
Abstract
Cancer is the second leading cause of death globally. Abnormity in gene expression regulation characterizes the trajectory of tumor development and progression. RNA-binding proteins (RBPs) are widely dysregulated, and thus implicated, in numerous human cancers. RBPs mainly regulate gene expression post-transcriptionally, but emerging studies suggest that many RBPs can impact transcription by acting on chromatin as transcription factors (TFs) or cofactors. Here, we review the evidence that RBM38, an intensively studied RBP, frequently plays a tumor-suppressive role in multiple human cancer types. Genetic studies in mice deficient in RBM38 on different p53 status also establish RBM38 as a tumor suppressor (TS). By uncovering a spectrum of transcripts bound by RBM38, we discuss the diversity in its mechanisms of action in distinct biological contexts. Examination of the genomic features and expression pattern of RBM38 in human tissues reveals that it is generally lost but rarely mutated, in cancers. By assessing future trends in the study of RBM38 in cancer, we signify the possibility of targeting RBM38 and its related pathways as therapeutic strategies against cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer, also known as a malignant tumor, is a generic term for a group of diseases that can affect any part of the body. It is a major public health problem and a leading cause of death globally. Based on the GLOBOCAN 2018 estimates of cancer incidence and mortality worldwide, there will be an estimated 18.1 million new cases and 9.6 million cancer deaths in 2018 [1]. In both sexes combined, lung cancer is the most commonly diagnosed cancer and the leading cause of cancer death, closely followed by female breast, male prostate, and colorectal cancer for incidence and colorectal, stomach, and liver cancer for mortality [1]. Unfortunately, the incidence and mortality are rapidly growing globally. In essence, cancer is the uncontrolled growth of abnormal cells in any tissues. Although it is conventionally believed that cancer is caused by genetic mutations, it is now clear that most human cancers are, in fact, induced by nongenetic factors [2], such as physical carcinogens (e.g., ultraviolet, ionizing radiation), chemical carcinogens (e.g., asbestos, components of tobacco smoke), and biological carcinogens (e.g., infections from viruses, bacteria, or parasites). Regardless of the origins in cancer initiation, these cancer-causing factors all converge, eventually, on gene expression regulation at versatile levels [3], in that gene expression is fundamentally the key determinant of cellular phenotypes.
RNA-binding proteins (RBPs) are a class of proteins that primarily bind to RNA to form ribonucleoprotein (RNP) complexes for the regulation of gene expression and exertion of their pleiotropic functions in physiological and pathological processes [4]. To date, over 1500 RBPs, representing ~ 7.5% of all protein-coding genes in the human genome, have been identified [4, 5], highlighting their functional diversity and importance. RBPs are virtually involved in all aspects of RNA metabolism, including pre-mRNA splicing, capping, and polyadenylation, as well as mRNA export, turnover, localization, and translation [6]. Mechanistically, many RBPs bind to sequence-specific motifs or RNA secondary structures, or a combination of both, through unique modular arrangements of individual RNA-binding domains (RBDs), together with an auxiliary module to mediate interplay between proteins [4, 7]. Currently, over 50 different RNA binding modules have been proposed, typified by RNA recognition motif (RRM), heterogeneous nuclear ribonucleoprotein (hnRNP) K-homology domain (KH), double-stranded RNA binding motif (dsRBM), zinc fingers (ZnF) of the CCHC/CCCH/ZZ types, arginine-glycine-glycine (RGG) motif, cold shock domain (CSD), and others [4, 5]. However, almost half of RBPs interact with RNA in the absence of specific motifs or structural domains [5], adding another layer of complexity within RNP interaction networks. Notably, and interestingly, previous studies have mainly focused RNPs on post-transcriptional events, a recent report showed that RBPs can prevalently exert their functions at the level of chromatin by regulating transcription as transcription factors (TFs) or cofactors [8]. Collectively, RBPs paly pivotal roles in RNA biogenesis and metabolism and, therefore, perturbations in RBP–RNA network activities have been, not surprisingly, causally associated with tumorigenesis [4]. There is already ample experimental evidence that altered RBP function has a significant impact on cancer phenotypes (summarized in [4]). For instance, HNRNPA2B1 is characterized as a novel oncogene in glioblastoma in that it is overexpressed in tumor versus (vs.) normal tissues and predicts poor prognosis [9]. ESRP1, an epithelial cell-specific splicing regulatory protein, enhances lung colonization of metastatic cancer cell via splicing isoform switching tumor suppressive CD44s to oncogenic CD44v [10]. Moreover, ELAVL1 (best known as HuR) promotes the migration of tumor cells and lung cancer metastasis by stabilizing and thus upregulating SNAI1 (i.e., Snail), a marker of epithelial-mesenchymal transition (EMT) [11].
RNA-binding motif protein 38 (RBM38, also dubbed RNPC1) belongs to RRM-containing RBP family including NCL (i.e., nucleolin), U2AF56 and HuR [12, 13]. Since the first discovery of RBM38 as a p53 family target gene transcriptionally [14], later studies have unveiled pleiotropic roles of RBM38 in diverse pathological conditions, via frequently forming negative feedback loop with tumor suppressors (TS) such as p53 family (i.e., p53, p63, p73) [12, 15, 16]. Many well-known transcripts involved in tumor development and progression have been identified as RBM38 targets, rendering RBM38 a relatively extensively investigated RBP in the cancer research field. In this review, we describe the genomic features and expression pattern of RBM38 in human tissues, provide evidence for a tumor-suppressive role in various cancer types, and emphasize the diversity in its mechanisms of action in distinct biological contexts. By surveying the mutational landscape of RBM38 across the spectrum of human cancers, we also assess future trends in the study of RBM38 in cancer.
