
Abstract
The sperm acrosome is a lysosome-related organelle that develops using membrane trafficking from the Golgi apparatus as well as the endolysosomal compartment. How vesicular trafficking is regulated in spermatids to form the acrosome remains to be elucidated. VPS13B, a RAB6-interactor, was recently shown involved in endomembrane trafficking. Here, we report the generation of the first Vps13b-knockout mouse model and show that male mutant mice are infertile due to oligoasthenoteratozoospermia. This phenotype was explained by a failure of Vps13b deficient spermatids to form an acrosome. In wild-type spermatids, immunostaining of Vps13b and Rab6 revealed that they transiently locate to the acrosomal inner membrane. Spermatids lacking Vps13b did not present with the Golgi structure that characterizes wild-type spermatids and showed abnormal targeting of PNA- and Rab6-positive Golgi-derived vesicles to Eea1- and Lamp2-positive structures. Altogether, our results uncover a function of Vps13b in the regulation of the vesicular transport between Golgi apparatus, acrosome, and endolysosome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Spermiogenesis is the complex morphogenetic differentiation that produces spermatozoa. In this process, post-meiotic male germ cells acquire the necessary structures to travel to and fertilize oocytes. Among the sperm-specific structures that are developed, the acrosome is of primary importance. It is the organelle that mediates the dispersion of cumulus cells and allows the male gamete nucleus to traverse the zona pellucida [1,2,3]. Rather than just preventing entry of the sperm nucleus within the oocyte, impaired acrosome biogenesis has a severe impact on sperm morphogenesis and causes infertility through globozoospermia [3,4,5].
Acrosome biogenesis occurs in spermatids and follows four consecutive phases: Golgi, cap, acrosome, and maturation phase. In Golgi and cap phase spermatids, proacrosomal vesicles are synthesized at the trans-Golgi network (TGN) [6,7,8] and transported to the anterior part of the nucleus [9,10,11] which is defined by the presence of a nuclear dense lamina (NDL). Recent and convergent studies have demonstrated that the acrosomal vesicle develops utilizing membrane trafficking other than from the Golgi apparatus [12,13,14]. West and Willison first showed that components of the plasma membrane were recruited to the acrosome via the endocytic trafficking [15]. Later, Sh3p13, a regulator of clathrin-coated vesicle endocytosis was found to be involved in acrosome biogenesis through its interaction with Dydc1 [16]. In addition, proper targeting and fusion of proacrosomal vesicles were found to be not solely dependent on Golgi-derived factors. Several endosomal, lysosomal, and autophagosomal components such as Vps54 [17, 18], SPE-39 [19], UBPy [20, 21] (also known as Usp8), and Atg7 [22] were found essential to this process. Overall, the maturation of Golgi-derived vesicles into effective proacrosomal vesicles requires the complex orchestration of anterograde trafficking from the Golgi apparatus and retrograde transport from the endolysosomal system.
Among the proteins with emerging roles in Golgi and endolysosomal trafficking is the Vacuolar Protein Sorting 13 family member VPS13B which associates with Cohen Syndrome [23,24,25,26,27,28,29,30] (CS, OMIM 216550). Domain homologies with Saccharomyces cerevisiae protein vps13p suggest a role of VPS13B in intracellular transport and vesicular sorting [23]. Recent studies have localized VPS13B to the Golgi apparatus membrane where it interacts with RAB6 [31, 32], a small GTPase that also locates to the rodent acrosome [10]. RAB6 is an interactor of TMF [33] (also known as ARA160), the conserved oligomeric Golgi (COG) [34] tethering complex, and the Golgi-associated retrograde protein complex (GARP) [35, 36]. All of these interactors were shown to be essential to the acrosome formation [17, 18, 37,38,39]. In addition, implication of VPS13B in the endosome-lysosome trafficking has already been suggested in a previous study on fibroblasts of CS patients [40] where we found abnormally enlarged lysosomes as well as absent early endosomes. Given the predicted function of VPS13B in intracellular trafficking and its interaction with RAB6, we hypothesized that it could be involved in the maturation of Golgi-derived vesicles into proacrosomal vesicles, their transport to the NDL, their fusion, and/or their docking onto the nuclear membrane. Further supporting the hypothesis that VPS13B is involved in spermiogenesis, another VPS13 family member (VPS13A) was recently shown to be essential for male fertility in mice [41].
We generated the first mouse model of CS (Vps13b∆Ex3/∆Ex3) and took the opportunity to investigate Vps13b functions in reproduction along with CS-related symptoms (unpublished observations). While female mutant mice were not affected with reduced fertility, males were infertile and presented with oligoasthenoteratozoospermia. We found that acrosome biogenesis is impaired in Vps13b∆Ex3/∆Ex3 males and leads to either spermatid degeneration or abnormal round-headed sperm cells. In wild-type spermatids, Vps13b locates with Rab6 to the acrosomal vesicle. In the absence of Vps13b, proacrosomal vesicles were targeted to the endolysosomal system. Altogether our data demonstrate that Vps13b is a necessary factor for the transport of proacrosomal vesicles to the NDL.
Results
Vps13b∆Ex3/∆Ex3 male mice are infertile
Generation of Vps13b∆Ex3/∆Ex3 mice lacking Vps13b exon 3 on a C57Bl/6N background was performed at the MCI/ICS (Mouse Clinical Institute-Institut Clinique de la Souris, Illkirch, France; http://www-mci.u-strasbg.fr) through Cre-LoxP recombination as described in the “Materials and methods” section and briefly summarized in Fig. 1a. Deletion of Vps13b exon 3 resulted in a reading-frame shift and introduced a premature stop codon in exon 4. The deletion was confirmed by PCR on tail genomic DNA (Fig. 1b), and transcript levels of Vps13b exon 1/2, 3/4, 4/5, 41/42, and 62/63 were measured by RT-qPCR. Apart from the PCR targeting exon 3, no significant difference with wild-type transcript levels was identified (Fig. 1c). This result suggests that the introduced deletion does not lead to nonsense-mediated decay of mutant Vps13b transcripts. Vps13b∆Ex3/∆Ex3 cells are, thus, likely to express truncated isoforms of Vps13b, lacking their Chorein domain [23].

Generation and reproductive tract of Vps13b∆Ex3/∆Ex3 mice. a Schematic representation of the recombination events targeting Vps13b exon 3. b PCR genotyping on tail DNA using primers Gf and Gr flanking exon 3. A 923 bp product is amplified from the wild-type allele, while a 307 bp product is amplified from the null allele. cVps13b relative mRNA expression levels from RT-qPCR targeting Vps13b exon 1/2, 3/4, 4/5, 41/42, and 62/63 on 10 ng of cDNA reverse-transcribed from Vps13b+/+ and Vps13b∆Ex3/∆Ex3 testis total RNA. N = 3 testes per group were analyzed. Values are normalized to Hprt expression levels and presented as the mean ± SD. d Photograph of wild-type and homozygous mutant reproductive tracts. No developmental issue affected the mutant tract. e Photograph of wild-type and homozygous mutant testes. f Boxplot displaying the distribution of the weight of wild-type and mutant testes normalized to total body weight. N = 12 testes per group were measured at 6 months of age. As per Welch two-sample t test, the mean value was significantly smaller in mutant mice compared to wild-types (p < 0.001)
To determine whether Vps13b possesses a function in fertility, both male and female Vps13b∆Ex3/∆Ex3 mice were crossed with heterozygous mice of the opposite sex. Mating started at 2 months of age and lasted for 4 months. Crosses involving Vps13b∆Ex3/∆Ex3 females gave regular monthly litters of ten pups on average. In contrast, Vps13b∆Ex3/∆Ex3 males were unable to sire offspring during the 4 months of mating. Vaginal plugs were observed in all crosses and, therefore, confirmed that Vps13b∆Ex3/∆Ex3 males mated. Dissections of 2-month-old Vps13b∆Ex3/∆Ex3 males allowed the observation of fully developed reproductive tracts (Fig. 1d), but significantly, though slightly, smaller testes compared to wild-type mice (Fig. 1e, f).
Vps13b∆Ex3/∆Ex3 mice are affected with oligoasthenoteratozoospermia
To identify whether mutant spermatozoa are affected with morphological and/or functional defects, they were collected through mechanical dilaceration of cauda epididymides and their concentration, mobility and structure were evaluated. The concentration of spermatozoa was about 6.5 times lower in mutants (1.7 × 106 spermatozoa/mL) compared to wild-types (23.5 × 106 spermatozoa/mL, Fig. 2a). In addition, mutant epididymides released a vast majority of immotile and abnormally shaped spermatozoa among cellular debris and nonflagellated cells (Fig. 2b–d, Supplemental Movies S1 and S2). Though nuclei of most mutant spermatozoa were condensed, none exhibited the characteristic sickle-shape of the wild-types. Instead, they were round-headed and 38.5% were nonflagellated, while 6.7% were multiflagellated. The absence of well-differentiated spermatozoa was confirmed on HE-stained paraffin sections of mutant cauda epididymides (Fig. 2e, right panel). TUNEL assay performed on these sections showed a majority of apoptotic/necrotic spermatozoa (Fig. 2e, left panel). Taken together, the data suggest that spermiogenesis is impaired in Vps13b∆Ex3/∆Ex3 mice, which, in turn, causes sperm apoptosis/necrosis and infertility through oligoasthenoteratozoospermia.

Oligoasthenoteratozoospermia in Vps13b∆Ex3/∆Ex3 mice. Dot plots describing sperm count (a) and percentage of motile sperm (b) from cauda epididymis dilacerations of wild-type and Vps13b∆Ex3/∆Ex3 mice (100 spermatozoa per mouse, N = 6 mice per group). Vps13b∆Ex3/∆Ex3 mice showed a significant decrease in sperm count (p = 0.006) and sperm motility (p = 0.003) compared to wild-type mice. Error bars represent the standard deviation. Significance was determined using Wilcoxon rank sum test. c Representative images of sperm cells collected from Vps13b∆Ex3/∆Ex3 cauda epididymides compared to a wild-type spermatozoon. Scale bars 50 μm. d Dot plots of the quantification of normal-appearing spermatozoa with sickle-shape head (1) and of the three observed morphological defects: round-headed (2), multiflagellated (3), and nonflagellated (4). Mutant mice had no spermatozoa with normal morphology. Instead, they were round-headed as well as more frequently multiflagellated (p = 0.003) or nonflagellated (p = 0.002). Error bars represent the standard deviation. Significance was determined using Wilcoxon rank sum test. e Representative images of hematoxylin/eosin-stained cauda epididymis sections (left panels). Scale bars 50 μm. TUNEL assay on cauda epididymis sections showing sperm cell death in Vps13b∆Ex3/∆Ex3 mice (right panels). Scale bars 50 μm
Structural abnormalities and degeneration of mutant spermatids
To decipher the defects affecting Vps13b∆Ex3/∆Ex3 spermiogenesis, testicular paraffin sections of 2-month-old mice were stained with hematoxylin and eosin (Fig. 3a). Though all the stages of the seminiferous tubule could be identified, drastic differences affected mutant spermatids from differentiation stage 9–16. Their nuclei condensed at stage 9, but failed to elongate and acquire the characteristic sickle-shape of late spermatids (stages 10–16). In addition, the number of late spermatids was reduced by half in Vps13b∆Ex3/∆Ex3 mice compared to wild-types (Fig. 3b). TUNEL assay confirmed that cell death occurred in spermatids from stage 9 to 16 (Fig. 3c, d).

