
Abstract
Cellular senescence is the final fate of most cells in response to specific stimuli, but is not the end. Indeed, it is the beginning of a singular life, with multiple side roads leading to diverse effects on the organism. Many studies have been done in the last few years to elucidate the intriguing role of senescent cells in the organism, demonstrating them as the cause of several age-related diseases. However, these cells are also positively implicated in other important pathways, such as embryogenesis and wound healing. It appears that the multiple effects are time-dependent: long-term senescence is mostly implicated in chronic inflammation and disease, whereas in the short term, senescent cells seem to be beneficial, being rapidly targeted by the innate immune system. The influence of senescent cells on their neighbors by paracrine factors, differential activity depending on developmental stage, and duration of the effects make the cellular senescent program a unique spatial–temporal mechanism. During pathological conditions such as progeroid syndromes, this mechanism is deregulated, leading to accelerated onset of some aging-related diseases and a shorter lifespan, among other physiological defects. Here, we review the three primary cell senescence programs described so far (replicative, stress-induced, and developmentally programmed senescence), their onset during development, and their potential roles in diseases with premature aging. Finally, we discuss the role of immune cells in keeping senescence burden below the threshold of disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The road so far: routes toward cellular senescence
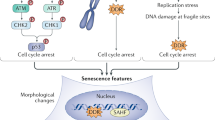
Cellular senescence is a specific form of irreversible growth arrest, coupled to stereotypical phenotypic changes including resistance to apoptosis, expression of anti-proliferative molecules, and activation of damage-sensing signaling routes, among others [1, 2]. Cellular senescence was first described by Leonard Hayflick and Paul Moorhead [3], whose pioneering work was based on cultured human fibroblast cells (WI-38 fibroblasts) that lost their ability to proliferate, reaching permanent arrest after about 50 population doublings (referred to as the Hayflick limit). Though met with some skepticism at the time, the study of cellular aging had begun, with three primary cellular senescence routes identified to date.
Replicative senescence (RS)
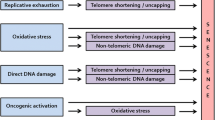
The first pathway of cellular senescence to be identified is related to the finite proliferation capacity of replication-competent cells, due to telomere shortening during each population doubling (the so-called “DNA end replication problem”) [4]. In this phenomenon, observed by Hayflick and Moorhead [3], shortened telomeres lead to chromosomal instability and pro-tumorigenic events if the cell continues dividing [5]. To avoid this process and safeguard genomic stability, the DNA damage response (DDR) mechanism is activated, triggering a cascade of events involving the ataxia telangiectasia mutated (ATM)/ATM and Rad3-related (ATR)-p53-p21CIP1 and p16Ink4a pathways, leading to permanent cell arrest and/or apoptosis [6]. RS is age-dependent and considered to be a tumor-suppression mechanism [7,8,9].
Stress-induced premature senescence (SIPS)
Cells can become senescent in response to a multitude of stressors that include, but are not limited to, DNA damage (independent of telomeric shortening). The first experimental evidence was obtained from hydrogen peroxide treatment and oncogene over-expression [10, 11]. Currently many stress inducers have been identified from oncogenic, oxidative, and genotoxic sources, which can lead to SIPS by activating the ATM/ATR-p53-p21CIP1 and p16Ink4a DDR pathways. This premature cell cycle arrest is also considered to be a tumor-suppression mechanism, by arresting pre-neoplastic (damaged) cells [7,8,9]. The nomenclature for SIPS is diverse, since it originated from the nature of the stressors, such as oncogene-induced senescence (OIS), chemotherapy-induced senescence (CIS), or epigenetically induced senescence (EIS) [11, 12]. The main differences between RS and SIPS are the shorter time required for cell cycle arrest in SIPS and the shorter telomeres in RS [13]. However, they share some phenotypic aspects and seem to depend on the same pathways, p53-p21CIP1 and p16Ink4a-retinoblastoma protein (pRB), which cause cell cycle arrest by inhibiting E2F [1, 14].
Developmentally programmed senescence (DPS)
The latest senescence pathway to be described, DPS, has been detected in the first days of organism development (embryogenesis) [15, 16]. Two ground-breaking studies reported that senescence occurs at multiple locations during mammalian embryogenesis [15, 16]. Interestingly, these studies did not detect DNA damage markers in the senescent cells, nor the activation of DNA damage-dependent kinases ATM and ATR; rather, DPS is dependent on p21CIP1 expression, and shares features with OIS. The biological function of these senescent cells seems to be the establishment of a correct balance between cell populations. The authors claimed that this new cell senescence program is a physiological process, orchestrated through developmental signals, which may have evolved to arrange tissue regeneration and healing upon damage in adult organisms [15, 16].
Other senescence processes have been reported, with characteristics similar to DPS. In the context of embryo development, senescence of placental cells displays some (though not all) features of DPS [17, 18]. An earlier study, in 2012, described senescence in natural killer (NK) cells during embryo implantation, the features of which also resembled DPS [19].
A recent study suggested that senescence may be part of normal maturation in pancreatic β cells, executing a programmed developmental role not triggered by stress, leading to insulin secretion and maintaining glucose homeostasis [20]. In contrast, another recent study showed that senescent β cells (positive for insulin growth factor 1 receptor, IGF1R, and p16Ink4a) secreted less insulin than non-senescent β cells [21]; more work is needed to resolve this controversy.
Another example of DPS-like cellular senescence is induced by integrin β3 (ITGB3), via the p21CIP1 and transforming growth factor β (TGFβ) pathways [22].
It remains to be seen if all these different mechanisms share the same pathway as DPS. The lack of DNA damage and highly orchestrated program seems to support this hypothesis, though some differences have been observed, such as the finding that ITGB3, placental, NK and embryogenesis-induced senescence are p21CIP1-dependent, whereas senescence in β cells is p16Ink4a-dependent.
Targets of cell senescence
All cells in an organism (either in adults or developing embryos) are exposed to numerous signals and stressors that might activate either RS, SIPS, or DPS [13].
Supporting this is the finding that cellular senescence occurs in numerous cell types from NK cells, T and B lymphocytes, placental syncytiotrophoblasts, fibroblasts, muscle cells, lens epithelial cells, endothelial cells, β cells, megakaryocytes, foam cells, and post-mitotic neurons to cells from different structures and organs in the developing embryo [23,24,25,26]. This is an example of the complexity of this cellular process, triggered by different stimuli and affecting many (if not most) cells.
Are cellular senescence pathways interconnected?
It has been suggested that senescence pathways evolved from tissue remodeling mechanisms to damage repair processes [15, 16]. This evolution had to deal with several types of environments to develop a suitable cellular response, thus their complexity is not surprising (Fig. 1). In fact, not all tissues accumulate senescent cells to the same degree; for example, in aged mice the liver, skin, lung, and spleen have more senescent cells than the heart, kidney, and skeletal muscle, which show no change between young and old mice [27]. Similarly, in non-human primates, the skin accumulates more senescent cells than skeletal muscle [28, 29].

Scheme of the different cellular senescence pathways. Asterisks indicate that the cell senescence program needs further confirmation. Green arrows indicate a unique route
Determination of which specific cellular senescence pathway is activated can follow a two-step process: (1) what type of stimulus (DNA damage or not) is the cause? and (2) in what cell type/tissue/organ does it occur?
Answering these two questions will determine whether the triggered cellular senescence pathway is DPS, SIPS, or RS. In the case of telomere attrition, the activated pathway is RS, independent of the cell type or tissue. On the other hand, cellular senescence detected in the embryos seems to exclusively correspond to DPS [15, 16]. In adult organisms, DPS has been also detected in placental syncytiotrophoblasts and megakaryocytes [23, 24].
Likewise, two other important factors define the final effect (output) of the “chosen” senescence pathway. (1) Type of response: most senescent cells express a complex senescence-associated secretory phenotype (SASP) [30], whose exact composition determines the influence of senescence on the surrounding environment/tissue. (2) Duration of response: this aspect is mainly related to the clearance efficiency of senescent cells (immunosurveillance) [9].
SASP
An important feature of cellular senescence is SASP, which involves a range of interleukins (ILs), inflammatory factors, chemokines, proteases and growth factors, among others [30, 31]. This secretome is fully active a few days after the persistent damage, and displays diverse functions dependent on both autocrine and paracrine signaling, such as pro-tumorigenic and tumor suppression, inductor or reinforcement of senescence, and pro- and anti-inflammatory effects. Interestingly, SASP composition is different between the senescence pathways, and follows a time-course with secretion of specific components [30, 32]. If this secretome remains chronically active, the effects on the surrounding cells can lead to organ dysfunction and disease. In fact, therapies based on cellular senescence with reduced SASP (via ectopic expression of p16Ink4a) have proven efficient to treat inflammatory diseases, such as rheumatoid arthritis [33].
