
Abstract
The protein kinase D (PKD) family of proteins are important regulators of tumor growth, development, and progression. CRT0066101, an inhibitor of PKD, has antitumor activity in multiple types of carcinomas. However, the effect and mechanism of CRT0066101 in bladder cancer are not understood. In the present study, we show that CRT0066101 suppressed the proliferation and migration of four bladder cancer cell lines in vitro. We also demonstrate that CRT0066101 blocked tumor growth in a mouse flank xenograft model of bladder cancer. To further assess the role of PKD in bladder carcinoma, we examined the three PKD isoforms and found that PKD2 was highly expressed in eight bladder cancer cell lines and in urothelial carcinoma tissues from the TCGA database, and that short hairpin RNA (shRNA)-mediated knockdown of PKD2 dramatically reduced bladder cancer growth and invasion in vitro and in vivo, suggesting that the effect of the compound in bladder cancer is mediated through inhibition of PKD2. This notion was corroborated by demonstrating that the levels of phospho-PKD2 were markedly decreased in CRT0066101-treated bladder tumor explants. Furthermore, our cell cycle analysis by flow cytometry revealed that CRT0066101 treatment or PKD2 silencing arrested bladder cancer cells at the G2/M phase, the arrest being accompanied by decreases in the levels of cyclin B1, CDK1 and phospho-CDK1 (Thr161) and increases in the levels of p27Kip1 and phospho-CDK1 (Thr14/Tyr15). Moreover, CRT0066101 downregulated the expression of Cdc25C, which dephosphorylates/activates CDK1, but enhanced the activity of the checkpoint kinase Chk1, which inhibits CDK1 by phosphorylating/inactivating Cdc25C. Finally, CRT0066101 was found to elevate the levels of Myt1, Wee1, phospho-Cdc25C (Ser216), Gadd45α, and 14-3-3 proteins, all of which reduce the CDK1-cyclin B1 complex activity. These novel findings suggest that CRT0066101 suppresses bladder cancer growth by inhibiting PKD2 through induction of G2/M cell cycle arrest, leading to the blockade of cell cycle progression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bladder cancer is the ninth most common cancer in the world [1, 2]. Of all newly diagnosed patients of bladder cancer, about 70–75% present as non-muscle-invasive bladder cancers (NMIBCs) and 25–30% are MIBCs [3]; as many as 50–70% of those NMIBCs will recur and approximately 10–20% will develop the ability to invade into the muscular wall of the bladder at which point the development of metastatic disease increases substantially [4]. For patients who develop metastatic disease, median survival is just over a year with current chemotherapy regimens [5]. Even the most effective form of therapy (cisplatin-based chemotherapy) produces objective responses in only one-fourth of patients [6]. Unfortunately, there have been few recent advances in the treatment of bladder cancer [7,8,9,10]. Therefore, there is an urgent need to develop therapeutic agents that can effectively treat this potentially deadly disease.
Protein kinase D (PKD) is a subfamily of serine/threonine kinases of the calcium/calmodulin-dependent kinase superfamily and is composed of three isoforms, PKD1, PKD2 and PKD3. PKD activity has been shown to be an important part of the regulation of diverse cellular processes, including cell migration/invasion [11, 12], proliferation [13], angiogenesis [14], apoptosis [15], epithelial to mesenchymal transition [16, 17], vesicular transport [18, 19], response to oxidative stress [20], response to serum growth factors [21], and gene transcription [15]. Changes in the expression of the various isoforms of PKD have been found in multiple cancer types [22,23,24], making it a potential chemotherapeutic target for the treatment of human cancers.
Several PKD inhibitors have been developed and have been shown to be effective in inhibiting cancer growth in vitro and in vivo [23, 25, 26]. Among them, CRT0066101 (2-[4-((2R)-2-aminobutyl-amino)-pyrimidin-2-yl]-4-(1-methyl-1H-pyrazol-4-yl)-phenol dihydrochloride) (Fig. 1) is a potent small molecule pan-PKD inhibitor that targets all three PKD isoforms and is orally bioavailable [23]. When used as a single agent, CRT0066101 has been demonstrated to block tumor growth in animal models of several types of carcinomas including breast, colorectal, and pancreatic cancer [23, 25, 27]. Results from these studies indicate that this compound is well tolerated in mice and produced no apparent side effects, thus making CRT0066101 an ideal candidate for clinical development. Additionally, in a combination treatment study, CRT0066101 has synergistic antitumor activity with regorafenib, a multi-kinase inhibitor, in human colorectal cancer cells [28].

Chemical structure of CRT0066101
The mechanisms of action of CRT0066101 in cancers are still elusive. Previous studies have shown that CRT0066101 triggers apoptosis in pancreatic and colorectal cancer, and the apoptosis induced by CRT0066101 is associated with increase in expression of cleaved poly ADP ribose polymerase (PARP) and activated caspase-3 in colorectal cancer and abrogating expression of NF-κB-dependent prosurvival proteins such as survivin and cellular inhibitor of apoptosis protein-1 (cIAP-1) in pancreatic cancer [23, 25]. In addition, a recent study on CRT0066101 shows that the induction of cell cycle arrest may account for its mechanism of action in colorectal cancer [23]; however, the molecular basis for cell cycle arrest by CRT0066101 remains undefined.
The eukaryotic cell cycle is regulated by cyclins, cyclin-dependent kinases (CDKs), and cyclin-dependent kinase inhibitors (CKIs) [29, 30]. CDK activity is modulated through three distinct mechanisms: (1) cyclin binding; (2) positive and negative phosphorylation events; and (3) interaction with CKIs [30, 31]. Cells pass through several checkpoints as they proceed through the cell cycle, and certain criteria must be met to pass each of these checkpoints. In the G2 checkpoint, CDK1 (also known as Cdc2) and cyclin B1 are the two major players for control of the G2/M transition in the cell cycle. Therefore, the kinase activity of CDK1-cyclin B1 is critical for the G2/M transition. The CDK-interacting protein/kinase inhibitory protein (CIP/KIP) p27Kip1 acts as an inhibitor of CDK1 [32, 33]. The activity of the CDK1-cyclin B1 complex is regulated, at least in part, by a positive regulator Cdc25C and two negative regulators Myt1 and Wee1 [34]. Cdc25C activity in turn can be inhibited by phosphorylation on Ser216 by the upstream cell cycle checkpoint kinase 1 (Chk1) [35,36,37]. The phosphatase Cdc25C regulates CDK1-cyclin B1 activity by dephosphorylation on Thr14 and Tyr15 of CDK1, leading to the activation of the CDK1-cyclin B1 complex [37, 38]; whereas the protein kinases Myt1 [39, 40] and Wee1 [41,42,43,44] phosphorylate CDK1 at Thr14 and Tyr15, thereby inactivating the CDK1-cyclin B1 complex and leading to G2/M arrest [34]. Not surprisingly, the loss or deactivation of either component of the CDK1-cyclin B1 complex will block cellular progression out of G2 [45].
To date, nothing is known about the effect and mechanism of the PKD inhibitor CRT0066101 in human bladder urothelial cell carcinoma. We undertook this study to assess the antitumor effect of CRT0066101 in urothelial bladder cancer and determine the underlying mechanisms. Our focus was on whether the regulation of cell cycle control is involved in the mechanisms for the antitumor activity of CRT0066101 in bladder cancer. Here we report that CRT0066101 blocked bladder tumor growth in vitro and in vivo via inhibition of PKD2 activity. CRT0066101 also had differential effects on cell survival between urothelial bladder carcinoma cells and human uroepithelial cells. Furthermore, we show that the effect of CRT0066101 on growth suppression of bladder carcinoma was associated with cell cycle arrest at the G2/M phase, as well as alterations in the expression and activation of key regulators of the G2/M transition, suggesting that the inhibitory activity of CRT0066101 on the growth and proliferation of bladder cancer cells is mediated, at least in part, through modulation of the cell cycle G2/M checkpoint leading to the blockade of cell cycle progression.
Materials and methods
Chemicals and reagents
The CellTiter-Glo Luminescent Cell Viability Assay was purchased from Promega Corp. (Madison, WI, USA). CRT0066101 was obtained from Cancer Research Technologies (Cambridge, UK). Ninety-six-well clear flat bottom ultra-low attachment microplates were purchased from Corning Incorp. (Oneonta, NY, USA). Polyvinylidene difluoride (PVDF) membranes and 4–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels were obtained from Life Technologies (Carlsbad, CA, USA). Monoclonal antibody against phospho-PKD2 (Ser876) was from Abcam (Cambridge, MA, USA). Monoclonal antibody against 14-3-3σ was bought from ThermoFisher Scientific (Waltham, MA, USA). Monoclonal or polyclonal antibodies against PKD1, PKD2, PKD3, phospho-PKD2 (Ser706/710), phospho-PKD2 (Ser916), c-Jun, phospho-c-Jun (Ser63), Gadd45α, p27Kip1, phospho-Chk1 (Ser317), phospho-Chk1 (Ser345), Cdc25C, phospho-Cdc25C (Ser216), Wee1, Myt1, cyclin B1, CDK1, phospho-CDK1 (Thr14), phospho-CDK1 (Tyr15), phospho-CDK1 (Thr161), pan-14-3-3, 14-3-3ε, and β-actin were purchased from Cell Signaling Technology (Danvers, MA, USA). Restore Western Blot Stripping Buffer was from Thermo Scientific (Rockford, IL, USA). Phorbol 12-myristate 13-acetate (PMA) and all other reagents were from Sigma-Aldrich (St. Louis, MO, USA).
Cell lines and cell proliferation assay
The human bladder carcinoma cell lines SCaBER, 5637, T24, UMUC3, and TCCSUP, and the SV40-transformed human uroepithelial cell line SV-HUC were purchased from the American Type Culture Collection (Manassas, VA, USA). UMUC1 and UMUC5 were kind gifts from Dr. David McConkey (University of Texas MD Anderson Cancer Center, Houston, TX, USA). T24T was a kind gift from Dr. Michael Nickerson (National Cancer Institute, National Institutes of Health, Frederick, MD, USA). Unless otherwise noted, all cell lines used in this study were maintained as a monolayer in minimum essential medium (MEM), supplemented with 10% fetal bovine serum (FBS), 50 IU/ml penicillin, and 50 µg/ml streptomycin (Life Technologies), at 37 °C in a humidified atmosphere with 5% CO2.