Spermatid abnormalities and death during Vps13b∆Ex3/∆Ex3 spermiogenesis. a Representative haematoxylin/eosin-stained sections of wild-type and Vps13b∆Ex3/∆Ex3 seminiferous tubules at stages II–III, V–VII, and X–XII. Nuclei of late spermatids failed to elongate during acrosome and maturation phases (arrows). Se sertoli cell, Spg spermatogonia, Spc spermatocyte, Spd(E) early spermatid, Spd(L) late spermatid, M meiosis metaphase II. Scale bars 50 µm. b Quantification of spermatid numbers in seminiferous tubules from spermatid stages 1–16 (N = 15 tubules per stage and per genotype; 5 mice per genotype were used and 3 tubules per stage and per mouse were counted). Spermatids were divided into five groups: stages 1–4, 5–8, 9–12, 13, 14, and 15, 16. From stage 9 onward, spermatids were significantly fewer in tubules of Vps13b∆Ex3/∆Ex3 mice than wild-type mice [p(9–12) = 0.004; p(13–14) < 0.001; p(15–16) < 0.001]. All values are presented as the mean ± SD. Significance was determined using Welch two sample t test. c Representative images of TUNEL staining on a Vps13b∆Ex3/∆Ex3 seminiferous tubule section at stages V–VII. Scale bars 50 µm. d Quantification of TUNEL-positive spermatids in stage groups 1–8, 9–12, and 13–16 (N = 21 tubules per stage and per genotype; 7 mice per genotype were used and 3 tubules per stage and per mouse were counted). Mutant sections showed significantly more TUNEL-positive spermatids, especially from stages 9 to 16 [p(1–8) = 0.03; p(9–12) = 0.002; p(13–16) = 0.002]. All values are presented as the mean ± SD. Significances were determined using Wilcoxon rank sum test
Acrosome formation is impaired in mutant spermatids
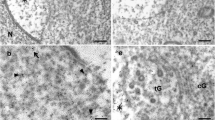
Impairment in nuclear morphogenesis being often associated with deficient acrosome biogenesis [5, 8, 17, 22, 42,43,44,45,46], we investigated whether the acrosome formation is impaired in mutant spermatids through Periodic Acid Schiff (PAS) staining of testicular sections. In Golgi phase, most mutant spermatids displayed a much weaker dot-like staining compared to wild-types (Fig. 4a, right panels). In addition, none of the PAS-positive structures that characterize cap, acrosomal, and maturation phases of wild-type spermatids were observed on mutant sections (Fig. 4a). Electron microscopy was conducted to determine whether the acrosomal vesicle was absent and to document the ultrastructural changes occurring during spermatid differentiation. Despite formation of the nuclear dense lamina (NDL) in early mutant spermatids, no acrosomal vesicle was ever found to associate with the nuclear membrane (Fig. 4b). Despite impaired acrosome formation and incomplete nuclear morphogenesis, surviving late spermatids retained the ability to form a flagellum and showed proper mitochondrial organization at the middle piece (Supplemental Fig. S1A, B). In contrast, highly vacuolated giant spermatids as well as bi-nucleated and multinucleated late spermatids were often found (Supplemental Fig. S1C, D). They likely result from disturbances in the spermatogenic syncytium (Supplemental Fig. S1E) and multinucleated/multiflagellated spermatozoa found in the epididymis likely develop from the observed multinucleated spermatids.

Acrosome formation is impaired during Vps13b∆Ex3/∆Ex3 spermiogenesis. a Representative images of PAS-stained testicular sections at tubule stages II, III, V–VII, and X–XII. Arrows point at PAS-positive acrosomal figures in wild-type spermatids and faint dot-like PAS signal in mutant spermatids. Spd(E) early spermatid, Spd(L) late spermatid. Scale bars 50 µm. b Electron micrographs of Golgi, cap, acrosome, and maturation phases of wild-type and mutant spermatids. G golgi, AV acrosomal vesicle, AG acrosomal granule, AC acrosome, N nuclei, CN condensed nuclei, NDL nuclear dense lamina, M mitochondria
Vps13b is not required for the transcription of major acrosomal genes
To determine whether transcription of acrosome-associated genes was affected in Vps13b∆Ex3/∆Ex3 testes and could account for the lack of acrosome, we performed RT-qPCR on the following genes: Dpy19l2, Spaca1, Acrosin, Acrbp, Sp56, Spaca7, Spata16, Zpbp1, and Zpbp2. Despite showing a tendency toward being reduced, none of the selected genes had a significantly different expression in Vps13b∆Ex3/∆Ex3 mice compared to wild-types (Supplemental Fig. S2A). More extensively, we assessed the transcript level of the following genes implicated in the regulation of spermiogenesis and in proacrosomal vesicular transport: Brdt, Cul4b, Dazap1, Ddx4, Hrb, Rfx2, Spata16, Tdrd6, and Vps54. Again, a small reduction in gene expression was measured, but likely resulted from the reduced number of late spermatids in mutant testes (Supplemental Fig. S2B). Since it was previously described that VPS13B participates to the regulation of the lysosomal structure [40], we suspected that the CB, spermatid-specific transcriptional control centre constituted of lysosomal components [47,48,49], may be structurally affected in mutant spermatids. Immunostaining of Ddx4 located the chromatoid body of mutant spermatids to the cytoplasm and no structural defect was observed (Supplemental Fig. S2C). On electron micrographs, we observed that the CB was normally associated with small vesicles and retained its ability to interact with the nuclear membrane in the absence of Vps13b (Supplemental Fig. S2D). Altogether, there is no evidence that neither the CB nor transcription of spermiogenesis-associated genes is affected in mutant spermatids.
Mutant spermatids display impaired Golgi dynamics
The acrosome results from intense vesicular transport from the Golgi apparatus [9, 15]. Since VPS13B mutations are known to cause disorganization of the Golgi apparatus [9, 15, 31], we looked at the Golgi structure in Vps13b∆Ex3/∆Ex3 spermatids. From stages 2 to 8, Golgi membrane stacks of wild-type spermatids were systematically located in close proximity (~ 500 nm) of the NDL (Fig. 5a, left panel) with the TGN facing the NDL. They displayed an open conformation by aligning with the nuclear membrane. In contrast, the Golgi apparatus of mutant spermatids appeared to be randomly located within the cytoplasm (Fig. 5a, middle panel). In occurrences where the Golgi apparatus was observed next to the NDL, membrane stacks did not align with the nuclear membrane and the TGN did not face the NDL. Instead, they harbored a closed conformation and randomly orientated (Fig. 5a, left panel). In addition, vesicular transport toward the NDL was not identified. During cap phase, wild-type spermatids showed large vesicles (~ 50 to 100 nm) leaving the TGN and fusing with the acrosomal sac, whereas, in mutant spermatids, smaller vesicles (~ 10 to 50 nm) were found at the TGN and scattered within the cytoplasm. To further examine the impact of Vps13b deficiency on the Golgi structure, testicular sections were stained against the Golgi protein Golga5. Immunofluorescence showed, in mutant spermatids, none of the rearrangements that occur during wild-type cap phase (Fig. 5b) and confirmed that the observations made on electron micrographs were systematic. The Golgi apparatus was misoriented, far apart from the nuclear membrane, and did not flatten to surround it. Measurement of the Golgi aspect ratio of both wild-type (R = 3.3) and mutant (R = 1.4) spermatids confirmed that they had significantly different structures.

Impaired Golgi organization in Vps13b∆Ex3/∆Ex3 spermatids. a Representative electron micrographs showing Golgi apparatus of wild-type and mutant spermatids. Golgi apparatus of wild-type spermatids displayed a conformation in which the membrane stacks organized in parallel to the nuclear membrane and the TGN faced the NDL. In mutant spermatids, the Golgi apparatus showed a random localization and orientation within the cytoplasm. It was often found at the posterior part of the nucleus rather than next to the NDL. In addition, the TGN was mostly enclosed within the membrane stacks that formed a semi-circular structure instead of the linear morphology seen in wild-type spermatids. b Images of wild-type and mutant cap phase spermatids stained against Golga5. Scale bar 5 µm. c Calculation of the aspect ratio (height [X] over width [Y]) of wild-type and mutant Golgi apparatuses based on measurements on Golga5 immunostaining. N = 54 apparatuses were measured per genotype (3 mice; 3 tubules per mouse; 6 Golgi per tubules). Based on the aspect ratio, mutant apparatuses had a significantly different structure (p < 0.001, as per Wilcoxon rank sum test)
Proacrosomal vesicles are not targeted to the NDL
To determine where proacrosomal vesicles are transported to within mutant spermatids, they were stained using Peanut Agglutinin-FITC (PNA). From stages 3 to 8, small and homogenous PNA-positive vesicles of about 0.5–1 µm accumulated in the cytoplasm of early mutant spermatids (Fig. 6a, b). During acrosome phase (stages 9–12), those vesicles were replaced by larger vesicular structures of lower PNA signal intensity and variable shapes (Fig. 6a, b). The vesicular structures and/or their PNA-binding content gradually disappeared through maturation phase (stages 13–16) and suggest the degradation of the abnormally targeted proacrosomal vesicles. In addition, some early mutant spermatids displayed higher PNA intensity at the plasma membrane than their wild-type counterparts (Fig. 6c). Abnormal export of proacrosomal vesicle to the plasma membrane may, therefore, occur. Finally, despite not being able to form an acrosome, early mutant spermatids sometimes displayed a PNA signal at the anterior part of their nucleus (Fig. 6c).

