
Abstract
Apoptosis is a form of directed programmed cell death with a tightly regulated signalling cascade for the destruction of single cells. MicroRNAs (miRNAs) play an important role as fine tuners in the regulation of apoptotic processes. MiR-493-3p mimic transfection leads to the induction of apoptosis causing the breakdown of mitochondrial membrane potential and the activation of Caspases resulting in the fragmentation of DNA in several ovarian carcinoma cell lines. Ovarian cancer shows with its pronounced heterogeneity a very high death-to-incidence ratio. A target gene analysis for miR-493-3p was performed for the investigation of underlying molecular mechanisms involved in apoptosis signalling pathways. Elevated miR-493-3p levels downregulated the mRNA and protein expression levels of Serine/Threonine Kinase 38 Like (STK38L), High Mobility Group AT-Hook 2 (HMGA2) and AKT Serine/Threonine Kinase 2 (AKT2) by direct binding as demonstrated by luciferase reporter assays. Notably, the protein expression of RAF1 Proto-Oncogene, Serine/Threonine Kinase (RAF1) was almost completely downregulated by miR-493-3p. This interaction, however, was indirect and regulated by STK38L phosphorylation. In addition, RAF1 transcription was diminished as a result of reduced transcription of ETS proto-oncogene 1 (ETS1), another direct target of miR-493-3p. Taken together, our observations have uncovered the apoptosis inducing potential of miR-493-3p through its regulation of multiple target genes participating in the extrinsic and intrinsic apoptosis pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Apoptosis is a form of directed programmed cell death for the destruction of only single cells without damaging surrounding tissues [1]. It is induced via two distinct but interrelated main signalling pathways, an intrinsic and an extrinsic one. The extrinsic pathway is initiated by ligand binding to a transmembrane receptor (i.e. via the tumour necrosis factor receptor (TNFR) [1]) or the vascular endothelial growth factor receptor (VEGFR) leading to the regulation of the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signalling pathway [2]. The intrinsic pathway is induced by the release of cytochrome C from the mitochondria and involves different non-receptor-mediated stimuli. The integrity of the mitochondria is mediated by different pro- and anti-apoptotic B cell lymphoma 2 (Bcl2) members such as Bcl-2-associated X protein (Bax) and Bcl-2 homologous antagonist killer (Bak) [3]. The release of cytochrome C activates the initiator Caspase 9, leading to the activation of effector Caspase 3 [3]. Caspase 3 is furthermore cleaving and thereby inactivating the DNA repair protein poly (ADP-ribose) polymerase (PARP) [4]. Finally, genomic DNA is cleaved by Caspase-activated deoxyribonucleases [5]. Apoptosis represents a tightly regulated signalling cascade culminating in activation of Caspases and subsequent specific morphological and biochemical changes resulting in the elimination of degenerated cells by phagocytosis [1].
Previous studies showed that microRNAs (miRNAs) play an important role as fine tuners in the regulation of physiological and pathological cellular processes such as apoptosis, proliferation or differentiation [6, 7]. MiRNAs are endogenous single-stranded small non-coding RNAs with a length of around 22 nucleotides [8]. They are transcribed by RNA-polymerase II and processed by the RNase-III enzymes DROSHA and DICER. The processed miRNAs are bound to Argonaut 2 proteins and guided to the RNA-induced silencing complex. The miRNA guide strand can bind to the 3 prime untranslated region (3′ UTR) of a target gene and thereby functions as post-transcriptional regulator [9]. With the imperfect base pairing of the miRNA to the target mRNA, one miRNA can target multiple genes and therefore interact in several signalling pathways [7]. Binding of a miRNA to the target mRNA typically leads to translational repression or mRNA decay by endonucleolytical cleavage [9].
Several miRNAs have already been reported to play a role in the regulation of apoptotic signalling pathways, for example, by targeting the mRNA of Bcl2 proteins, Caspases or members of the p53 network [10, 11]. This may lead to cancer development or treatment resistance [12]. The identification of death inducing miRNAs might influence the outcome of treatment therapies [13], overcome treatment resistances and help to cure cancer (CA).
Ovarian CA is a common human CA with poor prognosis and the highest death-to-incidence ratio. For 2017, the American Cancer Society estimates 22,440 new cases and 14,080 deaths due to ovarian CA in the United States [14]. Ovarian CA refers to a highly heterogeneous type of CA including the subgroup epithelial ovarian carcinoma [15]. Patients are treated with chemotherapy i.e. with Carboplatin and Paclitaxel for 3–6 cycles [16]. However, due to the often arising resistances researchers are focusing on oncogenes as well as cell signalling pathways exploring their role in tumour progression [17] to overcome these obstacles. As miRNAs target multiple genes and signalling pathways [7], they are highly interesting molecules for the generation of novel anticancer therapeutics. However, signalling pathways involved in miRNA-mediated apoptosis need to be further investigated.
Based on a previous cellular high-throughput screening [18], we identified miR-493-3p to strongly induce apoptosis in the ovarian CA cell line SKOV3. The aim of this study was to identify the role of miR-493-3p in programmed cell death signalling of various ovarian CA cell lines. In particular, target genes regulated by miR-493-3p resulting in altered signalling pathways that lead to cellular responses of cell death were investigated.
Results
Apoptosis screening identifies miR-493-3p as a novel pro-apoptotic miRNA in ovarian carcinoma cells
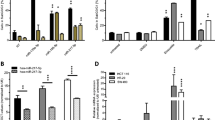
MicroRNAs are increasingly gaining interest as tumour suppressor genes with their potential to regulate important cellular processes such as apoptosis [11]. To identify novel pro-apoptotic miRNAs, we previously transfected 188 potential pro-apoptotic miRNA mimics into the human CA cell lines SKOV3, HCT 116 and T98G as well as in the preadipocytes SGBS and measured hallmarks of apoptosis induction by quantitative flow cytometry [18]. In the current study, we focused on miRNA-493-3p, which we found to induce significant apoptosis in SKOV3 cells (5.4 fold ± 0.5 fold increase compared to NT) as well as in HCT 116 cells (3.2 fold ± 0.4 fold increase compared to NT; Fig. 1a). The previously described pro-apoptotic miR-183-5p [19] induced apoptosis up to 6.6 fold ± 0.5 fold in SKOV3 cells compared to a non-targeting control (NT). As miR-493-3p showed the highest fold changes in the ovarian CA cell line SKOV3, we further investigated the influence of this miRNA in various ovarian CA cell lines.

MiRNA screen validation and miR-493 expression in ovarian cell lines. For validation screening T98G, HCT 116, SKOV3 and SGBS cells were seeded 24 h before transfection with miRNA mimics (50 nM and 0.4 µl ScreenFect A) or non-targeting siRNA (NT, negative control for cell death) control. Apoptosis rates 72 h after transfection were analysed by Nicoletti staining followed by flow cytometric analysis (a). The location of miRNA-493 is in chromosome 14 in a miRNA cluster between Meg3 and RTL1 (b). QRT-PCR of untreated ovarian cells for miR-493-3p and -5p expression (c). For determination of miR-493 expression after induction of apoptosis, SKOV3 (d) and OVCAR3 (e) cells were treated with 25 µM Etoposide, 80 µM Carboplatin or 0.25 µM Paclitaxel. The miRNA expression of miR-493 was normalized to the CT value of U6 snRNA and the untreated control (d, e) or the expression in HOSE 2170 cells (c) employing the Livak method [71]. Statistical analyses were performed by one-way ANOVA (a, d, e) followed by Bonferroni post-test. For part (c) an unpaired t test was used [n = 3 replicates; mean ± SD, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001]
Apoptosis induction increases aberrant miR-493-3p expression
MicroRNA-493 is known as a tumour suppressor in lung CA [20], human bladder CA [21] and gastric CA [22]. It is encoded on the human chromosome 14 (14q32) in the Delta Like Non-Canonical Notch Ligand 1 (DLK1)—Deiodinase, Iodothyronine Type III (DIO3) genomic region in an pattern of imprinted genes, long noncoding RNAs like maternally expressed 3 (Meg3) and Meg8 and several miRNAs [23, 24] (Fig. 1b).
To further assess the functions of miR-493 in the regulation of apoptosis in ovarian CA, the expression of miR-493 was examined in six different ovarian CA cell lines, SKOV3, OVCAR3, TOV21G, TOV112D, A2780 and A2780-cis (Cisplatin resistant cells). Comparing the expression of both miRNA strands to normal human ovarian surface epithelial cells (HOSE 2170 cells) [25], the expression of the 3p and 5p strand of hsa-miR-493 was reduced to at least 0.03 fold ± 0.001 fold in TOV21G cells (Fig. 1c). After induction of apoptosis by treating the cells for 48 h with the clinically relevant chemotherapeutic drugs Carboplatin, Paclitaxel [16] and Etoposide as positive controls, the expression of hsa-miR-493 increased for both strands in both cell lines SKOV3 (Fig. 1d) and OVCAR3 (Fig. 1e). In general, the previously as potentially pro-apoptotic identified 3p strand of hsa-miR-493 was more consistently and higher induced by pro-apoptotic stimuli than the 5p strand, again suggesting an involvement in the initiation or progression of the apoptotic pathway.