The antiproliferative effect of CRT0066101 was assessed using an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)-based assay, as described previously [46]. In brief, bladder carcinoma cells were evenly seeded in 96-well plates with 100 µl of MEM medium and cells were allowed to attach for 24 h during day 0. The medium was then removed and replenished with 200 µl fresh medium containing CRT0066101 with increasing concentrations (0, 0.625, 1.25, 2.5, 5, 10, and 20 µM). Cell numbers were quantified at days 0, 2, and 4 by incubating cells in 0.5 mg/ml of MTT (Sigma-Aldrich) in Opti-MEM medium at 37 °C and 5% CO2 for 1 h. Finally, the optical density at 570 nm was determined using an iMark™ Microplate Reader (Bio-Rad Laboratories; Hercules, CA, USA). Proliferation rates were calculated from the optical densities of CRT0066101-treated cells relative to the optical density of untreated control cells with no CRT0066101 exposure (control value, 100%). The half-maximal inhibitory concentration (IC50) values for CRT0066101 at day 4 in the 4 cell lines (T24, T24T, TCCSUP, and UMUC1) were calculated using GraphPad Prism version 6.01 software (GraphPad Software; La Jolla, CA, USA). IC50 was considered as the drug concentration that decreases the cell count by 50%.
Celigo cell viability assay
Bladder cancer cells (T24, T24T, TCCSUP, and UMUC1) and human uroepithelial cells (SV-HUC) were trypsinized and placed in 96-well plates (Greiner Bio One, Monroe, NC, USA; Cat# 655090) at a concentration of 1 × 104 cells per well, and the cells were incubated overnight at 37 °C. CRT0066101 at 5 µM was added to those wells to a final volume of 200 µl per well. Cells were incubated with the compound for 6 days, and then simultaneously stained with a mixture of calcein AM, propidium iodide, and Hoechst 33342 reagents for respective staining of live, dead, and all cells following the manufacturer’s protocol (Nexcelom Bioscience LLC, Lawrence, MA, USA; Cat# CSK-V0001-1), and cell viability was quantified with a Celigo® S Imaging Cell Cytometer. Live and dead cell counts as well as the percentage of viable cells were calculated with the Celigo software program.
Growth in low attachment (GILA) assay
In this modified colony formation assay [47], we plated 3 × 103 bladder cancer cells per well in a 96-well clear flat bottom ultra-low attachment microplate (Corning #3474) in 200 µl of complete medium with increasing concentrations of CRT0066101 (0, 0.625, 1.25, 2.5, 5, 10, and 20 µM). Cells without treatment with CRT0066101 were left as the untreated controls. After 5 days of incubation at 37 °C in 5% CO2, all the cells were transferred to a 96-well clear flat bottom white polystyrene microplate (Corning #3610) for measuring cell viability by using the CellTiter-Glo Luminescent Cell Viability Assay (Promega). The CellTiter-Glo Luminescent Cell Viability Assay is a homogeneous method to determine the number of viable cells in culture based on quantitation of the ATP present, which signals the presence of metabolically active cells. The amount of ATP is directly proportional to the number of live cells present in culture. The plate was read on EnSpire Multimode Plate Reader (Perkin Elmer, Waltham, MA, USA), and each condition was measured in triplicate.
In vitro cell migration and invasion assay
To assess the effect of CRT0066101 on the invasion capability of bladder tumor cells, a two-chamber cell invasion assay (Corning #3422 and #3458) was set up with 20% medium in the outer chamber and 1% medium in the inner chamber. Bladder carcinoma cells were first treated with CRT0066101 at the indicated concentrations (0, 1, 2, and 3 µM), and then 2 × 105 cells were resuspended in the inner chamber in 200 µl of fresh MEM medium containing 1% FBS. After 2 days of incubation at 37 °C in 5% CO2 in air, the residual cells in the inner chamber were removed and the outer membrane of the two-chamber system was fixed with 100% methanol and stained with 1% crystal violet (Sigma-Aldrich). Excess crystal violet solution was washed using distilled water, and cell migration/invasion was assessed under microscopy at 10× magnification. Each experiment was performed in triplicate.
Study of bladder cancer growth in xenograft flank mouse models
Athymic NCr-nu/nu mice were raised at the Experimental Animal Center of the National Cancer Institute under specific pathogen-free conditions and were cared for in an environmentally controlled room with a 12-h light/12-h dark cycle and a constant temperature of 22 °C. The mice were provided with a standard diet and water in accordance with the guidelines for the treatment of experimental animals at the National Institutes of Health. For investigating the role of PKD2 in bladder tumor growth, ten female athymic NCr-nu/nu mice were injected subcutaneously in the right flank with UMUC1 cells that were transfected with either control vector or PKD2 vector. Each mouse received 2 × 106 UMUC1 cells resuspended in 100 µl of 50% Matrigel (Corning) in phosphate-buffered saline (PBS). The mice were euthanized using CO2 14 days after UMUC1 tumor cell implantation, and the tumors were dissected and weighed.
For assessing the effect of CRT0066101 on bladder cancer growth in an in vivo mouse model, 20 athymic NCr-nu/nu mice (6-week-old) were implanted subcutaneously with 3 × 105 UMUC1 cells, as described above. The treatment started 8 days after UMUC1 cells had been injected, and mice were randomized into two groups (n = 10 mice/group) and administered with 100 µl of 5% dextrose (Vehicle) or with CRT0066101 (120 mg/kg/day) dissolved in 100 µl of 5% dextrose (CRT0066101), respectively. Treatment was administered 3 days per week by oral gavage for 25 days. The tumors were measured using external calipers three times a week, and the mean tumor volume was calculated by taking tumor length (mm) × tumor breadth2 (mm2) × 0.52. All measurements were performed in a coded, blinded fashion. Mice were euthanized 2 h after the last dose of the therapeutic regimen, and tumors were harvested and resected to determine protein level and activity by immunoblotting. The above two animal studies were conducted in strict compliance with the recommendations set forth in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (eighth version, 2010). The animal use protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the National Institutes of Health (Bethesda, MD, USA).
Western blot analysis
The cell cultures of bladder cancer cell lines were harvested and lysed with 200 µl of RIPA lysis buffer containing 50 mM Tris–HCl (pH 7.4), 1.0% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM aprotinin, 1 µg/ml PMSF, leupeptin, and pepstatin. The protein concentration was quantified by BCA assay. In a separate experiment, UMUC1 xenograft tumor tissues from the vehicle-treated and CRT0066101-treated mice were cut into small (≤ 10 mg) pieces and collected in a tube. Then a tiny amount of glass beads and 200–300 µl of RIPA lysis buffer were added to each tube. The tumor tissues were sonicated using a bead beater and centrifuged at maximal speed for 15 min. Supernatants were collected and the protein concentration was quantified by BCA assay. Equal amounts of protein (60–80 µg) from the cell line cultures or the vehicle-treated and CRT0066101-treated tumor explant tissues were boiled for 5 min in Laemmli buffer, separated by 4–12% SDS–polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to a polyvinylidene difluoride (PVDF) membrane (Life Technologies). The blots were probed with primary antibodies against PKD1, PKD2, PKD3, phospho-PKD2 (Ser706/710), phospho-PKD2 (Ser876), phospho-PKD2 (Ser916), c-Jun, phospho-c-Jun (Ser63), Gadd45α, p27Kip1, phospho-Chk1 (Ser317), phospho-Chk1 (Ser345), Cdc25C, phospho-Cdc25C (Ser216), Myt1, Wee1, cyclin B1, CDK1, phospho-CDK1 (Thr14), phospho-CDK1 (Tyr15), phospho-CDK1 (Thr161), pan-14-3-3, 14-3-3ε, 14-3-3σ, and β-actin, followed by secondary AP-conjugated antibodies. Some blots were stripped with Restore Western Blot Stripping Buffer, and reprobed with an anti-β-actin antibody as the loading control. The immunoreactivity was visualized by enhanced chemiluminescence (Life Technologies).
Evaluation of PKD gene expression in urothelial bladder carcinoma
Data from The Cancer Genome Atlas (TCGA) project were downloaded from the TCGA website (http://cancergenome.nih.gov/) [48]. Level 3 RNA-Seq (RNA sequencing) count data were downloaded for all patients with bladder cancer, and the data were transformed for linear modeling using the voom function in the limma R package [49]. We included 131 patients in the initial comprehensive characterization of urothelial carcinoma [48]. We also included gene expression data for all 19 normal urothelial samples included in the TCGA bladder cancer provisional dataset as a control reference. Gene expression was evaluated within each of four subtypes as defined in the original TCGA publication—we matched the TCGA identifiers in the original TCGA publication to samples in the downloaded data in order to define the subtypes. Differences in mean gene expression between the bladder cancer tissues and the normal urothelium tissues were analyzed by Student’s t test, while differences in mean gene expression between the four subtypes were evaluated with analysis of variance (ANOVA).
For determining mRNA expression of PKD in bladder tumor tissues, we analyzed the bladder cancer data from the Oncomine database (https://www.oncomine.org). Three independent bladder cancer datasets from three different clinical studies were used in this analysis. The first study contained 14 normal biopsies and 46 cancer samples, where cancer tissues were tumor biopsies from 28 superficial bladder tumors (Ta and T1), 13 invasive carcinomas (T2–T4), and 5 biopsies from patients with carcinoma in situ (CIS) [50]. The second study contained 48 normal urothelium and 109 cancer samples. Among the cancer tissues, 28 samples were from superficial bladder tumors (Ta–Tis–T1), and 81 samples were from invasive carcinoma tissues (T2–T4) [51]. The third study contained 3 normal bladder tissues and 80 bladder tumor samples, the latter include 17 Ta, 10 T1, 15 T2, 25 T3, and 13 T4 tumors. Of the 80 bladder tumors, 74 were transitional cell carcinoma (TCC) and 6 were squamous cell carcinoma (SCC) [52]. Differences in mean mRNA expression between the bladder cancer tissues and the normal bladder tissues were analyzed by Student’s t test.
Generation of stable PKD2 depletion in bladder carcinoma cells
RNA interference-mediated silencing of the PKD2 gene was performed using a replication-incompetent lentiviral plasmid vector (PLKO.l-hygro) with a hygromycin resistance marker (Addgene™, Cambridge, MA, USA). The short hairpin RNA (shRNA) against PKD2 (PLKO.1-hygro-shPKD2) and non-target control (PLKO.l-hygro-shNTC) was constructed with the following target sequences: 5′-CACGACCAACAGATACTATAA-3′ for shPKD2#1, 5′-GTTGGGTGGTTCATTACAGCA-3′ for shPKD2#2, and 5′-CAACAAGATGAAGAGCACCAA-3′ for shNTC.
To produce lentivirus for shPKD2 and the control shNTC, 1.5 µg of the purified DNA (shPKD2 or shNTC plasmid vector) and 1 µg Packaging Mix (pMD2.G and psPAX2) were suspended in 600 µl of opti-MEM reduced serum medium (Life Technologies), and then 10 µl of TurboFect transfection reagent (ThermoFisher Scientific) was added and incubated for 25 min at room temperature. At the completion of incubation, the DNA mixture was added to HEK-293FT cells (ThermoFisher Scientific) in 60 mm dishes at 80–90% confluence, and 6 ml of fresh medium was added the following day. Viruses were harvested from the culture media at 2 days post transfection, and the virus-containing medium was filtered through a 0.45 µ filter. Lentiviral stocks were titrated using the Lenti-X™ qRT-PCR Titration Kit (Clontech Laboratories, Inc., Mountain View, CA, USA).