Impaired proacrosomal vesicular transport in Vps13b∆Ex3/∆Ex3 spermatids. a–c Acrosome and proacrosomal vesicle staining using PNA-FITC. a Images of wild-type and mutant spermatids were taken from stages 3 to 16. PNA staining showed accumulation of small vesicles within the cytoplasm of early mutant spermatids (stages 3–8). In late spermatids, PNA signal was found within and at the membrane of larger vesicular structures. Vesicular PNA signal gradually decreased from the beginning of acrosome phase (stage 9) to the end of maturation phase (stage 16). Scale bar 20 µm. b Close-up images of PNA-positive vesicles found in the early and late mutant spermatids. In addition, PNA signal was also found to be higher at the plasma membrane of some mutant spermatids than usually seen in wild-type spermatids. Scale bars 2 µm. c Images with higher brightness to clearly observe membranous PNA signals (arrows). Interestingly, despite the absence of acrosomes in mutant spermatids, a weak PNA signal similar to that observed at the plasma membrane was often detected at the anterior part of the nuclear membrane. Scale bars 10 µm. d Electron micrographs showing vesicular accumulation in the early mutant spermatids at the TGN (upper left panel), close to the NDL (upper middle panel) and enclosed within larger vesicular structures (upper right panel). Lower panels show large and electron dense membranous structures into an endomembrane compartment reminiscent of the lysosome. Arrows point at abnormally accumulated vesicular and membranous structures in mutant spermatids. G golgi, NDL nuclear dense lamina, E endomembrane compartment
To confirm abnormal targeting of acrosomal proteins, we immunodetected Spaca1 in both wild-type and mutant spermatids (Supplemental Fig. S3). In contrast with wild-type spermatids, where Spaca1 localized to the acrosomal inner membrane, mutant spermatids displayed a dispersed staining in cap, acrosome, and maturation phase.
Electron micrographs of early mutant spermatids showed that, in addition to vesicles accumulating at the TGN, small vesicles (< 10 nm) clustered into structures of about 0.5–1 µm at random location within the cytoplasm (Fig. 6d). Flat and small vesicles also associated with the NDL and likely account for the PNA signal detected at this location (Fig. 6d, upper middle panel). Structures reminiscent of endosomes/lysosomes were also found to contain membranes that are likely remnants of mistargeted proacrosomal vesicles (Fig. 6d, upper right panel). Such membrane-containing structures were much more prominent in late spermatids (Fig. 6d, lower right panel).
Proacrosomal vesicles are targeted to the endosome and lysosome in Vps13b∆Ex3/∆Ex3 spermatids
Considering the recent evidences that the endolysosomal pathway is implicated in the acrosome formation [16, 18, 19, 21, 22, 42, 50] and that VPS13B mutations affect this endomembrane compartment [40], we suspected that it could be targeted by proacrosomal vesicles in spermatids lacking Vps13b. To verify this hypothesis, we performed immunostainings against Eea1 and Lamp2. In early wild-type spermatids, we observed the systematic presence of a large and intensely stained Eea1-positive structure (Fig. 7a, left panels). Initially close to proacrosomal vesicles during Golgi phase (stages 1 and 2), it migrated to the posterior part of the nucleus during cap phase (stages 3–7). During this phase, a faint PNA signal colocalized with this Eea1 structure and suggested that a subset of PNA-binding proteins is targeted to the early endosome of wild-type spermatids. In contrast, PNA signal greatly accumulated within and around Eea1-positive vesicles in early mutant spermatids (Fig. 7a, right panels). Colocalization of PNA-binding proteins and Eea1 was confirmed on a confocal microscope (Supplemental Fig. S4). This observation could result from an abnormal targeting of proacrosomal vesicles toward the early endosome or a lack of transport from the endosome to the nuclear membrane. The early endosomal strcture disappeared from both wild-type and mutant spermatids during acrosome phase. During this phase, large lysosomal structures appeared at the spermatid's posterior part.

Proacrosomal vesicles are targeted to the endosome and lysosome in Vps13b∆Ex3/∆Ex3 spermatids. a Representative images of wild-type and mutant spermatids stained using anti-Eea1 antibody and PNA-FITC. PNA-positive vesicles scattered within mutant spermatids were frequently Eea1-positive. During cap phase, Eea1 signal in wild-type spermatids systematically colocalized with a small and faint PNA signal, while it was associated with larger and multiple vesicular structures in mutant spermatids. Left panels of cap phase wild-type spermatids were over-exposed to distinguish PNA signal at the endosomal structure. Images of cap phase mutant spermatids did not require over exposition to detect PNA signal at this structure. Scale bar 10 µm. b Representative images of wild-type and mutant spermatids stained using anti-Lamp2 antibody and PNA-FITC. In cap phase, mutant spermatids displayed larger Lamp2-positive structures than wild-type spermatids. In addition, PNA signal at and around these structures was prominently detected in mutant but not wild-type spermatids. Scale bar 10 µm. c Schematic representation of PNA-binding vesicle trafficking in developing mutant spermatids compared to the wild-type situation. In green is depicted the location of PNA-binding proteins. A acrosome, EE early endosome, G golgi, L lysosome
Lamp2 staining showed a gradual increase from cap to acrosome phase in wild-type spermatids. Small lysosomes observed in cap phase did not contain PNA-positive vesicles (Fig. 7b, upper panels). During acrosome phase, multiple large lysosomes were observed per spermatids and all contained a faint PNA signal, suggesting that a recycling of some acrosomal components through the lysosome could occur. In contrast, mutant spermatids contained large Lamp2-positive structures during cap phase (stages 5–7) and abnormally targeted PNA-positive vesicles were found to colocalize with Lamp2 (Fig. 7b, lower panels). Despite showing high content of membranes when observed through electron microscopy, lysosomes of late spermatids (stages 12–16) no longer contained PNA signal. A faint PNA signal was detected at the lysosomal membrane but not in the lumen. This observation could be due to the degradation of acrosomal protein glycosylation by lysosomal hydrolases. We confirmed the targeting of acrosomal proteins to the lysosomes by co-localizing Spaca1 with Lamp2 by confocal microscopy (Supplemental Fig. S5).
Altogether, the results suggest that Vps13b∆Ex3/∆Ex3 spermatids lack proper vesicular targeting to the NDL and that PNA-binding vesicles are mainly transported to the endolysosomal system. In addition, acrosomal content at the plasma membrane may be transported from the endosome. Dynamic and relation of the endomembrane compartments involved in acrosomogenesis are depicted for both wild-type and mutant spermiogenesis in Fig. 7c.
Formation of actin filaments at the ectoplasmic specialization is impaired in Vps13b∆Ex3/∆Ex3 spermatids
Due to the incomplete mutant sperm head morphogenesis, we suspected that actin remodeling at the Sertoli cell ectoplasmic specialization [51] may not occur. To test this hypothesis, testicular sections were stained with Phalloidin-488. In wild-type seminiferous tubules, the distribution of actin filaments followed a specific pattern through the tubule stages (Fig. 8a). At the beginning of the acrosome phase (stage 9), actin filaments at the Sertoli membrane formed the ectoplasmic specialization that surrounds the spermatid’s acrosome. Actin filaments surrounded the acrosome until the end of maturation phase (stage 16). Then, they disassembled and reassembled around the acrosome of newly formed acrosome phase spermatids. In mutant seminiferous tubules, actin filaments appeared to mostly organize around the plasma membrane of cap and acrosome phase spermatids (stages 4–12, Supplemental Fig. S6A–C) and dissociated from most spermatids entering maturation phase. The early organization of actin filaments around mutant spermatids may be the result of the presence of acrosomal proteins at the plasma membrane that possibly signal the presence of a fully developed acrosomal vesicle to the surrounding Sertoli cell. Using electron microscopy, we investigated another actin-based structure participating to nuclear morphogenesis, the acroplaxome marginal ring [52], and could not identify intermediate filaments at this location in mutant spermatids (Supplemental Fig. S6D).

Vps13b and Rab6 locate to the acrosome. a Representative images of isolated wild-type spermatids in Golgi, cap, acrosome, and maturation phases stained against an epitope of Vps13b within amino acids 64–412 (upper panels) and an epitope at the C-terminus end of the Vps13b Chorein domain (amino acids 103–121, lower panels) and counterstained with PNA-FITC and DAPI. Overlay images include a DIC picture. Using both antibodies, results showed that Vps13b expression was not detectable at the Golgi apparatus of spermatogonia and spermatocytes. Expression of Vps13b increased in early spermatids. The protein essentially localized to proacrosomal vesicles in Golgi phase and to the acrosomal inner membrane (aa64–412) and acrosomal granule (aa103–121) in acrosome phase. Signals detected with Vps13baa64–412 gradually left the acrosome through maturation phase, while signals detected with Vps13baa103–121 remained at the acrosome. b Images of Rab6 staining on cap phase wild-type and mutant spermatids. Rab6 located to the Golgi apparatus of all spermatogenic cells. In addition, Rab6 followed the sequential acrosomal localization displayed by the Vps13b epitope comprised between amino acids 64 and 412 in wild-type spermatids. In mutant spermatids, it mainly remained at the Golgi apparatus and was sometimes found in mislocalized PNA-positive vesicles during cap phase. Scale bars 10 µm
Differentiation events subsequent to the acrosome formation do occur in Vps13b∆Ex3/∆Ex3 spermatids
We then wanted to assess whether events that are subsequent to the acrosome formation were also affected in Vps13b∆Ex3/∆Ex3 spermatids. More specifically, we addressed whether Vps13b loss of function would also affect basal plate formation at the posterior part of the nucleus and manchette formation. To this end, we stained the basal plate component Spata6 in both mutant and wild-type mice. In spite of the absence of acrosomes, we found Spata6 to be properly assembled at the nuclear membrane of Vps13b∆Ex3/∆Ex3 spermatids (Supplemental Fig. S7A). In addition, β-tubulin staining of testicular sections showed that polymerization of the manchette microtubule tracks occurred at the correct stages, but that it failed to surround the nucleus (Supplemental Fig. S7B). This observation likely results from the lack of acrosome, thereby preventing newly formed microtubules to anchor onto the nuclear membrane, rather than from a direct effect of the loss of Vps13b function.
Vps13b and Rab6 locate to the acrosome in wild-type mice
To comprehend the role of Vps13b in targeting proacrosomal vesicles to the NDL, immunostainings against Vps13b and its interactor Rab6 were performed on wild-type testicular sections. To detect Vps13b, two antibodies were used. One, hereby name Vps13baa103–121, was raised against the C-terminus part of the Chorein domain of the human VPS13B. The other one was raised using a larger antigen and is referred to as Vps13baa64–412. The two antibodies used in this study show a Golgi localization in cultured fibroblasts as previously reported (data not shown). However, unlike previously reported in other cell types [31], Vps13b was not detected at the Golgi apparatus of spermatogenic cells. In seminiferous tubules, the expression of Vps13b was first detectable in Golgi phase spermatids where it located to the pre-acrosome (Fig. 8a). Throughout cap, acrosome, and maturation phase, Vps13b staining was located to various positions at the acrosome depending on the epitope of Vps13b that is targeted and the differentiation stage. Vps13baa64–412 was found to localize to proacrosomal vesicles in Golgi phase and to the inner acrosomal membrane in cap and acrosome phase. Through maturation phase, Vps13baa64–412 staining was found to delocalize from the acrosome and was no longer detectable at the end of maturation phase. The Vps13baa103–121 antibody was found at proacrosomal vesicles in Golgi phase and at the acrosomal granule in cap and acrosome phase. In addition, unlike Vps13baa64–412 staining, Vps13baa103–121 staining remained at the acrosome throughout maturation phase. Both antibodies against Vps13b gave no staining in mutant spermatids (Supplemental Fig. S8).