Molecular structure of RBM38
RBM38 is located on chromosome 20q13.31 and is highly expressed in a variety of human tissues (RPKM > 4 in 19 of 27 tissues; Fig. 1). Comparative phylogenic analysis of RBM38 at both mRNA and protein sequence levels reveals an evolutionary conservation across vertebrate species [13]. These results, together, signify a functional importance of RBM38 in development. Previous studies have all discovered that RBM38 has 4 exons and is expressed as two isoforms: RBM38a with 239 amino acids (aa) and RBM38b with 121aa identical to the N-terminal region of RBM38a (Fig. 2A). Genomically, RBM38b is the result of exon 4 skipping through alternative splicing. Interestingly, based on the recent NCBI annotation of Reference Sequences (RefSeq; updated on Mar 13, 2020), a new and the longest isoform RBM38c encoding 271aa is reported (Fig. 2A). Alignment of these three isoforms suggests the existence of an RRM domain consisting of two RNP sub-motifs (Fig. 2B). Notably, due to the inclusion of a cryptic short exon 2, the RNP1 motif is disrupted by an insertion of extra 32aa in RBM38c (Fig. 2B). Whether this novel isoform also functions normally, as does RBM38a, is unclear and requests a future investigation. Most of the previous studies have experimentally characterized isoforms a and b, with RBM38a being the predominant isoform that preserves the RBP properties in term of modulating mRNA post-transcriptionally [12, 14, 15]. Therefore, the evidence presented in this review is mainly attributed to RBM38a’s function. Notably, a paralogue of RBM38 located on chromosome 6p22.3, namely RBM24, is reported to preferentially express in striated muscles [17, 18]. RBM24 gene encodes 236aa and shares a high degree of sequence similarity with that of RBM38a. Particularly, both of them have almost an identical RRM [17, 19], indicating functional similarity and the possible interplay between them (discussed below).

Expression of RBM38 in human tissues. Reads per kilobase million (RPKM) values, derived from RNA-seq analysis of tissue samples from 95 human individuals representing 27 different tissues, were extracted from NCBI with Gene ID: 55544 (accessed on Mar 18, 2020)

Gene structure of RBM38. A Scheme depicting the central dogma (DNA/RNA/Protein) information of RBM38 locus. Previous studies have all discovered two isoforms (a, b) of RBM38 encoded by 4 exons (exon 1, 3, 4, 5). However, a novel and the longest isoform c is reported, due to an inclusion of cryptic exon 2, by recently updated NCBI RefSeq (Gene ID: 55544; accessed on Mar 18, 2020). The numbers underneath denote genomic coordinates corresponding to either DNA or RNA sequences. B Alignment of three isoforms highlighting the RNA recognition motif (RRM) domain consisting of two ribonucleoprotein (RNP) motifs. The consensus sequence of RNP1 (8 aa) and RNP2 (6 aa) are shown. Please see more details in the text
RBM38 mainly functions as a TS in diverse human cancers
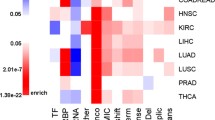
Due to the earlier observations that chromosome 20q13 is often amplified in multiple cancers including breast [20], prostate [21], ovarian [22], and colorectal cancer [23, 24], and chronic lymphocytic leukemia [23], and that RBM38 is overexpressed in dog lymphomas [12], human colorectal [24] and esophageal cancer [25], RBM38 has, therefore, been proposed as a putative oncogene in these cancers. However, we performed a close examination of the aforementioned literature and found no direct evidence supporting the amplification of 20q13.31 region specifically, where RBM38 resides, due to the low resolution of earlier mapping technologies. Consistently, little functional evidence was reported to implicate RBM38 as an oncogenic factor. To address this discrepancy, we surveyed the mutational landscape of RBM38 across 32 human cancer types offered by current The Cancer Genome Atlas (TCGA) DNA sequencing effort through cBioPortal [26]. Results clearly showed that, although amplification represents the main form of alterations in multiple cancers (e.g., colon, breast, ovary, sarcoma), the frequency is very low (generally ≤ 5.31%) (Fig. 3). Notably, the average mutation rate for RBM38 in total is 2.4% (Fig. 3), suggesting that genomic amplification is a rare event at the population level and thus should not be perceived as a causal rationale for proposing RBM38 for an oncogene. In fact, the vast majority of studies have established RBM38 as a TS in various human cancers (Table 1), including breast, hepatocellular, colorectal, renal, gastric, and other cancers.

Mutational landscape of RBM38 in 32 human cancers. Data derived from TCGA pan-cancer analysis was viewed through cBioPortal (accessed on Mar 17, 2020)
RBM38 in breast cancer
Breast cancer (BCa) is the most frequently diagnosed cancer and ranked as the second leading cause of death in women worldwide [27]. Study of RBM38 in BCa is relatively intensive, we thus separate it from other cancer types for reviewing the role of RBM38 in cancers in detail. In 2014, Xue et al. first reported that RBM38 is downregulated in multiple BCa lines compared with non-transformed mammary epithelial cells (e.g., MCF-10A), as well as in 121 human BCa vs. adjacent normal tissues, at both mRNA (by qPCR) and protein (by western blot) levels [28]. Experimentally, overexpression of RBM38a inhibits cell proliferation and migration and invasion in both estrogen receptor positive (ER+) MCF7 and triple-negative MDA-MB-231 cells in vitro by arresting cells at G1 phase [28]. Consistently, exogenous RBM38a expression also dampens MDA-MB-231 tumor development in vivo [28]. Of clinical relevance, RBM38 is positively correlated with the longer relapse-free and overall survival [29]. Furthermore, immunohistochemistry (IHC) analysis of 90 BCa samples revealed that RBM38a protein expression is significantly positively correlated with ERα [30] and progesterone receptor (PR) [31]. RBM38 increases the expression of PR [31] and ERα, but not ERβ, by stabilizing their mRNAs in ER+/PR+ BCa cells; however, ERα reversely regulates the expression of RBM38 in response to estrogen stimulation (Fig. 4A) [30]. MYC (i.e., c-Myc) is a notorious oncogenic TF that overexpresses in numerous human cancers and promotes G1/S cell cycle progression [32]. Interestingly, a recent report showed that, in a tissue microarray containing 162 BCa samples, RBM38 expression is negatively correlated with MYC at the protein level [33]. Although MYC is generally considered as a transcription activator, it binds to an E-box region in the promoter of RBM38 to inhibit transcription and RBM38, in turn, binds to and destabilizes MYC mRNA in ER+ BCa cells [33]. PTEN is a well-characterized TS and an inhibitor of the phosphatidylinositol-3-kinase (PI3K)/AKT pathway, which is frequently overactivated in many cancers and is associated with metastasis and cancer stemness [34]. Another IHC examination of a cohort of 77 BCa revealed that the expression of RBM38 and PTEN are positively correlated [34]. Experimentally, RBM38 upregulates PTEN expression by stabilizing its mRNA in ER+/HER2+ BT474 and triple negative MDA-MB-453 cells (Fig. 4A) [34], underscoring a possible positive feedback loop between these two TSs.