For transduction of the cell using lentivirus with shRNA targeting PKD2 and the control, bladder cancer cells were plated on two 10 cm dishes and grown to 50–75% confluence in complete medium. The medium was then replaced with fresh complete medium, and polybrene (Sigma-Aldrich) was added at a final concentration of 8 µg/ml. One ml of virus-containing medium with a multiplicity of infection (MOI) of 12–16 was then added to the tumor cell monolayers of different bladder carcinoma cell lines. These MOIs were based on the viral titer for inoculation. Unless otherwise specified, these MOIs were used for lentivirus with shRNA targeting PKD2 and the control shNTC throughout the study. Two days after transduction, cells with stable expression of shPKD2 and shNTC were selected by incubation in a medium containing hygromycin (200 µg/ml; ThermoFisher Scientific) for 4 days. Depletion of PKD2 protein from the bladder cancer cells (shPKD2#1 and shPKD2#2) was validated by western blot analysis.
Cell cycle analysis by flow cytometry
One million UMUC1 or T24T cells were plated in 10 cm dishes and allowed to grow until they were 80–90% confluent. Cells were treated with CRT0066101 at the indicated concentrations (0, 0.5, 1, 2, 2.5, and 3 µM) for 3 days. The medium was then replaced with fresh complete medium containing 10 µM of 5-bromo-2′-deoxyuridine (BrdU), and cells were incubated at 37 °C for 2 h. At the end of incubation, cells were converted to single cell suspension using trypsin (Life Technologies). Cells were then fixed with 70% chilled ethanol, and the nuclei were extracted by digesting the cells with 3 ml of 0.08% pepsin in 0.1 N HCl. The nuclei were washed with 2 N HCl followed by 0.1 M sodium borate. The BrdU-containing DNA within the nuclei was analyzed using an anti-BrdU FITC-conjugated antibody (Life Technologies). The DNA content was analyzed by staining the nuclei with 1 µl of FxCycle Violet Stain (ThermoFisher Scientific), and cell cycle analysis was performed using a BD FACSCanto II Flow Cytometer. Data were analyzed using FlowJo v14.0 software (Tree Star, Inc., Ashland, OR, USA).
Statistical data analysis
Analysis of variance (ANOVA) was used for categorical variables. Student’s t test was used to analyze the statistical significance of differences between the control groups and the experimental groups. Statistical analyses were performed using R version 3.2.1, GraphPad Prism version 6.01, and SPSS 18.0 (SPSS Inc., Chicago, IL, USA) for Windows. All P values were determined using a two-sided test, and P < 0.05 was considered statistically significant.
Results
CRT0066101 inhibits proliferation, anchorage-independent growth, and invasion of urothelial bladder carcinoma cells in vitro
To test the effect of the PKD inhibitor CRT0066101 on bladder cancer cell growth and proliferation, we chose the human bladder cancer cell lines T24, T24T, TCCSUP, and UMUC1, which are commonly used tumor cell lines as models for studying urothelial bladder carcinoma. The time-course of CRT0066101 suppression of the proliferation of the four cell lines was characterized in vitro using the MTT assay. All of the four cell lines examined showed a maximal inhibitory effect at day 4 following CRT0066101 treatment (Fig. 2a). Similar data were observed in UMUC3 cells (data not shown). Our dose–response studies of CRT0066101 at concentrations of 0.625–20 µM revealed that the compound caused dose-dependent inhibition of the growth of the four cell lines at days 2 and 4. As seen in Fig. 2b, the half-maximal inhibitory concentration (IC50) values of CRT0066101 at day 4 were 0.3333, 0.4782, 0.4796, and 1.4300 µM for T24T, T24, UMUC1, and TCCSUP, respectively. These results indicate that CRT0066101 potently inhibits cell proliferation and induces cell toxicity in bladder cancer cells. However, 2-day treatment did not have a dramatic effect on cell viability (see Fig. 2a), suggesting that extended exposure to CRT0066101 is required for it to exert its activity on cell growth and death.


CRT0066101 suppresses proliferation, anchorage-independent growth, and migration of bladder carcinoma cells in vitro. a, b Bladder cancer cells (T24, T24T, TCCSUP, and UMUC1) were treated with CRT0066101 at the indicated concentrations. Cell viability was determined at days 0, 2, and 4 by the MTT assay (a). Dose–response effect of CRT0066101 at 4-day treatment (b). Cell viability values are expressed relative to those for cells with no drug exposure. The IC50 values for CRT0066101 at day 4 in the four cell lines were calculated using GraphPad Prism version 6.01 software. c Differential effect of CRT0066101 on cell survival between human uroepithelial cells and bladder tumor cells as determined by using a Celigo image cytometer. The four bladder cancer cell lines and the human uroepithelial cell line SV-HUC were incubated with CRT0066101 at 5 µM for 6 days. Cell viability was simultaneously stained with a mixture of calcein AM, propidium iodide, and Hoechst 33342 reagents for respective staining of live, dead, and all cells, and the percentage of viable cells was quantified with the Celigo imaging cytometer. *P < 0.01 versus SV-HUC cells. d CRT0066101 abrogates bladder carcinoma cell growth in low attachment (GILA). The four bladder cancer cell lines were distributed in a 96-well flat bottom ultra-low attachment microplate and treated with CRT0066101 at the indicated concentrations. After 5 days of incubation, cell viability was measured as described in “Materials and methods”. NC no cells. *P < 0.01 and **P < 0.05 vs. the untreated control group. e, f CRT0066101 reduces the invasive ability of bladder cancer cells. A two-chamber cell invasion assay was used for assessing the effect of CRT0066101 on the invasion capability of bladder carcinoma cells. TCCSUP cells (e) and UMUC1 cells (f) were treated with CRT0066101 at the indicated concentrations, and cell migration/invasion was then assessed after 2 days of incubation at 37 °C in the chamber, as described in “Materials and methods”. The numeric data for the two cell lines are shown in the two column graphs, respectively. *P < 0.01 and **P < 0.05 vs. the untreated control group
To confirm the antitumor effect of CRT0066101 in bladder cancer cells, we assessed cell viability by staining cells with live and dead cell-specific dyes using the Celigo® Image Cytometry System. Treatment with the PKD inhibitor significantly reduced cell survival as assessed by cell viability staining. The percentages of live cells were 41.41, 51.36, 20.90, and 11.85% in the T24, T24T, TCCSUP, and UMUC1 groups, respectively, as compared with the untreated control group of the respective cell lines (Fig. 2c).
However, the clinical use of CRT0066101 will be linked to its safety, particularly the lack of toxicity in normal cells. Therefore, we evaluated the effect of CRT0066101 on cell viability in a nontumorigenic human uroepithelial cell line, SV-HUC. The cell viability data showed that the percentages of viable cells for SV-HUC were 92.13% (Fig. 2c), which was 2.23-, 1.79-, 4.41-, and 7.78-fold higher than those for the bladder cancer cell lines T24, T24T, TCCSUP, and UMUC1, respectively (P < 0.01), indicating a differential effect of CRT0066101 on cell viability between the human epithelial bladder carcinoma cell lines and the human uroepithelial cell line SV-HUC.
Since anchorage-independent growth is a hallmark of cancer cells but not normal cells, we performed the GILA (growth in low attachment) assay [47] to investigate the effect of CRT0066101 on anchorage-independent growth of bladder tumor cells using T24, T24T, TCCSUP, and UMUC1 cell lines. Our data showed that CRT0066101 was effective in inhibition of cell growth in low attachment conditions, with the concentrations of 2.5 and 5 µM effective in all four bladder cancer cell lines (Fig. 2d). Similar results were obtained in UMUC3 cells (data not shown).
Invasion through the extracellular matrix is a crucial step in tumor cell metastasis. To determine the effect of CRT0066101 on the migration and invasiveness of bladder carcinoma cells, we used a Boyden-chamber assay. As shown in Fig. 2e, f, treatment with CRT0066101 profoundly inhibited in vitro invasion of TCCSUP and UMUC1 cells in a concentration-dependent manner. The number of migrating cells was markedly decreased in CRT0066101-treated TCCSUP and UMUC1 cells as compared with those of the untreated control cells (Fig. 2e, f), suggesting that CRT0066101 reduces the invasive ability of bladder cancer cells.
CRT0066101 suppresses bladder cancer growth in an in vivo tumor xenograft model
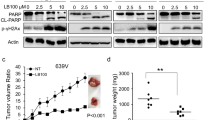
Given that cell survival and proliferation were reduced in bladder carcinoma cells in vitro following treatment with CRT0066101 (Fig. 2), we next evaluated the effect of the compound on human bladder cancer growth in an in vivo mouse model. The administration of CRT0066101 significantly blocked tumor growth of xenograft bladder cancer implants. There was a substantial difference in tumor size between the CRT0066101-treated group and the vehicle-treated control group even on day 18 of the therapy; at the termination of the study (day 25 post-treatment), the difference in tumor volume between the CRT0066101 group and the control group was significant (P < 0.0001). The average tumor volumes for bladder cancer on day 25 of the treatment were 835.83 and 1943.08 mm3 in the CRT0066101-treated group and the vehicle-treated control group, respectively (Fig. 3). These data suggest that CRT0066101 has a potential therapeutic value for patients with bladder cancer and that its targets, PKDs, may play an important role in the survival of bladder tumors.

Antitumor effect of CRT0066101 on the growth of human bladder cancer in nude mice. Athymic NCr-nu/nu mice (6-week-old) were implanted subcutaneously with 3 × 105 UMUC1 cells. The treatment started after the tumors were established in the mice, and mice were randomized into two groups (n = 10 mice/group) and administered with 100 µl of 5% dextrose (Vehicle) or with CRT0066101 (120 mg/kg/day) dissolved in 100 µl of 5% dextrose (CRT0066101), respectively. Treatment was administered 3 days per week by oral gavage for 25 days. Tumor volumes were recorded three times a week, as described in “Materials and methods”. Mice were euthanized 2 h after the last dose of the therapeutic regimen. P < 0.0001 for the CRT0066101-treated group vs. the vehicle-treated group at day 25 of the treatment
Evaluation of PKD gene expression in urothelial bladder carcinoma
Given that CRT0066101 is a pan-PKD inhibitor and that the PKD isoform targeted by CRT0066101 in the bladder cancer cell is unclear, we investigated the role of the PKD isoforms in urothelial cell carcinoma in order to elucidate the anticancer mechanism of CRT0066101 in this tumor. To this end, we first determined the protein expression of PKD isoforms in several different bladder carcinoma cell lines. We found that PKD2 protein was highly expressed in all bladder cancer cell lines except for SCaBER, which presented with slightly lower PKD2 expression levels (Fig. 4b), whereas the levels of PKD3 protein in the eight bladder carcinoma cell lines were distinctly variable and considerably lower than those of PKD2 (Fig. 4b). We also tested the levels of PKD1 in four bladder tumor cell lines (5637, SCaBER, UMUC1, and UMUC5). We only measured low levels of PKD1 in 5637 cells; however, PKD1 protein was not detectable in SCaBER, UMUC1, and UMUC5 cell lines by immunoblotting (Fig. 4a). The levels of PKD1, PKD2, and PKD3 expression in our bladder cancer cell lines were similar to those detected in human colorectal cancer cell lines [23].