Concerning Rab6, staining was found at the Golgi apparatus of all spermatogenic cells (Supplemental Fig. S9). In addition, an acrosomal staining similar to that of Vps13baa64–412 was detected in spermatids during Golgi, cap, and acrosome phases (Fig. 8b, right panels and Supplemental Fig. S9). In contrast, there was no detectable Rab6 at the nuclear membrane of mutant spermatids (Fig. 8b, left panels) despite showing Golgi localization in all spermatogenic cells. Instead, Rab6 colocalized with some abnormally accumulated PNA vesicles. We conclude that Vps13b is not necessary to the Golgi and, to some extent, the proacrosomal vesicular localization of Rab6 but to its transport toward the NDL.
Discussion
The implication of VPS13B in protein sorting and trafficking has long been hypothesized based on domain homologies with the yeast Vps13 protein [23], cellular phenotypes of VPS13B-deficient cell models [40], and functions of VPS13B interactor RAB6 [32, 33, 35, 53,54,55,56]. In this study, we provide evidences that support this hypothesis and demonstrate that Vps13b is necessary for the proper trafficking of the subset of vesicles that constitutes the acrosome. Golgi fragmentation, lack of early endosomal structures, and defective protein glycosylation led to consider that VPS13B is implicated in trafficking within the Golgi–endosome–lysosome membrane system [31, 40]. Here, we show that, in the absence of Vps13b, Golgi-derived vesicles that should constitute the acrosome are targeted to early endosomes prior to be degraded within lysosomes. In addition, we show that other differentiation events that occur in late spermatids after the acrosomal vesicle formation are not directly affected by loss of Vps13b function. Those events include connecting piece, manchette, and flagellum formation.
The acrosome is a germ cell-specific organelle that forms in spermatids through the synthesis, trafficking, and fusion of Golgi-derived vesicles [7,8,9]. Proacrosomal vesicles receive content from the endolysosomal pathway [20,21,22, 42, 50], traffic toward the NDL [11, 44, 57], and anchor onto the nuclear membrane via a cytoskeletal scaffold called the acroplaxome [52, 58]. In this study, we found Vps13b to be strongly present at the pre-acrosome during Golgi phase. Later, in cap and acrosome phases, it localized together with Rab6 to the acrosomal inner membrane. In the absence of Vps13b, and in spite of normal expression of acrosomal genes, the acrosomal sac was missing at the NDL. In contrast, proacrosomal vesicles were found to be scattered within the spermatid cytoplasm. They accumulated with Eea1-positive vesicles in early spermatids and within lysosomes in later stages. In addition, PNA signal was sometimes increased at the cell surface of round spermatids, thus, suggesting that an abnormal trafficking of acrosomal membrane proteins toward the plasma membrane occurs. Altogether our results show that Vps13b is necessary to transport proacrosomal vesicles to the NDL. Similarly to other models with deficiencies in acrosome formation [33, 35, 43,44,45,46], we show that Vps13b∆Ex3/∆Ex3 mice are affected with globozoospermia. Although Vps13b∆Ex3/∆Ex3 mice display round-headed spermatozoa, they are also largely affected with oligospermia. In addition to be round-headed, mutant spermatozoa were often multiflagellated and multinucleated. Polynucleation could be observed as early as in acrosome phase. We suspect that the presence of acrosomal proteins at the plasma membrane may reach sufficient levels to promote spontaneous spermatid fusion and lead to multinucleated/multiflagellated sperm cells.
We provide in Fig. 9, our working hypothesis for the potential functions of Vps13b in spermatids. Rab6, the small GTPase that interacts with Vps13b [32], plays a role in retrograde transport and actin-based processes, two essential functions to the acrosomogenesis. First, Rab6 is involved in endosome-to-Golgi retrograde transport and is found in larger complexes with Tmf [33, 59] and Vps54 [35], two proteins also involved in this transport [36, 56] and essential to the acrosome formation [17, 37, 38, 42]. Tmf is a Golgi-associated protein whose the absence in mice causes globozoospermia due to impaired homing of proacrosomal vesicles [37, 38]. Vps54 is part of the Golgi-associated retrograde protein complex (GARP) that interacts with Rab6 through Vps52 [35]. Vps54 was found to locate to the acrosome of mouse spermatids [21]. Germ cells expressing the Vps54–L967Q variant are unable to develop an acrosome due to impaired retrograde transport of UBPy-sorted endosomal cargoes [20]. In Vps13b∆Ex3/∆Ex3 mice, Rab6 was no longer found at the NDL. It may be that Vps13b is necessary to Rab6-dependent tethering functions of proteins such as Tmf [60], GARP [61], or other tethering complexes not yet described in acrosomogenesis. In turn, the absence of Vps13b in proacrosomal vesicles could prevent their maturation through lack of incorporation of proteins from the endolysosomal pathway (Fig. 9a, mechanism 2) and lead to their abnormal targeting to the plasma membrane and other endomembrane compartments [62] (Fig. 9b, mechanism 2).

Working hypothesis for the role of the Vps13b-Rab6 complex in acrosome biogenesis. a Schematic illustration of the mechanisms which are possibly dependent on the presence of the Vps13b-Rab6 complex at proacrosomal vesicles. b Schematic illustration depicting how those mechanisms are affected in the absence of Vps13b
The formation of the acrosomal vesicle over the nuclear membrane was shown to depend on microtubule [21, 57, 63] and actin filament [51, 64, 65] tracks. Both Golgi- and endosome-derived factors may recruit proacrosomal vesicles onto cytoskeletal tracks leading to the NDL. For instance, the endosomal deubiquitinase UBPy and the Golgi-associated protein Tmf, possess a microtubule interacting and trafficking (MIT) domain that may directly link proacrosomal vesicles to microtubule tracks [21, 37, 66] (Fig. 9a, mechanism 3). It is, therefore, possible that lack of recruitment of such factors at proacrosomal vesicles may prevent their interaction with microtubules in Vps13b∆Ex3/∆Ex3 spermatids (Fig. 9b, mechanism 3). In addition, Rab6 was shown to mediate recruitment of Dynein–Dynactin complexes to the membrane of Golgi-derived vesicles [67,68,69] (Fig. 9a, mechanism 3). Lack of Rab6 in some Vps13b∆Ex3/∆Ex3 proacrosomal vesicles may prevent their linkage to microtubules via the Dynein–Dynactin complex (Fig. 9b; mechanism 3). Alternatively, the actin-based motor Myosin Va that localizes to the acroplaxome [58] and interacts with Rab6 may no longer allow the docking of proacrosomal vesicles [70] in absence of the Vps13b–Rab6 complex [53] (Fig. 9a, b, mechanism 4).
Interestingly, actin dynamics at the Sertoli cell ectoplasmic specialization was also affected in Vps13b∆Ex3/∆Ex3 testes [71]. It would be interesting to determine the actin dynamic in testes of other acrosome-less models to determine whether actin polymerization at the ectoplasmic specialization is generally dependent on the presence of the acrosome or whether VPS13B is essential to an actin-based mechanism that affects both the acrosome and the ectoplasmic specialization. Another actin-dependent process that we found to be affected in Vps13b∆Ex3/∆Ex3 spermatids is the Golgi organization (Fig. 9b, mechanism 1). Once again, loss of interaction of Rab6 with Vps13b could explain this observation due to Rab6 function in regulating KIF1C motor domain [72].
In this study, we demonstrate that Vps13b is a necessary factor for the targeting of Golgi-derived vesicles to the spermatid NDL. It is therefore likely that in other cell types of tissues affected in Cohen syndrome, a subset of vesicles leaving the TGN also requires Vps13b for their transport toward their proper target. Instead of reaching the nuclear membrane, proacrosomal vesicles of Vps13b∆Ex3/∆Ex3 spermatids end up enclosed into the lysosome (Fig. 9b, mechanism 2′). Similarly, vesicles in other cell types may be abnormally targeted and accumulate into the lysosome. This might explain the presence of enlarged lysosomal structures previously described in fibroblasts derived from patient biopsies.
The Vps13b∆Ex3/∆Ex3 mouse line is the second animal model reported for Cohen syndrome. Shearman and Wilton published in 2011 a Border collie breed with a 4-bp deletion in Vps13b exon 19 [73]. This breed was affected with clinical features of Cohen syndrome including neonatal hypotonia, developmental delay, microcephaly, and neutropenia. Unfortunately, the phenotypes were not studied at the cellular and molecular levels in this model. In conclusion, considering that Vps13b∆Ex3/∆Ex3 mice present with molecular aspects of Cohen syndrome, it is a promising model for the investigation of phenotypes that could potentially be treated such as progressive retinal dystrophy, cyclic neutropenia, and metabolic symptoms. Last but not least, male infertility in Vps13b∆Ex3/∆Ex3 mice suggests that (1) male individuals with Cohen syndrome may be infertile and (2) novel mouse models of intellectual disability may be good candidates to investigate the production of germ cells.
Materials and methods
Animals
Experiments were conducted in accordance with the FELASA guidelines for the care and use of laboratory animals (FELASA category B accreditation for R.D.C.) and the French legislation (animal quarters agreement number 821231010 EA), after approval by the local ethics committee (#105 Comité d’Ethique de l’Expérimentation Animale Grand Campus Dijon) and the French Ministry of Higher Education, Research and Innovation (agreement number APAFIS#9524-2017040712176062). Animals were kept in animal quarters under controlled temperature (21 ± 1 °C) and light conditions (12-h/12-h light/dark cycle). Animals were fed ad libitum with standard laboratory chow (Safe #A04) and 0.2 µm filtered water.