Role and mechanism of RBM38 in inhibiting BCa cell growth and metastasis. A RBM38 suppresses cell proliferation via destabilizing oncogene MYC and stabilizing TS PTEN through AREs in their 3′-UTRs. It is currently unclear how RBM38 maintains its tumor suppressive function while enhancing the expression, and likely, function of ER and PR, both of which are oncogenic in BCa. Notably, both MYC and ERα transcriptionally inhibits RBM38. B EMT-specific TF Snail halts RBM38 transcription, leading to its loss during EMT. Functionally, ectopic expression of RBM38 enhances ZO-1 expression via mRNA stability, which eventually causes pause in metastatic process of BCa cells. C RBM38 binds to, and stabilizes, several ceRNAs for STARD13, thus avoiding it from degradation mediated by oncogenic miRNAs. STARD13-ceRNA network inhibits cancer cell metastatic properties. Lines with arrow and perpendicular denote a promotion or an inhibition effect, respectively. Please see more details in the text. BCa breast cancer, AREs AU/U-rich or CU-rich elements, EMT epithelial–mesenchymal transition, TF transcriptional factor
EMT confers polarized epithelial cells with mobility and certain stem cell (SC) properties, aiding metastasis and drug resistance [6]. Interestingly, RBM38 is lost during TGF-β induced EMT in ER+ BCa cells via TF SNAI1-mediated transcriptional repression through the E-box elements in the promoter of RBM38 [35]. Biologically, RBM38 blocks EMT via upregulation of an epithelial marker ZO-1 by enhancing its mRNA stability (Fig. 4B) [35]. This observation has clinical implications because the expression of RBM38 and ZO-1 is positively correlated in human BCa specimens by IHC staining [35]. Besides modulating EMT, RBM38 might halts metastasis by interfering the competing endogenous RNAs (ceRNAs) network. The ceRNA hypothesis postulates that RNA transcripts sharing common sequences called microRNA (miRNA) recognition elements (MREs) may crosstalk and indirectly regulate the expression of each other by competing for binding of the same pool of miRNAs [36]. This strategy has emerged as an important mechanism of post-transcriptional gene expression regulation, and is proposed as a therapeutic target for treating cancers [36]. It was reported that STARD13 and several other transcripts (i.e., CDH5, HOXD10 and HOXD1) forms a ceRNA network to suppress BCa EMT and metastasis (Fig. 4C) [37]. Through binding to and stabilizing CDH5 and HOXD10 mRNAs, RBM38 facilitates STARD13-ceRNA network development in both ER+ and triple-negative BCa cell lines by alleviating the attack of metastasis-promoting miRNAs (i.e., miR-9, miR-10b, and miR-125b) to tumor suppressive STARD13 [29].
Collectively, RBM38 is a TS in BCa and can participate in either oncogenic or tumor-suppressive pathways to exert its anticancer functions (Fig. 4). In support, pharmacological targeting either MYC [33] or PTEN [34] attenuates RBM38-mediated growth suppression in cancer cells given that RBM38 negatively and positively regulates MYC and PTEN, respectively.
RBM38 in other cancer types
Similar to BCa, IHC analysis also revealed a common downregulation (vs. normal tissues) of RBM38 in hepatocellular carcinoma (HCC) [38], renal cell carcinoma (RCC) [39], gastric cancer [40], colorectal cancer [41], lung cancer [42], and leukemia [43], highlighting potentially a tumor-suppressive role for RBM38 (Table 1). In support, the RBM38 mRNA level is negatively correlated with the overall survival in patients with RCC [39], gastric [40] and colorectal cancer [13]. Biologically, forced expression of RBM38 induces apoptosis, senescence, and cell cycle arrest, leading to inhibition of proliferation and invasion in vitro and tumorigenesis in vivo in multiple tumor systems [38, 39, 41, 42]. Mechanistically, overexpression of RBM38 restores the expression of wild-type (WT), but not mutant, p53 via binding to, and thus destabilizing, the p53 inhibitor MDM2 transcript in HCC [38, 44]. Notably, previous studied has established that RBM38 binds to and suppresses p53 mRNA translation [12] (see below). These seemingly opposite results signify the differential regulatory effect of RBM38 on p53 expression in a context-specific manner. Furthermore, RBM38 increases the expression of tumor-suppressive long non-coding RNA CASC2 by competitively binding to its mRNA with oncogenic miR-181a in non-small cell lung cancer (NSCLC) cells [42]. RBM38 also upregulates p21 and DND1, two TSs in leukemia [43]. In aggregate, these evidences, again, establish RBM38 as a TS in a number of human cancers. Importantly, it is worth noting that there exist controversial results indicating that RBM38 might have oncogenic potential in certain cancer types, given the fact that RBM38 is amplified (Fig. 3; albeit at low frequency) and might be overexpressed in particular pathological contexts. For example, it was reported that RBM38a is frequently overexpressed, along with downregulation of p53, in dog lymphoma, indicative of an oncogenic role in lymphomagenesis [12]. In NSCLC cells under hypoxic stress in vitro, RBM38 inhibits the expression of miR-34a (a well-known TS [45]), leading to upregulation of SIRT1 protein which deacetylases and inhibits the transcriptional activity of p53 [46]. Further characterization of RBM38 in a context-dependent manner is required to reconcile the discrepancy.
