue is unpractical, the strategy regulated macrophages’ polarization under ER stress might be more preferable and is worthy of further investigation.
To date, the underlying mechanisms of how ER stress is transferred among cells remain elusive. The molecules produced by ER stress transmission were suggested as a considerable participant that can facilitate the tumor cell progression and survival in the microenvironmental surroundings [41]. The cellular ER stress induced in HepG2 cells promotes the release of DAMPs to enable the death signals [37]. ICD elicited by UPR enables the release of critical danger signals (normally DAMP) and propagates immunity to augment therapeutic efficacy eventually to anti-neoplastic immunity [38, 54]. Emerging evidence revealed that the cancer cells could communicate with non-cancer cells involving ER stress via secreted factors, such as tumor-derived extracellular vesicles (exosomes) [55, 56], secreted proteins (Golgi protein 73) [57] even the lactic acid [41]. Chemotherapeutics and radiotherapy have been reported to induce ER stress with the emission of DAMPs to efficiently stimulate the immune system and critically provoke immunogenic cell death (ICD) [38, 54]. S100 proteins are believed to express the hallmark of DAMPs and play a crucial role in regulating the immune responses. In fact, DAMPs, especially S100 proteins were reported to be able to excrete extracellularly freely or in exosomes [58]. Free S100 proteins could bind to cell membrane receptors (RAGE or TLRs) to initiate the inflammatory response [59], however, the mechanism of S100 proteins within EVs remains to be investigated. In the current study, we identified S100A8 and S100A9 in the ER-stressed CM using both ELISA and Western blot; therefore, it is likely that the released S100 proteins are freely exist extracellularly, however, whether they are also in EVs require the further studies. Nevertheless, the release of more S100A8 and S100A9 by the ER-stressed M1-like macrophage could trigger the MAPK signaling in tumor cells, which is in synergy with amplified transferred ER stress leading to significant tumor death. Further studies identifying the whole spectrum of necessary and sufficient secreted factors mediating ER stress transmission may constitute key points to elucidate the pro- and anti-cancer mechanisms. The deeper understanding of ER stress mediated complex intertumoral cell communications might contribute to further clinical therapies to cancer patients.
Data availability
The data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE193669. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE193669).
Abbreviations
- ER:
-
Endoplasmic reticulum
- TME:
-
Tumor microenvironment
- UPR:
-
Unfolded protein response
- DC:
-
Dendritic cell
- TLR:
-
Toll-like receptor
- M-CSF:
-
Macrophage colony-stimulating factor
- GM-CSF:
-
Granulocyte–macrophage colony-stimulating factor
- TAM:
-
Tumor-associated macrophage
- GM-BMDM:
-
GM-CSF cultured bone marrow-derived macrophage
- M-BMDM:
-
M-CSF cultured bone marrow-derived macrophage
- DAMP:
-
Damage-associated molecular pattern
- MAPK:
-
Mitogen-activated protein kinase
- Tm:
-
Tunicamycin
- Tg:
-
Thapsigargin
- CM:
-
Conditioned media
- Grp78:
-
Glucose-regulated protein-78
- sXbp1:
-
Spliced X-box binding protein 1
- CHOP:
-
C/EBP homologous protein
- IL:
-
Interleukin
- CXCL1:
-
C–X–C motif chemokine ligand 1
- cPARP:
-
Cleaved Poly (adp-ribose) polymerase
- p-JNK:
-
Phosphorylated c-Jun N-terminal kinase
- p-ERK:
-
Phosphorylated extracellular signal-regulated kinase
- NT:
-
No treatment
- DEG:
-
Differentially expressed gene
- GO:
-
The Gene Ontology
- IFN:
-
Interferon
- IPA:
-
Ingenuity pathway analysis
- EV:
-
Extracellular vesicle
- TNF:
-
Tumor necrosis factor
- ICD:
-
Immunogenic cell death
- DMEM:
-
Dulbecco's modified eagle medium
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- FC:
-
Fold change
References
Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420:860–867
Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 454:436–444
Diakos CI, Charles KA, McMillan DC, Clarke SJ (2014) Cancer-related inflammation and treatment effectiveness. Lancet Oncol 15:e493-503
Bettigole SE, Glimcher LH (2015) Endoplasmic reticulum stress in immunity. Annu Rev Immunol 33:107–138
Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, Zanetti M (2011) Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc Natl Acad Sci USA 108:6561–6566
Xiong Z, Jiang R, Zhang P, Han X, Guo FJ (2015) Transmission of ER stress response by ATF6 promotes endochondral bone growth. J Orthop Surg Res 10:141
Doron B, Abdelhamed S, Butler JT, Hashmi SK, Horton TM, Kurre P (2019) Transmissible ER stress reconfigures the AML bone marrow compartment. Leukemia 33:918–930