The type and duration of response are directly linked; for example, in RAS-induced senescence (a type of OIS), there are two types of SASP responses over time [32]. The first is immunosuppressive and profibrotic, whereas the second is pro-inflammatory and fibrolytic [32]. Therefore, in this scenario if the senescent cells are not cleared, there are two antagonistic types of responses, ending in a chronic inflammatory response [8]. On the contrary, quick clearance is followed by an immunosuppressive response. It remains to be elucidated which of these two scenarios are most desirable for proper tissue function, and may be tissue specific. The precise balance in the response (type and timing) is the difference between health and disease (Fig. 1).
Cellular senescence onset: from embryo implantation to old age
The in vivo detection of cellular senescence is a challenge due to the lack of universal markers. The earliest studies showed increasing senescent cells in aged tissues [28, 34], as well as SIPS in several murine and human tissues [35, 36]. Currently, there are many senescence-associated markers, including short telomeres, persistent DNA damage, lack of proliferation, lipofuscin accumulation, altered morphology, loss of lamin B1, SA-β-Gal activity, senescence-associated heterochromatin foci (SAHF), SASP (expression of > 50 factors), and expression of p16Ink4a, ARF, p53, p21CIP1, among others [2]. Improvements in current technology make the detection of senescent cells in tissues more reliable [37, 38].
Paradoxically, a cellular pathway that has been associated with aging is detectable in the first moments of embryonic development. However, the landscape is complex, and the three cellular senescence programs may be active from embryonic stages to old age (Fig. 2), with variable effects on the organism depending on how long the senescent cells exist in specific organs/tissues. This represents a complex equilibrium that separates health and longevity from disease and aging.

The onset of cellular senescence occurs throughout organism development. Acute senescence is represented by “waves”, assuming an effective immunosurveillance process that clears senescent cells. Chronic senescence appears when the burden of old cells increases due to either immunosenescence or the downregulation of immunosurveillance-dependent antigens. Asterisk indicates that the cell senescence pathway is still unknown for some physiological processes. DPS developmentally programmed senescence, SIPS stress-induced premature senescence, RS replicative senescence
Senescent NK cells have been suggested to play an important role in the embryo implantation mechanism [19, 39], initiated by activation of NK receptor CD158d by the fetal trophoblast component, HLA-G, triggering a p21CIP1-dependent DDR signaling pathway in the absence of apparent DNA damage. It appears that this NK senescence could be part of an orchestrated senescence pathway (DPS), where SASP plays a role remodeling the decidua to promote the vascularization required for fetus implantation [40].
Senescence of syncytiotrophoblasts has been shown to play a pivotal role during placental formation. This cell-fusion mechanism is important to maintain the viability of the placenta, and is dependent on syncytin-1’s function, with up-regulation of senescence markers such as SA-β-gal, p16Ink4a, p21CIP1, and p53 [18]. The fusion induces, among other things, cytoskeletal changes which activate proteins upstream of p53 and high reactive oxygen species (ROS) levels, suggesting a SIPS-related process. Although DDR is activated, specific DNA damage has not been described to date [18]. At this stage, failure or deregulation of the cell-fusion process severely compromises the viability of the embryo [17, 18].
During the first weeks of embryo formation, there is an acute accumulation of short-term DPS cells that are rapidly eliminated by macrophages once they have executed their patterning task [15, 16]. These senescent cells are dependent on p21CIP1 activation, from TGFβ/mothers against decapentaplegic (SMAD) and phosphatidylinositol 3-kinase (PI3K)/forkhead box proteins O (FOXO) signaling branches. Interestingly, blocking senescence (i.e., p21 null mice) led to increased incidence (up to 15%) of vaginal septa that could compromise the fertility of females, as well as defects in the apical ectodermal ridge (AER) and patterning. To compensate, an apoptotic program is activated in the absence of senescent cells [15, 16].
What is the trigger for these senescent cells? Neurogenic locus notch (NOTCH) signaling plays a pivotal role in embryogenesis [41], making it a promising candidate, though its role as the trigger for DPS is still undetermined [32]. Several components of NOTCH signaling are upregulated in SIPS, and inhibition of NOTCH prevents or delays senescence. Moreover, ectopic expression of intracellular domains of NOTCH receptors induces senescence. The expression pattern of NOTCH encompasses very early stages of development [42], supporting a possible causative role for DPS.
After birth and during maturation, various stimuli activate the programs of cellular senescence. In this scenario it is plausible that in the first year of life, processes such as wound healing and tissue repair are quite normal, triggered by different types of injuries (stressors).
The acute onset of short-term senescent cells plays an important role in the wound-healing process [43,44,45]. Senescent cells appear at wound sites a few days after the injury, promoting optimal healing by secreting the SASP factor platelet-derived growth factor subunit A (PDGFA) [45]. These senescent cells have been proposed to be part of an orchestrated DNA damage-independent DPS pathway, executing an evolved embryogenic-related role [15]. However, cell senescence in the wound is triggered by CCN1 (also called CYR61, a matricellular cell-adhesion protein highly expressed during wound healing) through the activation of internal DDR mechanisms after accumulation of ROS [44, 46]. Another difference between DPS and senescence in wound repair is the presence of SASP factors (including IL-6 and IL-8) in the latter, which have not been detected in embryos [16, 44]. In this regard, wound-induced cell senescence can be considered a short-term SIPS mechanism, instead of DPS. Perhaps depending on the site of the injury (specific organ/tissue), different factors could be involved, triggering different cellular senescence pathways [47].
Interestingly, senescent cells also secrete plasminogen activator inhibitor 1 (PAI-1) [30], which reduces the capacity for wound healing, thus exerting the opposite effects of PDGFA. The expression level and timing of each SASP factor has been suggested to differ by tissue, depending on short or long-term action (acute vs. chronic) for differential final effects in surrounding cells [32, 48].
A lack of senescent cells in wounds, such as in p16Ink4a/p21CIP1 double-knockout mice, leads to increased fibrosis and less efficient repair, demonstrating that although important, they are not essential [45]. In this sense, a lack of CCN1 also leads to an increase in fibrosis, which persists for a few weeks after wound closure [44] and an absence of senescent cells at the wound sites. Interestingly, CCN1 expression is elevated in several human pathologies, associated with the occurrence of senescent cells in chronologically aged human skin [44, 49].
During repair of skeletal muscle injury, local transient senescent cells promote (facilitate) repair mainly through the SASP factor IL-6 [43]. Injury activates reprogramming of muscle stem cells, and this mechanism is directly related to the onset of neighboring senescent cells. These are short-term (acute) senescent cells, mainly dependent on p16Ink4a and p19Arf activation with an active SASP program (IL-6 secretion) [43]. Thus, similar to the wound-healing mechanism, this pathway can be considered short-term SIPS, though additional studies are necessary to resolve this issue.
When the functions of senescent cells are inhibited, injury-dependent reprogramming is compromised, leading to chronic muscle deterioration [43]. To understand the precise mechanism and cells involved in this repair process, it is very important to consider the injury inductor, because the trajectories of the regenerative process could differ considerably [50].
In youth, depending on the lifestyle, environment, and genetic background, senescent cells arise mainly from two mechanisms: RS and SIPS. Whereas RS is a cell duplication (time)-dependent process involving mainly proliferative tissues through telomere attrition, SIPS is time-independent, leading to cell cycle arrest regardless of telomere length. Both processes are triggered by damage, and are p53-p21CIP1/p16Ink4a-pRB-dependent in most experimental settings [8]. Activation of senescence impairs the ability of damaged cells to proliferate, and thereby acts as a brake for cancer progression (tumor-suppressor mechanism).
In young organisms with fully functional immune systems, senescent cells are effectively cleared by macrophages and NK cells [51]. Thus, at this point there is a continuous onset of short-term and “potentially” long-term senescent cells, effectively removed by the innate immune system. The tumor-suppressor function of senescence is properly balanced within functional immune systems. It is important to note that it isn’t known whether RS cells are cleared by the immune system, unlike the SIPS and DPS-derived ones.