Targets of RBM38 unveil pleiotropic mechanisms
As one of intensively investigated RBPs, RBM38 is reported to binds to the AU/U- or CU-rich elements (AREs) in 3′-UTR of transcripts to eventually modulate cellular behavior through, mainly, three types of mechanisms (i.e., stabilization, or destabilization, and/or translational regulation, of its cargo mRNAs). For example, RBM38 targets transcripts of p53-related genes (e.g., p21 [12, 14], p73 [47], GFD15 [48]), nuclear receptors (e.g., ER [30], PR [31]), RBP ELAVL1 [49], TS PTEN [34], and metastasis-suppressive genes (e.g., ZO-1 [35] and STARD13-ceRNAs (CDH5 and HOXD10) [29]) for stabilization. Oppositely, mRNAs encoding oncogene MYC [33] and MDM2 [44] and, likely oncogenic, ΔNp63α [15] are targeted for degradation. Besides the post-transcriptional regulation, RBM38 also modulates gene expression at the translation level. By preventing or recruiting the eIF4E, the translation initiation complex, from binding its targets, RBM38 reduces protein level of p53 [12] and oncogenic HIFα [50], but increases PPM1D synthesis [51]. Table 2 summarizes so far the reported targets of RBM38 with their-related pathways. Highlighted by these three mechanisms underpinning RBM38′s actions, an intriguing, and obviously important, question is how and what determines the fate of a mRNA upon bound by RBM38.
The most well-characterized mechanism that explains the TS properties of RBM38 is the constitution of a negative feedback loop between RBM38 and multiple TSs (e.g., p53 family; Fig. 5) or oncogenes (e.g., MYC and E2F1), respectively. As a direct transcriptional target of the p53 family (but not the mutant R249S p53) in both normal and genotoxic conditions [14], RBM38 destabilizes p63 (mainly the epithelium-expressing N-terminal truncated isoform ΔNp63α), and stabilizes p73, but minimally affects p53, transcripts [12]. Instead, RBM38 represses p53 translation by abolishing cap-binding protein eIF4E from binding its mRNA via its C-terminal domain for physical interaction with eIF4E and its N-terminal RRM domain for binding p53 mRNA [12]. The full-length TAp63α is tumor suppressive, but the ΔNp63α is oncogenic in squamous cell carcinoma [52]. The functional consequence of RBM38′s implication in p53 pathway is the upregulation of antiproliferative genes such as p21 (an effector of p53) through RBM38-mediated mRNA stability, together with transcriptional output in both p53-dependent and -independent manner [14, 53, 54]. Notably, RBM38 and HuR coordinately regulate the stability of p21 mRNA but have converse effects on p53 translation [12, 55]. The E2F family of TFs plays a pivotal role in cell-cycle progression and is a critical downstream target of the TS pRB. The pRB/E2F pathway is defective in most human tumors [54]. Interestingly, RBM38 is transcriptionally upregulated by E2F1 in an p53-independent manner and in turn restricts E2F1-mediated proliferation in ovarian cancer cells [54]. Alternatively, MYC decreases RBM38 expression via binding to its promoter; RBM38 also abolishes MYC activity by destabilizing its transcript [33]. Similarly, ER negatively regulates RBM38 transcription, but RBM38 stabilizes ER transcript in BCa cells [30].

Intricate RBM38-p53 family regulatory network. Transcriptionally targeted by p53 family, RBM38 in turn destabilizes p63 but stabilizes p73, and suppresses p53 translation. GSK3β phosphorylates RBM38 at S195, which converts RBM38 from a repressor to an activator of p63 mRNA degradation and p53 translation. Notably, genetic PTEN loss-mediated PI3K-Akt hyperactivation in cancers can phosphorylates GSK3β, leading to attenuated kinase activity and thus RBM38 phosphorylation. RBM38 stabilizes PTEN transcript in PTEN-expressing cancer cells. PPM1D, a target of p53 and RBM38 via transcription and mRNA translation, respectively, dephosphorylates RBM38 at S195 to restrain p53 upregulation and thus exerts oncogenic function in many cancers. Moreover, it has also been reported that PPM1D can dephosphorylates p53 at S15 to impair p53′s function in cell cycle suppression [70]. Lines with arrow and perpendicular denote a promotion or an inhibition effect, respectively
How does RBM38 promote the stability of one transcript over the other? An area of interest is that RBM38 can restrict the accessibility of a selective set of miRNAs to its targets. For example, RBM38 can block miRNA-mediated repression of p53 target genes such as RBM38 itself, p21, DDIT4 and LATS2 [56]. By loading into the RNA-induced silencing complex (RISC), miRNAs usually interact with mRNAs to induce degradation and/or translational repression [45, 56]. Ago2, an essential RISC component, possesses the endonuclease activity. Interestingly, RBM38 interacts with Ago2 to protect p21 3′-UTR from miR17-mediated decay [56], but conversely, to recruit miR-203 to target p63 mRNA for degradation [57]. Intriguingly, the RBM38-Ago2 interplay is disrupted by its Serine-195 (S195) phosphorylation (see below), yielding reduced accessibility of miR-203 to, and thus upregulation of, p63 mRNA [57]. The difference in selectivity of targets and their corresponding miRNAs by RBM38 for miRNA-mediated decay not only highlights the complexity of RBM38-centered regulatory network but also signifies a future research direction.
S195 phosphorylation rewires RBM38’s biological activity
Post-translational modifications greatly impact protein functions, and phosphorylation is the most prevalent one [57, 59]. Inspired by an observation that RBM38 appears as two bands in SDS-PAGE gels, Dr. Chen’s group has made a significant discovery that RBM38 can be phosphorylated by GSK3β kinase at Serine-195 (S195) and this phosphorylation converts RBM38 from a repressor to an activator of p53 mRNA translation via altered interaction with eIF4E [59]. Similarly, they recently reported that S195 phosphorylation can turn RBM38 from a repressor to an activator of p63 mRNA stability by disassociating with Ago-miR203 complex (also see above) [57]. Subsequently, in a search for phosphatases that regulate RBM38, Zhang et al. found that PPM1D phosphatase, a target of p53 and RBM38, interacts directly with and dephosphorylates RBM38 at S195, leading to suppression of p53 translation and thus p53-dependent growth inhibition [51]. An oncogenic activity has been observed for PPM1D [51]. Leveraging the large-scale DNA sequencing data, we examined the S195-site mutation across the TCGA 32 human cancer types (Fig. 3) and revealed only 2 out of 10,967 samples bearing a non-phosphorylatable S195L mutation, indicating the importance of GSK3β-mediated S195 phosphorylation in balancing RBM38′s functions.