MiR-493 induces apoptosis in ovarian carcinoma cells
To investigate the apoptotic effect of hsa-miR-493-3p in more detail, miR-493-3p mimic or a non-targeting control (NT) was individually transfected into ovarian CA cell lines. To verify elevated intracellular levels of the miRNAs after transfection, the amounts were assessed via qPCR revealing an increase of miR-493-3p by 3400 fold in SKOV3 or 6000 fold in OVCAR3 cells when compared to NT transfected cells (Supplementary Fig. 1), confirming a generally high enrichment after transfection. Several methods were applied for the detection of various apoptosis stages to a panel of ovarian carcinoma cell lines with different genetic background including SKOV3p53null, OVCAR3p53R248Q [26], TOV21G, TOV112Dp53R175H [27], A2780 and A2780-cisp53K351N [28, 29]. Regarding cell growth, hsa-miR-493-3p led to a significant reduction of cell confluence in the different ovarian cell lines. The highest reduction in cell confluence was detected in hsa-miR-493-3p transfected A2780 cells when compared to NT transfected cells (39.9% ± 0.8% vs 68.5% ± 0.3% cell confluence, respectively). The chemotherapeutic drugs Paclitaxel, Carboplatin and Etoposide as well as the death inducing siRNA control (DT) were applied as controls known to reduce cell confluence after treatment (Fig. 2a). As cytochrome C is released from the mitochondria into the cytoplasm after induction of apoptosis, its release as well as alterations in mitochondrial membrane potential (ΔΨm) were analysed by flow cytometry employing the potential-sensitive fluorescent dye tetramethylrhodamine ethyl ester (TMRE). Comparing the different ovarian CA cell lines, the highest release of cytochrome C was detected in miR-493-3p transfected TOV21G cells when compared to the control (NT) (90.0% ± 1.4% vs 11.7% ± 3.8%, respectively; Fig. 2b). Further, compared to NT transfected cells, a high amount of miR-493-3p transfected TOV21G cells showed a low ∆Ψm (10.6% ± 1.3% vs 59.9% ± 3.9%, respectively). In addition, also a large number of hsa-miR-493-3p transfected SKOV3 and OVCAR3 cells showed a low ∆Ψm compared to the control (NT) (55.3% ± 2.3% vs 68.1% ± 4.5%, respectively; Fig. 2c). The DT control as well as the chemotherapeutic drug Paclitaxel led to the strongest loss of ∆Ψm in these cell lines. At the level of fragmented DNA, high apoptosis induction was detected in hsa-miR-493-3p transfected SKOV3 and TOV21G cells (48.3% ± 0.7% vs. 17.2% ± 0.3% in NT transfected SKOV3 cells, 50.6% ± 0.3% vs. 17.8% ± 1.4% in NT transfected TOV21G cells; Fig. 2d).

Analysis of apoptosis induction by miR-493-3p. The ovarian CA cell lines SKOV3, OVCAR3, TOV21G, TOV112D, A2780 as well as A2780-cis were seeded 24 h prior transfection with 62.5 nM miR-493-3p mimics, non-targeting siRNA (NT, negative control for cell death), cell death inducing siRNA (DT, positive control for cell death) or treatment with 25 µM Etoposide, 80 µM Carboplatin or 0.25 µM Paclitaxel. 48 h after treatment, the cells were analysed for their release of cytochrome C (b) as well as the loss of ∆Ψm (c). 72 h after treatment the cell confluency (a) and the fragmentation of DNA (d) was analysed. 5 µM CCCP served as a positive control for the breakdown of ∆Ψm (c). Statistical analysis was performed by one-way ANOVA followed by Bonferroni post-test. [n = 3 replicates; mean ± SD, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001]
To obtain more detailed data about the time course of apoptosis induced by miR-493-3p, long-term video-microscopy using the IncuCyte ZOOM Live-Cell Analysis System was applied. SKOV3 cells were analysed as they previously showed the highest induction of DNA fragmentation. Cells were grown for 72 h and stained with IncuCyte AnnexinV Red Reagent for apoptosis. The number of AnnexinV-positive cells after treatment with miR-493-3p or Carboplatin steadily increased and reached a 1.8 and 2.2 fold higher level when compared to NT or Dimethyl sulfoxide (DMSO) treated cells, respectively. A significant increase of p < 0.05 was analysed by two-way ANOVA followed by Bonferroni post-test. It showed for Carboplatin-treated cells after 49 h and for hsa-miR-493-3p transfected cells after 55 h significant differences compared to NT-transfected cells (Fig. 3a).

Activation of Caspase 3/-7 by miR-493. For long-term analysis, SKOV3 cells were seeded and transfected as described in Fig. 2. To detect apoptosis induction by miR-493, the cells were stained with IncuCyte AnnexinV Red Reagent (a) or IncuCyte Caspase-3/7 Green Apoptosis Assay Reagent (b). The cells were automatically photographed every hour by the IncuCyte ZOOM System. The amount of AnnexinV-positive cells or cells with activated Caspase 3 or -7 was calculated by the IncuCyte ZOOM Software. For Western Blot analysis, the cells were harvested 72 h after treatment. ß-Actin served as a loading control. Cells were treated with 30 µM zVAD to inhibit Caspase 3 activity (c). For measuring motility, cells were seeded, scratched with a 1000 µl pipet tip and transfected with the negative control siRNA (NT), miR-493-3p or the cell death positive control (DT). 0 h and 24 h after transfection images of the cell layer were taken by the automated single well microscope NyOne (d). The distance between the gap was measured and plotted on a box-and-whisker diagram (e). Statistical analyses were performed by one-way ANOVA followed by Bonferroni post-test. [n = 3 replicates; mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001]
A further hallmark of apoptosis is the activation of the effector Caspases 3 and -7 that play important roles in the intrinsic as well as extrinsic apoptotic pathways [30]. Therefore, the analysis of Caspase 3 and -7 activity was performed to examine the molecular changes induced after miR-493-3p transfection. The activation of Caspase 3 and -7 was detected by long-term video-microscopy using the IncuCyte ZOOM Live-Cell Analysis System together with the IncuCyte Caspase-3/7 Green Apoptosis Assay Reagent. Caspase activity significantly (p < 0.05; analysed by two-way ANOVA followed by Bonferroni post-test) increased 43 h after cell transfection with miR-493-3p, and 49 h after treatment with Carboplatin, while in NT- and DMSO-treated cells nearly no increase in Caspase activity was detectable (Fig. 3b). To confirm the Caspase activation by Western Blotting, protein lysates were prepared 72 h after treatment or transfection. 30 µM zVAD was used as Caspase inhibitor [31]. After treating the cells with the chemotherapeutic drug Carboplatin, as well as after transfection with death control siRNA (DT) or hsa-miR-493-3p, an induction of Caspase 3 and PARP cleavage was observed. The amount of cleaved PARP was reduced in cells treated with zVAD and the pattern of cleaved Caspase 3 was altered as the fragment of 12 kDa was undetectable (Fig. 3c).
Besides apoptosis induction cell motility after transfection was analysed by cell motility assay. 24 h after transfection, miR-493-3p transfected SKOV3 cells left a 1.5 broader gap than NT or untreated cells (miR-493-3p: 297 µm ± 131 µm, NT: 188 µm ± 70 µm, untreated: 114 µm ± 65 µm). Cells transfected with the death control siRNA (DT) hardly grew into the gap (Fig. 3d, e).
Altogether, these data strongly suggest a potential role of miR-493-3p in the regulation of cellular growth in different ovarian CA cells. However, only little is known about miR-493-3p regulated target genes which are involved in signalling pathways leading to the detected effects of apoptosis.
Gene and pathway regulation by miR-493-3p through direct binding partners
To investigate involved signalling pathways and critical downstream effectors leading to apoptosis after transient transfection of miR-493 in ovarian CA cells, we aimed to identify potential miR-493-3p target genes. First an in silico target gene prediction analysis for miR-493-3p using six different prediction databases was performed followed by a functional clustering analysis using Ingenuity Pathway Analysis (Qiagen Bioinformatics). Focusing on survival promoting or anti-apoptotic functions as well as ovarian CA signalling, the four target genes including Signal Transducer And Activator Of Transcription 3 (STAT3), High Mobility Group AT-Hook 2 (HMGA2), Mitogen-Activated Protein Kinase Kinase 5 (MAP2K5) and AKT Serine/Threonine Kinase 2 (AKT2) were selected for further analysis (Table 1, upper part). In addition, the data set for “Ovarian serous cystadenocarcinoma” from the cancer genome atlas (TCGA) data base served for negative correlation of miR-493 expression and potential target mRNAs as well as protein expression. In this study, a correlation with a p value lower than 0.1 was considered as significant. The number of potential targets was reduced to twelve by in silico binding site prediction and clustering using PANTHER analysis [32] (Table 1, middle and lower part).
To analyse the potential regulation of those identified sixteen target genes (Table 1), SKOV3 and OVCAR3 cells were transiently transfected with miR-493-3p or NT and the mRNA expression analysed by qRT-PCR 48 h later. Four putative target genes were confirmed to be significantly downregulated in both cell lines after miR-493-3p transfection (Fig. 4a), including Forkhead Box M1 (FOXM1), Fragile X Mental Retardation 1 (FMR1P), RAF1 Proto-Oncogene, Serine/Threonine Kinase (RAF1) and Serine/Threonine Kinase 38 Like (STK38L). Four additional putative targets were significantly downregulated in at least one cell line, including HMGA2, Mitogen-Activated Protein Kinase 1 (MAPK1), AKT2 and STAT3.