Cell senescence and physiological aging
As we age, the efficiency of our immune cells decays in the face of continuous senescence, leading to the accumulation of aged cells [52] via DPS, RS, and SIPS. Megakaryocytes and NK cells are the only cell type where DPS occurs in adults [23], whereas telomere attrition in mitotic cells leads to RS [5]. Shortened telomeres are associated with some age-related pathologies and increased mortality in elderly people [53], and deletion of telomerase in mice results in premature aging [54]. SIPS can be induced by numerous stressors, and OIS/CIS have been detected in human and murine tissues [12, 35].
While there is no universal marker for senescence, most accepted markers are present in DPS, RS, and SIPS, such as p16Ink4a, p21CIP1, and SA-β-Gal expression. The detection of these markers in aged tissues correlates with the onset of age-related diseases, although in some cases it is still unknown if the effect of senescent cells is beneficial or detrimental. Studies in mice where senescent cells have been cleared by either genetic or pharmacological approaches have shed light on the role of these cells at the onset of age-related diseases such as cardiovascular disease, atherosclerosis, cancer, diabetes, and neurodegenerative disease [23, 53], which represent ~ 30% of all deaths [55].
Cell senescence has been associated with neurodegenerative disorders, such as Alzheimer’s and Parkinson’s diseases [56, 57]. Importantly, some neurological functions were improved when senescent cells were removed either by genetic depletion or senolytics [58, 59], suggesting a detrimental role in the onset of neurodegenerative diseases. Senescence plays a causative role in atherosclerosis and cardiovascular disease, supported not only by the detection of these cells in atherosclerotic plaques but also when they are in vivo removed [23, 60].
In the case of type 2 diabetes (DM2), it seems that senescent cells may play a causative role [53]; however, more work is necessary to confirm the role of senescent β cells in glucose homeostasis, as indicated above. In cancer, the primary role of the senescent pathway is beneficial, as a brake for the progression of the disease [7,8,9]. However, there is strong evidence indicating that chronic senescent cells, through SASP, provide the appropriate environment for cancer development [30]. Senescent cells have been also associated as the cause of other age-related disorders, such as liver cirrhosis, idiopathic pulmonary fibrosis (IPF), cachexia, sarcopenia, osteoarthritis, cataracts, and glomerulosclerosis [23, 53].
In the physiological aging process, there is a moment when the balance between generation and clearance of senescent cells shifts toward their chronic presence, leading to dysfunction and disease in tissues and organs [1, 2, 52] (Fig. 2). It would be interesting to determine what percentages of senescent cells present in old organisms correspond to RS, DPS, or SIPS, and the influence of each on the indicated diseases.
Cell senescence and pathogenesis: lessons from progeroid syndromes
One scenario where cellular senescence programs are likely affected is in premature aging diseases, rare disorders with a prevalence from < 1 to 9 cases per million births, depending on the disease [61,62,63]. A hallmark of most of these diseases is premature/accelerated senescence of cells derived from patients, triggered by accelerated telomere erosion and/or accelerated accumulation of DNA damage [1].
This review will focus on three well-known progeroid syndromes: Werner syndrome (WS), Rothmund–Thomson syndrome (RTS), and Hutchinson–Gilford progeria (HGP), which share premature senescence of cells though differing in lifespan and other clinical features [63,64,65,66,67]. These diseases are also called segmental progerias, as they are associated with many, but not all, of the clinical characteristics seen in the normal aging process [68]. Could some of the clinical features of these diseases be explained by a defective/dysfunctional specific senescence program(s)?
Embryogenesis and DPS
To date, no data exist regarding senescence either from embryos of patients with progeroid syndromes, or from embryos of mouse models mimicking these diseases. Thus, possible dysfunctional DPS during embryogenesis can only be speculated based on the clinical features displayed in progeroid syndrome patients.
In WS, RTS, and HGP, senescence is not inhibited but accelerated, leading to an accumulation of aged cells. However, as DPS is a special pathway that is DNA damage-independent [15, 16], both the timing of onset and the specific SASP components of DPS in progeroid syndromes embryos remain to be elucidated. Defects in developmental pathways could be masked (or attenuated) because embryos are particularly resilient, able to adapt to alterations. For example, and as explained above, a lack of senescence (p21 null mice) activates an apoptotic programme [15, 16]. In this sense, children with WS, HGP, and RTS usually appear healthy at birth, and in some cases develop normally until adolescence [64].
Common clinical features of WS, RTS, and HGP include short stature [average of two standard deviations below the mean (− 2 SD) and − 4.3 SD in WS/RTS and HGP, respectively], skeletal abnormalities, and micrognathia [65, 69, 70]. Interestingly, haploinsufficiency in the homeobox gene SHOX has been implicated in idiopathic short stature (average of − 2.2 SD), skeletal malformations, and micrognathia among other clinical manifestations [71, 72]. SHOX is regulated by signals from the AER, including those from fibroblast growth factor (FGF) and bone morphogenic proteins (BMPs) [72]. AER is an embryonic structure implicated in the proper development of the limbs, and DPS seems to play an important role there [15, 16]. Consistent with this, FGF and BMP-2 are SASP factors [30, 52]. It is tempting to speculate that defects in AER-dependent SHOX signaling due to altered DPS (from a disease-specific gene mutation) contributes to the clinical manifestations indicated above. In this hypothetical scenario, senescent cells carrying mutations in WRN, RecQ4L or LMNA genes, could display a specific phenotype affecting SASP expression.
In this sense, a dysfunctional SASP has been hypothesized in cells lacking lamin A/C (in cases of HGP); these cells fail to activate NF-κB, a transcription factor important in SASP expression [32, 73,74,75]. Conversely, an up-regulation of NF-κB pathways has been detected in Wrn-mutant mice [76]. This situation may be a conserved characteristic of the WS phenotype, where over-expression of pro-inflammatory factors is normal not only in WS senescent cells but also present in WS fibroblasts [77]. Altogether, these results reinforce the hypothesis that progeroid syndromes have an altered SASP, which is already present in the embryo.
Altered SASP could also be responsible for reduced recruitment of macrophages, leading to a continuous presence of senescent cells in the AER due to a clearance deficiency (“chronic-like” SASP signals). On the other hand, a similar situation could occur if the expression of antigens recognized by macrophages was downregulated in progeroid syndrome cells.
SASP expression starts a few days after senescence induction [30], and senescent cells have been detected in the AER within a 3- to 5-day window of mouse embryonic development (7- to 8-day window for the whole embryo) [15, 16]. Thus, the time-course of embryo-related senescence supports the scenario presented here.
As indicated above, NOTCH signaling plays an essential role in embryogenesis [32]. Importantly, cells from transgenic mice mimicking HGP (LMNA c.1824C>T, p.G608G) show downregulation in the NOTCH pathway [73]. In contrast, cells overexpressing progerin, a mutant form of lamin A/C (LMNA gene), activate downstream effectors in the NOTCH signaling pathway (without activation of upstream components), leading to dysregulation of this pathway [78]. While the difference in regulation of NOTCH signaling could be explained by the different experimental systems, it is clear that the NOTCH pathway was altered in both cases.
Wound healing and tissue injuries
One important clinical feature of WS is impaired wound healing, resulting in chronic ulcers, often located around the Achilles tendons and elbows, displaying subcutaneous calcifications and often leading to amputation of feet or lower extremities [64, 79]. WS fibroblasts express higher levels of PAI-1 than normal fibroblasts [80], which as indicated previously, reduces the capacity for wound healing. Importantly, this process can be partially reverted by topical PDGF [81].
The signaling molecule nitric oxide (NO) plays an important role in wound healing, and its deficiency leads to impaired healing [82, 83]. High concentrations of NO induce both cell cycle arrest and growth inhibition [82, 84], which resembles cellular senescence; interestingly, senescent fibroblasts express high levels of NO [30]. In this sense, WS fibroblasts show a premature replicative senescence, leading to an accumulation of these cells in vivo. It is plausible to hypothesize that in WS tissues and in response to injuries, there is a higher level of PAI-1 and NO, which together with the pro-inflammatory environment would lead to cytotoxic effects and impaired wound healing.
It is not clear whether there is a deficiency in wound healing in HGP patients (chronic wounds), although this is likely due to the short life expectancy. However, mouse models of HGP carrying either LMNA mutations or Zmpste24 deficiency display impaired and delayed wound healing, respectively [73, 85].
One of the clinical features in HGP and WS patients is atherosclerosis, which is directly related to injuries in vascular smooth muscle and endothelial cells, leading to coronary lesions [86]. Moreover, and as indicated above, WS fibroblasts express high levels of PAI-1 [80], which is a risk factor for atherosclerosis. In fact, the main cause of death in HGP and WS patients is myocardial infarction. Thus, a role of senescent cells in atherogenesis mainly through SASP has been hypothesized [9].