Genetic evidence for a tumor-inhibiting role
To definitively establish RBM38 as a TS (or an oncogene), a systematic RBM38-null or -knockout (KO) mouse model has been generated by Dr. Chen’s group (Table 3). Based on the evidence that mice deficient in RBM38 exhibit signs of accelerated aging, via partial upregulation of p53, and are prone to hematopoietic defects and spontaneous tumors [60], a TS-like property has been proposed for RBM38. Interestingly, the young RBM38−/− mice (< 12 months) were largely fine but elder ones (~ 18 months) developed a broad spectrum of tumors, including lymphoma, hemangiosarcoma, and HCC [60]. Consistent with the hematopoietic defects, RBM38 is highly expressed in mature blood cell types (Fig. 1) and has been reported to promote erythroid differentiation [61], with its loss of expression often observed in acute myeloid leukemia [43]. Importantly, RBM38 deficiency markedly decreases the tumor penetrance in heterozygous p53+/− mice via enhanced p53 expression. In p53-null mice, loss of RBM38 shortens the animal lifespan and accelerates lymphomagenesis due to ineffectiveness of RBM38-KO mediated p53 upregulation [60], consistent with its tumor-suppressive role. Interestingly, the overall tumor burden was markedly reduced in RBM38−/−; p53−/− mice compared with p53−/− mice [60]. This is seemingly counterintuitive as one would expect an accelerated tumorigenesis in double-KO of TSs [60]. The discrepancy could be potentially explained by the p53-independent functions of RBM38 and the possibility that tumor development in double KO mice takes longer but the hosts possess a shorter lifespan compared with single KO mice. Also, a “intergenic suppression” effect might exist (see below). Besides the genetic loss, p53 is frequently altered as missense mutations, typified by hotspot mutations (e.g., R175H, R248W, and R273H) in human cancers [62]. These mutations often confer gain-of-functions to convert p53 to an oncogenic driver of metastasis [58, 62]. To determine the role of RBM38-mutant p53 axis in tumorigenesis in vivo, mutant p53 knock-in (KI) mice were generated and crossed with RBM38-KO mice. Results showed that ablation of RBM38 shortens the lifespan of, and alters tumor incidence in, both p53R270H/− and p53R172H/− (equivalent to human R273H and R175H, respectively) mice [58], again establishing RBM38 as a TS. Although the overall tumor spectrum remained similar, the incidence of T cell lymphomas was significantly higher in RBM38−/−;p53KI/− mice than that in p53KI/− mice, along with increased mutant p53 translation and decreased stability of PTEN [58].
RBM38 and p63 also form a negative feedback loop [15] and mice deficient in TAp63 are prone to enhanced aging and spontaneous tumors [63]. Similar to RBM38−/−;p53+/− mice, RBM38-KO extends the lifespan, reduces tumor penetrance and liver steatosis in TAp63+/− mice, probably due to upregulation of tumor-suppressive TAp63 [16]. Biologically, loss of RBM38 or TAp63 alone results in an increase in cellular senescence, but their combined loss reduces, surprisingly, senescence, as seen in RBM38−/−;TAp63+/− mice [16]. The “intergenic suppression” is proposed for the effect of RBM38-null in a p53−/− or TAp63+/− background [16]. Intergenic suppression denotes a second mutation that relieves or reverts the phenotypic effects of an already existing mutation within the genome. Altogether, the negative feedback loops between RBM38 and p53, p63, and likely other cancer drivers, are not linear but rather intricate, especially in tumors with diverse genetics backgrounds. Future investigation is warranted to uncover the detailed molecular mechanisms.
RBM38 as a therapeutic target
RBPs control many, if not all, biological processes and their dysregulation is prevalent in human cancers, and thus have been emerged as a novel class of therapeutic targets for cancer prevention and treatment [4, 5]. In light of being a TS in numerous cancers by often forming negative feedback loops with cancer drivers, RBM38 presents itself as a promising therapeutic target. Restoration of WT p53 has been shown in animal models to be an effective avenue to suppress tumor growth [64]. As a p53 target, RBM38 inhibits, but its S195 phosphorylation enhances, p53 translation via interaction with eIF4E [12]. Therefore, blocking RBM38-mediated translational suppression would be a strategy to restore p53 protein expression in p53-WT and RBM38-expressing tumors. In support, it was recently reported that a synthetic 8aa peptide (Pep8), derived from RBM38 and corresponding to the binding interface between RBM38 and eIF4E, is effective in relieving RBM38-mediated repression of p53 [65]. Functionally, Pep8 alone or together with a low dose of doxorubicin potently induces p53 expression and impedes tumor growth in vitro and in vivo in a RBM38- and p53-dependent manner [65], establishing therapeutic potential for modulation of the RBM38-eIF4E complex in cancers carrying WT p53. Alternatively, the PI3K/AKT pathway is often overactivated in a number of cancers and it phosphorylates GSK3β at S9 to inhibit its kinase activity [66]. The GSK3β can phosphorylate and promote degradation of oncogenic CCND1 and MYC [66]. In this regard, blocking PI3K/AKT activity to de-phosphorylate GSK3β might represent a way to enhance S195 phosphorylation of RBM38 and thus upregulate p53 translation in tumors positive for both RBM38 and p53. Encouragingly, treatment with AKT kinase inhibitor, MK2206, increases the level of phosphorylated RBM38 and p53 (via a RBM38-dependent manner) in a dose-dependent manner [59]. Clinically, p53 is frequently lost genomically and RBM38 is rarely mutated in human cancers (Fig. 3). We thus postulate that the loss of RBM38 expression is largely attributed to epigenetic mechanisms, and in such scenarios, epigenetic modulation may convey an approach to boost RBM38 expression. In support, RBM38 is silenced in BCa due to, at least partially, hypermethylation of its promoter CpG island [56]. Treatment with DNA demethylating agents such as 5-aza-2′-deoxycytidine (5-Aza or Decitabine) induces upregulation of RBM38 in triple negative BCa cells and thus inhibition of cell proliferation [56].