Immunoblot analysis of PKD1, PKD2, and PKD3 protein levels in human urothelial bladder carcinoma cell lines. Four bladder cancer cell lines (SCaBER, UMUC5, UMUC1, and 5637) were used for analysis of PKD1 protein expression, while 8 bladder tumor cell lines (TCCSUP, T24T, T24, UMUC3, SCaBER, UMUC5, UMUC1, and 5637) were utilized for analysis of PKD2 and PKD3 protein levels. Total cellular protein was extracted from these cell lines, and a total of 80 µg cell extract protein was subjected to SDS-PAGE and immunoblotted with an antibody against PKD1, PKD2, or PKD3, as described in Materials and Methods. β-Actin was used as the control to verify equal protein loading and transfer
To evaluate the PKD gene expression in urothelial carcinoma tissues, we analyzed the levels of PKD gene expression using a level 3 RNA-Seq count dataset in the TCGA database, which includes 131 patient tumor samples and 19 normal urothelial samples. The mean expression of PKD2 gene was found significantly higher in the urothelial carcinoma tissues than that of the normal urothelial tissues (P = 0.003; Fig. 5a). We also showed that PKD2 gene expression was clearly upregulated in the bladder tumor tissues of the four molecular subtypes of bladder cancer patients (P = 0.011; Fig. 5b), whereas the levels of PKD3 expression were only moderately higher in the bladder carcinoma tissues as compared to the normal bladder tissues in the TCGA bladder cancer provisional dataset (P = 0.769; Fig. 5d). Intriguingly, the levels of PKD1 expression were obviously downregulated in the bladder tumor tissues than in the normal bladder tissues (P < 0.001; Fig. 5c), which is consistent with other studies demonstrating downregulation of PKD1 in several other human cancers including carcinoma of the prostate, breast, and stomach. These results of the protein expression of the three PKD isoforms in bladder tumor tissues are largely consistent with those found in our bladder cancer cell lines (see Fig. 4).

Evaluation of PKD isoform expression in urothelial bladder carcinoma tissues using the TCGA bladder cancer dataset. PKD gene expression was evaluated using the level 3 RNA-Seq count dataset from the TCGA bladder cancer database, which includes 131 patients with urothelial carcinoma and 19 normal urothelial samples. Mean expression of PKD2 gene is significantly upregulated in bladder tumor tissues (a) and in the four molecular subtypes of bladder cancer tissues (b). P < 0.003 for the bladder cancer group vs. the normal urothelium group. Mean expression of PKD1 gene is clearly downregulated in the four molecular subtypes of bladder cancer tissues (c). P < 0.001 for the bladder cancer group vs. the normal urothelium group. PKD3 gene expression is only moderately upregulated in the tumor tissues of the four molecular subtypes of bladder cancer patients (d). P = 0.769 for the bladder cancer group vs. the normal urothelium group
To validate the expression of PKD isoforms in bladder carcinoma tissues, we also analyzed the mRNA expression of PKD1 and PKD2 by cDNA microarray using bladder cancer data from three independent tumor datasets on the Oncomine database. As seen in Fig. 6a, the levels of PKD1 mRNA in bladder tumor tissues were markedly lower than those in the normal bladder samples in both Blaveri et al. [52] and Sanchez-Carbayo et al. [51] studies (P < 0.0001). In contrast, the mean mRNA expression of PKD2 isoform was significantly upregulated in the bladder carcinoma tissues compared with the normal bladder tissues in both Dyrskjot et al. [50] and Sanchez-Carbayo et al. [51] studies (P < 0.0001; Fig. 6b).

PKD2 mRNA expression is upregulated and PKD1 mRNA expression is downregulated in urothelial bladder cancer tissues. a PKD1 mRNA expression was analyzed using the bladder cancer datasets from two independent clinical studies by Blaveri et al. [52] and Sanchez-Carbayo et al. [51], as described in “Materials and methods”. The mRNA expression levels of PKD1 are significantly decreased in the bladder tumor samples of both studies. P < 0.0001 for the tumor tissue groups vs. the normal tissue groups. b PKD2 mRNA expression was assessed using the bladder cancer datasets from two independent clinical studies by Dyrskjot et al. [50] and Sanchez-Carbayo et al. [51] downloaded from the Oncomine database. The mRNA expression levels of PKD2 are markedly increased in the bladder tumor samples of both studies. P < 0.0001 for the tumor tissue groups vs. the normal tissue groups
Depletion of PKD2 abrogates proliferation, anchorage-independent growth, and migration of bladder cancer cells in vitro
Because PKD2 expression was upregulated in both bladder carcinoma cell lines and bladder tumor tissues, we wanted to understand the role of PKD2 in the proliferation and migration of bladder cancer cells. Thus, we generated stable derivatives of 6 bladder carcinoma cell lines (T24, T24T, TCCSUP, UMUC1, UMUC3, and UMUC5) under-expressing PKD2 (shPKD2#1 and shPKD2#2) and their corresponding controls (shNTC). As shown in Fig. 7a, b, the levels of PKD2 protein in both shPKD2#1 and shPKD2#2 were decreased by about 70–80% compared to the control shNTC in a particular cell line and was consistent across cell lines.

Knockdown of PKD2 gene expression in urothelial bladder carcinoma cells. a UMUC1 cells stably transduced with lentivirus coding for human shRNA sequences for PKD2 and non-target control were used for analysis of PKD2 expression. Total cellular protein was extracted from these cells, and a total of 80 µg cell extract protein was analyzed by SDS-PAGE and immunoblotting with an antibody against PKD2 or β-actin to verify equal loading. b Quantitation of the PKD2 protein levels in shPKD2#1 and shPKD2#2 transduced cells comparing cells transduced with shNTC, n = 3. Error bars denote variation in biological replicates. c Validation of PKD2 knockdown efficiency in shPKD2 transduced cells. UMUC1 cells transduced with shPKD2#1 or shNTC were incubated with or without 200 nM PMA for 30 min and lysed. Total protein was extracted, and PKD2 and c-Jun protein level and activity were analyzed by western blotting with the anti-PKD2, phospho-PKD2 (Ser876), c-Jun, phospho-c-Jun (Ser63), or β-actin antibodies, respectively. shNTC, non-target control; shPKD2#1, PKD2 clone 1; shPKD2#2, PKD2 clone 2; PMA, Phorbol 12-myristate 13-acetate
To validate the PKD2 depletion efficiency, we treated shPKD2#1 of UMUC1 with PMA, a known activator of the protein kinase C/PKD pathway, and found that PMA markedly increased PKD2 (Ser876) phosphorylation without affecting PKD2 protein expression (Fig. 7c). We also showed that stimulation of shPKD2#1 with PMA dramatically enhanced phosphorylation of c-Jun (Ser63), a substrate of PKD2, without significantly changing the levels of c-Jun protein (Fig. 7c). To further verify c-Jun activation in PKD2 knockdown UMUC1 cells following PMA stimulation, we used a Human Phospho-Kinase Antibody Array Kit (R&D Systems, Inc., Minneapolis, MN, USA). UMUC1 shNTC and shPKD2#1 were incubated with 200 nM of PMA for 30 min, and we showed that c-Jun (Ser63) phosphorylation was significantly increased in shNTC cells, but remained unaltered in shPKD2#1 cells (data not shown), indicating a decent PKD2 silencing efficiency in UMUC1 cells.
Next, we examined cell growth and proliferation in the PKD2-depleted bladder cancer cells. Interestingly, cellular proliferation of shPKD2#1 and shPKD2#2 was significantly reduced in all six bladder tumor cell lines as compared with those of shNTC in proliferation assays (P < 0.01; Fig. 8a). We also determined the ability of the bladder carcinoma T24T and UMUC1 cells under-expressing PKD2 for growth in low attachment. Cell viability was significantly decreased in shPKD2#1 and shPKD2#2 compared with that of shNTC (P < 0.01 or P < 0.05; Fig. 8b). Finally, we assessed the invasive ability of the bladder cancer TCCSUP and UMUC1 cells with PKD2 gene knockdown. The numbers of migrating and invading cells in the PKD2 knockdown (shPKD2#1 and shPKD2#2) TCCSUP cells (Fig. 8c) and UMUC1 cells (Fig. 8d) were significantly decreased as compared with those transfected with the control shNTC vector (P < 0.05 or P < 0.01). Taken together, the findings from both bladder carcinoma cell lines and bladder tumor tissues suggest that PKD2 isoform may play an essential role in the growth, invasion, and proliferation of bladder cancer cells and may be a promising therapeutic target.


Knockdown of PKD2 expression decreases proliferation, anchorage-independent growth, and invasion of bladder carcinoma cells in vitro. a Six cell lines (T24, T24T, TCCSUP, UMUC1, UMUC3, and UMUC5) were transduced with lentivirus coding for human shRNA sequences for PKD2 (shPKD2#1 and shPKD2#2) or for the control (shNTC). Cell growth and proliferation were assessed by MTT assay at days 0, 2, 4, and 6 after cell plating. P < 0.01 for the shPKD2 groups vs. the control shNTC group at day 6 in all the six cell lines tested. b Depletion of PKD2 abrogates bladder cancer cell growth in low attachment. T24T and UMUC1 cell lines were transduced with lentivirus coding for human shRNA sequences for PKD2 (shPKD2#1 and shPKD2#2) or for the control (shNTC). Cell growth in low attachment was assessed by GILA assay at day 5 after cell plating, as described in “Materials and methods”. *P < 0.01 and **P < 0.05 for the shPKD2 groups vs. the control shNTC group. c, d Knockdown of PKD2 expression compromises the invasive ability of bladder carcinoma cells. TCCSUP cells (c) and UMUC1 cells (d) were transduced with lentivirus coding for human shRNA sequences for PKD2 (shPKD2#1 and shPKD2#2) or for the control (shNTC). A two-chamber cell invasion system was set up, as described in “Materials and methods”, and the number of invading cells was counted in ten random microscopic fields (×40). The numeric data for the two cell lines are shown in the two column graphs, respectively. Values are expressed as mean ± SD. **P < 0.05 and *P < 0.01 for the shPKD2 groups vs. the shNTC group in TCCSUP and UMUC1 cells, respectively
Knockdown of PKD2 expression reduces tumor growth in a xenograft mouse model of bladder cancer
Since PKD2 plays a vital part in bladder cancer cell proliferation in vitro, we next determined the role of PKD2 in human bladder tumor growth in an in vivo mouse model. For this end, we stably silenced PKD2 in UMUC1 cells using two shRNAs targeting PKD2 (shPKD2#1) and the control (shNTC). A total of 2 × 106 UMUC1 cells was implanted subcutaneously into athymic BCr-nu/nu mice on the right flank. The tumors in the shPKD2 group grew very slowly in vivo, with dimensions considerably smaller than those in the shNTC control group (Fig. 9). There was a substantial difference in tumor mass between the shPKD2 group and the shNTC group as early as day 7 of the study; at the termination of the study (day 14), the difference in tumor weight between the shPKD2 group and the shNTC control group was significant (P = 0.0194; Fig. 9). Together, these data suggest that PKD2 plays a pivotal role in the growth, proliferation, migration, and invasion of bladder carcinoma cells in vitro and culminating in significantly slower growth rate in vivo.