Altogether these data strongly suggest that following injury to the heart, accelerated accumulation of senescent cells contributes to cardiovascular disease in HGP and WS.
RS/SIPS and cataracts
Cataracts are an age-related disease characterized by opacity of the lens of the eye [87]. The lens is in a constant oxidative environment (high levels of H2O2), which is one of the main causes of cataracts. As human lens epithelial cells (HLECs) are sensitive to oxidative stress, they acquire markers of senescence (SIPS) [88,89,90]. Importantly, lens capsules from patients suffering cataracts show an age-dependent increase of senescent HLECs [91], raising the possibility of a causal role. The strongest experimental evidence for a causative role of senescence in cataract formation is derived from mouse studies, where the specific removal of p16Ink4a-dependent senescent cells decreased incidence of cataracts in old mice (more than a 50% decrease in 2-year-old C57BL/6 mice) [58]. If so, patients with progeroid syndromes would be expected to have an increased incidence of cataracts early in life. Consistent with this, bilateral cataracts are present in 99 and 50% of WS and RTS cases, respectively. The age of onset varies from 30 years in WS to 5–7 years in RTS, in both cases being sub-capsular (lens located) cataracts [62, 64].
Altogether these results suggest than an accelerated RS or SIPS process in WS and RTS would lead to premature senescence in HLECs, triggering cataract formation. Supporting this hypothesis is the finding that WS patients show an in vivo pro-oxidant state [92], and WS fibroblasts display a differential response to H2O2, leading to accumulation of damaged (oxidized) cells [93, 94]. It would be interesting to investigate the response to oxidative stress of HLECs from patients with progeroid syndromes. HGP patients do not show increased incidence of cataracts [95].
Cellular senescence and diabetes
DM2 is an age-related disease that usually develops over the course of years [96, 97], but approximately 70% of WS patients show early onset (at 30 years of age) [64]. Interestingly, RTS and HGP do not show diabetes as a main clinical phenotype [65, 98], in the case of HGP probably due to the short life expectancy.
Cell senescence in insulin-sensitive tissues (such as the adipose tissue) leads to insulin resistance and an increase in β cell mass, which requires high proliferation rates [96, 97]. If this response is not functional, DM2 will progress. It has been suggested that the premature and accelerated senescence of WS β cells contributes to the high rates of DM2 in WS patients, and their failure to respond to increasing insulin demands finally leads to disease [96]. Importantly, the double-null WS mouse model (Terc−/−; Wrn−/−) showed symptoms of DM2 [99], and Wrn−/− mice fed a diabetogenic diet had insulin resistance and hyperinsulinemia [100]. Altogether these results support the hypothesis that a deregulated β cell senescence, due to the lack of WRN protein, is the cause of DM2 in WS patients. However, the precise role of senescent β cells needs further confirmation, as indicated by the contradiction between studies indicated above [20, 21].
Cellular senescence and cancer
Initially, senescence was identified as a tumor-suppression mechanism [8], so why do some progeroid syndromes (premature senescence-prone diseases) show high cancer incidence? In this scenario there are two aspects to consider: (1) the cellular function of the mutated gene; and (2) the influence of the accumulated (premature) senescent cells on neighboring cells (pro-tumorigenic environment).
WS and RTS patients show higher rates of cancer incidence compared to age-matched controls, especially tumors of mesenchymal origin [62, 64]. The mutated genes in both WS and RTS belong to the RecQ helicase family (Wrn and RecQ4L, respectively), implicated in genomic stability [66]. Thus, genomic instability from a lack of these genes, together with the pro-carcinogenic environment of increasing populations of RS cells, is a combination of factors that greatly increases probability of new tumor formation. Data from mice mimicking these diseases support this hypothesis. G1–G3 Terc−/− and Wrn−/− mice die prematurely due to osteosarcomas and soft tissue sarcomas (like WS patients) [99]. Interestingly, G4–G6 Terc−/− and Wrn−/− mice are not cancer-prone [99], probably related to premature death, a situation that resembles HGP. The in vivo accumulation of senescent cells has also been demonstrated in a mouse model of RTS [67].
The most common neoplasms in WS are thyroid carcinoma, melanoma, meningioma, soft tissue sarcoma, primary bone tumors, and leukemia/myelodysplasia, with incidence being 2- to 60-fold higher than age-matched controls [64]. In the case of RTS, the most common cancers are osteosarcoma and skin carcinoma (basal and squamous) [62], which are the main causes of death. Cancer incidence is not increased in HGP. The mutated gene (LMNA) has an important structural function which affects many cellular processes [61], but the short life expectancy of the patients may be responsible for this low incidence.
Altogether these scenarios strongly suggest that continuous defects in the senescence programs, caused by mutations in Wrn, RecQ4L, or LMNA, can lead to some clinical characteristics of WS, RTS, and HGP, respectively (Table 1). Whether efficient clearance of these senescent cells could serve as a potential therapy for reducing cancer incidence in these patients remains to be determined.
Longevity: clearance efficiency of senescent cells matters
Both senescent and immune cells (mainly macrophages and NK cells) must be coordinated. Senescent cells, besides other physiological processes, “call” to immune cells through SASP, and the immune cells clear them in order to avoid their accumulation (Fig. 3). As we age, our immune system weaken, thus we accumulate more senescent cells. Once this balance has been upset, the risk for age-related diseases increases [1, 2] (Fig. 3).

Onset of cellular senescence and time-dependent effects. After persistent stimuli, a normal cell becomes senescent. The first days show a beneficial cellular response, affecting several physiological processes (represented in 1). Subsequently, senescent cells attract immune cells (mainly macrophages) through SASP, for proper and controlled clearance (represented in 2). When the immune system is not functioning efficiently (e.g., immunosenescence), the accumulation of senescent cells increases (as in a flood caused by a dam eliminating water in an uncontrolled manner), leading to undesirable affects (represented in 3)
Immunosenescence is the decline of functions of the immune system driven by age. In this age-related decline, there are two main causes: hematopoietic stem cell (HSC) exhaustion and cellular senescence of immune cells [101].
HSC exhaustion leads to diminished immunity and reduced NK cell activity, among other effects [53, 102]. NK cells, as explained before, are involved in clearance of senescent cells. Thus, in this scenario, aging of HSCs leads to reduced NK cell activity, which in turn contributes to the accumulation of senescent cells. Remarkably, healthy elderly individuals and centenarians show an increase of total NK cells [103]. It would be interesting to correlate these data with accumulation of senescent cells in different tissues.
NK cells recognize the receptor CD58/ICAM1 present in senescent cells, which in turn express IL-15, among other factors, which up-regulate NK cells receptors important for killing senescent cells [40]. Interestingly in a mouse model of lung metastasis, inhibition of NK cells led to increased cancer metastasis. Perhaps in this scenario senescent cells become chronic, leading to cancer progression [104].
Senescence of immune cells has not been as explored as in other cell types. Some markers are not indicative of senescent cells, but perhaps a new subset of active “aged” immune cells [51], and there are studies of aged organisms showing decreased activity in specific immune cell types.
B and T lymphocytes are key players of the adaptive immune response. A decline of their functions could explain some age-related pathologies. Elderly individuals show (1) decrease antibody responses, leading to increase susceptibility to infectious diseases, (2) reduction in B cell repertoire diversity, and (3) low plasma cells in the bone marrow [105]. At the cellular level it has been described that some B-cells subsets display senescent markers, as is the case of late/exhausted memory (LM) B cells. LM B-cells have shorter telomeres, increased p16Ink4a expression and over-expression of pro-inflammatory cytokines [106]. Moreover, switched memory B-cells, implicated in the antibody responses, decrease with age [105].
T cells are generated in the thymus, and this organ decays with age, in a process that remains to be understood [105, 107]. Elderly individuals show (1) reduced T-cell responses to neoantigens, (2) increased terminally differentiated T lymphocytes, (3) decreased T-cell receptor rearrangement, and iv) increased senescent T-cells [105]. The accumulation of terminally differentiated T cells is related to hyper-inflammatory status. Senescent T cells display surface senescent markers, short telomeres and express a SASP involve in immune functions. Intriguingly, in some T-cells subsets, senescence (independent of telomeric shortening) may be triggered by ROS [108]. Some of these age-associated characteristics can be found in young individuals after excessive T-cell proliferation (e.g., chronic infections). Importantly, the senescence status of T-cells is reversible, opening new avenues for therapies enhancing immune responses [108].