Conclusions and future perspectives
The involvement of RBM38 in multiple key cancer-related pathways (Table 2) has placed RBM38 in a hub position within the RBP-RNA regulatory networks. In vitro studies have revealed a limited number of transcripts targeted by RBM38 for mRNA stability and translation modulations. However, to uncover the full picture of RBM38′s function as an RBP, the identification of its targets on a global scale via high throughput techniques is necessary. Mechanistically, it would be interesting to examine how RBM38 recruits other partners to fulfill pleotropic impacts on mRNA fate in diverse contexts. Could one of them be HuR? It was reported that RBM38 interacts and cooperates with HuR to regulate p21 mRNA stability [53], but RBM38 and HuR have opposite effects on (i.e., RBM38 decreases [12] while HuR stimulates [55]) p53 mRNA translation. As a RBM38 closely related protein, RBM24 is also reported to be a p53 target and positively regulate p21 mRNA stability [17]. Interestingly, RBM24 binds to the same regions of p63 transcript as that of RBM38 (likely due to their identical RRM), and it can negatively regulate p63 expression in the absence of RBM38 [19], suggesting possible cooperative or antagonistic mechanisms between these two RBPs in regulating the similar set of transcripts. Like RBM38, RBM24 also forms a negative feedback loop with p53 by suppressing its mRNA translation [67]. In the context of cancer development, RBM24 was found frequently downregulated in human nasopharyngeal carcinoma, with its overexpression inhibiting cell proliferation and invasion in vitro and metastatic colonization in vivo [68]. Collectively, it appears that RBM38 and RBM24 function similarly in terms of RBP properties and tumor suppressive role in human cancers. The detailed interplay between them remains to be elucidated. RBM38 localizes in both nucleus and, largely, cytosol [14, 34]. Therefore, besides the post-transcriptional regulation, another emerging question is whether RBM38 also regulates transcription, like other RBPs reported recently [8].
Animal studies have uncovered differential effects of RBM38 deficiency on tumorigenesis depending on p53 status [58, 60], signifying a consideration of p53 when exploring RBM38 as a target for cancer treatment. However, both p53-dependent and -independent mechanisms have been reported to downregulate RBM38 in a spectrum of human cancers bearing diverse genetic backgrounds [14, 54]. It is, therefore, necessary to investigate that whether RBM38 is also transcriptionally regulated by other driver TFs. Based on a simple motif analysis of RBM38 promoter sequence, several TFs (e.g., SOX5, RUNX3, CUTL1, PPARγ2, ATF6) are identified to potentially regulate RBM38 expression [13], likely in distinct cancer contexts.
RBM38 is obviously a splicing regulator. However, such an RBP property has been overlooked in previous cancer studies. Currently, there is only one report that has examined the role of RBM38 in alternative splicing during human erythropoiesis [61]. Using a splicing-detecting microarray, Heinicke et al. has identified a subset of RBM38-regulated splicing events and determined that RBM38 regulates activation of EPB41 exon-16 during late erythroid differentiation [61]. In our recent work, we discovered that RBM38 might function as a splicing inhibitor in aggressive prostate cancer [69]. Subsequently, we are now characterizing the roles and mechanisms of RBM38 in human prostate tumorigenesis and leukemia. In aggregate, further understanding of detailed RBM38 biology will shed fresh lights on the aetiology of, and developing novel therapeutic strategies against, human cancers.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68(2018):394–424
Blackadar CB (2016) Historical review of the causes of cancer. World J Clin Oncol 7:54–86
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Pereira B, Billaud M, Almeida R (2017) RNA-binding proteins in cancer: old players and new actors. Trends Cancer 3:506–528
Hong S (2017) RNA binding protein as an emerging therapeutic target for cancer prevention and treatment. J Cancer Prev 22:203–210
Aparicio LA, Abella V, Valladares M, Figueroa A (2013) Posttranscriptional regulation by RNA-binding proteins during epithelial-to-mesenchymal transition. Cell Mol Life Sci 70:4463–4477
Glisovic T, Bachorik JL, Yong J, Dreyfuss G (2008) RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett 582:1977–1986
Xiao R, Chen JY, Liang Z, Luo D, Chen G, Lu ZJ, Chen Y, Zhou B, Li H, Du X, Yang Y, San M, Wei X, Liu W, Lecuyer E, Graveley BR, Yeo GW, Burge CB, Zhang MQ, Zhou Y, Fu XD (2019) Pervasive chromatin-RNA binding protein interactions enable RNA-based regulation of transcription. Cell 178:107–121
Golan-Gerstl R, Cohen M, Shilo A, Suh SS, Bakacs A, Coppola L, Karni R (2011) Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res 71:4464–4472
Yae T, Tsuchihashi K, Ishimoto T, Motohara T, Yoshikawa M, Yoshida GJ, Wada T, Masuko T, Mogushi K, Tanaka H, Osawa T, Kanki Y, Minami T, Aburatani H, Ohmura M, Kubo A, Suematsu M, Takahashi K, Saya H, Nagano O (2012) Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun 3:883
Wang X, Liu R, Zhu W, Chu H, Yu H, Wei P, Wu X, Zhu H, Gao H, Liang J, Li G, Yang W (2019) UDP-glucose accelerates SNAI1 mRNA decay and impairs lung cancer metastasis. Nature 571:127–131
Zhang J, Cho SJ, Shu L, Yan W, Guerrero T, Kent M, Skorupski K, Chen H, Chen X (2011) Translational repression of p53 by RNPC1, a p53 target overexpressed in lymphomas. Genes Dev 25:1528–1543