shRNA-mediated depletion of PKD2 dramatically reduces bladder tumor growth in xenograft nude mice. UMUC1 cells were transduced with lentivirus coding for human shRNA sequences for PKD2 (shPKD2#1) and the control (shNTC). A total of 2 × 106 UMUC1 human bladder carcinoma cells was implanted subcutaneously into athymic BCr-nu/nu mice on the right flank, as described in “Materials and methods”. Mice were sacrificed humanely 14 days later, and tumors were resected. The tumors on mice and excised tumor images are shown. P = 0.0194 for the shPKD2 group vs. the shNTC group at day 14. shRNA short hairpin RNA
Administration of CRT0066101 reduces the activity of PKD2 protein and its substrate c-Jun in urothelial bladder carcinoma cells
We showed that PKD2 plays an essential role in bladder cancer cell growth and proliferation (Figs. 8, 9), and that the PKD inhibitor CRT0066101 markedly suppresses anchorage-dependent and -independent proliferation of bladder tumor cells in vitro and in vivo (Figs. 2, 3), suggesting that CRT0066101 may exert its anticancer activity in bladder carcinoma through inhibition of PKD2. To verify this notion, we performed western blot analysis of PKD2 protein in cell lysates from the UMUC1 tumor explants treated with CRT0066101 or with vehicle (5% dextrose) as the control. Our data showed that CRT0066101 dramatically decreased phosphorylation on Ser706/710, Ser876, and Ser916 of PKD2 protein without affecting PKD2 protein expression in the tumor cells as compared with the vehicle-treated control cells (Fig. 10a). Since c-Jun is a substrate of PKD2, it is instructive to assess whether inhibition of PKD2 results in the inhibition of its direct target in our bladder cancer cells, we analyzed c-Jun and phospho-c-Jun (Ser63) in the same model system, and found that the levels of phospho-c-Jun (Ser63) were strikingly reduced in CRT0066101-treated UMUC1 cells, while c-Jun protein levels remained unchanged as compared with the vehicle-treated tumor cells (Fig. 10b), confirming that suppression of PKD2 isoform as well as its downstream signaling is associated with the growth inhibitory activity of CRT0066101 in urothelial bladder carcinoma.

CRT0066101 treatment diminishes the activity of PKD2 protein and its substrate c-Jun in xenograft bladder cancer tumor explants. Immunoblot analysis of PKD2, phospho-PKD2 (Ser706/710), phospho-PKD2 (Ser876), phospho-PKD2 (Ser916), c-Jun, and phospho-c-Jun (Ser63) in cellular lysates from vehicle-treated (5% dextrose) and CRT0066101-treated UMUC1 tumor explants, as described in “Materials and methods”. A total of 60 or 80 µg cell extract protein was subjected to SDS-PAGE and immunoblotted with antibodies against the indicated proteins, or with a β-actin antibody to verify equal protein loading. The levels and activities of PKD2 protein in both vehicle-treated group and CRT0066101-treated group are shown in a, while the expression and activity of c-Jun protein in vehicle-treated and CRT0066101-treated groups are presented in b
Silencing and pharmacologic inhibition of PKD2 cause bladder cancer cell cycle arrest at the G2/M phase
It is well-established that uncontrolled cell proliferation is a hallmark of cancer, and CRT0066101 was shown to effectively inhibit bladder carcinoma cell growth in vitro and in vivo (Figs. 2, 3), we reasoned that this inhibitory activity of CRT0066101 may be due to interference with the cell cycle. Therefore, we examined the cell cycle perturbation after exposure of bladder cancer cells to CRT0066101. To this end, we treated UMUC1 cells with CRT0066101 at 0, 0.5, 1, 2, 2.5, and 3 µM for 3 days, and performed cell cycle analysis by flow cytometry. Figure 11a shows that CRT0066101 arrested UMUC1 cells at the G2/M phase in a dose-dependent manner with all the concentrations examined. Similar data were also obtained in T24T cells (Fig. 11b) and UMUC3 cells (data not shown). These results indicate that CRT0066101 suppresses the growth and proliferation of bladder tumor cells by controlling the G2/M checkpoint and inducing a specific block in cell cycle progression.

CRT0066101 blocks bladder carcinoma cells at G2/M phase as analyzed by flow cytometry. UMUC1 and T24T cell lines were exposed to increasing concentrations (0, 0.5, 1, 2, 2.5, and 3 µM) of CRT0066101 for 3 days. Cells were then harvested, stained, and the propidium iodide-labeled cells were analyzed for their cell cycle status by flow cytometry, as described in “Materials and methods”. The percentage of UMUC1 cells in each phase of the cell cycle was calculated using FlowJo v14.0 and is indicated in each cell cycle profile (a). Each assay was performed twice and one profile for each treatment is shown in a. The average numeric data of the two biological replicates for both UMUC1 and T24T cells are presented in b. Note the accumulation of the cells in the G2/M phase in both UMUC1 and T24T cell lines following CRT0066101 treatment (a, b)
To ascertain the specific role of PKD2 in G2/M phase arrest by CRT0066101, we utilized the UMUC1 cells under-expressing PKD2 (shPKD2#1 and shPKD2#2), PKD3 (shPKD3#1 and shPKD3#2), and the corresponding control (shNTC). The proportions of cells in different phases of the cell cycle were analyzed by flow cytometry using propidium iodide staining. As seen in Fig. 12a, b, there were substantial increases in the fraction of cells in the G2/M phase in PKD2-knockdown UMUC1 cells, but not in PKD3-knockdown UMUC1 cells. These data suggest that the effect of CRT0066101 on G2/M phase arrest is specifically mediated through inhibition of PKD2 in our bladder cancer cells.

shRNA-mediated silencing of PKD2 causes G2/M cell cycle arrest in bladder cancer cells. UMUC1 cells were transduced with lentivirus coding for human shRNA sequences for either PKD2 (shPKD2#1 and shPKD2#2), PKD3 (shPKD3#1 and shPKD3#2), or the control (shNTC). Cells were harvested after transduction and stained with propidium iodide. The cell cycle distribution of the propidium iodide-labelled cells was analyzed by flow cytometry, as described in “Materials and methods”. The percentage of cells in each phase of the cell cycle was calculated using FlowJo v14.0 and is indicated in each cell cycle profile (a). The assay was performed twice and one profile for each treatment is shown in (a). The average numeric data of the two biological replicates for both PKD2-depleted and PKD3-depleted UMUC1 cells are presented in b. Note the accumulation of the cells in the G2/M phase in PKD2-knockdown UMUC1 cells, but not in PKD3-knockdown UMUC1 cells (b). shRNA short hairpin RNA
CRT0066101 causes alterations in the level and activity of cell cycle G2/M transition regulatory proteins in xenograft bladder cancer tumor explants
Eukaryotic cell cycle progression is regulated by a series of CDKs, their activity being positively regulated by cyclins and negatively regulated by cyclin-dependent kinase inhibitors (CKIs). CRT0066101 was found to block cell cycle progression and arrest UMUC1 and T24T cells at the G2/M phase (Fig. 11a, b), and these findings prompted us to investigate whether inhibition of cell cycle progression by the drug was associated with alterations in key cell cycle regulatory molecules in bladder cancer cells. To analyze the expression of intracellular proteins regulating cell cycle progression at the G2/M boundary, we first assessed the effect of CRT0066101 on cyclin B1, CDK1, and p27kip1 expression in UMUC1 tumor explants. As seen in Fig. 13a, the levels of cyclin B1 and CDK1 were decreased but the levels of p27kip1 were increased following treatment of UMUC1 cells with CRT0066101. Since the CDK1-cyclin B1 complex is maintained in an inactive form by negative phosphorylation of the residues Thr14 and Tyr15 on CDK1, and CDK1-cyclin B1 activity is increased by phosphorylation of CDK1 at Thr161, we examined CDK1 phosphorylation status by immunoblotting and found that the phosphorylation of CDK1 at Thr14 and Tyr15 was increased but the phosphorylation of CDK1 at Thr161 was decreased after CRT0066101 treatment (Fig. 13a). These data suggest that reduced CDK1-cyclin B1 activity is responsible for CRT0066101-induced G2/M arrest in our model system.

Effects of CRT0066101 on the level and activity of key regulators of the cell cycle G2/M transition in xenograft bladder carcinoma tumor explants. Western blot analysis of proteins in cellular lysates from vehicle-treated (5% dextrose) and CRT0066101-treated UMUC1 tumor explants, as described in “Materials and methods”. A total of 60 or 80 µg cell extract protein was subjected to SDS-PAGE and immunoblotted with an antibody against cyclin B1, CDK1, phospho-CDK1 (Thr161, Thr14, Tyr15), p27kip1, Gadd45α, 14-3-3σ, 14-3-3ε, phospho-Chk1 (Ser317/Ser345), pan-14-3-3, Cdc25C, phospho-Cdc25C (Ser216), Myt1, and Wee1, or with a β-actin antibody as the loading control. The levels and activities of cyclin B1, CDK1, p27kip1, Gadd45α, and 14-3-3σ/ε proteins in both vehicle-treated group and CRT0066101-treated group are shown in a, while the expression and activation of Chk1, pan-14-3-3, Cdc25C, Wee1, and Myt1 proteins in vehicle-treated and CRT0066101-treated groups are presented in b
The kinases Myt1 and Wee1 are responsible for phosphorylation of CDK1 on Thr14 and Tyr15 and lead to the inhibition of CDK1. Conversely, the protein phosphatase Cdc25C activates CDK1 by dephosphorylation of the residues Thr14 and Tyr15. Since CDK1 phosphorylation at Thr14 and Tyr15 was enhanced by CRT0066101 treatment, we examined the effect of the compound on Cdc25C, Myt1, and Wee1 expression by western blotting. As shown in Fig. 13b, the administration of the bladder tumor with CRT0066101 markedly reduced Cdc25C protein levels but elevated the Myt1 and Wee1 protein levels. Additionally, Cdc25C is kept inactive and the activity of Cdc25C in the nucleus is reduced through negative phosphorylation at Ser216 by upstream Chk1 kinase. Therefore, we tested the activity of Chk1 and Cdc25C and found that the levels of phospho-Chk1 (Ser317) and phospho-Chk1 (Ser345) as well as Cdc25C phosphorylation at Ser216 were all increased in CRT0066101-treated UMUC1 tumor cells, indicating that enhanced Cdc25C phosphorylation at Ser216 is attributable to the increased Chk1 activity (Fig. 13b). Since there was an upregulation of Myt1 and Wee1 expression following CRT0066101 treatment (Fig. 13b), which was positively correlated with the G2/M cell proportion (Fig. 11) and negatively correlated with the CDK1-cyclin B1 complex activity (Fig. 13a), this suggests that CDK1 inactivation is due not only to increased phosphorylation by Myt1 and Wee1, but also to decreased dephosphorylation by Cdc25C in UMUC1 tumor explant tissues.