The Vps13b∆Ex3/∆Ex3 mouse line carrying a constitutive deletion of Vps13b exon 3 was established at the MCI/ICS. The targeting vector was constructed as follows. A 710-bp fragment encompassing Vps13b exon 3 (ENSMUSE00000999073) was amplified by PCR from BAC RP24-286C24 genomic DNA and subcloned in an MCI proprietary vector. This vector contains an LoxP site as well as a floxed and flipped Neomycin resistance cassette. A 3.7 kb fragment corresponding to the 3′ homology arm and 4.1 kb fragment corresponding to the 5′ homology arms were amplified by PCR and subcloned in a step 1 plasmid to generate the final targeting construct. The linearized construct was electroporated in C57BL/6N mouse embryonic stem (ES) cells. After selection, targeted clones were identified by PCR using external primers and further confirmed by Southern blot analysis on 5′ and 3′ digests with a Neo probe as well as a 5′ external probe. Two positive ES clones were injected into BALB/cN blastocysts. Resulting male chimeras were bred with Flp deleter C57Bl/6N females [74]. Germline transmission of the conditional allele was achieved and constitutive Vps13b exon 3 deletion was obtained by crossing Vps13b+/Flox-Ex3 males with Cre deleter C57Bl/6N females.
Genotyping PCR
Genomic DNA was extracted from tail and embryo biopsies through an overnight incubation in lysis buffer (0.2% SDS, 5 mM EDTA, 100 mM Tris pH 8.5, and 200 mM NaCl) supplemented with 100 µg/mL of Proteinase K (Promega, Charbonnières-les-Bains, France) at 56 °C. The extract was centrifuged at 13,200g for 5 min following inactivation of the Proteinase K at 80 °C for an hour. A 1:100 dilution of the supernatant was used for genotyping by PCR analysis using GoTaq G2 Flexi DNA polymerase (Promega) according to the manufacturer’s instruction using forward primer Gf: GCTAGATTGGCTGTCATGAAGCAC and reverse primer Gr: CTAACAGTTGACTGAGGAAGCAGCAATG to target a genomic fragment spanning Vps13b exon 3 in wild-type mice and including the LoxP sites in mutants.
Reverse transcription and real-time qPCR analysis
Total RNA from testis biopsies was isolated using TRIzol reagent (Invitrogen, Courtaboeuf, France) and reverse-transcribed using the iScript Reverse Transcription Supermix (Bio-Rad #1708891, Marnes-la-Coquette, France) in accordance with the manufacturers’ instructions. mRNA levels of Vps13b and 9 acrosomal-associated genes were measured by performing real-time qPCR (RT-qPCR) on a CFX-96 thermal cycler (Bio-Rad), using the iTaq Universal SYBR Green Supermix (Bio-Rad), 0.125 µM of primers each, and 2 ng of cDNA per reaction. Ct values were normalized against hypoxanthine phosphoribosyltransferase (Hprt). Primer sequences are available in supplemental table S1.
Mating phenotypes and embryonic viability
To assess mating performances of Vps13b∆Ex3/+ and Vps13b∆Ex3/∆Ex3 mutants, [♂+/∆Ex3 × ♀+/∆Ex3], [♂+/∆Ex3 × ♀∆Ex3/∆Ex3], and [♂∆Ex3/∆Ex3 × ♀+/∆Ex3] crosses utilizing 2-month-old animals were set. Number of pups and their genotypes were recorded over a 4-month period.
Spermatozoa phenotyping
Prior to collect their sperm, 2 month-old males were isolated in individual cages for 3 days and sacrificed through cervical dislocation. Their cauda epididymides were collected and dilacerated in 500 µL of M16 medium (Sigma, Saint-Quentin-Fallavier, France) at 37 °C. Sperm motility was assessed after 2–4 h of incubation at 37 °C. To determine the percentage of motile spermatozoa, two drops of 10 µL each were placed between slide and cover glass, and a duplicate counting of motile spermatozoa among 100 sperm cells was performed. To evaluate sperm concentration, 10 µL of collection medium was placed on a MAKLER cell and fixed by adding 10 µL of 4% PFA (VWR) prior to counting spermatozoa. To document abnormalities in the shape of mutant spermatozoa, 10 µL drops of M16 medium were smeared on slides, stained in Gill’s Hematoxylin II (Merck), stained in Shorr solution (Merck), dried and mounted under cover glass with EUKITT (Kindler). Structural observations were conducted on 100 spermatozoa per mouse.
Histology
Testes of 2-month-old Vps13b+/+ and Vps13b∆Ex3/∆Ex3 males were collected along with their epididymis, fixed in 4% PFA for 48 h, and embedded in paraffin. To observe the testicular structure, 5-µm-thick sections were deparaffinized and stained with Harris hematoxylin:eosin Y reagents (Leica). To observe the acrosomal structure, sections were stained with Periodic acid Schiff reagents. Nuclear counterstain was obtained by staining with Methyl green (Merck) at pH4.0. Stained slides were dried and mounted in organic mounting medium. Observations were conducted on Axioscope microscope (Zeiss).
Transmission electron microscopy
Testes were fixed for 48 h at 4 °C in 4% paraformaldehyde (EMS) and 2.5% of glutaraldehyde (EMS) in Sorensen phosphate buffer (0.1 M, pH 7.3). After fixation, samples were washed in Sorensen phosphate buffer. Post-fixation treatment was done through incubation in 1% osmium tetroxide (EMS) at room temperature for 1 h. Dehydration and resin impregnation of the samples were performed with a Leica EM AMW automatic microwave tissue processor. Dehydration was done through consecutive incubation in ethanol baths of increasing concentration followed by incubation in an acetone bath. Infiltration was done through three consecutive incubations in acetone:Embed-812 resin mixtures and a final incubation in Embed-812 resin. Sample polymerization was performed using 3% BDMA at 60 °C for 48 h. Blocks were cut on a Reichert Ultracut E ultramicrotome and 60-nm-thick sections were deposited on copper/palladium grids. After drying, sections were contrasted with uranyl acetate and lead citrate. Observations were conducted on an HITACHI H-7500 transmission electron microscope operating at 80 kV.
Isolation of spermatogenic cells
Spermatogenic cells were isolated from testes of 2-month-old mice. Testicular capsules were cut and seminiferous tubules placed in 2 ml of RPMI medium (ThermoFisher Scientific #11554516, Illkirch, France) supplemented with 0.1% collagenase I (ThermoFisher Scientific #10114532) at 37 °C for 15 min. Tubules-containing tubes were inverted ten times to separate interstitial cells from the tubules. Tubules were let to decent and supernatants were removed. Tubules were washed once in PBS before being dilacerated and passed through a 23G needle to release spermatogenic cells that were then fixed in 4% PFA for 15 min. Cells were then washed in PBS, then in deionized water, and finally spotted onto Superfrost Plus slides (ThermoFisher Scientific #10149870).
TUNEL assay and immunofluorescence
TUNEL assays were performed using the In Situ Cell Death Detection Kit (Roche, Boulogne-Billancourt, France) according to the manufacturer’s instruction. Briefly, rehydrated paraffin sections were permeabilized in a 0.3% triton X-100 PBS solution prior to being incubated for 1 h at 37 °C in a solution containing a terminal deoxynucleotidyl transferase (EC 2.7.7.31) and fluorescein-labeled dNTPs.
Immunostainings were performed on frozen sections. Testes were fixed in 4% PFA for 48 h, subsequently incubated in a 30% sucrose solution for 24 h, and embedded in Frozen Section Compound (Leica, Nanterre, France). Sections of 5 µm thickness were incubated in 100% methanol for 5 min at − 20 °C and rehydrated in PBS. A 30 min heat-mediated antigen retrieval step was performed at 80 °C in 1× citric acid-based antigen unmasking solution (Vector Laboratories #H-3300, Burlingame, CA, USA). Sections were blocked for 30 min at room temperature in a PBS solution containing 10% fetal calf serum (Hyclone, ThermoFisher Scientific) and 0.3% Triton X-100. Sections were then incubated overnight at 4 °C with rabbit primary antibodies diluted in blocking solution, washed in PBS supplemented with 0.1% Tween-20, incubated for 2 h at room temperature with anti-rabbit or anti-rat Alexa Fluor 488/568 secondary antibody (ThermoFisher Scientific, 1:500), washed, counterstained with FITC-labeled Peanut Agglutinin (Sigma-Aldrich #L7381, PNA, 2 µg/mL) for 2 h and DAPI (Sigma-Aldrich #D9542, 2 µg/mL) for 5 min, washed again, and mounted in ProLong™ Diamond Antifade Mountant (ThermoFisher Scientific #P36961). Primary antibodies used in this study were: anti-Golga5 (Genetex #GTX104255, 1:200, Hsinchu City, Taiwan), anti-Vps13b (Vps13baa64–412, Proteintech #24505-1-AP, 1:50, Manchester, United Kingdom), anti-Rab6 (Genetex #GTX110646, 1:200), anti-Spaca1 (Abcam #191843, 1:200, Cambridge, United Kingdom) anti-Eea1 (Cell Signaling #3288, 1:200, Leiden, The Netherlands), and anti-Lamp2 (Santa-Cruz #19991, 1:50, Dallas, USA). A custom-made antibody targeting VPS13B amino acids 103–121 was generated by Covalab (Vps13baa103–121, Rabbit #1329007, 1:50, Villeurbanne, France).
Microscopy
Immunostainings on isolated spermatogenic cells were documented using an SP8 confocal microscope (Leica, Wetzlar, Germany) and the Leica Application Suite X. Immunostainings on testis sections and TUNEL assays were observed using the SP8 confocal microscope or the Axio Vert.A1 microscope (Carl Zeiss Meditec, Marly-le-roi, France) equipped with a Retiga R3 camera (QImaging, Surrey, BC, Canada) and a X-Cite series 120 fluorescence lamp (Excelitas, Waltham, MA, USA). Epifluorescence images were documented using ImageJ and the µManager software package.
Statistical analysis
Statistical analyses were performed using R software 3.4.4. The data are either presented as boxplots, dot plots ± SD, or the mean ± SD. Boxplots and dot plots were drawn using R library “easyGgplot2”. To test for the difference between two means, normal distribution and equal variances of the samples were first determined using a Shapiro–Wilk test and an F test, respectively. Whenever sample groups had a normal distribution and equal variances, equality of the means was assessed using an unpaired two-sample t test. Otherwise, a Wilcoxon rank sum test was performed.