Macrophages are present in all vertebrate tissues [109], and studies in aged organisms have identified senescent foamy macrophages as playing a causative role in atherosclerosis [60]. Moreover, there is a shift to type M2 macrophages during aging, which correlates with reduced immune response, tumor promotion, and impaired phagocytosis and chemotaxis, among other physiological consequences [109, 110]. It is tempting to speculate that reduced chemotaxis is involved in impaired capacity to migrate to the places where senescent cells accumulate (e.g., impaired response to SASP factors). Thus, in this scenario macrophages senescence would contribute to senescent cell accumulation.
As macrophages are key immune cells for the clearing of senescent cells, their absence should lead to high levels of senescence markers (unless other compensatory pathways are activated) and/or the failure of senescence-related mechanisms. Consistent with this, depletion of macrophages in MaFIA-transgenic mice showed increased levels of SASP pro-inflammatory cytokines, such as IL-1β, IL-6, IL-8, tumor necrosis factor (TNF), and granulocyte colony-stimulating factor (GCSF) [111], as well as an impairment in some tissue repair processes [112].
A recent study suggested that elimination of senescent cells by macrophages may play a role in tissue/organ regeneration. This mechanism could explain why some organisms, such as salamanders, are able to undergo indefinite rounds of regeneration [113]. Another study focused on the importance of macrophages in epimorphic regeneration in African spiny mice [114]. Depletion of macrophages during limb and fin regeneration resulted in the failure of this process [113, 115, 116].
Interestingly, macrophage self-renewal is suggested to be a relevant parameter of aging, with the SIRT1 protein acting as a key factor for this process [117]. SIRT1 belongs to the sirtuin family of NAD-dependent protein deacetylases and ADP ribosyltransferases, and has been implicated in genomic stability and enhanced metabolic efficiency, leading to healthy aging [1].
Macrophages have several functional states, with M1 and M2 as the extremes. M1 macrophages are implicated in inflammation, tumor destruction and tissue damage, whereas M2 macrophages are associated with tumor promotion and tissue remodeling [118]. Cytokines and cellular environment are key aspects that determine M1/M2 polarization. For example, IL-13 and interferon gamma (IFN-γ) are triggers for the M2 and M1 phenotype, respectively [118]. SASP is a very heterogeneous system and, as indicated above, its composition is dependent on the particular cellular senescence pathway [30]. Cells undergoing RS express IL-13 but not IFN-γ, while SIPS is associated with both, in addition to other cytokines [30]. Thus, it is a plausible scenario in which RS cells accumulate and promote tumorigenesis, whereas accumulation of SIPS cells could be both pro-inflammatory (atherosclerosis-prone) and pro-tumorigenic, depending on the time-course of specific cytokines secreted. Moreover, the presence of other immune cells can influence the M1/M2 polarization process [118], suggesting a finely balanced regulation of macrophage functions that leads ultimately to either tissue remodeling or destruction.
Interestingly, WS cells display premature RS and SIPS, and some of the main clinical features of WS patients are high tumor incidence, chronic ulcers and atherosclerosis [64].
It is plausible that organisms with an efficient mechanism to clear senescent cells better deal with aging-related processes. In this regard, it has been hypothesized that long-lived organisms are equipped with an immune system that efficiently eliminates these types of cells [119]. Such a process would enable them to avoid or delay aging-related diseases and to live longer. By contrast, a dysfunctional immune system has been detected in a progeria mouse model, leading to immunosenescence, susceptibility to infection [120], and defects in B-cell development [121].
Concluding remarks
Senescent cells are considered a hallmark of aging [1]. Far from being inactive, senescent cells are at the crossroads of several cellular pathways, and act as a driver for either disease or health (Fig. 4). The onset of the different cellular senescence pathways occurs throughout development, in response to a plethora of stimuli. Deregulation of this process, as in some pathological conditions, has consequences that can be fatal for the organism. In addition, senescent cells are the final outcome of some anti-cancer therapies (e.g., TIS), although their accumulation (as occurs during normal aging) is deleterious for the organism unless a robust immunosurveillance mechanism remains active (Fig. 4).

Old cells to hyperactive ones: senescent cells at the crossroads of several pathways and therapies
Interestingly, senescent cells play a key role in in vivo reprogramming, which could affect tissue responses to damage [122]. This indicates a key role of epigenetic remodeling during aging, opening new avenues for anti-aging therapies. Current therapies are also focused on targeting senescent cells by: (1) selectively killing them by senolytics; (2) inhibiting SASP functions; or (3) improving recognition and clearance by immune cells [25, 59, 123, 124].
The list of senolytics is continuously growing, with several compounds that specifically kill senescent cells by targeting specific cellular pathways and proteins, such as apoptosis (e.g., dasatinib, quercetin, ABT 737 and 263, A1331852, A1155463), chaperone protein HSP90 (e.g., alvespimycin, tanespimycin, geldanamycin), histone deacetylase (panobinostat), or the FOXO4-p53 interaction (DRI), among others [25, 59, 124]. Some clinical trials are currently ongoing, using a combination of dasatinib and quercetin (D + Q) [25, 59].
SASP inhibitors are drugs that reduce the deleterious effects of chronic senescent cells. These inhibitors mainly target NF-κB (SB203586, UR-135756 and BIRB 796), p38MAPK (resveratrol, apigenin, wogonin, kaempferol), IL-1A (cortisol/corticosterone) and mTOR (rapamycin) functions. The drug BIRB 796 is currently in phase III trials [101, 123].
Other approaches are based on the improvement of NK cells, macrophages, and CD4+ T cells to specifically recognize and remove senescent cells. This strategy would counteract the decline of the immune system by age [123].
Altogether, these scenarios place senescence as a key mechanism in the aging process and longevity. According to some theories of aging, the balance between energy used on different processes could define the longevity of the organism [125]. In this sense, an effective senescent cell clearance mechanism is linked to health, and in contrast, accumulation of senescent cells leads to disease.
There are several questions regarding cellular senescence programs that remain to be answered. For example, could an RS cell be converted into a SIPS cell (in terms of SASP expression) following peroxide exposure? Over time, if RS cells (with their specific SASP) become damaged by insults other than telomere attrition, could that trigger the activation of a new secretome? What percentages of senescent cells present in old organisms correspond to RS, DPS or SIPS? It is also important to keep in mind that in some diseases, cellular senescence may be a consequence and not the cause of the clinical features; in these situations, targeting senescent cells would not alleviate the symptoms.
Future work is required to shed light on the exact role(s) of cellular senescence in the organism, from the developing embryo through old age.