Lastly, we investigated the involvement of Gadd45α and 14-3-3 proteins in cell cycle G2/M block in CRT0066101-treated UMUC1 bladder carcinoma cells. Gadd45α and 14-3-3 proteins are other regulators of CDK1-cyclin B1 activity in the G2/M transition of the cell cycle. Gadd45α is involved in the G2 checkpoint through disrupting the interaction between CDK1 and cyclin B1 [53]. 14-3-3 proteins activate kinases Chk1 [54] and Wee1 [55, 56]. Moreover, 14-3-3 proteins such as 14-3-3ε and 14-3-3γ also bind to phospho-Cdc25C (Ser216) and anchor Cdc25C in the cytoplasm where it cannot activate CDK1, while the protein 14-3-3σ can bind to CDK1-cyclin B1 and sequester it in the cytoplasm where it cannot induce mitosis. Our western blot analysis of extracts from UMUC1 tumor explants after 25-day treatment with CRT0066101 showed increases in Gadd45α, 14-3-3ε, 14-3-3σ, and pan-14-3-3 protein levels (Fig. 13a, b). These results suggest that the increased expression of Gadd45α, 14-3-3ε, 14-3-3σ, and other isoforms of 14-3-3 proteins by CRT0066101 may account for a large part of the reduction in CDK1-cyclin B1 activity and the control of the G2/M checkpoint in bladder cancer cells.
Discussion
PKDs are involved in the regulation of a variety of cellular processes including cell survival, differentiation, proliferation, and motility. Aberrant expression of PKD isoforms has been reported in multiple types of carcinomas, making it a promising target in the development of novel therapies for human cancers. In the current study, we show that the PKD inhibitor CRT0066101 suppressed bladder tumor growth and invasion in vitro and in an in vivo xenograft mouse model. We also show that CRT0066101 had differential cytotoxic activity between urothelial bladder cancer cells and nontumorigenic human uroepithelial cells. Furthermore, we found that shRNA-mediated knockdown of PKD2 expression compromised bladder carcinoma growth and migration in vitro and in vivo, suggesting that the antitumor activity of CRT0066101 is mediated through inhibition of PKD2 in bladder cancer. This notion was verified through the demonstration that the activity of PKD2 was dramatically reduced in CRT0066101-treated bladder tumor explants. These data are consistent with the effects of CRT0066101 in several other types of human cancers including carcinoma of the breast, colon, and pancreas [23, 25, 27]. In addition, our cell cycle analysis revealed that CRT0066101 treatment induced bladder carcinoma cell cycle arrest at the G2/M phase. Further experiments showed that depletion of PKD2 also caused cell cycle G2/M phase arrest in the same tumor cell model. Taken together, these results indicate that CRT0066101-blocked bladder cancer growth is through induction of G2/M phase arrest of the cell cycle, and that the effect of the compound on cell cycle arrest is mediated through inhibition of PKD2 in bladder urothelial cell carcinoma.
Recently, several small molecule inhibitors targeting PKD2 including CRT0066101 have been identified, and the latter is currently under intensive investigation for preclinical development as an anticancer agent. Harikumar and colleagues first show that CRT0066101 given orally (80 mg/kg/day) for 24 and 21 days abrogate pancreatic cancer growth in vivo in xenograft model and orthotopic model, respectively [25]. While Wei et al. also examine the effect of CRT0066101 on in vivo tumor growth and find that daily administration of the compound orally at 40, 80, and 120 mg/kg/day for 21 days all results in significantly inhibition of colorectal cancer growth in xenograft nude mice [23]. Consistently, we show in this study that CRT0066101 at 120 mg/kg/day orally for 25 days (3 days/week) in UMUC1 subcutaneous xenograft model potently blocked bladder cancer growth in vivo (Fig. 3). These data from our three independent studies strongly support the development of CRT0066101 for clinical use for different cancers, and demonstrate that daily administration of CRT0066101 orally at 40–120 mg/kg/day for 3–4 weeks is both effective and safe in animal studies for different types of human carcinomas.
Although we have all shown that CRT0066101 is an effective drug in our own tumor models, the focus of our studies on the mechanisms of the compound differs from each other. Harikumar et al. demonstrate that CRT0066101 increases apoptosis in pancreatic cancer cells, and that the effect of the drug on apoptosis is mediated through abrogating the expression of NF-κB-dependent proliferative and prosurvival proteins including cyclin D1, survivin, and cIAP-1 [25]. In Wei and coworkers’ investigation, they find that in addition to induction of apoptosis in human colorectal cancer cells, CRT0066101 also induces G2/M phase arrest in their tumor models. However, they fail to explore the mechanism for G2/M blockade by CRT0066101 in their studies [23]. Whereas our group show that CRT0066101 had similar effect on G2/M phase accumulation in bladder cancer cells, and we take a step further to elucidate the molecular mechanisms underlying the effect of the compound on G2/M cell cycle arrest in our bladder tumor model system (Figs. 13, 14). Altogether, the data from our three groups suggest that the effect of CRT0066101 on tumor cell death and growth inhibition is mediated via modulating cell apoptosis and cell cycle progression in different types of cancers.

Proposed possible mechanism by which the PKD inhibitor CRT0066101 suppresses bladder cancer growth through blocking cell cycle progression at the G2/M phase. This schematic diagram shows that PKD2 inhibition by CRT0066101 treatment activates the kinase Chk1 through a yet-to-be-defined mechanism, thereby preventing from the activation of the downstream stimulatory phosphatase Cdc25C leading to decreased the CDK1-cyclin B1 complex activity. On the other hand, Chk1 kinase also reduces CDK1-cyclin B1 activity by activating the inhibitory kinases Myt1 and Wee1 through phosphorylating CDK1 at Thr14 and Tyr15. In addition, CRT0066101 modulates several other regulators of the G2/M transition involving regulation of CDK1, including p27Kip1, Gadd45α, 14-3-3σ, and other isoforms of 14-3-3 proteins. All of the regulators may act cooperatively and collaboratively to suppress the proliferation of bladder tumor cells through regulating CDK1 and the cell cycle G2 checkpoint. Such a mechanism may serve to integrate the roles of these regulators in culmination in G2/M arrest and cell cycle block, which may underlie the mechanism of CRT0066101-mediated tumor growth suppression of bladder urothelial cell carcinoma. See “Discussion” for further elaboration. T14 threonine 14, Y15 tyrosine 15
It is widely accepted that loss of cell cycle checkpoint control is a hallmark of human cancers. The development of new antitumor drugs that target cell cycle regulatory molecules such as CDKs is being currently explored as a therapeutic approach for cancers [35, 57]. Cell cycle progression is regulated primarily by protein kinase complexes, each of which contains a cyclin and a CDK. CDKs usually exist as phosphorylated proteins, and their activation is associated with a cyclin partner and other regulatory proteins [33, 58, 59]. At the G2 checkpoint, cyclin B1 regulates cell cycle progression and provides a substrate specific to its partner, CDK1 (Cdc2) [60]. The loss or deactivation of either cyclin B1 or CDK1 will block progression to G2 phase [45].
In this study, we found that, in the CRT0066101-treated UMUC1 bladder tumor, the levels of CDK1, phospho-CDK1 (Thr161), and cyclin B1 were decreased, whereas the levels of phospho-CDK1 (Thr14) and phospho-CDK1 (Tyr15) were increased, all these leading to reduced activity of the CDK1-cyclin B1 complex. These data suggest that the G2/M arrest by CRT0066101 in our tumor model is due to a decrease in CDK1 phosphorylation on Thr161 by CDK-activating kinase (CAK), an increase in CDK1 phosphorylation on Thr14 and Tyr15 by Myt1 and Wee1, and a reduction in CDK1 and cyclin B1 protein levels.
Additionally, we showed that CRT0066101 downregulated the expression of Cdc25C, which dephosphorylates/activates CDK1, but increased the activity of the cell cycle checkpoint kinase Chk1, which phosphorylates/inactivates Cdc25C and leads to enhanced Cdc25C phosphorylation at the Ser216 residue in our model system. The activity of Cdc25C, which is essential for progression into mitosis, is regulated by changes in protein level, subcellular localization, and phosphorylation state [61]. For instance, G2/M cell cycle-blocking agents have been shown to inhibit Cdc25C activity through phosphorylation of the Ser216 by upstream Chk1 kinase [62, 63]. Therefore, the effect of CRT0066101 on G2/M arrest in UMUC1 cells may be mediated by Chk1 activation through a p53-independent Chk1/Cdc25C/CDK1 signaling pathway.
Finally, we demonstrate that CRT0066101 induced increases in the levels of p27kip1, Gadd45α, 14-3-3ε, 14-3-3σ, and other isoforms of 14-3-3 proteins in UMUC1 bladder cancer cells. p27kip1 is a CIP/KIP family inhibitor of CDK1 kinase, but is not a target gene of p53. While Gadd45α is also implicated in regulation of the G2/M transition, and its expression is mediated by both p53-dependent and -independent mechanisms. The effect of Gadd45 on the G2/M transition is due to its ability to dissociate complexes of CDK1 and cyclin B1 [64, 65]. Addition of recombinant Gadd45 releases CDK1 from the CDK1-cyclin B1 complex, suggesting that Gadd45 inhibits CDK1 by blocking its binding to cyclin B1 [64, 65]. Upon phosphorylation by Chk1 kinase, phospho-Cdc25C (Ser216) binds to 14-3-3 proteins (14-3-3ε/γ), which anchor Cdc25C in the cytoplasm where it cannot activate CDK1. Other isoforms of 14-3-3 proteins, such as 14-3-3β [55] and 14-3-3ζ [56], also control the cell cycle by directly affecting Wee1 enzymatic activity. The association of 14-3-3 proteins to Wee1 enhances its activation [55, 56, 66]. Activated Wee1 inhibits CDK1 phosphorylation and blocks cell cycle progression [66]. For CDK1 to induce mitosis, it must enter the nucleus, a process mediated by binding of cyclin B1 to importin β which, along with importin α, translocates the CDK1-cyclin B1 complex into the nucleus [67, 68]. The protein 14-3-3σ can bind to CDK1-cyclin B1 and sequester it in the cytoplasm causing cells to arrest in G2 [69], suggesting that the binding of 14-3-3σ blocks entry into mitosis by anchoring the CDK1-cyclin B1 complex in the cytoplasm.