Post-transcriptional regulations of miR-493 target genes. To validate miR-493 mediated regulation of potential target genes, SKOV3 and OVCAR3 cells were transfected with miR-493 mimic or non-targeting siRNA (NT) as described in Fig. 2. After 48 h, potential target gene expression was analysed by qRT-PCR. The relative mRNA expression of potential target genes was normalized to PPIA and NT (a) employing the Livak method [71]. The significance was determined using unpaired t test. Significantly downregulated potential targets were further analysed by Western Blotting (b). GAPDH was used as loading control. Downregulated potential targets were further analysed by Luciferase Reporter Assay (c, d) to proof direct binding of the miRNA to its predicted target sequence. Anti-miR-493 was used to confirm the binding sites of miR-493 to the target mRNAs (c). The binding sites in the 3′ UTR with (c) and without seed sequence (d, Table 2), based on data of microRNA.org [34] and TargetScanHuman [33], were cloned into a pMirGLO vector. Relative luciferase activity was measured 72 h after co-transfection of the pMirGLO vector with miR-493 mimic, miRNA inhibitor anti-miR-493 or non-targeting siRNA (NT) in HEK293T cells. The relative luciferase expression was normalized to the expression after co-transfection with NT. The significance was determined using unpaired t test. [n = 3 replicates; mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001]
Focusing on these eight downregulated genes, we investigated the regulatory activity of miR-493-3p at protein expression level by Western Blotting. Protein lysates were prepared 60 h after transient transfection of miR-493-3p in SKOV3 and OVCAR3 cells. As shown in Fig. 4b, a major downregulation was observed for RAF1, AKT2 and HMGA2 in both cell lines followed by FOXM1, FMR1P and STK38L. No regulation on protein level was observed for MAPK1 and STAT3.
Next, we tested whether miR-493 directly modulated the downregulated genes via miRNA/mRNA interaction. Thus, AKT2, HMGA2, STK38L, FOXM1, RAF1 and FMR1P were assessed for direct interactions with hsa-miR-493-3p. Predicted binding sites in the 3′ UTR of the mRNAs (Table 2 upper part, TargetScanHuman [33] and microRNA.org [34]) were cloned into the 3′ UTR of a luciferase reporter gene of the pmirGLO Dual-Luciferase miRNA target expression vector and transfected together with miR-493-3p mimic, miRNA inhibitor anti-miR-493-3p or a non-targeting siRNA control into HEK293T cells as they represent an easy to transfect cell system for plasmids [35]. Luciferase activity was determined 72 h after transfection. The activity was significantly reduced with the binding site of AKT2 as well as with each of the two binding sites for HMGA2 and STK38L. The latter one revealed luciferase activity which was reduced to a minimum of 0.5 fold ± 0.04 fold for its second binding site. Anti-miR-493-3p had no or only a marginal effect on luciferase activity (Fig. 4c). To confirm the binding sites, mutations lacking the seed sequence of the predicted binding site (lacking red nucleotides in Table 2 upper part) were cloned into the 3′UTR of a luciferase reporter gene and transfected into HEK293T cells together with miR-493-3p or NT. As shown in Fig. 4d the previously detected reduction in luciferase activity was abolished. Taken together, our data indicate that AKT2, HMGA2 and STK38L are direct targets of miR-493-3p leading to apoptosis in ovarian carcinoma cell lines, while RAF1, FOXM1 and FMR1P are regulated in an indirect manner.
Analysis of downstream signalling induced by direct targets of miR-493-3p leading to apoptosis
To examine underlying signalling pathways induced by miR-493-3p leading to apoptosis in more detail, we examined possible pro-apoptotic signal cascades downstream of the identified direct targets of miR-493-3p. As shown above, miR-493-3p transfected cells showed a decreased expression of AKT2. The analysis of myeloid cell leukaemia 1 (MCL1), a signalling molecule downstream of AKT2, showed a reduced expression together with a higher rate of cleavage when analysed by Western Blotting after transfection of miR-493-3p (Fig. 5a). It is known that MCL1 binds to Bak and Bax and, therefore, no longer inhibits the formation of pores in the mitochondrial membrane [36]. Western Blot analysis of these proteins showed that the expression of the pro-apoptotic Bak was induced, whereas the expression of the anti-apoptotic B-cell lymphoma-extra large (Bcl-XL) was reduced (Fig. 5a). These data may explain the observed release of cytochrome C (Fig. 2b), the reduction of mitochondrial potential (Fig. 2c) and the detected cleavage of Caspase 3 together with the activation of PARP induced by transfection of miR-493-3p.

Downstream signalling induced by miR-493-3p. To detect the apoptosis signalling pathway after transfection of miR-493 leading to apoptosis Western Blotting were conducted (a). ß-Actin served as a loading control. Kaplan–Meier Blots of old (older than 67.5 years) ovarian serous cystadenocarcinoma patients (data from TCGA) with a low and high expression (mean cutoff) of AKT2 (b) or STK38L (c) were performed to demonstrate median survival. Prolonged survival time of ovarian CA patients expressing low levels (blue) of AKT2 or STK38L compared to high expressers (red line)
To further assess the significance of the identified direct binding partners of miR-493 in ovarian CA, TCGA data of ovarian serous cystadenocarcinoma patients were subsequently employed to analyse the survival time of high and low expressers of mRNA/miR-493-3p binding partners. For AKT2 and STK38L, older patients (older than 67.5 years) showed a longer median survival. Patients expressing low levels of AKT2 lived 11.5 months longer than high expressers (Fig. 5b), whereas patients with a low expression of STK38L lived 7.4 months longer compared to high expressers (Fig. 5c).
Analysis of indirect regulation of RAF1 expression by miR-493-3p
The analysis of target gene regulation by miR-493-3p revealed a pronounced downregulation of RAF1 mRNA and protein levels (Fig. 4a, b). However, no direct binding of miR-493-3p to identified binding sites in RAF1 mRNA was observed (Fig. 4c, d). Since these data point towards an efficient but indirect regulation of RAF1 by miR-493-3p, it was of interest to identify underlying regulatory pathways. Therefore, a transcription analysis using the GeneChip Human Gene ST 2.0 Array was performed. Comparing RNA expression of miR-493-3p and NT transfected SKOV3 cells, 390 annotated genes were significant reduced in their expression after miR-493-3p transfection. Of these, 30 genes were coding for transcription factors and twelve of these genes were predicted by microRNA.org [34] to have binding sites for miR-493-3p (Table 3). Out of these, ETS proto-oncogene 1 and 2 (ETS1 and ETS2), E2F transcription factor 5 (E2F5) and QKI, KH domain containing RNA binding (QKI) had previously been reported in connection with RAF1 [37,38,39,40]. To verify potential regulation by miR-493-3p, qRT-PCR was carried out 48 h after miRNA transfection into SKOV3 cells and the highest downregulation was observed for ETS1 mRNA (0.1 fold ± 0.05 fold), followed by E2F5, ETS2 and QKI (0.5 fold ± 0.05 fold; Fig. 6a). To assess the downregulation on protein level, Western Blotting was performed with protein lysates 60 h after miR-493-3p transfection into SKOV3 cells. As shown in Fig. 6b, ETS1 and QKI protein expression was reduced, while ETS2 and E2F5 protein expression was not influenced by miR-493-3p overexpression. To test whether miR-493 directly regulates ETS1, ETS2, QKI or E2F5 on transcript level, predicted binding sites in the 3′ UTR of the mRNAs (Table 2 lower part, TargetScanHuman [33] and microRNA.org [34]) were cloned into the 3′ UTR of a luciferase reporter gene of the pmirGLO Dual-Luciferase miRNA target expression vector. The vectors were transfected together with miR-493-3p mimic, miRNA inhibitor anti-miR-493-3p or a non-targeting siRNA control into HEK293T cells. The luciferase activity was significantly reduced for all three binding sites of ETS1 (ETS1_1 0.7 fold ± 0.04 fold, ETS1_2 0.7 fold ± 0.1 fold and for ETS1_3 0.8 fold ± 0.04 fold) as well as for the binding site of E2F5 with a fold change of 0.8 fold ± 0.03 fold. In contrast, QKI- and ETS2 binding sites as well as the anti-miR-493-3p had no influence on the reduction of luciferase activity (Fig. 6c). To confirm the binding sites, mutations lacking the seed sequence of the predicted binding site (lacking red nucleotides in Table 2 upper part), were cloned into the 3′ UTR of a luciferase reporter gene. As shown in Fig. 6d, the previously detected reduction in luciferase activity was abolished for ETS1 and E2F5.