References
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153:1194–1217. https://doi.org/10.1016/j.cell.2013.05.039
He S, Sharpless NE (2017) Senescence in health and disease. Cell 169:1000–1011. https://doi.org/10.1016/j.cell.2017.05.015
Hayflick L, Moorhead PS (1961) The serial cultivation of human diploid cell strains. Exp Cell Res 25:585–621
Olovnikov AM (1996) Telomeres, telomerase, and aging: origin of the theory. Exp Gerontol 31:443–448
Greider CW (1998) Telomeres and senescence: the history, the experiment, the future. Curr Biol 8(5):178–181
Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, Bucci G, Dobreva M, Matti V, Beausejour CM, Herbig U, Longhese MP, d’Adda di Fagagna F (2012) Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol 14:355–365. https://doi.org/10.1038/ncb2466
Campisi J, d’Adda di Fagagna F (2007) Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8:729–740. https://doi.org/10.1038/nrm2233
Collado M, Blasco MA, Serrano M (2007) Cellular senescence in cancer and aging. Cell 130:223–233. https://doi.org/10.1016/j.cell.2007.07.003
Childs BG, Durik M, Baker DJ, van Deursen JM (2015) Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 21(12):1424–1435. https://doi.org/10.1038/nm.4000
Chen Q, Ames BN (1994) Senescent-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc Natl Acad Sci USA 91:4130–4134
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997) Oncogenic ras provokes premature senescence associated with accumulation of p53 and p16Ink4a. Cell 88:593–602
Hernández-Segura A, Nehme J, De Maria M (2018) Hallmarks of cellular senescence. Trends Cell Biol. https://doi.org/10.1016/j.tcb.2018.02.001
Bielak-Zmijewska A, Mosieniak G, Sikora E (2017) Is DNA damage indispensable for stress-induced senescence? Mech Ageing Dev. https://doi.org/10.1016/j.mad.2017.08.004
Ott C, Jung T, Grune T, Höhn A (2017) SIPS as a model to study age-related changes in proteolysis and aggregate formation. Mech Ageing Dev. https://doi.org/10.1016/j.mad.2017.07.007
Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, Rodríguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, Serrano M (2013) Programmed cell senescence during mammalian embryonic development. Cell 155:1–15. https://doi.org/10.1016/j.cell.2013.10.019
Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, Keyes WM (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155:1119–1130. https://doi.org/10.1016/j.cell.2013.10.041
Cox LS, Redman C (2017) The role of cellular senescence in ageing of the placenta. Placenta 52:139–145
Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, Krizhanovsky V (2013) Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev 27:2356–2366. https://doi.org/10.1101/gad.227512.113
Rajagopalan S, Long EO (2012) Cellular senescence induced by CD158d reprograms natural killer cells to promote vascular remodeling. Proc Natl Acad Sci USA 109:20596–20601
Helman A, Klochendler A, Azazmeh N, Gabai Y, Horwitz E, Anzi S, Swisa A, Condiotti R, Granit RZ, Nevo Y, Fixler Y, Shreibman D, Zamir A, Tornovsky-Babeay S, Dai C, Glaser B, Powers AC, Shapiro AM, Magnuson MA, Dor Y, Ben-Porath I (2016) p16Ink4a-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med 22(4):412–422. https://doi.org/10.1038/nm.4054
Aguayo-Mazzucato C, van Haaren M, Mruk M, Lee TB Jr, Crawford C, Hollister-Lock J, Sullivan BA, Johnson JW, Ebrahimi A, Dreyfuss JM, Van Deursen J, Weir GC, Bonner-Weir S (2017) β cell aging markers have heterogeneous distribution and are induced by insulin resistance. Cell Metab 25:898–910. https://doi.org/10.1016/j.cmet.2017.03.015
Rapisarda V, Borghesan M, Miguela V, Encheva V, Snijders AP, Lujambio A, O’Loghlen A (2017) Integrin beta 3 regulates cellular senescence by activating the TGF-β pathway. Cell Rep 18:2480–2493. https://doi.org/10.1016/j.celrep.2017.02.012
Muñoz-Espín D, Serrano M (2014) Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15:482–496. https://doi.org/10.1038/nrm3823
Besancenot R, Chaligne R, Tonetti C, Pasquier F, Marty C, Lecluse Y, Vainchenker W, Constantinescu SN, Giraudier S (2010) A senescence-like cell-cycle arrest occurs during megakaryocytic maturation: implications for physiological and pathological megakaryocytic proliferation. 8(9):e1000476. https://doi.org/10.1371/journal.pbio.1000476
Katsuumi G, Shimizu I, Yoshida Y, Minamino T (2018) Vascular senescence in cardiovascular and metabolic diseases. Front Cardiovasc Med 5:18. https://doi.org/10.3389/fcvm.2018.00018
Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, Gonos ES, Thrasivoulou C, Saffrey MJ, Cameron K, von Zglinicki T (2012) Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11:996–1004. https://doi.org/10.1111/j.1474-9726.2012.00870.x
Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T (2009) DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 8:311–323. https://doi.org/10.1111/j.1474-9726.2009.00481.x
Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM (2006) Cellular senescence in aging primates. Science 311:1257
Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U (2007) Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev 128(1):36–44
Coppe JP, Desprez PY, Krtolica A, Campisi J (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5:99–118
Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6(12):2853–2868. https://doi.org/10.1371/journal.pbio.0060301
Ito Y, Hoare M, Narita M (2017) Spatial and temporal control of senescence. Trends in Cell Biol. https://doi.org/10.1016/j.tcb.2017.07.004
Taniguchi K, Kohsaka H, Inoue N, Terada Y, Ito H, Hirokawa K, Miyasaka N (1999) Induction of the p16INK4a senescence gene as a new therapeutic strategy for the treatment of rheumatoid arthritis. Nat Med 5:760–767. https://doi.org/10.1038/10480
Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medranos EE, Linskens M, Rubelj I, Pereire-Smith O, Peacocke M, Campisi J (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 92:9363–9367
Sharpless NE, DePinho R (2005) Crime and punishment. Nature 436:636. https://doi.org/10.1038/436636a
Salama R, Sadaie M, Hoare M, Narita M (2014) Cellular senescence and its effector programs. Genes Dev 28(2):99–114. https://doi.org/10.1101/gad.235184.113
Evangelou K, Lougiakis N, Rizou SV, Kotsinas A, Kletsas D, Muñoz-Espín D, Kastrinakis NG, Pouli N, Marakos P, Townsend P, Serrano M, Bartek J, Gorgoulis VG (2017) Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 16(1):192–197. https://doi.org/10.1111/acel.12545
Galbiati A, Beauséjour C, d’Adda di Fagagna F (2017) A novel single-cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues. Aging Cell 16(2):422–427. https://doi.org/10.1111/acel.12573
Rajagopalan S (2014) HLA-G-mediated NK cell senescence promotes vascular remodeling: implications for reproduction. Cell Mol Immunol 11:460–466. https://doi.org/10.1038/cmi.2014.53
Vicente R, Mausset-Bonnefont AL, Jorgensen C, Louis-Plence P, Brondello JM (2016) Cellular senescence impact on immune cell fate and function. Aging Cell 15:400–406. https://doi.org/10.1111/acel.12455
Louvi A, Artavanis-Tsakonas S (2006) Notch signalling in vertebrate neural development. Nat Rev 7:93–102
Cormier S, Vandormael-Pournin S, Babinet C, Cohen-Tannoudji M (2004) Developmental expression of the Notch signaling pathway genes during mouse preimplantation development. Genes Expr Patterns 4:713–717. https://doi.org/10.1016/j.modgep.2004.04.003
Chiche A, Le Roux I, von Joest M, Sakai H, Aguín SB, Cazin C, Salam R, Fiette L, Alegria O, Flamant P, Tajbakhsh S, Li H (2017) Injury-induced senescence enables in vivo reprogramming in skeletal muscle. Cell Stem Cell 20:407–414. https://doi.org/10.1016/j.stem.2016.11.020
Jun JI, Lau LF (2011) Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov 10(12):945–963
Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dollé ME, Hoeijmakers JH, de Bruin A, Hara E, Campisi J (2014) An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31(6):722–733. https://doi.org/10.1016/j.devcel.2014.11.012
Borkham-Kamphorst E, Schaffrath C, Van de Leur E, Haas U, Tihaa L, Meurer SK, Nevzorova YA, Liedtke C, Weiskirchen R (2014) The anti-fibrotic effects of CCN1/CYR61 in primary portal myofibroblasts are mediated through induction of reactive oxygen species resulting in cellular senescence, apoptosis and attenuated TGF-β signaling. Biochim Biophys Acta 1843:902–914. https://doi.org/10.1016/j.bbamcr.2014.01.023