Although CRT0066101 can modulate the activation and expression of several G2/M transition regulators in our bladder tumor model, the ability of CRT0066101 to upregulate 14-3-3σ in UMUC1 cells was unexpected, because 14-3-3σ has been demonstrated to be a transcriptional target of p53, and we show by DNA sequencing that UMUC1 cells express a mutated form of p53 (data not given). This indicates that UMUC1 bladder cancer cells possess alternative pathways for p53-independent 14-3-3σ induction. Our data are consistent with the literature reporting upregulation of 14-3-3σ in several pancreatic carcinoma cell lines with mutated p53 phenotype is controlled by an alternative p53-independent mechanism [70]. It could be speculated that other p53 family members such as p63 or p73 [71] might be responsible for the enhanced expression of 14-3-3σ in CRT0066101-treated UMUC1 bladder tumor.
A proposed potential mechanism that is consistent with the evidence is presented in Fig. 14. In this model, PKD2 inhibition by CRT0066101 treatment regulates the G2/M transition via modulation of the cyclin dependent kinase CDK1, which is essential for entry into mitosis. Binding to cyclin B1 and phosphorylation at Thr161 by CDK-activating kinase (CAK) are required to activate CDK1. Following treatment of bladder tumor cells with CRT0066101, the activity of Chk1 kinase is increased, which activates the inhibitory kinases Myt1 and Wee1, which then phosphorylate CDK1 at Thr14 and Tyr15, keeping the CDK1-cyclin B1 complex inactive during G2. At the onset of mitosis, both of these residues (Thr14/Tyr15) are dephosphorylated by the activating phosphatase Cdc25C [72]. CDK1-cyclin B1 can then phosphorylate Cdc25C, further activating it and initiating a positive feedback loop [73]. Besides targeting Myt1 and Wee1, the enhanced Chk1 kinase also phosphorylates Cdc25C at Ser216 and reduce the activity of the phosphatase for activation of the CDK1-cyclin B1 complex. Moreover, upon phosphorylation by Chk1, phospho-Cdc25C (Ser216) binds to 14-3-3 proteins such as 14-3-3ε and 14-3-3γ, which anchor Cdc25C in the cytoplasm where it cannot activate CDK1. Some isoforms of 14-3-3 proteins (14-3-3β [55] and 14-3-3ζ [56]) activate Wee1 tyrosine kinase as well, the latter inhibits CDK1 by phosphorylation on Tyr15. Additionally, several other regulators of the G2/M transition can inhibit CDK1, including p27Kip1, which inhibits CDK1 directly, Gadd45α, which dissociates CDK1 from cyclin B1, and 14-3-3σ, which anchors CDK1 in the cytoplasm where it cannot induce mitosis. Together, the concerted actions of all these regulators contribute to the cell cycle G2/M arrest, leading to inhibition of urothelial bladder carcinoma cell growth and proliferation.
Lastly, we show in this study a significant reduction in migration and invasion of bladder cancer cells in response to silencing and pharmacological inhibition of PKD2. The mechanisms accounting for the reduced invasive ability of bladder tumor cells by CRT0066101 treatment or PKD2 depletion are not clear. Proteins of the matrix metalloproteinase (MMP) family and cell adhesion molecules such as integrins are well known for their important roles in tumor progression and metastasis. Urokinase-type plasminogen activator (uPA) and urokinase-type plasminogen activator receptor (uPAR) are also known to promote invasion, migration, and metastasis in cancer cells. Recent evidence shows that CRT0066101 treatment or PKD2 silencing markedly decreases tumor cell migration and invasion, prevents PKD2 and c-Jun phosphorylation, and inhibits downstream activation of p44/42 MAPK, p54/46 JNK, p38 MAPK, and NF-κB signaling [74, 75]. Silencing of PKD2 also downregulates the expression of integrin α2/4, uPA, uPAR, and MMP1, but increases plasminogen activator inhibitor-2 (PAI-2) expression in cancer cells [74, 75]. Further, PKD2 is found to enhance the activity of uPA and MMP9, whereas depletion of PKD2 significantly impairs uPA and MMP9 activity in prostate carcinoma cells [75]. These findings suggest that the distinct reduction in tumor cell migration and invasion by pharmacologic inhibition and silencing of PKD2 is mediated via regulating expression of the genes involved in invasion and metastasis in the uPA–uPAR and MMP pathways. In the present study, we also show that pharmacological inhibition of PKD2 by CRT0066101 or knockdown of PKD2 by RNA interference profoundly diminished phosphorylation of PKD2 and its substrate c-Jun in our bladder tumor model system (Figs. 7, 10). However, the mechanism for how CRT0066101 reduces bladder carcinoma cell migration and invasion via inhibition of PKD2 remains experimentally determined.
Collectively, we have shown that the PKD inhibitor CRT0066101 suppressed bladder cancer cell growth and invasion in vitro and in xenografts through inhibition of PKD2. Moreover, CRT0066101 had differential effects on cell survival between human urothelial bladder cancer cells and nontumorigenic human uroepithelial cells. The suppressive effect of the compound on cell proliferation within the bladder tumor was apparently due to enhanced cell cycle arrest at G2/M, a process mediated by modulation of the level and activity of the key G2/M transition regulators. However, further investigation is required to examine whether CRT0066101-induced G2/M arrest links to apoptosis, delineate the p53-dependent signaling pathway by which CRT0066101 triggers cell cycle arrest, as well as elucidate the mechanism underlying the inhibitory effect of CRT0066101 on cancer cell migration and metastatic progression. This report represents a pioneering study on the mechanistic actions of PKD inhibitors in bladder carcinoma. These findings also provide a scientific basis for developing the use of CRT0066101 as a novel intravesical agent or a combinatorial treatment for use with other drugs (e.g., cell cycle checkpoint inhibitors [76], histone deacetylase inhibitors [77], and heat shock protein 90 inhibitors [78]) as a potentially more effective chemotherapeutic regimen for treatment of bladder tumors, and suggest that further clinical study on PKD inhibition-mediated therapy in patients with bladder cancer is warranted.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A (2015) Global cancer statistics, 2012. CA Cancer J Clin 65:87–108
Torre LA, Siegel RL, Ward EM, Jemal A (2016) Global cancer incidence and mortality rates and trends—an update. Cancer Epidemiol Biomark Prev 25:16–27
Sanli O, Dobruch J, Knowles MA, Burger M, Alemozaffar M, Nielsen ME, Lotan Y (2017) Bladder cancer. Nat Rev Dis Prim 3:17022
Kaufman DS, Shipley WU, Feldman AS (2009) Bladder cancer. Lancet 374:239–249
Fletcher A, Choudhury A, Alam N (2011) Metastatic bladder cancer: a review of current management. ISRN Urol 2011:545241
Grossman HB, Natale RB, Tangen CM, Speights VO, Vogelzang NJ, Trump DL, deVere White RW, Sarosdy MF, Wood DP Jr, Raghavan D, Crawford ED (2003) Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer. N Engl J Med 349:859–866
Dovedi SJ, Davies BR (2009) Emerging targeted therapies for bladder cancer: a disease waiting for a drug. Cancer Metastasis Rev 28:355–367
Bellmunt J, Petrylak DP (2012) New therapeutic challenges in advanced bladder cancer. Semin Oncol 39:598–607
Weintraub MD, Li QQ, Agarwal PK (2014) Advances in intravesical therapy for the treatment of non-muscle invasive bladder cancer (review). Mol Clin Oncol 2:656–660
Mohammed AA, El-Tanni H, El-Khatib HM, Mirza AA, Mirza AA, Alturaifi TH (2016) Urinary bladder cancer: biomarkers and target therapy, new era for more attention. Oncol Rev 10:320
Alpsoy A, Gunduz U (2015) Protein kinase D2 silencing reduced motility of doxorubicin-resistant MCF7 cells. Tumour Biol 36:4417–4426
Zhu Y, Cheng Y, Guo Y, Chen J, Chen F, Luo R, Li A (2016) Protein kinase D2 contributes to TNF-α-induced epithelial mesenchymal transition and invasion via the PI3 K/GSK-3β/β-catenin pathway in hepatocellular carcinoma. Oncotarget 7:5327–5341
Wong C, Jin ZG (2005) Protein kinase C-dependent protein kinase D activation modulates ERK signal pathway and endothelial cell proliferation by vascular endothelial growth factor. J Biol Chem 280:33262–33269
Ha CH, Wang W, Jhun BS, Wong C, Hausser A, Pfizenmaier K, McKinsey TA, Olson EN, Jin ZG (2008) Protein kinase D-dependent phosphorylation and nuclear export of histone deacetylase 5 mediates vascular endothelial growth factor-induced gene expression and angiogenesis. J Biol Chem 283:14590–14599
Dequiedt F, Van Lint J, Lecomte E, Van Duppen V, Seufferlein T, Vandenheede JR, Wattiez R, Kettmann R (2005) Phosphorylation of histone deacetylase 7 by protein kinase D mediates T cell receptor-induced Nur77 expression and apoptosis. J Exp Med 201:793–804
Bastea LI, Doppler H, Balogun B, Storz P (2012) Protein kinase D1 maintains the epithelial phenotype by inducing a DNA-bound, inactive SNAI1 transcriptional repressor complex. PLoS One 7:e30459
Zheng H, Shen M, Zha YL, Li W, Wei Y, Blanco MA, Ren G, Zhou T, Storz P, Wang HY, Kang Y (2014) PKD1 phosphorylation-dependent degradation of SNAIL by SCF-FBXO11 regulates epithelial-mesenchymal transition and metastasis. Cancer Cell 26:358–373
Baron CL, Malhotra V (2002) Role of diacylglycerol in PKD recruitment to the TGN and protein transport to the plasma membrane. Science 295:325–328
Hausser A, Storz P, Martens S, Link G, Toker A, Pfizenmaier K (2005) Protein kinase D regulates vesicular transport by phosphorylating and activating phosphatidylinositol-4 kinase IIIβ at the Golgi complex. Nat Cell Biol 7:880–886
Waldron RT, Rozengurt E (2000) Oxidative stress induces protein kinase D activation in intact cells. Involvement of Src and dependence on protein kinase C. J Biol Chem 275:17114–17121
Zugaza JL, Sinnett-Smith J, Van Lint J, Rozengurt E (1996) Protein kinase D (PKD) activation in intact cells through a protein kinase C-dependent signal transduction pathway. EMBO J 15:6220–6230