References
Jin M, Fujiwara E, Kakiuchi Y, Okabe M, Satouh Y, Baba SA, Chiba K, Hirohashi N (2011) Most fertilizing mouse spermatozoa begin their acrosome reaction before contact with the zona pellucida during in vitro fertilization. Proc Natl Acad Sci USA 108:4892–4896. https://doi.org/10.1073/pnas.1018202108
Kornbluth S, Fissore R (2015) Vertebrate Reproduction. Cold Spring Harb Perspect Biol 7:a006064. https://doi.org/10.1101/cshperspect.a006064
Foster JA, Gerton GL (2016) The acrosomal matrix. Adv Anat Embryol Cell Biol 220:15–33. https://doi.org/10.1007/978-3-319-30567-7_2
Yan W (2009) Male infertility caused by spermiogenic defects: lessons from gene knockouts. Mol Cell Endocrinol 306:24–32. https://doi.org/10.1016/j.mce.2009.03.003
Dam AH, Feenstra I, Westphal JR, Ramos L, van Golde RJ, Kremer JA (2007) Globozoospermia revisited. Hum Reprod Update 13:63–75. https://doi.org/10.1093/humupd/dml047
Tang XM, Lalli MF, Clermont Y (1982) A cytochemical study of the Golgi apparatus of the spermatid during spermiogenesis in the rat. Am J Anat 163:283–294. https://doi.org/10.1002/aja.1001630402
Yao R, Ito C, Natsume Y, Sugitani Y, Yamanaka H, Kuretake S, Yanagida K, Sato A, Toshimori K, Noda T (2002) Lack of acrosome formation in mice lacking a Golgi protein, GOPC. Proc Natl Acad Sci USA 99:11211–11216. https://doi.org/10.1073/pnas.162027899
Han F, Liu C, Zhang L, Chen M, Zhou Y, Qin Y, Wang Y, Duo S, Cui X, Bao S, Gao F (2017) Globozoospermia and lack of acrosome formation in GM130-deficient mice. Cell Death Dis 8:e2532. https://doi.org/10.1038/cddis.2016.414
Moreno RD, Ramalho-Santos J, Sutovsky P, Chan EK, Schatten G (2000) Vesicular traffic and golgi apparatus dynamics during mammalian spermatogenesis: implications for acrosome architecture. Biol Reprod 63:89–98
Ramalho-Santos J, Moreno RD, Wessel GM, Chan EK, Schatten G (2001) Membrane trafficking machinery components associated with the mammalian acrosome during spermiogenesis. Exp Cell Res 267:45–60. https://doi.org/10.1006/excr.2000.5119
Yang WX, Sperry AO (2003) C-terminal kinesin motor KIFC1 participates in acrosome biogenesis and vesicle transport. Biol Reprod 69:1719–1729. https://doi.org/10.1095/biolreprod.102.014878
Berruti G, Paiardi C (2011) Acrosome biogenesis: revisiting old questions to yield new insights. Spermatogenesis 1:95–98. https://doi.org/10.4161/spmg.1.2.16820
Berruti G (2016) Towards defining an ‘origin’—the case for the mammalian acrosome. Semin Cell Dev Biol 59:46–53. https://doi.org/10.1016/j.semcdb.2016.01.013
Tanii I, Toshimori K, Araki S, Oura C (1992) Extra-Golgi pathway of an acrosomal antigen during spermiogenesis in the rat. Cell Tissue Res 270:451–457
West AP, Willison KR (1996) Brefeldin A and mannose 6-phosphate regulation of acrosomic related vesicular trafficking. Eur J Cell Biol 70:315–321
Li S, Qiao Y, Di Q, Le X, Zhang L, Zhang X, Zhang C, Cheng J, Zong S, Koide SS, Miao S, Wang L (2009) Interaction of SH3P13 and DYDC1 protein: a germ cell component that regulates acrosome biogenesis during spermiogenesis. Eur J Cell Biol 88:509–520. https://doi.org/10.1016/j.ejcb.2009.05.001
Paiardi C, Pasini ME, Gioria M, Berruti G (2011) Failure of acrosome formation and globozoospermia in the wobbler mouse, a Vps54 spontaneous recessive mutant. Spermatogenesis 1:52–62. https://doi.org/10.4161/spmg.1.1.14698
Schmitt-John T, Drepper C, Mussmann A, Hahn P, Kuhlmann M, Thiel C, Hafner M, Lengeling A, Heimann P, Jones JM, Meisler MH, Jockusch H (2005) Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat Genet 37:1213–1215. https://doi.org/10.1038/ng1661
Zhu GD, Salazar G, Zlatic SA, Fiza B, Doucette MM, Heilman CJ, Levey AI, Faundez V, L’Hernault SW (2009) SPE-39 family proteins interact with the HOPS complex and function in lysosomal delivery. Mol Biol Cell 20:1223–1240. https://doi.org/10.1091/mbc.E08-07-0728
Berruti G, Paiardi C (2015) USP8/UBPy-regulated sorting and the development of sperm acrosome: the recruitment of MET. Reproduction 149:633–644. https://doi.org/10.1530/REP-14-0671
Berruti G, Ripolone M, Ceriani M (2010) USP8, a regulator of endosomal sorting, is involved in mouse acrosome biogenesis through interaction with the spermatid ESCRT-0 complex and microtubules. Biol Reprod 82:930–939. https://doi.org/10.1095/biolreprod.109.081679
Wang H, Wan H, Li X, Liu W, Chen Q, Wang Y, Yang L, Tang H, Zhang X, Duan E, Zhao X, Gao F, Li W (2014) Atg7 is required for acrosome biogenesis during spermatogenesis in mice. Cell Res 24:852–869. https://doi.org/10.1038/cr.2014.70
Kolehmainen J, Black GC, Saarinen A, Chandler K, Clayton-Smith J, Traskelin AL, Perveen R, Kivitie-Kallio S, Norio R, Warburg M, Fryns JP, de la Chapelle A, Lehesjoki AE (2003) Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet 72:1359–1369
Seifert W, Holder-Espinasse M, Kuhnisch J, Kahrizi K, Tzschach A, Garshasbi M, Najmabadi H, Kuss AW, Kress W, Laureys G, Loeys B, Brilstra E, Mancini GMS, Dollfus H, Dahan K, Apse K, Hennies HC, Horn D (2009) Expanded mutational spectrum in cohen syndrome, tissue expression, and transcript variants of COH1. Hum Mutat 30:E404–E420. https://doi.org/10.1002/humu.20886
Bugiani M, Gyftodimou Y, Tsimpouka P, Lamantea E, Katzaki E, d’Adamo P, Nakou S, Georgoudi N, Grigoriadou M, Tsina E, Kabolis N, Milani D, Pandelia E, Kokotas H, Gasparini P, Giannoulia-Karantana A, Renieri A, Zeviani M, Petersen MB (2008) Cohen syndrome resulting from a novel large intragenic COH1 deletion segregating in an isolated Greek island population. Am J Med Genet A 146A:2221–2226. https://doi.org/10.1002/ajmg.a.32239
Seifert W, Holder-Espinasse M, Spranger S, Hoeltzenbein M, Rossier E, Dollfus H, Lacombe D, Verloes A, Chrzanowska KH, Maegawa GHB, Chitayat D, Kotzot D, Huhle D, Meinecke P, Albrecht B, Mathijssen I, Leheup B, Raile K, Hennies HC, Horn D (2006) Mutational spectrum of COH1 and clinical heterogeneity in Cohen syndrome. J Med Genet. https://doi.org/10.1136/jmg.2005.039867
Mochida GH, Rajab A, Eyaid W, Lu A, Al-Nouri D, Kosaki K, Noruzinia M, Sarda P, Ishihara J, Bodell A, Apse K, Walsh CA (2004) Broader geographical spectrum of Cohen syndrome due to COH1 mutations. J Med Genet. https://doi.org/10.1136/jmg.2003.014779
Hennies HC, Rauch A, Seifert W, Schumi C, Moser E, Al-Taji E, Tariverdian G, Chrzanowska KH, Krajewska-Walasek M, Rajab A, Giugliani R, Neumann TE, Eckl KM, Karbasiyan M, Reis A, Horn D (2004) Allelic heterogeneity in the COH1 gene explains clinical variability in Cohen syndrome. Am J Hum Genet 75:138–145. https://doi.org/10.1086/422219
Falk MJ, Feiler HS, Neilson DE, Maxwell K, Lee JV, Segall SK, Robin NH, Wilhelmsen KC, Traskelin AL, Kolehmainen J, Lehesjoki AE, Wiznitzer M, Warman ML (2004) Cohen syndrome in the Ohio Amish. Am J Med Genet A 128A:23–28. https://doi.org/10.1002/ajmg.a.30033
El Chehadeh S, Aral B, Gigot N, Thauvin-Robinet C, Donzel A, Delrue MA, Lacombe D, David A, Burglen L, Philip N, Moncla A, Cormier-Daire V, Rio M, Edery P, Verloes A, Bonneau D, Afenjar A, Jacquette A, Heron D, Sarda P, Pinson L, Doray B, Vigneron J, Leheup B, Frances-Guidet AM, Dienne G, Holder M, Masurel-Paulet A, Huet F, Teyssier JR, Faivre L (2010) Search for the best indicators for the presence of a VPS13B gene mutation and confirmation of diagnostic criteria in a series of 34 patients genotyped for suspected Cohen syndrome. J Med Genet 47:549–553. https://doi.org/10.1136/jmg.2009.075028
Seifert W, Kuhnisch J, Maritzen T, Horn D, Haucke V, Hennies HC (2011) Cohen syndrome-associated protein, COH1, is a novel, giant Golgi matrix protein required for Golgi integrity. J Biol Chem 286:37665–37675. https://doi.org/10.1074/jbc.M111.267971
Seifert W, Kuhnisch J, Maritzen T, Lommatzsch S, Hennies HC, Bachmann S, Horn D, Haucke V (2015) Cohen syndrome-associated protein COH1 physically and functionally interacts with the small GTPase RAB6 at the Golgi complex and directs neurite outgrowth. J Biol Chem 290:3349–3358. https://doi.org/10.1074/jbc.M114.608174
Fridmann-Sirkis Y, Siniossoglou S, Pelham HR (2004) TMF is a golgin that binds Rab6 and influences Golgi morphology. BMC Cell Biol 5:18. https://doi.org/10.1186/1471-2121-5-18
Fukuda M, Kanno E, Ishibashi K, Itoh T (2008) Large scale screening for novel Rab effectors reveals unexpected broad Rab binding specificity. Mol Cell Proteomics 7:1031–1042. https://doi.org/10.1074/mcp.M700569-MCP200
Liewen H, Meinhold-Heerlein I, Oliveira V, Schwarzenbacher R, Luo G, Wadle A, Jung M, Pfreundschuh M, Stenner-Liewen F (2005) Characterization of the human GARP (Golgi associated retrograde protein) complex. Exp Cell Res 306:24–34. https://doi.org/10.1016/j.yexcr.2005.01.022
Bonifacino JS, Hierro A (2011) Transport according to GARP: receiving retrograde cargo at the trans-Golgi network. Trends Cell Biol 21:159–167. https://doi.org/10.1016/j.tcb.2010.11.003
Elkis Y, Bel S, Rahimi R, Lerer-Goldstein T, Levin-Zaidman S, Babushkin T, Shpungin S, Nir U (2015) TMF/ARA160 governs the dynamic spatial orientation of the Golgi apparatus during sperm development. PLoS One 10:e0145277. https://doi.org/10.1371/journal.pone.0145277