Childs BG, Gluscevic M, Baker DJ, Laberge RM, Marquess D, Dananberg J, van Deursen JM (2017) Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov 16(10):718–735. https://doi.org/10.1038/nrd.2017.116
Rao SG, Jackson JG (2016) SASP: tumor suppressor or promoter? Yes! Trends Cancer 2(11):676–687
Du J, Klein JD, Hassounah F, Zhang J, Zhang C, Wang XH (2014) Aging increases CCN1 expression leading to muscle senescence. Am J Physiol Cell Physiol 306:28–36. https://doi.org/10.1152/ajpcell.00066.2013
Hardy D, Besnard A, Latil M, Jouvion G, Briand D, Thépenier C, Pascal Q, Guguin A, Gayraud-Morel B, Cavaillon JM, Tajbakhsh S, Rocheteau P, Chrétien F (2016) Comparative study of injury models for studying muscle regeneration in mice. PLoS One. https://doi.org/10.1371/journal.pone.0147198
Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin I, Leonova K, Polinsky A, Chernova OB, Gudkov AV (2016) Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging 8(7):1294–1315. https://doi.org/10.18632/aging.100991
Burton DGA, Krizhanovsky V (2014) Physiological and pathological consequences of cellular senescence. Cell Mol Life Sci 71:4373–4386. https://doi.org/10.1007/s00018-014-1691-3
McHugh D, Gil J (2017) Senescence and aging: causes, consequences, and therapeutic avenues. J Cell Biol 217(1):65–77. https://doi.org/10.1083/jcb.201708092
Lee HW, Blasco MA, Gottlieb GJ, Horner JW, Greider CW, DePinho RA (1998) Essential role of mouse telomerase in highly proliferative organs. Nature 392:569–574. https://doi.org/10.1038/33345
World Health Organization (2018) Global Health Observatory (GHO) data. https://www.who.int/gho/mortality_burden_disease/en/
Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, Johnson FB, Trojanowski JQ, Sell C, Torres C (2012) Astrocyte senescence as a component of Alzheimer’s disease. PLoS One 7(9):e45069. https://doi.org/10.1371/journal.pone.0045069
Chinta SJ, Lieu CA, Demaria M, Laberge RM, Campisi J, Andersen JK (2013) Environmental stress, ageing and glial cell senescence: a novel mechanistic link to Parkinson’s disease? J Intern Med 273(5):429–436. https://doi.org/10.1111/joim.12029
Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM (2016) Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530(7589):184–1899. https://doi.org/10.1038/nature16932
Kirkland JL, Tchkonia T (2017) Cellular senescence: a translational perspective. EBiomedicine 21:21–28. https://doi.org/10.1016/j.ebiom.2017.04.013
Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM (2016) Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354(6311):472–477. https://doi.org/10.1126/science.aaf6659
Ahmed MS, Ikran S, Bibi N, Mir A (2017) Hutchinson–Gilford progeria syndrome: a premature aging disease. Mol Neurobiol. https://doi.org/10.1007/s12035-017-0610-7
Larizza L, Roversi G, Volpi L (2010) Rothmund–Thomson syndrome. Orphanet J Rare Dis. https://doi.org/10.1186/1750-1172-5-2
Coppede F (2012) Premature aging syndrome. Neurodegenerative Dis. Landes Bioscience and Springer Science + Business Media, New York, pp 317–331
Oshima J, Sidorova JM, Monnat RJ Jr (2017) Werner syndrome: clinical features, pathogenesis and potential therapeutic interventions. Ageing Res Rev 33:105–114. https://doi.org/10.1016/j.arr.2016.03.002
Halaschek-Wiener J, Brooks-Wilson A (2007) Progeria of stem cells: stem cell exhaustion in Hutchinson–Gilford progeria syndrome. J Gerontol A Biol Sci Med Sci 1:3–8
Muftuoglu M, Oshima J, von Kobbe C, Cheng WH, Leistritz DF, Bohr VA (2008) The clinical characteristics of Werner syndrome: molecular and biochemical diagnosis. Hum Genet 124:369–377. https://doi.org/10.1007/s00439-008-0562-0
Lu H, Fang EF, Sykora P, Kulikowicz T, Zhang Y, Becker KG, Croteau DL, Bohr VA (2014) Senescence induced by RECQL4 dysfunction contributes to Rothmund–Thomson syndrome features in mice. Cell Death Dis. https://doi.org/10.1038/cddis.2014.168
Opresko PL, Cheng WH, von Kobbe C, Harrigan JA, Bohr VA (2003) Werner syndrome and the function of the Werner protein; what they can teach us about the molecular aging process. Carcinogenesis 24(5):791–802
Hilhorst-Hofstee Y, Shah N, Atherton D, Harper JI, Milla P, Winter RM (2000) Radial aplasia, poikiloderma and auto-immune enterocolitis–new syndrome or severe form of Rothmund–Thomson syndrome? Clin Dysmorphol 9(2):79–85
Gordon CM, Gordon LB, Snyder BD, Nazarian A, Quinn N, Huh S, Giobbie-Hurder A, Neuberg D, Cleveland R, Kleinman M, Miller DT, Kieran MW (2011) Hutchinson–Gilford progeria is a skeletal dysplasia. J Bone Miner Res 26(7):1670–1679. https://doi.org/10.1002/jbmr.392
Yu L, Liu H, Yan M, Yang J, Long F, Muneoka K, Chen Y (2007) Shox2 is required for chondrocyte proliferation and maturation in proximal limb skeleton. Dev Biol 306(2):549–559. https://doi.org/10.1016/j.ydbio.2007.03.518
Tiecke E, Bangs F, Blaschke R, Farrell ER, Rappold G, Tickle C (2006) Expression of the short stature homeobox gene Shox is restricted by proximal and distal signals in chick limb buds and affects the length of skeletal elements. Dev Biol 298:585–596. https://doi.org/10.1016/j.ydbio.2006.07.008
Rosengardten Y, McKenna T, Grochová D, Eriksson M (2011) Stem cell depletion in Hutchinson–Gilford progeria syndrome. Aging Cell 10:1011–1020. https://doi.org/10.1111/j.1474-9726.2011.00743.x
Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT (2004) Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Investig 113(3):370–378. https://doi.org/10.1172/JCI19670
Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, Kogan SC, Lowe SW (2011) Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev 25:2125–2136. https://doi.org/10.1101/gad.17276711
Turaga RV, Paquet ER, Sild M, Vignard J, Garand C, Johnson FB, Masson JY, Lebel M (2009) The Werner syndrome protein affects the expression of genes involved in adipogenesis and inflammation in addition to cell cycle and DNA damage responses. Cell Cycle 8(13):2080–2092. https://doi.org/10.4161/cc.8.13.8925
Davis T, Kipling D (2006) Werner Syndrome as an example of inflamm-aging: possible therapeutic opportunities for a progeroid syndrome? Rejuvenation Res. 9(3):402–407. https://doi.org/10.1089/rej.2006.9.402
Scaffidi P, Misteli T (2008) Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol 10(4):452–459. https://doi.org/10.1038/ncb1708
Yeong EK, Yang CC (2004) Chronic ulcers in Werner’s syndrome. Br Assoc Plast Surg 57:86–88. https://doi.org/10.1016/j.bjps.2003.10.011
Goldstein S, Moerman EJ, Fujii S, Sobel BE (1994) Overexpression of plasminogen activator inhibitor type-1 in senescent fibroblasts from normal subjects and those with Werner syndrome. J Cell Physiol 161(3):571–579
Wollina U, Gruner M, Koch A, Köstler E, Hubl W, Hanson NB, Oshima J (2004) Topical PDGF-BB results in limited healing in a patient with Werner’s syndrome and chronic leg ulcers. J Wound Care 13(10):415–416. https://doi.org/10.12968/jowc.2004.13.10.26690
Park JH, Kim JY, Kim DJ, Kim M, Chang M, Chuck RS, Park CY (2017) Effect of nitric oxide on human corneal epithelial cell viability and corneal wound healing. Sci Rep 7:8093. https://doi.org/10.1038/s41598-017-08576-9
Yamasaki K, Edington HD, McClosky C, Tzeng E, Lizonova A, Kovesdi I, Steed DL, Billiar TR (1998) Reversal of impaired wound repair in iNOS-deficient mice by topical adenoviral-mediated iNOS gene transfer. J Clin Investig 101(5):967–971. https://doi.org/10.1172/JCI2067
Kim JC, Cheong TB, Park GS, Park MH, Kwon NS, Yoon HY (2002) The role of nitric oxide in ocular surface diseases. Adv Exp Med Biol 506:687–695
Butala P, Szpalski C, Soares M, Davidson EH, Knobel D, Warren SM (2012) Zmpste24−/− mouse model for senescent wound healing research. Plast Reconstr Surg 130(6):788–798. https://doi.org/10.1097/PRS.0b013e31826d102b
Huang S, Chen L, Libina N, Janes J, Martin GM, Campisi J, Oshima J (2005) Correction of cellular phenotypes of Hutchinson–Gilford Progeria cells by RNA interference. Hum Genet 118:444–450. https://doi.org/10.1007/s00439-005-0051-7
Spector A (1995) Oxidative stress-induced cataract: mechanism of action. FASEB J 9(12):1173–1182