Ding Z, Yang HW, Xia TS, Wang B, Ding Q (2015) Integrative genomic analyses of the RNA-binding protein, RNPC1, and its potential role in cancer prediction. Int J Mol Med 36:473–484
Shu L, Yan W, Chen X (2006) RNPC1, an RNA-binding protein and a target of the p53 family, is required for maintaining the stability of the basal and stress-induced p21 transcript. Genes Dev 20:2961–2972
Zhang J, Jun Cho S, Chen X (2010) RNPC1, an RNA-binding protein and a target of the p53 family, regulates p63 expression through mRNA stability. Proc Natl Acad Sci U S A 107:9614–9619
Jiang Y, Xu E, Zhang J, Chen M, Flores E, Chen X (2018) The Rbm38-p63 feedback loop is critical for tumor suppression and longevity. Oncogene 37:2863–2872
Jiang Y, Zhang M, Qian Y, Xu E, Zhang J, Chen X (2014) Rbm24, an RNA-binding protein and a target of p53, regulates p21 expression via mRNA stability. J Biol Chem 289:3164–3175
Yang J, Hung LH, Licht T, Kostin S, Looso M, Khrameeva E, Bindereif A, Schneider A, Braun T (2014) RBM24 is a major regulator of muscle-specific alternative splicing. Dev Cell 31:87–99
Xu E, Zhang J, Zhang M, Jiang Y, Cho SJ, Chen X (2014) RNA-binding protein RBM24 regulates p63 expression via mRNA stability. Mol Cancer Res 12:359–369
Ginestier C, Cervera N, Finetti P, Esteyries S, Esterni B, Adelaide J, Xerri L, Viens P, Jacquemier J, Charafe-Jauffret E, Chaffanet M, Birnbaum D, Bertucci F (2006) Prognosis and gene expression profiling of 20q13-amplified breast cancers. Clin Cancer Res 12:4533–4544
Bar-Shira A, Pinthus JH, Rozovsky U, Goldstein M, Sellers WR, Yaron Y, Eshhar Z, Orr-Urtreger A (2002) Multiple genes in human 20q13 chromosomal region are involved in an advanced prostate cancer xenograft. Cancer Res 62:6803–6807
Tanner MM, Grenman S, Koul A, Johannsson O, Meltzer P, Pejovic T, Borg A, Isola JJ (2000) Frequent amplification of chromosomal region 20q12-q13 in ovarian cancer. Clin Cancer Res 6:1833–1839
Korn WM, Yasutake T, Kuo WL, Warren RS, Collins C, Tomita M, Gray J, Waldman FM (1999) Chromosome arm 20q gains and other genomic alterations in colorectal cancer metastatic to liver, as analyzed by comparative genomic hybridization and fluorescence in situ hybridization. Genes Chromosomes Cancer 25:82–90
Carvalho B, Postma C, Mongera S, Hopmans E, Diskin S, van de Wiel MA, van Criekinge W, Thas O, Matthai A, Cuesta MA (2009) Multiple putative oncogenes at the chromosome 20q amplicon contribute to colorectal adenoma to carcinoma progression. Gut 58:79–89
Hotte GJ, Linam-Lennon N, Reynolds JV, Maher SG (2012) Radiation sensitivity of esophageal adenocarcinoma: the contribution of the RNA-binding protein RNPC1 and p21-mediated cell cycle arrest to radioresistance. Radiat Res 177:272–279
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:1
Siegel RL, Miller KD, Jemal A (2020) Cancer statistics. CA Cancer J Clin 70(2020):7–30
Xue JQ, Xia TS, Liang XQ, Zhou W, Cheng L, Shi L, Wang Y, Ding Q (2014) RNA-binding protein RNPC1: acting as a tumor suppressor in breast cancer. BMC Cancer 14:322
Zheng L, Zhang Z, Zhang S, Guo Q, Zhang F, Gao L, Ni H, Guo X, Xiang C, Xi T (2018) RNA binding protein RNPC1 inhibits breast cancer cell metastasis via activating STARD13-correlated ceRNA network. Mol Pharm 15:2123–2132
Shi L, Xia TS, Wei XL, Zhou W, Xue J, Cheng L, Lou P, Li C, Wang Y, Wei JF, Ding Q (2015) Estrogen receptor (ER) was regulated by RNPC1 stabilizing mRNA in ER positive breast cancer. Oncotarget 6:12264–12278
Lou P, Li C, Shi L, Xia TS, Zhou W, Wu J, Zhou X, Li X, Wang Y, Wei JF, Ding Q (2017) RNPC1 enhances progesterone receptor functions by regulating its mRNA stability in breast cancer. Oncotarget 8:16387–16400
Dang CV (2012) MYC on the path to cancer. Cell 149:22–35
Li XX, Shi L, Zhou XJ, Wu J, Xia TS, Zhou WB, Sun X, Zhu L, Wei JF, Ding Q (2017) The role of c-Myc-RBM38 loop in the growth suppression in breast cancer. J Exp Clin Cancer Res 36:49
Zhou XJ, Wu J, Shi L, Li XX, Zhu L, Sun X, Qian JY, Wang Y, Wei JF, Ding Q (2017) PTEN expression is upregulated by a RNA-binding protein RBM38 via enhancing its mRNA stability in breast cancer. J Exp Clin Cancer Res 36:149
Wu J, Zhou XJ, Sun X, Xia TS, Li XX, Shi L, Zhu L, Zhou WB, Wei JF, Ding Q (2017) RBM38 is involved in TGF-beta-induced epithelial-to-mesenchymal transition by stabilising zonula occludens-1 mRNA in breast cancer. Br J Cancer 117:675–684
Wang Y, Hou J, He D, Sun M, Zhang P, Yu Y, Chen Y (2016) The emerging function and mechanism of ceRNAs in cancer. Trends Genet 32:211–224
Li X, Zheng L, Zhang F, Hu J, Chou J, Liu Y, Xing Y, Xi T (2016) STARD13-correlated ceRNA network inhibits EMT and metastasis of breast cancer. Oncotarget 7:23197–23211
Ye J, Liang R, Bai T, Lin Y, Mai R, Wei M, Ye X, Li L, Wu F (2018) RBM38 plays a tumor-suppressor role via stabilizing the p53-mdm2 loop function in hepatocellular carcinoma. J Exp Clin Cancer Res 37:212
Huang W, Wei XL, Ni W, Cao M, Meng L, Yang H (2017) The expression of RNA-binding protein RBM38 decreased in renal cell carcinoma and represses renal cancer cell proliferation, migration, and invasion. Tumour Biol 39:1010428317701635
Wang P, Gu J, Li X, Wang Q, Ding Y (2017) RNA-binding protein RBM38 acts as a tumor suppressor in gastric cancer. Int J Clin Exp Pathol 10:11130–11136
Cheng G, Ji C, Yang N, Meng L, Ding Y, Wei J (2016) RNA-binding protein RBM38: Acting as a tumor suppressor in colorectal cancer. Int J Clin Exp Med 9:7115–7126
Yang L, Zhang Y, Ling C, Heng W (2018) RNPC1 inhibits non-small cell lung cancer progression via regulating miR-181a/CASC2 axis. Biotechnol Lett 40:543–550
Wampfler J, Federzoni EA, Torbett BE, Fey MF, Tschan MP (2016) The RNA binding proteins RBM38 and DND1 are repressed in AML and have a novel function in APL differentiation. Leuk Res 41:96–102