Transcriptome analysis detecting regulated transcription factors. To validate the results of the transcriptome analysis (Table 3), a qRT-PCR was carried out to analyse the expression of ETS1 and ETS2, E2F5 and QKI. The cells were transfected with miR-493 mimic or non-targeting siRNA (NT) as described in Fig. 2. The relative mRNA expression of potential target genes was normalized to PPIA and NT (a, e) employing the Livak method [71]. The significance was determined using unpaired t test. The transcription factors were further analysed by Western Blotting (b). GAPDH was used as loading control. To proof direct binding of the miRNA to its predicted target sequence luciferase assays were performed as described in Fig. 4. The binding sites in the 3′ UTR with (c) and without seed sequence (d, Table 2), based on data of microRNA.org [34] and TargetScanHuman [33], were cloned into a pMirGLO vector. Anti-miR-493 was used to confirm the binding sites of miR-493 to the target mRNAs (c). The relative luciferase expression was normalized to the expression after co-transfection with NT. The significance was determined using unpaired t test. To demonstrate the influence of ETS1 as transcription factor of RAF1, a qRT-PCR was performed with miRNA-493, NT- and siRNA-transfected cells (e). Further, transfected cell lysates were used for Western Blotting to observe effects on protein level. GAPDH served as loading control (f). Additionally, the fragmentation of DNA was measured by Nicoletti assay 72 h after transfection (g). Statistical analysis was performed by one-way ANOVA followed by Bonferroni post-test. [n = 3 replicates; mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001]
Taken together, our data demonstrate that miR-493-3p directly regulated ETS1 and E2F5 mRNA expression. However, since E2F5 does not seem to be regulated on protein level, ETS1 is the most relevant transcription factor candidate for the observed regulation of RAF1 transcription.
To further investigate the possible regulation of RAF1 by ETS1, SKOV3 cells were transfected with siRNA against ETS1 or miR-493-3p. 48 h after transfection, RNA was isolated and qRT-PCR performed against ETS1 and RAF1. As shown in Fig. 6e, transfection of siRNA against ETS1 led to a knockdown of the ETS1 mRNA and reduction of the RAF1 expression to 0.7 fold ± 0.08 fold. Western Blotting revealed a significant reduction of ETS1 protein expression after siRNA transfection. Further, the expression of RAF1 was reduced both after transfection of the ETS1 siRNA or miR-493 (Fig. 6f). Taken together, our data indicate that miR-493-3p directly downregulates the transcription factor ETS1 leading to reduced transcription and expression of RAF1. To underscore the obtained data, we next analysed whether the ETS1 siRNA might phenocopy the observed induction of apoptosis by miR-493. Therefore, SKOV3 cells were transfected with ETS1 siRNA and the amount of apoptosis measured after 72 h by Nicoletti assay, demonstrating a reduction of apoptosis of 34.6% ± 1.6%, as measured by cells with fragmented DNA (Fig. 6g).
Regulation of RAF1 by interaction with STK38L
As described above, the expression of RAF1 mRNA and protein was clearly reduced after miR-493-3p transfection. One regulatory mechanism can be accomplished via the transcription factor ETS1; nonetheless, also protein:protein interaction followed by degradation can be an underlying regulation mechanism. An interaction of RAF1 and STK38L, (the latter one a direct binding partner of miR-493 as shown above), was predicted by PIPs: Human protein–protein interactions prediction [41]. To assess the possible interaction of both proteins, co-immunoprecipitations were performed to verify this prediction. RAF1 and STK38L were immunoprecipitated and blotted for both proteins as well as their interaction in untreated SKOV3 cells (Fig. 7a). Immunoprecipitation with anti-RAF1 followed by Western Blotting with anti-STK38L revealed a moderate interaction of both proteins which was increased when anti-STK38L precipitates were blotted with RAF1 antibodies (Fig. 7a). This latter observation might be due to the different affinities and avidities of both antibodies. Further, to test whether STK38L also phosphorylates RAF1, Flag-tagged STK38L wild-type (Flag-STK38L WT) as well as a kinase dead version (Flag-STK38L KD) [42] were transfected into HEK293T cells. 48 h after transfection cells were immunoprecipitated with anti-RAF1. Blotting with anti-Flag antibody showed comparable expression of STK38L (Fig. 7b, right panel). Cell lysates blotted with anti-GAPDH revealed equal sample loading. However, blotting with anti-RAF1 or anti-phospho RAF1 serine 621 (Ser621) revealed that transfection with Flag-STK38L WT increased RAF1 expression and phosphorylation at Ser621 compared to untransfected cells or Flag-STK38L KD transfectants (Fig. 7b, left panel). As shown in Fig. 7b, RAF1 phosphorylation at Ser621 is less in cells transfected with Flag-STK38L KD when compared to untransfected or Flag-STK38L WT overexpressing cells. These data point to a regulation of RAF1 protein level via interaction with STK38L which upon downregulation by miR-493-3p might lead to less phosphorylation at Ser621 and subsequent proteosomal degradation [43].

STK38L is phosphorylating RAF1. To demonstrate an interaction of STK38L with RAF1, immunoprecipitations were carried out. Protein lysates of SKOV3 cells were precipitated for RAF1 or STK38L and blotted against both proteins (a). To obtain more information about the interaction between STK38L and RAF1, HEK293T cells were transfected with overexpression plasmids of Flag tagged STK38L Wild Type (Flag-STK38L WT) or a kinase dead version of STK38L (Flag-STK38L KD). The protein lysates were precipitated for RAF1 and blotted for total RAF1 and phosphorylated RAF1 (Ser621) (b, left part). The lysates were blotted against the Flag tag to demonstrate equal STK38L overexpression (b, right part). GAPDH served as loading control. [n = 2 replicates] The regulation of RAF1 by STK38L and ETS1 is illustrated schematically in part d. Furthermore, the signalling pathways leading to apoptosis in ovarian CA cell lines are shown schematically in part c
Discussion
Performing a high-throughput screen of 188 miRNAs in four different cell lines, we identified several novel pro-apoptotic miRNAs [18] including miR-493-3p. A potential downregulation of miR-493 in ovarian CA cell lines is supported by Wyman et al. [44] describing a downregulation of hsa-miR-493 in stage III/IV epithelial ovarian CA compared to normal HOSE cells. MiR-493 is also downregulated in other tumour types, for example, in lung or gastric CA leading to apoptosis [20, 22]. However, the function and regulation of apoptosis by miR-493 in ovarian cancer cells still remain unknown. Hence, the pro-apoptotic effect of miR-493 was determined by flow cytometric measurements and potential targets of miR-493 were identified.
Transfection of miR-493 in the various ovarian carcinoma cell lines resulted in cell responses such as changes in the percentage of cell confluency and an increase in DNA laddering (cells in Sub G0/G1 phase). Long-term measurements revealed a clear induction of apoptotic markers like AnnexinV and the activation of Caspase 3. The observed apoptotic effects might be p53-independent due to the fact that the tumour suppressor, p53 is mutated in these cell lines (SKOV3p53null, OVCAR3p53R248Q [26], TOV112Dp53R175H [27], A2780-cisp53K351N [28, 29]). MiRNAs might offer the opportunity to overcome p53-dependent treatment resistance by triggering the cells with miRNA-493-3p.
Since miRNAs are known to have the potential to control a multiplicity of target genes [45], the aim of this study was to identify the network of target genes that is regulated by miR-493-3p and causative for the observed pro-apoptotic functions. In silico target prediction in several databases (as described above) was used to find potential mRNA targets with a binding site for direct interaction with miR-493.
In this study, we demonstrate that miR-493 overexpression resulted in diminished FOXM1 and AKT2 protein expression (Fig. 4b). AKT2 was found to be a potential direct target of miR-493 by luciferase assay. Activation of the MAPK/ERK pathway leads to cell survival and proliferation by stimulating transcription factors such as proto-oncogene c-myc or Ets like protein 1 (Elk1) [2]. Downregulation of proteins involved in both, the MAPK/ERK as well as the PI3-kinase–AKT pathway leads to activation of the transcription factor FOXO3 [46]. Phosphorylation of FOXO3 causes its inhibition and translocation from the nucleus into the cytoplasm. FOXO3 is involved in the regulation of metabolism, apoptosis and DNA repair [47]. Reduction of AKT2 together with the activation of FOXO3 results in diminished FOXM1 activity. FOXM1 activates the transcription of mRNAs needed for cell cycle regulation or DNA repair [47] (Fig. 7c). MiR-493 was shown to bind directly to the 3′ UTR of AKT2 (Fig. 4c, d). AKT2, the key downstream effector of the PI3-kinase pathway, is best known for its anti-apoptotic effects [48]. Downregulation of AKT2 in non-small cell lung cancer (NSCLC) leads to cleavage of the induced myeloid leukaemia cell differentiation protein MCL1. MCL1 belongs to the Bcl-2 family and by binding to Bak and Bax it inhibits the formation of pores in the mitochondrial membrane [36]. Hence, a decrease of MCL1 mediated by decreased AKT2 levels resulted in a loss of mitochondrial potential and release of cytochrome C [49]. Further, the release of cytochrome C (Fig. 2b) and the alteration of mitochondrial membrane permeability modulate proteins of the Bcl2 family like Bak leading to apoptosis via the activation of effector Caspases (Fig. 7c) [1].
Kaplan–Meier Blots with ovarian serous cystadenocarcinoma patients (data from TCGA) suggested that patients with lower AKT2 expression had a longer median survival (Fig. 5b). In line with that, Zhu et al. [50] observed in osteosarcoma patients that a low expression level of AKT2 correlates with a better prognosis.
With the reduced expression of AKT2 after miR-493 transfection, the amount of FMR1P protein was reduced (Fig. 4b). Mechanistic Target of Rapamycin (mTOR), a major regulator of protein synthesis, is activated by AKT-mediated phosphorylation. Activated mTOR phosphorylates downstream ribosomal protein S6 kinase beta-1 (S6K1) at threonine 389 leading to the activation of FMR1P [51]. FMR1P acts as a shuttle by transporting mRNA from the nucleus to the cytoplasm. Diminished expression of FMR1P is described to lead to mental retardation [52].