Zhang H, Yue Y, Sun T, Wu X, Xiong S (2017) Transmissible endoplasmic reticulum stress from myocardiocytes to macrophages is pivotal for the pathogenesis of CVB3-induced viral myocarditis. Sci Rep 7:42162
Tirosh A, Tuncman G, Calay ES, Rathaus M, Ron I, Tirosh A, Yalcin A, Lee YG, Livne R, Ron S et al (2021) Intercellular transmission of hepatic ER stress in obesity disrupts systemic metabolism. Cell Metab 33(319–333):e316
Rodvold JJ, Chiu KT, Hiramatsu N, Nussbacher JK, Galimberti V, Mahadevan NR, Willert K, Lin JH, Zanetti M (2017) Intercellular transmission of the unfolded protein response promotes survival and drug resistance in cancer cells. Sci Signal 10
Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L (2019) Macrophages and metabolism in the tumor microenvironment. Cell Metab 30:36–50
Dehne N, Mora J, Namgaladze D, Weigert A, Brune B (2017) Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr Opin Pharmacol 35:12–19
Zhu XD, Zhang JB, Zhuang PY, Zhu HG, Zhang W, Xiong YQ, Wu WZ, Wang L, Tang ZY, Sun HC (2008) High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J Clin Oncol 26:2707–2716
Qian BZ, Pollard JW (2010) Macrophage diversity enhances tumor progression and metastasis. Cell 141:39–51
Wu K, Lin K, Li X, Yuan X, Xu P, Ni P, Xu D (2020) Redefining tumor-associated macrophage subpopulations and functions in the tumor microenvironment. Front Immunol 11:1731
Condeelis J, Pollard JW (2006) Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124:263–266
Lichtnekert J, Kawakami T, Parks WC, Duffield JS (2013) Changes in macrophage phenotype as the immune response evolves. Curr Opin Pharmacol 13:555–564
Ma J, Liu L, Che G, Yu N, Dai F, You Z (2010) The M1 form of tumor-associated macrophages in non-small cell lung cancer is positively associated with survival time. BMC Cancer 10:112
Hamilton TA, Zhao C, Pavicic PG Jr, Datta S (2014) Myeloid colony-stimulating factors as regulators of macrophage polarization. Front Immunol 5:554
Zhang L, Pavicic PG Jr, Datta S, Song Q, Xu X, Wei W, Su F, Rayman PA, Zhao C, Hamilton T (2019) Unfolded protein response differentially regulates TLR4-induced cytokine expression in distinct macrophage populations. Front Immunol 10:1390
Lee GY, Kenny PA, Lee EH, Bissell MJ (2007) Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods 4:359–365
Zoetemelk M, Rausch M, Colin DJ, Dormond O, Nowak-Sliwinska P (2019) Short-term 3D culture systems of various complexity for treatment optimization of colorectal carcinoma. Sci Rep 9:7103
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12:357–360
Shahriyari L (2019) Effect of normalization methods on the performance of supervised learning algorithms applied to HTSeq-FPKM-UQ data sets: 7SK RNA expression as a predictor of survival in patients with colon adenocarcinoma. Brief Bioinform 20:985–994
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Brief Bioinform 15:550
Lomax J (2005) Get ready to GO! A biologist’s guide to the Gene Ontology. Brief Bioinform 6:298–304
Orlando N, Babini G, Chiusolo P, Valentini CG, De Stefano V, Teofili L (2020) Pre-exposure to defibrotide prevents endothelial cell activation by lipopolysaccharide: an ingenuity pathway analysis. Front Immunol 11:585519
Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10:1794–1805
Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, Bassez A, Decaluwe H, Pircher A, Van den Eynde K et al (2018) Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 24:1277–1289
Ma L, Hernandez MO, Zhao Y, Mehta M, Tran B, Kelly M, Rae Z, Hernandez JM, Davis JL, Martin SP et al (2019) Tumor cell biodiversity drives microenvironmental reprogramming in liver cancer. Cancer Cell 36(418–430):e416
Jiang M, Li X, Zhang J, Lu Y, Shi Y, Zhu C, Liu Y, Qin B, Luo Z, Du Y et al (2021) Dual inhibition of endoplasmic reticulum stress and oxidation stress manipulates the polarization of macrophages under hypoxia to sensitize immunotherapy. ACS Nano 15:14522–14534
Wu J, Chen S, Liu H, Zhang Z, Ni Z, Chen J, Yang Z, Nie Y, Fan D (2018) Tunicamycin specifically aggravates ER stress and overcomes chemoresistance in multidrug-resistant gastric cancer cells by inhibiting N-glycosylation. J Exp Clin Cancer Res 37:272
Song S, Tan J, Miao Y, Li M, Zhang Q (2017) Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J Cell Physiol 232:2977–2984
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, He C, Pan H (2014) p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett 344:174–179
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Tyanova S, Temu T, Cox J (2016) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 11:2301–2319
Andersohn A, Garcia MI, Fan Y, Thompson MC, Akimzhanov AM, Abdullahi A, Jeschke MG, Boehning D (2019) Aggregated and hyperstable damage-associated molecular patterns are released during ER stress to modulate immune function. Front Cell Dev Biol 7:198
Garg AD, Martin S, Golab J, Agostinis P (2014) Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ 21:26–38