Lerer-Goldshtein T, Bel S, Shpungin S, Pery E, Motro B, Goldstein RS, Bar-Sheshet SI, Breitbart H, Nir U (2010) TMF/ARA160: a key regulator of sperm development. Dev Biol 348:12–21. https://doi.org/10.1016/j.ydbio.2010.07.033
Fari K, Takacs S, Ungar D, Sinka R (2016) The role of acroblast formation during Drosophila spermatogenesis. Biol Open 5:1102–1110. https://doi.org/10.1242/bio.018275
Duplomb L, Duvet S, Picot D, Jego G, El Chehadeh-Djebbar S, Marle N, Gigot N, Aral B, Carmignac V, Thevenon J, Lopez E, Riviere JB, Klein A, Philippe C, Droin N, Blair E, Girodon F, Donadieu J, Bellanne-Chantelot C, Delva L, Michalski JC, Solary E, Faivre L, Foulquier F, Thauvin-Robinet C (2014) Cohen syndrome is associated with major glycosylation defects. Hum Mol Genet 23:2391–2399. https://doi.org/10.1093/hmg/ddt630
Nagata O, Nakamura M, Sakimoto H, Urata Y, Sasaki N, Shiokawa N, Sano A (2018) Mouse model of chorea-acanthocytosis exhibits male infertility caused by impaired sperm motility as a result of ultrastructural morphological abnormalities in the mitochondrial sheath in the sperm midpiece. Biochem Biophys Res Commun 503:915–920. https://doi.org/10.1016/j.bbrc.2018.06.096
Gioria M, Pasini ME, Berruti G (2017) Dynamic of contribution of UBPy-sorted cargo to acrosome biogenesis: effects of its derailment in a mouse model of globozoospermia, the infertile Vps54 (L967Q) mutant. Cell Tissue Res 369:413–427. https://doi.org/10.1007/s00441-017-2592-1
Doran J, Walters C, Kyle V, Wooding P, Hammett-Burke R, Colledge WH (2016) Mfsd14a (Hiat1) gene disruption causes globozoospermia and infertility in male mice. Reproduction 152:91–99. https://doi.org/10.1530/REP-15-0557
Funaki T, Kon S, Tanabe K, Natsume W, Sato S, Shimizu T, Yoshida N, Wong WF, Ogura A, Ogawa T, Inoue K, Ogonuki N, Miki H, Mochida K, Endoh K, Yomogida K, Fukumoto M, Horai R, Iwakura Y, Ito C, Toshimori K, Watanabe T, Satake M (2013) The Arf GAP SMAP2 is necessary for organized vesicle budding from the trans-Golgi network and subsequent acrosome formation in spermiogenesis. Mol Biol Cell 24:2633–2644. https://doi.org/10.1091/mbc.E13-05-0234
Fujihara Y, Satouh Y, Inoue N, Isotani A, Ikawa M, Okabe M (2012) SPACA1-deficient male mice are infertile with abnormally shaped sperm heads reminiscent of globozoospermia. Development 139:3583–3589. https://doi.org/10.1242/dev.081778
Pierre V, Martinez G, Coutton C, Delaroche J, Yassine S, Novella C, Pernet-Gallay K, Hennebicq S, Ray PF, Arnoult C (2012) Absence of Dpy19l2, a new inner nuclear membrane protein, causes globozoospermia in mice by preventing the anchoring of the acrosome to the nucleus. Development 139:2955–2965. https://doi.org/10.1242/dev.077982
Haraguchi CM, Mabuchi T, Hirata S, Shoda T, Hoshi K, Akasaki K, Yokota S (2005) Chromatoid bodies: aggresome-like characteristics and degradation sites for organelles of spermiogenic cells. J Histochem Cytochem 53:455–465. https://doi.org/10.1369/jhc.4A6520.2005
Meikar O, Da Ros M, Korhonen H, Kotaja N (2011) Chromatoid body and small RNAs in male germ cells. Reproduction 142:195–209. https://doi.org/10.1530/REP-11-0057
Kotaja N, Sassone-Corsi P (2007) The chromatoid body: a germ-cell-specific RNA-processing centre. Nat Rev Mol Cell Biol 8:85–90. https://doi.org/10.1038/nrm2081
Li YC, Hu XQ, Zhang KY, Guo J, Hu ZY, Tao SX, Xiao LJ, Wang QZ, Han CS, Liu YX (2006) Afaf, a novel vesicle membrane protein, is related to acrosome formation in murine testis. FEBS Lett 580:4266–4273. https://doi.org/10.1016/j.febslet.2006.06.010
Sun X, Kovacs T, Hu YJ, Yang WX (2011) The role of actin and myosin during spermatogenesis. Mol Biol Rep 38:3993–4001. https://doi.org/10.1007/s11033-010-0517-0
Kierszenbaum AL, Rivkin E, Tres LL (2003) Acroplaxome, an F-actin-keratin-containing plate, anchors the acrosome to the nucleus during shaping of the spermatid head. Mol Biol Cell 14:4628–4640. https://doi.org/10.1091/mbc.e03-04-0226
Lindsay AJ, Jollivet F, Horgan CP, Khan AR, Raposo G, McCaffrey MW, Goud B (2013) Identification and characterization of multiple novel Rab-myosin Va interactions. Mol Biol Cell 24:3420–3434. https://doi.org/10.1091/mbc.E13-05-0236
Miserey-Lenkei S, Bousquet H, Pylypenko O, Bardin S, Dimitrov A, Bressanelli G, Bonifay R, Fraisier V, Guillou C, Bougeret C, Houdusse A, Echard A, Goud B (2017) Coupling fission and exit of RAB6 vesicles at Golgi hotspots through kinesin-myosin interactions. Nat Commun 8:1254. https://doi.org/10.1038/s41467-017-01266-0
Patwardhan A, Bardin S, Miserey-Lenkei S, Larue L, Goud B, Raposo G, Delevoye C (2017) Routing of the RAB6 secretory pathway towards the lysosome related organelle of melanocytes. Nat Commun 8:15835. https://doi.org/10.1038/ncomms15835
Yamane J, Kubo A, Nakayama K, Yuba-Kubo A, Katsuno T, Tsukita S (2007) Functional involvement of TMF/ARA160 in Rab6-dependent retrograde membrane traffic. Exp Cell Res 313:3472–3485. https://doi.org/10.1016/j.yexcr.2007.07.010
Kierszenbaum AL, Rivkin E, Tres LL, Yoder BK, Haycraft CJ, Bornens M, Rios RM (2011) GMAP210 and IFT88 are present in the spermatid golgi apparatus and participate in the development of the acrosome-acroplaxome complex, head-tail coupling apparatus and tail. Dev Dyn 240:723–736. https://doi.org/10.1002/dvdy.22563
Kierszenbaum AL, Rivkin E, Tres LL (2003) The actin-based motor myosin Va is a component of the acroplaxome, an acrosome-nuclear envelope junctional plate, and of manchette-associated vesicles. Cytogenet Genome Res 103:337–344. https://doi.org/10.1159/000076822
Siniossoglou S (2005) Affinity purification of Ypt6 effectors and identification of TMF/ARA160 as a Rab6 interactor. Methods Enzymol 403:599–607. https://doi.org/10.1016/S0076-6879(05)03052-1
Miller VJ, Sharma P, Kudlyk TA, Frost L, Rofe AP, Watson IJ, Duden R, Lowe M, Lupashin VV, Ungar D (2013) Molecular insights into vesicle tethering at the golgi by the conserved oligomeric golgi (COG) complex and the golgin TATA element modulatory factor (TMF). J Biol Chem 288:4229–4240. https://doi.org/10.1074/jbc.M112.426767
Makaraci P, Kim K (2018) trans-Golgi network-bound cargo traffic. Eur J Cell Biol 97:137–149. https://doi.org/10.1016/j.ejcb.2018.01.003
Hirata T, Fujita M, Nakamura S, Gotoh K, Motooka D, Murakami Y, Maeda Y, Kinoshita T (2015) Post-Golgi anterograde transport requires GARP-dependent endosome-to-TGN retrograde transport. Mol Biol Cell 26:3071–3084. https://doi.org/10.1091/mbc.E14-11-1568
Moreno RD, Palomino J, Schatten G (2006) Assembly of spermatid acrosome depends on microtubule organization during mammalian spermiogenesis. Dev Biol 293:218–227. https://doi.org/10.1016/j.ydbio.2006.02.001
Su W, Mruk DD, Cheng CY (2013) Regulation of actin dynamics and protein trafficking during spermatogenesis–insights into a complex process. Crit Rev Biochem Mol Biol 48:153–172. https://doi.org/10.3109/10409238.2012.758084
Rogat AD, Miller KG (2002) A role for myosin VI in actin dynamics at sites of membrane remodeling during Drosophila spermatogenesis. J Cell Sci 115:4855–4865
Row PE, Liu H, Hayes S, Welchman R, Charalabous P, Hofmann K, Clague MJ, Sanderson CM, Urbe S (2007) The MIT domain of UBPY constitutes a CHMP binding and endosomal localization signal required for efficient epidermal growth factor receptor degradation. J Biol Chem 282:30929–30937. https://doi.org/10.1074/jbc.M704009200
Wanschers B, van de Vorstenbosch R, Wijers M, Wieringa B, King SM, Fransen J (2008) Rab6 family proteins interact with the dynein light chain protein DYNLRB1. Cell Motil Cytoskelet 65:183–196. https://doi.org/10.1002/cm.20254
Kaufmann M, Bilbilis K, Kail M, Barnekow A (2005) Dynein light chain 2A–A link between the small GTPase Rab6 and the motor protein dynein. Eur J Cell Biol 84:73
Short B, Preisinger C, Schaletzky J, Kopajtich R, Barr FA (2002) The Rab6 GTPase regulates recruitment of the dynactin complex to Golgi membranes. Curr Biol 12:1792–1795
Kierszenbaum AL, Tres LL, Rivkin E, Kang-Decker N, van Deursen JM (2004) The acroplaxome is the docking site of Golgi-derived myosin Va/Rab27a/b-containing proacrosomal vesicles in wild-type and Hrb mutant mouse spermatids. Biol Reprod 70:1400–1410. https://doi.org/10.1095/biolreprod.103.025346
Kierszenbaum AL, Tres LL (2004) The acrosome-acroplaxome-manchette complex and the shaping of the spermatid head. Arch Histol Cytol 67:271–284
Lee PL, Ohlson MB, Pfeffer SR (2015) The Rab6-regulated KIF1C kinesin motor domain contributes to Golgi organization. Elife. https://doi.org/10.7554/eLife.06029
Shearman JR, Wilton AN (2011) A canine model of Cohen syndrome: trapped Neutrophil Syndrome. BMC Genom 12:258. https://doi.org/10.1186/1471-2164-12-258
Birling MC, Dierich A, Jacquot S, Herault Y, Pavlovic G (2012) Highly-efficient, fluorescent, locus directed cre and FlpO deleter mice on a pure C57BL/6N genetic background. Genesis 50:482–489. https://doi.org/10.1002/dvg.20826
Acknowledgements