Another target directly regulated by miR-493-3p is High Mobility Group AT-Hook 2 (HMGA2). HMGA2 co-localizes with key replication factors and therefore, stabilizes branched DNA structures in vitro. Yu et al. [53] demonstrated that HMGA2 prevents double strand brakes at stalled replication forks and enhances cell survival. Consequently, reduced HMGA2 levels, induced by miR-493-3p, led to apoptosis and DNA fragmentation as described in this study (Fig. 7c). However, the underlying mechanisms of HMGA2-mediated apoptosis are still unknown. Strikingly, although there was obviously no interaction of miR-493 and RAF1 binding site, a significant downregulation on mRNA and protein level was detected. ETS1, a direct target of miR-493-3p, was identified as a potential transcription factor for RAF1. Silencing ETS1 significantly reduces the transcription of RAF1. Although the effects of ETS1 on apoptosis are reported to be controversial [54], in our system knockdown of ETS1 clearly revealed pro-apoptotic effects (Fig. 6g). Furthermore, ETS1 might also regulate RAF1 expression via the regulation of VEGF [54] that is modulating the MAPK/ERK pathway (Fig. 7d).
STK38L also known as Nuclear Dbf2-related (NDR) protein kinase (NDR2) was identified as another direct miR-493-3p binding partner showing reduced protein levels after miR-493 transfection. Furthermore, the predicted interaction (PIPs: Human protein–protein interactions prediction [41]) between the serine/threonine kinase STK38L and RAF1 was experimentally confirmed. It is reported that knockdown of STK38L results in cell cycle arrest and reduction of cell proliferation as well as upregulation of apoptosis [55, 56]. Notably, we observed that STK38L phosphorylates RAF1 at Ser621. Autophosphorylation at Ser621 is a key step in stabilizing the kinase by binding to 14-3-3 proteins and prevention of proteasomal degradation [43]. STK38L-mediated RAF1 phosphorylation at this site might augment these effects since a kinase dead form of STK38L reduced RAF1 phosphorylation at Ser621 resulting in reduced RAF1 protein level. Due to the fact that miR-493-3p diminished the expression of STK38L, RAF1 phosphorylation at Ser621 is diminished, making the kinase potentially more prone for degradation (Fig. 7d) [43].
E2F5 was found to be another transcription factor modulated by miR-493. E2F5 binds to the promoters of target genes involved in cell cycle control, thereby regulating cell growth and proliferation [57]. Downregulation of E2F5 via miR-493 or other miRNAs is reported to influence apoptosis as well as cell proliferation, invasion and cell motility in ovarian cancer cells [57].
In summary, miR-493-3p was validated to induce apoptosis in different ovarian CA cell lines. AKT2, HMGA2, ETS1, E2F5 and STK38L were identified as novel players, contributing to our understanding of the molecular mechanisms of network regulation by miR-493-3p mediated apoptosis. ETS1 as well as STK38L was found as potentially new regulators for transcription and phosphorylation of RAF1, respectively. Our findings that miR-493 regulates certain signalling pathways leading to apoptosis may have clinical relevance with regard to the development of new therapy strategies for ovarian cancer patients.
Methods
Cell culture
T98G (CRL-1690, LGC Standards, Wesel, Germany), HCT116 (CCL-247, LGC Standards), OVCAR3, TOV21G and TOV112D [27] were cultured in RPMI-1640 medium (Th Geyer, Renningen, Germany). HOSE 2170 [25], SGBS [58], HEK293T (CRL-3216, LGC Standards) and SKOV3 (HTB-77, LGC Standards) cells were cultured in DMEM high glucose medium (Th Geyer). The media contained 4 mM stable glutamine and were supplemented with 10% (v/v) FBS (Sigma-Aldrich, München, Germany). A2780 and A2780-cis cells were grown in RPMI-1640 media with 20% (v/v) FBS. All cells were grown at 37 °C and 5% CO2. The phenotype as well as the cell density of the adherent cell culture was proofed with the automated single well microscope NyOne (SynenTec Bio Services, Münster, Germany).
SKOV3 cells are p53 null and were also used in the previous cellular high-throughput screening [18]; therefore, the assays were focused on this cell line. Additionally, the high invasive OVCAR3p53R248Q cells [26] were used. Furthermore, the cell lines TOV21G without p53 mutation or TOV112Dp53R175H with p53 mutation [27] as well as A2780-cisp53K351N with Cisplatin resistance or A2780 [28, 29] without Cisplatin resistance were included for apoptosis assays. HEK293T cells were used for luciferase assays and immunoprecipitations as they are an easy to transfect cell system for plasmids [35].
Transfection with miRNA mimics
The cells were seeded and transfected with 62.5 nM miRNA mimic (Qiagen, Hilden, Germany), non-targeting siRNA (AllStars Neg. control, order number: 1027281, negative control for cell death; NT), human cell death control siRNA (AllStars Cell Death control, order number: 1027299, positive control for cell death; DT; Qiagen) or siRNA against ETS1 (siRNA ID: VH S40614, Thermo Fisher Scientific, Darmstadt, Germany). The miRNA or siRNA was transfected with ScreenFect A reagent (ScreenFect, Eggenstein-Leopoldshafen, Germany) as described before [59, 60]. As controls, the cells were treated with 25 µM Etoposide, 80 µM Carboplatin or 0.25 µM Paclitaxel (all reagents from Enzo Life Sciences, Lörrach, Germany). The transfection efficiency was surveyed by a fluorescent-labelled non-targeting siRNA. After transfection, about 95–100% cells showed red fluorescence.
Analysis of apoptotic cells, detection of mitochondria potential and cytochrome C release
Apoptotic cells were identified by flow cytometry measuring the amount of cells with reduced DNA content (sub G0/G1) as previously described by Rudner et al. [61]. For detection of the mitochondrial potential of the cells, a TMRE assay was carried out as described by Flum et al. [60]. To stain for free cytochrome C after mitochondrial damage, the cells were detached, fixed with 4% (w/v) paraformaldehyde for 15 min, permeabilised with 0.1% (v/v) Triton X-100 and stained with an Alexa Fluor 488 labelled antibody (BLD-612308; Biozol, Eching, Germany) against cytochrome C for 1 h in the dark. The antibody was diluted 1:1,500 in 5% BSA/TBST solution.The quantitative analysis was performed with the MACSQuant Analyser using fluorescence channel B1 (525/50 nm filter).
Cell motility assay
For the analysis of cell motility 40,000 cells/ml were seeded into 96 well plates 24 h before transfection. Immediately before transfection, a scratch was performed using 1000 µl pipet tips on a multi-channel pipette. To remove scratched cells, the wells were washed with PBS three times, after the transfection mix was added. Immediately after transfection, the cells were analysed with the automated single well microscope NyOne.
RT-PCR
The extraction of total RNA was done using the miRNeasy Mini Kit (Qiagen, Hilden, Germany). 1000 ng of isolated RNA was transcribed via the miScript II RT Kit (Qiagen) using 5 × miScript HiSpec Buffer and an incubation time of 60 min at 37 °C. The 1:30 diluted cDNA was further analysed in the Roche Light Cycler 480 using GreenMasterMix (Genaxxon Bioscience, Ulm, Germany). For the qRT-PCR, the primers for hsa-miR-493-3p (5′- TGAAGGTCTACTGTGTGCCAGG-3′) or hsa-miR-493-5p (5′- TTGTACATGGTAGGCTTTCATT-3′) were used together with the universal reverse primer from the miScript PCR Starter Kit (Qiagen). U6 snRNA primer forward (5′-CTCGCTTCGGCAGCACA-3′) and U6 snRNA primer reverse (5′-AACGCTTCACGAATTTGCGT-3′) served for control loading. To analyse the expression of potential target genes, 1000 ng of isolated RNA was transcribed using the Transcriptor High Fidelity cDNA Synthesis Kit from Roche (Penzberg, Germany). For detection of mRNA expression of potential targets, the following primers were used: ADAR1_FW (5′-CTGGATTCCACAGGGATTGT-3′), ADAR1_RV (5′-TTCGAGAATCCCAAACAAGG-3′), AKT2_FW (5′-TGGGTCTGGAAGGCATACTT-3′), AKT2_RV (5′-CTCACACAGTCACCGAGAGC-3′), ALKBH3_FW (5′-TCCCATGATCCAAGGGTATC-3′), ALKBH3_RV (5′-CACGCACATTTGAGATGAGAA-3′), E2F5_FW (5′-CGGCGTTCTGGATCTCAA-3′), E2F5_RV (5′-CAATTCCCTCTAAGACATTGGTG-3′), EEF2K_FW (5′-GCGCGAGCTTTTGACTCT-3′), EEF2K_RV (5′-AGGGCCTCTAGCCAGTCTTG-3′), ETS1_FW (5´-CCATCATCAAGACGGAAAAAG-3′), ETS1_RV (5′-GGGACATCTGCACATTCCATA-3′), ETS2_FW (5′-CAGCGTCACCTACTGCTCTG-3′), ETS2_RV (5′-AGTCGTGGTCTTTGGGAGTC-3′), FMR1P_FW (5′-AATCCAAAAGAACAGTGGCATT-3′), FMR1P_RV (5′-GGAATCCCAGAAACCTGAACT-3′), FOXM1_FW (5′-CCACTGGATGTTGGATAGGC-3′), FOXM1_RV (5′-AGAAACGGGAGACCTGTGC-3′), GAB2_FW (5′-AGGGGCAGGACTGTTCGT-3′), GAB2_RV (5′-CGAAGAGAACTATGTCCCTATGC-3′), HMGA2_FW (5′-TCCCTCTAAAGCAGCTCAAAA-3′), HMGA2_RV (5′-ACTTGTTGTGGCCATTTCCT-3′), JAK2_FW (5′-CAGGAACAAGATGTGAACTGTTTC-3′), JAK2_RV (5′-CCCATGCAGAGTCTTTTTCAG-3´), MAP2K5_FW (5′-TCAGGGGAGCAGTATGGAAT-3′), MAP2K5_RV (5′-AAACCTCCCAAGAGCAAGC-3′), MAPK1_FW (5′-TCTGCACCGTGACCTCAA-3′), MAPK1_RV (5′-GCCAGGCCAAAGTCACAG-3′), PEA15_FW (5′-GTCCCGTACTCAGCCATGA-3′), PEA15_RV (5′-TTAGGAACCGGGGACTCA-3′), PIK3R3_FW (5′-GGCTTAGGTGGCTTTGGTG-3′), PIK3R3_RV(5′-TGATGCCCTATTCGACAGAA-3′), PPIA_FW (5′-ATGCTGGACCCAACACAAAT-3′), PPIA_RV (5′-TCTTTCACTTTGCCAAACACC-3′), QKI_FW (5′-CCAACTTCTGCGGGATCTT-3′), QKI_RV (5′-TGTCATTGTACATGTCTTTCCGTA-3′), RAF1_FW (5′-TGGGAAATAGAAGCCAGTGAA-3′), RAF1_RV (5′-CCTTTAGGATCTTTACTGCAACATC-3′), STAT3_FW (5′-CCCTTGGATTGAGAGTCAAGA-3′), STAT3_RV (5′-AAGCGGCTATACTGCTGGTC-3′), STK38L_FW (5′-CAAAGACCACCAGTCACACAA-3′) and STK38L_RV (5′-GAAGAAGAACAGGAGACAACTGG-3′).