Averill MM, Kerkhoff C, Bornfeldt KE (2012) S100A8 and S100A9 in cardiovascular biology and disease. Arterioscler Thromb Vasc Biol 32:223–229
Chen X, Cubillos-Ruiz JR (2021) Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer 21:71–88
Jiang Z, Zhang G, Huang L, Yuan Y, Wu C, Li Y (2020) Transmissible endoplasmic reticulum stress: a novel perspective on tumor immunity. Front Cell Dev Biol 8:846
Mahadevan NR, Anufreichik V, Rodvold JJ, Chiu KT, Sepulveda H, Zanetti M (2012) Cell-extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8(+) T cell priming. PLoS ONE 7:e51845
Zhang M, He Y, Sun X, Li Q, Wang W, Zhao A, Di W (2014) A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J Ovarian Res 7:19
Barros MH, Segges P, Vera-Lozada G, Hassan R, Niedobitek G (2015) Macrophage polarization reflects T cell composition of tumor microenvironment in pediatric classical Hodgkin lymphoma and has impact on survival. PLoS ONE 10:e0124531
Boutilier AJ, Elsawa SF (2021) Macrophage polarization states in the tumor microenvironment. Int J Mol Sci 22
Wei Z, Zhang X, Yong T, Bie N, Zhan G, Li X, Liang Q, Li J, Yu J, Huang G et al (2021) Boosting anti-PD-1 therapy with metformin-loaded macrophage-derived microparticles. Nat Commun 12:440
Silini AR, Magatti M, Cargnoni A, Parolini O (2017) Is immune modulation the mechanism underlying the beneficial effects of amniotic cells and their derivatives in regenerative medicine? Cell Transplant 26:531–539
Hu G, Su Y, Kang BH, Fan Z, Dong T, Brown DR, Cheah J, Wittrup KD, Chen J (2021) High-throughput phenotypic screen and transcriptional analysis identify new compounds and targets for macrophage reprogramming. Nat Commun 12:773
Szomolay B, Eubank TD, Roberts RD, Marsh CB, Friedman A (2012) Modeling the inhibition of breast cancer growth by GM-CSF. J Theor Biol 303:141–151
Pei BX, Sun BS, Zhang ZF, Wang AL, Ren P (2014) Interstitial tumor-associated macrophages combined with tumor-derived colony-stimulating factor-1 and interleukin-6, a novel prognostic biomarker in non-small cell lung cancer. J Thorac Cardiovasc Surg 148(1208–1216):e1202
Chen YC, Lai YS, Hsuuw YD, Chang KT (2021) Withholding of M-CSF supplement reprograms macrophages to M2-like via endogenous CSF-1 activation. Int J Mol Sci 22(7):3532. https://doi.org/10.3390/ijms22073532
Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I et al (2014) Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 25:846–859
Yan WL, Shen KY, Tien CY, Chen YA, Liu SJ (2017) Recent progress in GM-CSF-based cancer immunotherapy. Immunotherapy 9:347–360
Fuchs Y, Steller H (2015) Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol 16:329–344
Javeed N, Sagar G, Dutta SK, Smyrk TC, Lau JS, Bhattacharya S, Truty M, Petersen GM, Kaufman RJ, Chari ST et al (2015) Pancreatic cancer-derived exosomes cause paraneoplastic beta-cell dysfunction. Clin Cancer Res 21:1722–1733
Wu Z, Wang L, Li J, Wang L, Wu Z, Sun X (2018) Extracellular vesicle-mediated communication within host-parasite interactions. Front Immunol 9:3066
Wei C, Yang X, Liu N, Geng J, Tai Y, Sun Z, Mei G, Zhou P, Peng Y, Wang C et al (2019) Tumor microenvironment regulation by the endoplasmic reticulum stress transmission mediator golgi protein 73 in mice. Hepatology 70:851–870
Abu N, Rus Bakarurraini NAA, Nasir SN (2021) Extracellular vesicles and DAMPs in cancer: a mini-review. Front Immunol 12:740548
Ma L, Sun P, Zhang JC, Zhang Q, Yao SL (2017) Proinflammatory effects of S100A8/A9 via TLR4 and RAGE signaling pathways in BV-2 microglial cells. Int J Mol Med 40:31–38
Acknowledgements
This work was supported by the Major Program of National Natural Science Foundation of China (81991525) and Key R&D Program of Shandong Province (2020CXGC010503).
Funding
This work was supported by the Major Program of National Natural Science Foundation of China (81991525) and Key R&D Program of Shandong Province (2020CXGC010503).
Author information
Authors and Affiliations
Contributions
All authors participated in designing the study as well as interpreting the whole data. CZ and JY supervised the study; WW, YZ, XZ, ZW, XX and YY performed and evaluated individual experiments; YX and QZ performed bioinformatical analyses; WW, XL, QS, CZ and JY wrote the manuscript with the contributions from all the authors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have declared no conflicts of interests.
Ethical approval
The study was approved by the Committee on the Ethics of Animal Experiments of the School of Life Sciences of Lanzhou University (EAF-2021026).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wei, W., Zhang, Y., Song, Q. et al. Transmissible ER stress between macrophages and tumor cells configures tumor microenvironment. Cell. Mol. Life Sci. 79, 403 (2022). https://doi.org/10.1007/s00018-022-04413-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-022-04413-z