This work from the FHU TRANSLAD is supported by the Conseil Régional de Bourgogne through the plan d’actions régional pour l’innovation (PARI) and the European Union through the PO FEDER-FSE Bourgogne 2014/2020 programs. The mouse mutant line was established at the Mouse Clinical Institute (Institut Clinique de la Souris, MCI/ICS) in the Genetic Engineering and Model Validation Department with funds from Fondation Maladies Rares. The UMR1231 CellImaP/DimaCell core facility that is supported by the Regional Council of Bourgogne-Franche Comté and the FEDER. We also thank Christine Arnould and Elodie Noirot from the Dimacell Imaging Facility (Agrosup Dijon, INRA, INSERM, University of Bourgogne Franche-Comté, F-21000 Dijon, France) for their support with confocal microscopy. The authors gratefully acknowledge the animal facility of Centre des Sciences du Goût et de l’Alimentation (INRA, Dijon, France) for animal care taking. Finally, we thank Gaëtan Jego for his comments on the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization, RC and LD; Funding acquisition, CT and LF; Resources, PF, CT, and LF; Investigation, RC, MB, MG, VC, VL, HC, AB, and AC; Validation, RC and MB; Formal analysis, RC; Writing—original draft, RC; Writing—review and editing, RC, LD, CB, PF, CT, and LF; Supervision, RC and LD.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
18_2019_3192_MOESM1_ESM.pdf
Supplementary material 1 (PDF 912 kb) Supplemental Figure 1. (A) Electron micrographs of a Vps13b+/+ (left panel) and a Vps13b∆Ex3/∆Ex3 (right panel) flagellum cross-section showing that the cytoskeletal structure of the flagellum is not affected by the lack of Vps13b. (B) Electron micrographs of a Vps13b+/+ (left panel) and a Vps13b∆Ex3/∆Ex3 (right panel) late spermatid cross-section at the developing middle piece showing that mitochondrial organization around the flagellum is not affected in mutant spermatids. (C) Hematoxylin/Eosin-stained testicular sections showing multinucleated giant late spermatids in Vps13b∆Ex3/∆Ex3 sections (right panel) but not Vps13b+/+sections (left panel). Those giant spermatids were sometimes highly vacuolated (arrow). Scale bar, 50 µm. (D) Electron micrographs of multinucleated early (left panel) and late (right panel) spermatids in a Vps13b∆Ex3/∆Ex3 ultra-thin section. The late spermatid presents once again with large vacuoles. (E) Electron micrographs showing unusually large syncytial pores (dashed squares) in the mutant testicular syncytium
18_2019_3192_MOESM2_ESM.pdf
Supplementary material 2 (PDF 656 kb) Supplemental Figure 2. Transcriptional activity related to acrosomogenesis is not severely affected in Vps13b∆Ex3/∆Ex3 mice. (A) Transcript levels of acrosomal genes Acrbp, Acrosin, Dpy19l2, Sp56, Spaca1, Spaca7, Zpbp1 and Zpbp2. (B) Transcript levels of factors required for proper acrosome biogenesis: Brdt, Cul4b, Dazap1, Ddx4, Hrb, Rfx2, Spata16, Tdrd6 and Vps54. As per Welch two sample t-test, no significant differences were measured between mutant and wild-type levels. Values are presented as the mean ± SD (N=3). (D) Ddx4 immunostaining on mutant and wild-type testicular sections. Scale bar, 10 µm. (E) Electron micrograph of a CB in a mutant spermatid. The CB is enlarged in the right panel
18_2019_3192_MOESM3_ESM.pdf
Supplementary material 3 (PDF 566 kb) Supplemental Figure 3. Acrosomal protein Spaca1 is not targeted to the anterior nuclear membrane in mutant spermatids. Spaca1 immunostaining on wild-type (A) and mutant (B) isolated spermatids. While Spaca1 displayed an inner acrosomal membrane localization throughout cap, acrosome and maturation phase, it appeared dispersed and in forms of vesicles within mutant spermatids. Spaca1 staining sometimes overlapped with PNA-positive vesicles thereby confirming the acrosomal content of those vesicles
18_2019_3192_MOESM4_ESM.pdf
Supplementary material 4 (PDF 377 kb) Supplemental Figure 4. Confirmation of the localization of PNA-positive vesicles to the endosome of cap phase Vps13b∆Ex3/∆Ex3 spermatids by confocal microscopy. Upper panels, wild-type spermatids. Lower panels, mutant spermatids. Scale bar, 10 µm
18_2019_3192_MOESM5_ESM.pdf
Supplementary material 5 (PDF 351 kb) Supplemental Figure 5. Confirmation of the targeting of an acrosomal protein (Spaca1) to the lysosome of cap phase Vps13b∆Ex3/∆Ex3 spermatids by confocal microscopy. Upper panels, wild-type spermatids. Lower panels, mutant spermatids. Scale bar, 10 µm
18_2019_3192_MOESM6_ESM.pdf
Supplementary material 6 (PDF 947 kb) Supplemental Figure 6. Impaired actin remodeling in Vps13b∆Ex3/∆Ex3 spermatids. (A) Images of testicular sections stained with F-actin, PNA and DAPI. In wild-type Sertoli cells, actin filaments organized at the ectoplasmic specialization over the spermatid acrosomal region in early acrosome phase (stage 9). Actin filaments remained associated with the spermatid head until the last stage of maturation phase (stage 16). Then, they dissociated from mature spermatids to reassemble over the acrosome of newly formed acrosome phase spermatids. This cyclic dynamic of actin filaments was not observed in mutant spermatids. Instead, they associated with the plasma membrane of early spermatids. Scale bar, 50 µm. Panel (B) provides a close-up image on early mutant spermatids. (C) Electron micrographs showing actin filaments associated with the plasma membrane of an early mutant spermatid (left panel) but not late mutant spermatid (right spermatid). (D) Electron micrograph of a perinuclear ring in a Vps13b+/+ early spermatid at the edge of the NDL. (E) Electron micrographs of an extremity of the NDL in a Vps13b∆Ex3/∆Ex3 early spermatid. Dashed squares highlight the NDL extremities and are enlarged in the right panels of (D) and (E). Cytoskeletal filaments constituting the perinuclear ring are missing at the NDL of Vps13b∆Ex3/∆Ex3 spermatids
18_2019_3192_MOESM7_ESM.pdf
Supplementary material 7 (PDF 710 kb) Supplemental Figure 7. Post-acrosome differentiation events still occur in Vps13b∆Ex3/∆Ex3 spermatids. (A) Spata6 staining of wild-type and mutant seminiferous tubules displaying spermatids in maturation phase. Though nuclear condensation was incomplete and acrosome was lacking, Vps13b∆Ex3/∆Ex3 spermatids displayed a perinuclear localization of Spata6 as in wild-type spermatids. This result suggests that expression and transport of middle piece proteins, unlike that of acrosomal proteins, are not entirely impaired in absence of Vps13b function. (B) β-Tubulin staining on wild-type and mutant testicular sections counterstained with PNA and DAPI. Both genotypes displayed microtubule polymerization during acrosome phase (stage9-12) and disassembly during maturation phase (stage 13-16). Microtubules in mutant spermatids failed to form the manchette structure seen in wild-type spermatids likely because of the lack of acrosomal components that allow their anchoring around the nucleus and not directly because of Vps13b function loss. In spite of the impaired actin remodeling seen in Supplemental Figure S4, Vps13b∆Ex3/∆Ex3 spermatids do not display impaired polymerization of microtubules during manchette formation. Scale bar, 10 µm
18_2019_3192_MOESM8_ESM.pdf
Supplementary material 8 (PDF 551 kb) Supplemental Figure 8. Loss of Vps13b detection in Vps13b∆Ex3/∆Ex3 spermatids. Using antibody Vps13baa64−412 to stain Vps13b∆Ex3/∆Ex3 isolated spermatids did not reveal any perinuclear staining but a faint background staining of the cell body remained. Using antibody Vps13baa103−121, no signal was detectable in Vps13b∆Ex3/∆Ex3 spermatids. This result suggests that, at least, Chorein domain-containing isoforms of Vps13b are lost in Vps13b∆Ex3/∆Ex3 spermatids
18_2019_3192_MOESM9_ESM.pdf
Supplementary material 9 (PDF 583 kb) Supplemental Figure 9. Vps13b and its interactor Rab6 locate to the acrosome. Representative images of wild-type spermatids in Golgi, cap, acrosome and maturation phases stained against Vps13b (A, antibody Vps13baa64−412) or Rab6 (B) and counterstained with PNA-FITC and DAPI. In contrast with Figure 8, stainings were performed on testis cryosections. Images of Rab6 staining on mutant spermatids are presented in the left panel of B. Results showed that Vps13b expression was increased in early spermatids compared to spermatogonia and spermatocytes and that the protein essentially localized to the pre-acrosome in Golgi phase and to the acrosomal inner membrane in cap and acrosome phase. Rab6 located to the Golgi apparatus of all spermatogenic cells. In addition, Rab6 followed the sequential acrosomal localization displayed by Vps13b in wild-type spermatids but not in mutants. In mutant testis sections, Rab6 was not detectable at displaced PNA-positive vesicles. It was only localized to the Golgi apparatus. This difference in staining may be due to differences in fixation and blocking which may prevent accessibility of the Rab6 epitopes at proacrosomal vesicles. Scale bars, 10 µm
Supplementary material 11 (MP4 1945 kb) Supplemental Movie 1. Recording of spermatozoa extracted from a Vps13b+/+ epididymis
Supplementary material 12 (MP4 2912 kb) Supplemental Movie 2. Recording of spermatozoa extracted from a Vps13b∆Ex3/∆Ex3 epididymis
Rights and permissions
About this article

Cite this article
Da Costa, R., Bordessoules, M., Guilleman, M. et al. Vps13b is required for acrosome biogenesis through functions in Golgi dynamic and membrane trafficking. Cell. Mol. Life Sci. 77, 511–529 (2020). https://doi.org/10.1007/s00018-019-03192-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03192-4