Ovadya Y, Krizhanovsky V (2014) Senescent cells: SASPected drivers of age-related pathologies. Biogerontology 15:627–642. https://doi.org/10.1007/s10522-014-9529-9
Li S, Chen X, Lai W, Hu M, Zhong X, Tan S, Liang H (2017) Downregulation of SMP30 in senescent human lens epithelial cells. Mol Med Rep 16(4):4022–4028. https://doi.org/10.3892/mmr.2017.7106
Zhang ZF, Zhang J, Hui YN, Zheng MH, Liu XP, Kador PF, Wang YS, Yao LB, Zhou J (2011) Up-regulation of NDRG2 in senescent lens epithelial cells contributes to age-related cataract in human. PLoS One 6(10):e26102. https://doi.org/10.1371/journal.pone.0026102
Fu Q, Qin Z, Yu J, Yu Y, Tang Q, Lyu D, Zhang L, Chen Z, Yao K (2016) Effects of senescent lens epithelial cells on the severity of age-related cortical cataract in humans: a case-control study. Medicine (Baltimore). https://doi.org/10.1097/MD.0000000000003869
Pagano G, Zatterale A, Degan P, d’Ischia M, Kelly FJ, Pallardó FV, Kodama S (2005) Multiple involvement of oxidative stress in Werner syndrome phenotype. Biogerontology 6(4):233–243. https://doi.org/10.1007/s10522-005-2624-1
von Kobbe C, May A, Grandori C, Bohr VA (2004) Werner syndrome cells escape hydrogen peroxide-induced cell proliferation arrest. FASEB J. https://doi.org/10.1096/fj.04-1895fje
von Kobbe C, Harrigan JA, May A, Opresko PL, Dawut L, Cheng WH, Bohr VA (2003) Central role for the Werner syndrome protein/poly(ADP-ribose) polymerase 1 complex in the poly(ADP-ribosyl)ation pathway after DNA damage. Mol Cell Biol 23(23):8601–8613
Coutinho HDM, Falcão-Silva VS, Gonçalves GF, da Nóbrega RB (2009) Molecular ageing in progeroid syndromes: Hutchinson–Gilford progeria syndrome as a model. Immun Ageing. https://doi.org/10.1016/j.arr.2016.03.002
Cox LS (2008) Hypothesis: causes of type 2 diabetes in progeroid Werner syndrome. Open Longev Sci 2:100–103
Kahn SE, Cooper ME, Del Prato S (2014) Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet 383:1068–1083. https://doi.org/10.1016/S0140-6736(13)62154-6
de Renty C, Ellis NA (2017) Bloom’s syndrome: why not premature aging? A comparison of the BLM and WRN helicases. Ageing Res Rev 33:36–51. https://doi.org/10.1016/j.arr.2016.05.010
Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, Lombard D, Pathak S, Guarente L, DePinho RA (2004) Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet 36(8):877–882. https://doi.org/10.1038/ng1389
Moore G, Knoblaugh S, Gollahon K, Rabinovitch P, Ladiges W (2008) Hyperinsulinemia and insulin resistance in Wrn null mice fed a diabetogenic diet. Mech Ageing Dev 129(4):201–206. https://doi.org/10.1016/j.mad.2007.12.009
Burton DGA, Stolzing A (2018) Cellular senescence: immunosurveillance and future immunotherapy. Aging Res Rev 48:17–25. https://doi.org/10.1016/j.arr.2018.02.001
Mocchegiani E, Malavolta M (2004) NK and NKT cell functions in immunosenescence. Aging Cell 3(4):177–184. https://doi.org/10.1111/j.1474-9728.2004.00107.x
Campos C, Pera A, Lopez-Fernandez I, Alonso C, Tarazona R, Solana R (2014) Proinflammatory status influences NK cells subsets in the elderly. Immunol Lett 162:298–302. https://doi.org/10.1016/j.imlet.2014.06.015
Shimaoka H, Takeno S, Maki K, Sasaki T, Hasegawa S, Yamashita Y (2017) A cytokine signal inhibitor for rheumatoid arthritis enhances cancer metastasis via depletion of NK cells in an experimental lung metastasis mouse model of colon cancer. Oncol Lett 14(3):3019–3027. https://doi.org/10.3892/ol.2017.6473
Pinti M, Appay V, Campisi J, Frasca D, Fülöp T, Sauce D, Larbi A, Weinberger B, Cossarizza A (2016) Aging of the immune system: focus on inflammation and vaccination. Eur J Immunol 46(10):2286–2301. https://doi.org/10.1002/eji.201546178
Frasca D, Diaz A, Romero M, Blomberg BB (2017) Human peripheral late/exhausted memory B cells express a senescent-associated secretory phenotype and preferentially utilize metabolic signaling pathways. Exp Gerontol 87:113–120. https://doi.org/10.1016/j.exger.2016.12.001
Gruver AL, Hudson LL, Sempowski GD (2007) Immunosenescence of ageing. J Pathol 211:144–156. https://doi.org/10.1002/path.2104
Akbar AN, Henson SM, Lanna A (2016) Senescence of T lymphocytes: implications for enhancing human immunity. Trends Immunol 37:866–876. https://doi.org/10.1016/j.it.2016.09.002
Gordon S, Plüddemann A (2017) Tissue macrophages: heterogeneity and functions. BMC Biol 15:53. https://doi.org/10.1186/s12915-017-0392-4
Rawji KS, Mishra MK, Michaels NJ, Rivest S, Stys PK, Yong VW (2016) Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain 139:653–661. https://doi.org/10.1093/brain/awv395
Wu CL, McNeill J, Goon K, Little D, Kimmerling K, Huebner J, Kraus V, Guilak F (2017) Conditional macrophage depletion increases inflammation and does not inhibit the development of osteoarthritis in obese macrophage Fas-induced apoptosis–transgenic mice. Arthritis Rheumatol 69(9):1772–1783. https://doi.org/10.1002/art.40161
Burnett SH, Beus BJ, Avdiushko R, Qualls J, Kaplan AM, Cohen DA (2006) Development of peritoneal adhesions in macrophage depleted mice. J Surg Res 131(2):296–301. https://doi.org/10.1016/j.jss.2005.08.026
Yun MH, Davaapil H, Brockes JP (2015) Recurrent turnover of senescent cells during regeneration of a complex structure. eLife. https://doi.org/10.7554/elife.05505
Simkin J, Gawriluk TR, Gensel JC, Seifert AW (2017) Macrophages are necessary for epimorphic regeneration in African spiny mice. eLife. https://doi.org/10.7554/elife.24623
Godwin JW, Pinto AR, Rosenthal NA (2013) Macrophages are required for adult salamander limb regeneration. Proc Natl Acad Sci USA 110(23):9415–9420. https://doi.org/10.1073/pnas.1300290110
Petrie TA, Strand NS, Tsung-Yang C, Rabinowitz JS, Moon RT (2014) Macrophages modulate adult zebrafish tail fin regeneration. Development 141:2581–2591. https://doi.org/10.1242/dev.098459
Imperatore F, Maurizio J, Vargas Aguilar S, Busch CJ, Favret J, Kowenz-Leutz E, Cathou W, Gentek R, Perrin P, Leutz A, Berruyer C, Sieweke MH (2017) SIRT1 regulates macrophage self-renewal. EMBO J 36(16):2353–2372. https://doi.org/10.15252/embj.201695737
Biswas SK, Mantovani A (2010) Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 11(10):889–896. https://doi.org/10.1038/ni.1937
Peeper DS (2011) Old cells under attack. Nature 479:186–187. https://doi.org/10.1038/479186a
Xin L, Jiang TT, Kinder JM, Ertelt JM, Way SS (2015) Infection susceptibility and immune senescence with advancing age replicated in accelerated aging LmnaDhe mice. Aging Cell 14:1122–1126. https://doi.org/10.1111/acel.12385
Liu B, Zhou S, Liu X, Zhou K, Zhang F, Zhou Z (2013) Accumulation of prelamin A compromises NF-κB-regulated B-lymphopoiesis in a progeria mouse model. Longev Healthspan. https://doi.org/10.1186/2046-2395-2-1
Mosteiro L, Pantoja C, de Martino A, Serrano M (2018) Senescence promotes in vivo reprogramming through p16INK4a and Il-6. Aging Cell 17:e12711. https://doi.org/10.1111/acel.12711
Velarde MC, DeMaria M (2016) Targeting senescent cells: possible implications for delaying skin aging: a mini-review. Gerontology 62(5):513–518. https://doi.org/10.1159/000444877
Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA, van der Pluijm I, Essers J, van Cappellen WA, van IJcken WF, Houtsmuller AB, Pothof J, de Bruin RWF, Madl T, Hoeijmakers JHJ, Campisi J, de Keizer PLJ (2017) Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169:132–147. https://doi.org/10.1016/j.cell.2017.02.031
Stearns SC (1992) The evolution of life histories. Oxford University Press, Oxford
Acknowledgements
I am grateful to Carlos López-Otín, Patricia L. Opresko, Ignacio Flores, and Jeanine Harrigan for their helpful comments on the manuscript. I also thank Adrián V. and Victoria C. for their productive discussions and support. The professional editing service NB Revisions was used for technical editing of the manuscript prior to submission.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that he has no conflict of interest.
Rights and permissions
About this article

Cite this article
von Kobbe, C. Cellular senescence: a view throughout organismal life. Cell. Mol. Life Sci. 75, 3553–3567 (2018). https://doi.org/10.1007/s00018-018-2879-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-018-2879-8