Hao Q, McKenzie R, Gan H, Tang H (2013) Protein kinases D2 and D3 are novel growth regulators in HCC1806 triple-negative breast cancer cells. Anticancer Res 33:393–399
Wei N, Chu E, Wipf P, Schmitz JC (2014) Protein kinase d as a potential chemotherapeutic target for colorectal cancer. Mol Cancer Ther 13:1130–1141
Liou GY, Storz P (2015) Protein kinase D enzymes: novel kinase targets in pancreatic cancer. Expert Rev Gastroenterol Hepatol 9:1143–1146
Harikumar KB, Kunnumakkara AB, Ochi N, Tong Z, Deorukhkar A, Sung B, Kelland L, Jamieson S, Sutherland R, Raynham T, Charles M, Bagherzadeh A, Foxton C, Boakes A, Farooq M, Maru D, Diagaradjane P, Matsuo Y, Sinnett-Smith J, Gelovani J, Krishnan S, Aggarwal BB, Rozengurt E, Ireson CR, Guha S (2010) A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther 9:1136–1146
Durand N, Borges S, Storz P (2016) Protein kinase D enzymes as regulators of EMT and cancer cell invasion. J Clin Med 5:20
Borges S, Perez EA, Thompson EA, Radisky DC, Geiger XJ, Storz P (2015) Effective targeting of estrogen receptor-negative breast cancers with the protein kinase D inhibitor CRT0066101. Mol Cancer Ther 14:1306–1316
Wei N, Chu E, Wu SY, Wipf P, Schmitz JC (2015) The cytotoxic effects of regorafenib in combination with protein kinase D inhibition in human colorectal cancer cells. Oncotarget 6:4745–4756
Verbon EH, Post JA, Boonstra J (2012) The influence of reactive oxygen species on cell cycle progression in mammalian cells. Gene 511:1–6
Lu Z, Hunter T (2010) Ubiquitylation and proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2) CDK inhibitors. Cell Cycle 9:2342–2352
McDonald ER 3rd, El-Deiry WS (2000) Cell cycle control as a basis for cancer drug development (review). Int J Oncol 16:871–886
Lacy ER, Wang Y, Post J, Nourse A, Webb W, Mapelli M, Musacchio A, Siuzdak G, Kriwacki RW (2005) Molecular basis for the specificity of p27 toward cyclin-dependent kinases that regulate cell division. J Mol Biol 349:764–773
Hu X, Moscinski LC (2011) Cdc2: a monopotent or pluripotent CDK? Cell Prolif 44:205–211
O’Connell MJ, Walworth NC, Carr AM (2000) The G2-phase DNA-damage checkpoint. Trends Cell Biol 10:296–303
Brezak MC, Quaranta M, Mondesert O, Galcera MO, Lavergne O, Alby F, Cazales M, Baldin V, Thurieau C, Harnett J, Lanco C, Kasprzyk PG, Prevost GP, Ducommun B (2004) A novel synthetic inhibitor of CDC25 phosphatases: bN82002. Cancer Res 64:3320–3325
Damia G, Broggini M (2004) Cell cycle checkpoint proteins and cellular response to treatment by anticancer agents. Cell Cycle 3:46–50
Zhang Y, Hunter T (2014) Roles of Chk1 in cell biology and cancer therapy. Int J Cancer 134:1013–1023
Perdiguero E, Nebreda AR (2004) Regulation of Cdc25C activity during the meiotic G2/M transition. Cell Cycle 3:733–737
Mueller PR, Coleman TR, Kumagai A, Dunphy WG (1995) Myt1: a membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science 270:86–90
Ruiz EJ, Vilar M, Nebreda AR (2010) A two-step inactivation mechanism of Myt1 ensures CDK1/cyclin B activation and meiosis I entry. Curr Biol 20:717–723
Den Haese GJ, Walworth N, Carr AM, Gould KL (1995) The Wee1 protein kinase regulates T14 phosphorylation of fission yeast Cdc2. Mol Biol Cell 6:371–385
Watanabe N, Broome M, Hunter T (1995) Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J 14:1878–1891
Parker LL, Sylvestre PJ, Byrnes MJ 3rd, Liu F, Piwnica-Worms H (1995) Identification of a 95-kDa WEE1-like tyrosine kinase in HeLa cells. Proc Natl Acad Sci USA 92:9638–9642
Kawabe T (2004) G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther 3:513–519
Heller JD, Kuo J, Wu TC, Kast WM, Huang RC (2001) Tetra-O-methyl nordihydroguaiaretic acid induces G2 arrest in mammalian cells and exhibits tumoricidal activity in vivo. Cancer Res 61:5499–5504
Li QQ, Wang G, Liang H, Li JM, Huang F, Agarwal PK, Zhong Y, Reed E (2013) β-Elemene promotes cisplatin-induced cell death in human bladder cancer and other carcinomas. Anticancer Res 33:1421–1428
Rotem A, Janzer A, Izar B, Ji Z, Doench JG, Garraway LA, Struhl K (2015) Alternative to the soft-agar assay that permits high-throughput drug and genetic screens for cellular transformation. Proc Natl Acad Sci USA 112:5708–5713
Cancer Genome Atlas Research N (2014) Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507:315–322
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47
Dyrskjot L, Kruhoffer M, Thykjaer T, Marcussen N, Jensen JL, Moller K, Orntoft TF (2004) Gene expression in the urinary bladder: a common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res 64:4040–4048
Sanchez-Carbayo M, Socci ND, Lozano J, Saint F, Cordon-Cardo C (2006) Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol 24:778–789
Blaveri E, Simko JP, Korkola JE, Brewer JL, Baehner F, Mehta K, Devries S, Koppie T, Pejavar S, Carroll P, Waldman FM (2005) Bladder cancer outcome and subtype classification by gene expression. Clin Cancer Res 11:4044–4055
Wang XW, Zhan Q, Coursen JD, Khan MA, Kontny HU, Yu L, Hollander MC, O’Connor PM, Fornace AJ Jr, Harris CC (1999) GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci USA 96:3706–3711
Jiang K, Pereira E, Maxfield M, Russell B, Goudelock DM, Sanchez Y (2003) Regulation of Chk1 includes chromatin association and 14-3-3 binding following phosphorylation on Ser-345. J Biol Chem 278:25207–25217
Wang Y, Jacobs C, Hook KE, Duan H, Booher RN, Sun Y (2000) Binding of 14-3-3β to the carboxyl terminus of Wee1 increases Wee1 stability, kinase activity, and G2-M cell population. Cell Growth Differ 11:211–219
Rothblum-Oviatt CJ, Ryan CE, Piwnica-Worms H (2001) 14-3-3 Binding regulates catalytic activity of human Wee1 kinase. Cell Growth Differ 12:581–589
Eastman A (2004) Cell cycle checkpoints and their impact on anticancer therapeutic strategies. J Cell Biochem 91:223–231
Sherr CJ (1996) Cancer cell cycles. Science 274:1672–1677
Dynlacht BD (1997) Regulation of transcription by proteins that control the cell cycle. Nature 389:149–152
Choi YH, Lee WH, Park KY, Zhang L (2000) p53-independent induction of p21 (WAF1/CIP1), reduction of cyclin B1 and G2/M arrest by the isoflavone genistein in human prostate carcinoma cells. Jpn J Cancer Res 91:164–173
Lopez-Girona A, Furnari B, Mondesert O, Russell P (1999) Nuclear localization of Cdc25 is regulated by DNA damage and a 14-3-3 protein. Nature 397:172–175
Furnari B, Rhind N, Russell P (1997) Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science 277:1495–1497
Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ (1997) Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 277:1497–1501
Zhan Q, Antinore MJ, Wang XW, Carrier F, Smith ML, Harris CC, Fornace AJ Jr (1999) Association with Cdc2 and inhibition of Cdc2/Cyclin B1 kinase activity by the p53-regulated protein Gadd45. Oncogene 18:2892–2900
Vairapandi M, Balliet AG, Hoffman B, Liebermann DA (2002) GADD45b and GADD45g are cdc2/cyclin B1 kinase inhibitors with a role in S and G2/M cell cycle checkpoints induced by genotoxic stress. J Cell Physiol 192:327–338
Lee J, Kumagai A, Dunphy WG (2001) Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol Biol Cell 12:551–563
Moore JD, Yang J, Truant R, Kornbluth S (1999) Nuclear import of Cdk/cyclin complexes: identification of distinct mechanisms for import of Cdk2/cyclin E and Cdc2/cyclin B1. J Cell Biol 144:213–224
Takizawa CG, Weis K, Morgan DO (1999) Ran-independent nuclear import of cyclin B1-Cdc2 by importin β. Proc Natl Acad Sci USA 96:7938–7943
Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B (1997) 14-3-3σ is a p53-regulated inhibitor of G2/M progression. Mol Cell 1:3–11
Guweidhi A, Kleeff J, Giese N, El Fitori J, Ketterer K, Giese T, Buchler MW, Korc M, Friess H (2004) Enhanced expression of 14-3-3σ in pancreatic cancer and its role in cell cycle regulation and apoptosis. Carcinogenesis 25:1575–1585
Courtois S, Caron de Fromentel C, Hainaut P (2004) p53 protein variants: structural and functional similarities with p63 and p73 isoforms. Oncogene 23:631–638
Draetta G, Eckstein J (1997) Cdc25 protein phosphatases in cell proliferation. Biochim Biophys Acta 1332:M53–M63
Izumi T, Maller JL (1993) Elimination of cdc2 phosphorylation sites in the cdc25 phosphatase blocks initiation of M-phase. Mol Biol Cell 4:1337–1350
Bernhart E, Damm S, Wintersperger A, DeVaney T, Zimmer A, Raynham T, Ireson C, Sattler W (2013) Protein kinase D2 regulates migration and invasion of U87MG glioblastoma cells in vitro. Exp Cell Res 319:2037–2048
Zou Z, Zeng F, Xu W, Wang C, Ke Z, Wang QJ, Deng F (2012) PKD2 and PKD3 promote prostate cancer cell invasion by modulating NF-κB- and HDAC1-mediated expression and activation of uPA. J Cell Sci 125:4800–4811
Visconti R, Della Monica R, Grieco D (2016) Cell cycle checkpoint in cancer: a therapeutically targetable double-edged sword. J Exp Clin Cancer Res 35:153
Li QQ, Hao JJ, Zhang Z, Hsu I, Liu Y, Tao Z, Lewi K, Metwalli AR, Agarwal PK (2016) Histone deacetylase inhibitor-induced cell death in bladder cancer is associated with chromatin modification and modifying protein expression: a proteomic approach. Int J Oncol 48:2591–2607
Li QQ, Hao JJ, Zhang Z, Krane LS, Hammerich KH, Sanford T, Trepel JB, Neckers L, Agarwal PK (2017) Proteomic analysis of proteome and histone post-translational modifications in heat shock protein 90 inhibition-mediated bladder cancer therapeutics. Sci Rep 7:201
Acknowledgements
This research was supported by the Intramural Research Program of the U.S. National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Li, Q.Q., Hsu, I., Sanford, T. et al. Protein kinase D inhibitor CRT0066101 suppresses bladder cancer growth in vitro and xenografts via blockade of the cell cycle at G2/M. Cell. Mol. Life Sci. 75, 939–963 (2018). https://doi.org/10.1007/s00018-017-2681-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2681-z