Western blotting
Protein analysis was conducted by Western Blotting as previously described [60]. The antibodies against Caspase-3 (cs#9662), cleaved Caspase-3 (cs#9664), PARP (cs#9542) and cleaved PARP (cs#9541) were from Cell Signaling Technology (Danvers, United States) and diluted 1:1,000. For analysis of molecular mechanisms, cells were treated with 30 µM zVAD (Enzo Life Sciences). For detection of target proteins, the following antibodies were used: ETS1 (1:500; sc-55581), QKI (1:500; sc-517305), E2F5 (1:500; sc-374268) from Santa Cruz Biotechnology (Heidelberg, Germany), Bak (1:1000; cs#12105T), Bcl-XL (1:1000; cs#2764T), MCL1 (1:1000; cs#94296), RAF1 (1:1000; cs#53745), FMR1P (1:1000; cs#7104), FOXM1 (1:1000; cs#5436), HMGA2 (1:1000; cs#5269), MAPK1 (1:1000; cs#9102S) and STAT3 (1:1000; cs#9139) from Cell Signaling Technology as well as ETS1 (1:1000; orb393050; Biozol; Echingen, Germany), ETS2 (1:1000; CSB-PA007853LA01HU; Biozol), AKT2 (1:500; 680202; BioLegend, Fell, Germany), and STK38L (1:500; ABIN3185788; antikoerper-online.de, Aachen, Germany). The antibody against Flag (1:1000; F3156; Sigma-Aldrich) was used to detect the Flag-tagged STK38L proteins.
Depending on the size of the other proteins, the antibodies against β-Actin (1:10,000; A5441; Sigma-Aldrich) or GAPDH (1:5000; MA5-15738; Thermo Fisher Scientific) served as loading control.
The secondary antibody, anti-rabbit IgG, HRP-linked (cs#7074, Cell Signaling Technology) or anti-mouse IgG, HRP linked (A4416, Sigma-Aldrich) was diluted 1:10,000.
Apoptosis assay with long-term video-microscopy
The activation of Caspases was measured by long-term video-microscopy. The cells were transfected or treated with chemotherapeutic drugs as described above. 1 h after treatment, IncuCyte Caspase-3/7 Green Apoptosis Assay Reagent (Sartorius, Goettingen, Germany) was added to a final concentration of 5 nM as well as IncuCyte AnnexinV Red reagent with a final dilution of 1:200. The cells were placed into the IncuCyte ZOOM Live-Cell Analysis System detecting red and green fluorescence. The imaging system is placed in an incubator with standard culture conditions. Images from each well at two different positions were taken automatically every hour with a 20x objective and analysed with the IncuCyte ZOOM Software for the amount of fluorescent stained cells. The data were visualized using GraphPad Prism 5 software.
TCGA-analysis correlation
To examine the correlation between the miR-493 expression and potential targets, level 3 miRNA expression data of the “Ovarian serous cystadenocarcinoma” data set were downloaded from the cancer genome atlas (TCGA) data base (www.tcga-data.nci.nih.gov/tcga) using the “firehose-get“command-line tool (https://confluence.broadinstitute.org/display/GDAC/Download). A detailed description of the clinical characteristics of the cohort can be found in a study by Cancer Genome Atlas Research Network [62]. Additionally, the mRNA as well as the protein expression of all these patients was downloaded and negatively correlated to the miRNA expression of each of the 530 patients.
For the delineation of the survival of ovarian serous cystadenocarcinoma patients affected by miR-493 binding partners (AKT2 and STK38L), different age groups (range 26–89 years, mean: 67.5 years) were analysed dividing the patients’ age in first quartile, mean, median or third quartile. For each age group, differential survival analysis for patients with high- versus low-expression of AKT2 and STK38L was conducted employing the median expression as threshold. For this, the log-rank test was applied on the resulting cox-proportional hazard models and for the purpose of visualization Kaplan–Meier Blots were generated.
Gene expression analysis
To analyse the differential gene expression after transfection, the GeneChip Human Gene ST 2.0 Array (Thermo Fisher Scientific) was used. Reverse Transcription and biotin labelling of the RNA were carried out with the GeneChip WT PLUS Reagent Kit (Thermo Fisher Scientific). Hybridization and data analysis were performed as described previously [63].
In silico target prediction
For identification of potential miR-493-3p target genes, the prediction tools TargetScanHuman [33], microRNA.org [34], Rna22 [64], DIANA TOOLS [65], miRDB [66] and miRWalk [67] were used. The online gene classification software Protein Analysis Through Evolutionary Relationships (PANTHER) [68] was used to cluster the potential targets for apoptotic functions. Ingenuity Pathway Analysis (IPA, Qiagen Bioinformatics) was employed to suggest potential target genes restricted to genes with anti-apoptotic or survival promoting functions in ovarian CA signalling. Already experimentally validated miR-493-3p target genes listed in miRTarBase [69] and miRWalk [67] were excluded from further investigations.
Potential miR-493-3p binding sites were obtained from the database microRNA.org [34] as well as from TargetScanHuman [33].
Luciferase reporter assay
Luciferase reporter constructs were generated with oligonucleotide cloning. Luciferase reporter assays were performed in HEK293T cells as described by Flum et al. [60].
Immunoprecipitation and immunoblotting
40,000 HEK293T cells were seeded into 6-well culture pates. 24 h later, the cells were transfected with 5 μg of STK38L WT (Addgene, Cambridge, USA) or 10 μg of STK38L KD plasmid [42]. 48 h after transfection, cells were lysed in ice-cold lysis buffer (1% Triton X-100, 20 mM Tris, 150 mM NaCl, pH 8.0) containing a cocktail of protein inhibitors (Merck, Darmstadt, Germany). Post-nuclear lysates were incubated for 1 h with the indicated antibody. 30 μl of Protein A Sepaharose beads (Sigma Aldrich, Darmstadt, Germany) were added and incubated for 1 h at 4 °C. Immunoprecipitates were washed four times with ice-cold lysis buffer and proteins were eluted by boiling for 5 min in SDS sample buffer, separated by SDS-PAGE and transferred onto PVDF membrane for immunoblotting. Membranes were blocked with 5% BSA in TBST (TBS, 1% Tween) and incubated with the indicated antibody for 1 h. Bound antibody was revealed with the appropriate secondary antibody and protein was visualized by chemiluminescence using Immobilon Western Chemiluminescent HRP substrate (Thermo Fisher Scientific).
Statistical analysis
Data in general were expressed as mean ± SD. Statistical analysis was carried out using GraphPad Prism Version 5.04. The corresponding statistical test and the level of significance are indicated in each figure legend. For TCGA analysis, all calculations were conducted employing the R statistical platform [70] using functions from the CRAN package survival (www.cran.r-project.org/web/packages/survival).