Xu E, Zhang J, Chen X (2013) MDM2 expression is repressed by the RNA-binding protein RNPC1 via mRNA stability. Oncogene 32:2169–2178
Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S, Wiggins JF, Bader AG, Fagin R, Brown D, Tang DG (2011) The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med 17:211–215
Lin QY, Yin HL (2020) RBM38 induces SIRT1 expression during hypoxia in non-small cell lung cancer cells by suppressing MIR34A expression. Biotechnol Lett 42:35–44
Yan W, Zhang J, Zhang Y, Jung YS, Chen X (2012) p73 expression is regulated by RNPC1, a target of the p53 family, via mRNA stability. Mol Cell Biol 32:2336–2348
Yin T, Cho SJ, Chen X (2013) RNPC1, an RNA-binding protein and a p53 target, regulates macrophage inhibitory cytokine-1 (MIC-1) expression through mRNA stability. J Biol Chem 288:23680–23686
Cho SJ, Jung YS, Zhang J, Chen X (2012) The RNA-binding protein RNPC1 stabilizes the mRNA encoding the RNA-binding protein HuR and cooperates with HuR to suppress cell proliferation. J Biol Chem 287:14535–14544
Cho SJ, Teng IF, Zhang M, Yin T, Jung YS, Zhang J, Chen X (2015) Hypoxia-inducible factor 1 alpha is regulated by RBM38, a RNA-binding protein and a p53 family target, via mRNA translation. Oncotarget 6:305–316
Zhang M, Xu E, Zhang J, Chen X (2015) PPM1D phosphatase, a target of p53 and RBM38 RNA-binding protein, inhibits p53 mRNA translation via dephosphorylation of RBM38. Oncogene 34:5900–5911
Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW (2006) p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 9:45–56
Cho SJ, Zhang J, Chen X (2010) RNPC1 modulates the RNA-binding activity of, and cooperates with, HuR to regulate p21 mRNA stability. Nucleic Acids Res 38:2256–2267
Feldstein O, Ben-Hamo R, Bashari D, Efroni S, Ginsberg D (2012) RBM38 is a direct transcriptional target of E2F1 that limits E2F1-induced proliferation. Mol Cancer Res 10:1169–1177
Mazan-Mamczarz K, Galban S, Lopez de Silanes I, Martindale JL, Atasoy U, Keene JD, Gorospe M (2003) RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci U S A 100:8354–8359
Leveille N, Elkon R, Davalos V, Manoharan V, Hollingworth D, Oude Vrielink J, le Sage C, Melo CA, Horlings HM, Wesseling J, Ule J, Esteller M, Ramos A, Agami R (2011) Selective inhibition of microRNA accessibility by RBM38 is required for p53 activity. Nat Commun 2:513
Zhang Y, Feng X, Sun W, Zhang J, Chen X (2019) Serine 195 phosphorylation in the RNA-binding protein Rbm38 increases p63 expression by modulating Rbm38's interaction with the Ago2-miR203 complex. J Biol Chem 294:2449–2459
Zhang J, Xu E, Ren C, Yang HJ, Zhang Y, Sun W, Kong X, Zhang W, Chen M, Huang E, Chen X (2018) Genetic ablation of Rbm38 promotes lymphomagenesis in the context of mutant p53 by downregulating PTEN. Cancer Res 78:1511–1521
Zhang M, Zhang J, Chen X, Cho SJ, Chen X (2013) Glycogen synthase kinase 3 promotes p53 mRNA translation via phosphorylation of RNPC1. Genes Dev 27:2246–2258
Zhang J, Xu E, Ren C, Yan W, Zhang M, Chen M, Cardiff RD, Imai DM, Wisner E, Chen X (2014) Mice deficient in Rbm38, a target of the p53 family, are susceptible to accelerated aging and spontaneous tumors. Proc Natl Acad Sci USA 111:18637–18642
Heinicke LA, Nabet B, Shen S, Jiang P, van Zalen S, Cieply B, Russell JE, Xing Y, Carstens RP (2013) The RNA binding protein RBM38 (RNPC1) regulates splicing during late erythroid differentiation. PLoS ONE 8:e78031
Muller PA, Vousden KH (2014) Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25:304–317
Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL, Leung ML, El-Naggar A, Creighton CJ, Suraokar MB, Wistuba I, Flores ER (2010) TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 467:986–990
Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T (2007) Restoration of p53 function leads to tumour regression in vivo. Nature 445:661–665
Lucchesi CA, Zhang J, Ma B, Chen M, Chen X (2019) Disruption of the Rbm38-eIF4E complex with a synthetic peptide Pep8 increases p53 expression. Cancer Res 79:807–818
Luo J, Manning BD, Cantley LC (2003) Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell 4:257–262
Zhang M, Zhang Y, Xu E, Mohibi S, de Anda DM, Jiang Y, Zhang J, Chen X (2018) Rbm24, a target of p53, is necessary for proper expression of p53 and heart development. Cell Death Differ 25:1118–1130
Hua WF, Zhong Q, Xia TL, Chen Q, Zhang MY, Zhou AJ, Tu ZW, Qu C, Li MZ, Xia YF, Wang HY, Xie D, Claret FX, Song EW, Zeng MS (2016) RBM24 suppresses cancer progression by upregulating miR-25 to target MALAT1 in nasopharyngeal carcinoma. Cell Death Dis 7:e2352
Zhang D, Hu Q, Ji Y, Chao HP, Liu Y, Tracz A, Kirk J, Buonamici S, Zhu P, Wang J, Liu S, Tang DG (2020) Intron retention is a hallmark and spliceosome represents a therapeutic vulnerability in aggressive prostate cancer. Nat Commun 11(1):2089
Lu X, Nannenga B, Donehower LA (2005) PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev 19:1162–1174
Acknowledgements
This project was supported by Grants from the National Natural Science Foundation of China 81972418 (to D. Z.) and 31871481 (to Z. D.), the Wuhan Frontier Science and Technology Program 2019020701011490 (to D. Z.) and the Fundamental Research Funds for the Central Universities 531119200130 (to D. Z.) and 2662017PY109 (to Z. D).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zou, C., Wan, Y., He, L. et al. RBM38 in cancer: role and mechanism. Cell. Mol. Life Sci. 78, 117–128 (2021). https://doi.org/10.1007/s00018-020-03593-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03593-w