References
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35(4):495–516. https://doi.org/10.1080/01926230701320337
Zhang W, Liu HT (2002) MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 12(1):9–18
Cory S, Adams JM (2002) The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2(9):647–656. https://doi.org/10.1038/nrc883
Chaitanya GV, Alexander JS, Babu PP (2010) PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun Signal CCS 8:31. https://doi.org/10.1186/1478-811X-8-31
Kaufmann SH, Mesner PW Jr, Samejima K, Tone S, Earnshaw WC (2000) Detection of DNA cleavage in apoptotic cells. Methods Enzymol 322:3–15
He L, Hannon GJ (2004) MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5(7):522–531
MacFarlane L-A, Murphy PR (2010) MicroRNA: biogenesis, function and role in cancer. Curr Genomics 11(7):537–561. https://doi.org/10.2174/138920210793175895
Kim VN, Han J, Siomi MC (2009) Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10(2):126–139. https://doi.org/10.1038/nrm2632
Filipowicz W, Bhattacharyya SN, Sonenberg N (2008) Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9(2):102–114. https://doi.org/10.1038/nrg2290
Lynam-Lennon N, Maher SG, Reynolds JV (2009) The roles of microRNA in cancer and apoptosis. Biol Rev Camb Philos Soc 84(1):55–71. https://doi.org/10.1111/j.1469-185X.2008.00061.x
Lima RT, Busacca S, Almeida GM, Gaudino G, Fennell DA, Vasconcelos MH (2011) MicroRNA regulation of core apoptosis pathways in cancer. Eur J Cancer 47(2):163–174. https://doi.org/10.1016/j.ejca.2010.11.005
Adams JM, Cory S (2007) The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26(9):1324–1337. https://doi.org/10.1038/sj.onc.1210220
Pal MK, Jaiswar SP, Dwivedi VN, Tripathi AK, Dwivedi A, Sankhwar P (2015) MicroRNA: a new and promising potential biomarker for diagnosis and prognosis of ovarian cancer. Cancer Biol Med 12(4):328–341. https://doi.org/10.7497/j.issn.2095-3941.2015.0024
Siegel RL, Miller KD, Jemal A (2017) Cancer statistics. CA Cancer J Clin 67(1):7–30. https://doi.org/10.3322/caac.21387
Karst AM, Drapkin R (2010) Ovarian cancer pathogenesis: a model in evolution. J Oncol 2010:932371. https://doi.org/10.1155/2010/932371
Della Pepa C, Tonini G, Pisano C, Di Napoli M, Cecere SC, Tambaro R, Facchini G, Pignata S (2015) Ovarian cancer standard of care: are there real alternatives? Chin J Cancer 34(1):17–27. https://doi.org/10.5732/cjc.014.10274
Chang L, Graham P, Hao J, Ni J, Deng J, Bucci J, Malouf D, Gillatt D, Li Y (2016) Cancer stem cells and signaling pathways in radioresistance. Oncotarget 7(10):11002–11017. https://doi.org/10.18632/oncotarget.6760
Kleemann M, Bereuther J, Fischer S, Marquart K, Hanle S, Unger K, Jendrossek V, Riedel CU, Handrick R, Otte K (2016) Investigation on tissue specific effects of pro-apoptotic micro RNAs revealed miR-147b as a potential biomarker in ovarian cancer prognosis. Oncotarget. https://doi.org/10.18632/oncotarget.13095
Li J, Liang S, Jin H, Xu C, Ma D, Lu X (2012) Tiam1, negatively regulated by miR-22, miR-183 and miR-31, is involved in migration, invasion and viability of ovarian cancer cells. Oncol Rep 27(6):1835–1842. https://doi.org/10.3892/or.2012.1744
Gu Y, Cheng Y, Song Y, Zhang Z, Deng M, Wang C, Zheng G, He Z (2014) MicroRNA-493 suppresses tumor growth, invasion and metastasis of lung cancer by regulating E2F1. PLoS One 9(8):e102602. https://doi.org/10.1371/journal.pone.0102602
Ueno K, Hirata H, Majid S, Yamamura S, Shahryari V, Tabatabai ZL, Hinoda Y, Dahiya R (2012) Tumor suppressor microRNA-493 decreases cell motility and migration ability in human bladder cancer cells by down-regulating RhoC and FZD4. Mol Cancer Ther 11(1):244–253. https://doi.org/10.1158/1535-7163.MCT-11-0592
Zhou W, Zhang C, Jiang H, Zhang Z, Xie L, He X (2015) MiR-493 suppresses the proliferation and invasion of gastric cancer cells by targeting RhoC. Iran J Basic Med Sci 18(10):1027–1033
Benetatos L, Hatzimichael E, Londin E, Vartholomatos G, Loher P, Rigoutsos I, Briasoulis E (2013) The microRNAs within the DLK1-DIO3 genomic region: involvement in disease pathogenesis. Cell Mol Life Sci CMLS 70(5):795–814. https://doi.org/10.1007/s00018-012-1080-8
Benetatos L, Vartholomatos G, Hatzimichael E (2014) DLK1-DIO3 imprinted cluster in induced pluripotency: landscape in the mist. Cell Mol Life Sci CMLS 71(22):4421–4430. https://doi.org/10.1007/s00018-014-1698-9
Choi P-W, Yang J, Ng S-K, Feltmate C, Muto MG, Hasselblatt K, Lafferty-Whyte K, JeBailey L, MacConaill L, Welch WR, Fong W-P, Berkowitz RS, Ng S-W (2016) Loss of E-cadherin disrupts ovarian epithelial inclusion cyst formation and collective cell movement in ovarian cancer cells. Oncotarget 7(4):4110–4121. https://doi.org/10.18632/oncotarget.6588
Kobayashi M, Salomon C, Tapia J, Illanes SE, Mitchell MD, Rice GE (2014) Ovarian cancer cell invasiveness is associated with discordant exosomal sequestration of Let-7 miRNA and miR-200. J Transl Med 12:4. https://doi.org/10.1186/1479-5876-12-4
Samouelian V, Maugard CM, Jolicoeur M, Bertrand R, Arcand SL, Tonin PN, Provencher DM, Mes-Masson AM (2004) Chemosensitivity and radiosensitivity profiles of four new human epithelial ovarian cancer cell lines exhibiting genetic alterations in BRCA2, TGFbeta-RII, KRAS2, TP53 and/or CDNK2A. Cancer Chemother Pharmacol 54(6):497–504. https://doi.org/10.1007/s00280-004-0843-9
Behrens BC, Hamilton TC, Masuda H, Grotzinger KR, Whang-Peng J, Louie KG, Knutsen T, McKoy WM, Young RC, Ozols RF (1987) Characterization of a cis-diamminedichloroplatinum(II)-resistant human ovarian cancer cell line and its use in evaluation of platinum analogues. Cancer Res 47(2):414–418
Masuda H, Ozols RF, Lai GM, Fojo A, Rothenberg M, Hamilton TC (1988) Increased DNA repair as a mechanism of acquired resistance to cis-diamminedichloroplatinum (II) in human ovarian cancer cell lines. Cancer Res 48(20):5713–5716
Strasser A, O’Connor L, Dixit VM (2000) Apoptosis signaling. Annu Rev Biochem 69:217–245. https://doi.org/10.1146/annurev.biochem.69.1.217
Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA (1998) Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem 273(49):32608–32613
Mi H, Muruganujan A, Thomas PD (2013) PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res 41(Database issue):D377–D386. https://doi.org/10.1093/nar/gks1118
Agarwal V, Bell GW, Nam JW, Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. eLife. https://doi.org/10.7554/elife.05005
Betel D, Koppal A, Agius P, Sander C, Leslie C (2010) Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol 11(8):R90. https://doi.org/10.1186/gb-2010-11-8-r90
Fischer S, Buck T, Wagner A, Ehrhart C, Giancaterino J, Mang S, Schad M, Mathias S, Aschrafi A, Handrick R, Otte K (2014) A functional high-content miRNA screen identifies miR-30 family to boost recombinant protein production in CHO cells. Biotechnol J 9(10):1279–1292. https://doi.org/10.1002/biot.201400306
Thomas LW, Lam C, Edwards SW (2010) Mcl-1; the molecular regulation of protein function. FEBS Lett 584(14):2981–2989. https://doi.org/10.1016/j.febslet.2010.05.061
Lassmann S, Weis R, Makowiec F, Roth J, Danciu M, Hopt U, Werner M (2007) Array CGH identifies distinct DNA copy number profiles of oncogenes and tumor suppressor genes in chromosomal- and microsatellite-unstable sporadic colorectal carcinomas. J Mol Med (Berlin, Germany) 85(3):293–304. https://doi.org/10.1007/s00109-006-0126-5
Zhang SX, Garcia-Gras E, Wycuff DR, Marriot SJ, Kadeer N, Yu W, Olson EN, Garry DJ, Parmacek MS, Schwartz RJ (2005) Identification of direct serum-response factor gene targets during Me2SO-induced P19 cardiac cell differentiation. J Biol Chem 280(19):19115–19126. https://doi.org/10.1074/jbc.M413793200
Iorns E, Turner NC, Elliott R, Syed N, Garrone O, Gasco M, Tutt AN, Crook T, Lord CJ, Ashworth A (2008) Identification of CDK10 as an important determinant of resistance to endocrine therapy for breast cancer. Cancer Cell 13(2):91–104. https://doi.org/10.1016/j.ccr.2008.01.001
Jain P, Fierst TM, Han HJ, Smith TE, Vakil A, Storm PB, Resnick AC, Waanders AJ (2017) CRAF gene fusions in pediatric low-grade gliomas define a distinct drug response based on dimerization profiles. Oncogene 36(45):6348–6358. https://doi.org/10.1038/onc.2017.276
McDowall MD, Scott MS, Barton GJ (2009) PIPs: human protein-protein interaction prediction database. Nucleic Acids Res 37(Database issue):D651–D656. https://doi.org/10.1093/nar/gkn870
Devroe E, Erdjument-Bromage H, Tempst P, Silver PA (2004) Human Mob proteins regulate the NDR1 and NDR2 serine-threonine kinases. J Biol Chem 279(23):24444–24451. https://doi.org/10.1074/jbc.M401999200
Noble C, Mercer K, Hussain J, Carragher L, Giblett S, Hayward R, Patterson C, Marais R, Pritchard CA (2008) CRAF autophosphorylation of serine 621 is required to prevent its proteasome-mediated degradation. Mol Cell 31(6):862–872. https://doi.org/10.1016/j.molcel.2008.08.026
Wyman SK, Parkin RK, Mitchell PS, Fritz BR, O’Briant K, Godwin AK, Urban N, Drescher CW, Knudsen BS, Tewari M (2009) Repertoire of microRNAs in epithelial ovarian cancer as determined by next generation sequencing of small RNA cDNA libraries. PLoS One 4(4):e5311. https://doi.org/10.1371/journal.pone.0005311
Fischer S, Handrick R, Aschrafi A, Otte K (2015) Unveiling the principle of microRNA-mediated redundancy in cellular pathway regulation. RNA Biol 12(3):238–247. https://doi.org/10.1080/15476286.2015.1017238
Roy SK, Srivastava RK, Shankar S (2010) Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J Mol Signal 5:10. https://doi.org/10.1186/1750-2187-5-10
Gomes AR, Zhao F, Lam EWF (2013) Role and regulation of the forkhead transcription factors FOXO3a and FOXM1 in carcinogenesis and drug resistance. Chin J Cancer 32(7):365–370. https://doi.org/10.5732/cjc.012.10277
Zhang D, Piao HL, Li YH, Qiu Q, Li DJ, Du MR, Tsang BK (2016) Inhibition of AKT sensitizes chemoresistant ovarian cancer cells to cisplatin by abrogating S and G2/M arrest. Exp Mol Pathol 100(3):506–513. https://doi.org/10.1016/j.yexmp.2016.05.003
Lee MW, Kim DS, Lee JH, Lee BS, Lee SH, Jung HL, Sung KW, Kim HT, Yoo KH, Koo HH (2011) Roles of AKT1 and AKT2 in non-small cell lung cancer cell survival, growth, and migration. Cancer Sci 102(10):1822–1828. https://doi.org/10.1111/j.1349-7006.2011.02025.x
Zhu Y, Zhou J, Ji Y, Yu B (2014) Elevated expression of AKT2 correlates with disease severity and poor prognosis in human osteosarcoma. Mol Med Rep 10(2):737–742. https://doi.org/10.3892/mmr.2014.2314
Darnell JC, Klann E (2013) The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci 16(11):1530–1536. https://doi.org/10.1038/nn.3379
Dahlhaus R (2018) Of men and mice: modeling the fragile X syndrome. Front Mol Neurosci 11:41. https://doi.org/10.3389/fnmol.2018.00041
Yu H, Lim Hong H, Tjokro Natalia O, Sathiyanathan P, Natarajan S, Chew Tian W, Klonisch T, Goodman Steven D, Surana U, Dröge P (2014) Chaperoning HMGA2 protein protects stalled replication forks in stem and cancer cells. Cell Rep 6(4):684–697. https://doi.org/10.1016/j.celrep.2014.01.014
Dittmer J (2003) The biology of the Ets1 Proto-Oncogene. Mol Cancer 2:29. https://doi.org/10.1186/1476-4598-2-29
Cornils H, Kohler RS, Hergovich A, Hemmings BA (2011) Human NDR kinases control G(1)/S cell cycle transition by directly regulating p21 stability. Mol Cell Biol 31(7):1382–1395. https://doi.org/10.1128/MCB.01216-10
Grant TJ, Mehta AK, Gupta A, Sharif AAD, Arora KS, Deshpande V, Ting DT, Bardeesy N, Ganem NJ, Hergovich A, Singh A (2017) STK38L kinase ablation promotes loss of cell viability in a subset of KRAS-dependent pancreatic cancer cell lines. Oncotarget 8(45):78556–78572. https://doi.org/10.18632/oncotarget.20833
Tian H, Hou L, Xiong Y-M, Huang J-X, Zhang W-H, Pan Y-Y, Song X-R (2016) miR-132 targeting E2F5 suppresses cell proliferation, invasion, migration in ovarian cancer cells. Am J Transl Res 8(3):1492–1501
Wabitsch M, Brenner RE, Melzner I, Braun M, Moller P, Heinze E, Debatin KM, Hauner H (2001) Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation. Int J Obes Relat Metab Disord 25(1):8–15
Fischer S, Wagner A, Kos A, Aschrafi A, Handrick R, Hannemann J, Otte K (2013) Breaking limitations of complex culture media: functional non-viral miRNA delivery into pharmaceutical production cell lines. J Biotechnol 168(4):589–600. https://doi.org/10.1016/j.jbiotec.2013.08.027
Flum M, Kleemann M, Schneider H, Weis B, Fischer S, Handrick R, Otte K (2017) miR-217-5p induces apoptosis by directly targeting PRKCI, BAG3, ITGAV and MAPK1 in colorectal cancer cells. J Cell Commun Signal. https://doi.org/10.1007/s12079-017-0410-x
Rudner J, Ruiner CE, Handrick R, Eibl HJ, Belka C, Jendrossek V (2010) The Akt-inhibitor Erufosine induces apoptotic cell death in prostate cancer cells and increases the short term effects of ionizing radiation. Radiat Oncol 5:108. https://doi.org/10.1186/1748-717X-5-108
Cancer Genome Atlas Research N (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474(7353):609–615. https://doi.org/10.1038/nature10166
Emmerling VV, Fischer S, Stiefel F, Holzmann K, Handrick R, Hesse F, Horer M, Kochanek S, Otte K (2016) Temperature-sensitive miR-483 is a conserved regulator of recombinant protein and viral vector production in mammalian cells. Biotechnol Bioeng 113(4):830–841. https://doi.org/10.1002/bit.25853
Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I (2006) A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 126(6):1203–1217. https://doi.org/10.1016/j.cell.2006.07.031
Vlachos IS, Paraskevopoulou MD, Karagkouni D, Georgakilas G, Vergoulis T, Kanellos I, Anastasopoulos IL, Maniou S, Karathanou K, Kalfakakou D, Fevgas A, Dalamagas T, Hatzigeorgiou AG (2015) DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res 43(Database issue):D153–D159. https://doi.org/10.1093/nar/gku1215
Wong N, Wang X (2015) miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res 43(Database issue):D146–D152. https://doi.org/10.1093/nar/gku1104
Dweep H, Sticht C, Pandey P, Gretz N (2011) miRWalk–database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J Biomed Inform 44(5):839–847. https://doi.org/10.1016/j.jbi.2011.05.002
Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A (2003) PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13(9):2129–2141. https://doi.org/10.1101/gr.772403
Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ, Tsai TR, Ho SY, Jian TY, Wu HY, Chen PR, Lin NC, Huang HT, Yang TL, Pai CY, Tai CS, Chen WL, Huang CY, Liu CC, Weng SL, Liao KW, Hsu WL, Huang HD (2016) miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res 44(D1):D239–D247. https://doi.org/10.1093/nar/gkv1258
R Development Core Team (2011) R: A Language and Environment for Statistical Computing. Vienna, Austria: the R Foundation for Statistical Computing. ISBN:3-900051-07-0. Available online at http://www.R-project.org/
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
Acknowledgements
This study was funded by the Postgraduate Scholarships Act of the Ministry for Science, Research and Arts of the Federal State Government of Baden-Wuerttemberg, Germany. Further acknowledgements address the International Graduate School in Molecular Medicine of Ulm University, Germany, for scientific encouragement and support to Michael Kleemann. Pamela Fischer-Posovszky receives funding from the Baden-Württemberg Stiftung, Germany. The results published here are based on data generated by the TCGA Research Network. In addition, the authors are grateful to Dr. Anne-Marie Mes-Masson (Centre Hospitalier de l´Université de Montréal, Canada) for providing us with the TOV21G and TOV112D cells as well as to Alex Shu-Wing Ng (Department of Obstetrics, Gynecology and Reproductive Biology, the Brigham and Women’s Hospital, Boston, USA) for the HOSE 2170 cells. The OVCAR3, A2780 and A2780-cis cells were kindly provided by Verena Jendrossek (Institute of Cell Biology (Cancer Research), University of Duisburg-Essen, Germany).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical standards
The authors declare that the experiments comply with the current laws of the Federal Republic of Germany.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
18_2018_2958_MOESM1_ESM.tif
Supplementary Figure 1 Determination of miR-493 expression after transfection. For determination of miR-493 expression after transfection, cells were seeded and transfected as described in Figure 2. The miRNA expression of miR-493 was normalized to the CT value of U6 snRNA and the NT-transfected control employing the Livak method [72]. Statistical significance was tested by an unpaired t test. [n = 3 replicates; mean ± SD, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001] (TIFF 70 kb)
Rights and permissions
About this article

Cite this article
Kleemann, M., Schneider, H., Unger, K. et al. Induction of apoptosis in ovarian cancer cells by miR-493-3p directly targeting AKT2, STK38L, HMGA2, ETS1 and E2F5. Cell. Mol. Life Sci. 76, 539–559 (2019). https://doi.org/10.1007/s00018-018-2958-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-018-2958-x