
Abstract
Profilins were discovered in the 1970s and were extensively studied for their significant physiological roles. Profilin1 is the most prominent isoform and has drawn special attention due to its role in the cytoskeleton, cell signaling, and its link to conditions such as cancer and vascular hypertrophy. Recently, multiple mutations in the profilin1 gene were linked to amyotrophic lateral sclerosis (ALS). In this review, we will discuss the physiological and pathological roles of profilin1. We will further highlight the cytoskeletal function and dysfunction caused by profilin1 dysregulation. Finally, we will discuss the implications of mutant profilin1 in various diseases with an emphasis on its contribution to the pathogenesis of ALS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Profilin is a ubiquitous cytosolic protein and was one of the first identified actin-binding proteins. Initial studies suggested that its main function was to sequester actin monomers in a 1:1 complex and then release actin in response to downstream cellular signals such as an increase in phosphatidylinositol (4,5)-bisphosphate (PIP2) [1, 2]. However, subsequent studies revealed that the basal cellular levels of profilin are rather insufficient to sequester abundant actin monomers. These studies showed that in addition to its sequestering function, profilin promotes the assembly of globular-actin monomers (G-actin) into filamentous-actin (F-actin) [3]. Profilin catalyzes actin polymerization in a concentration-dependent manner. It serves as a catalyst at lower concentrations and an inhibitor at higher levels [4]. Furthermore, profilin interacts with multiple ligands and binding partners implicating it in signaling processes beyond its actin-binding function.
Four isoforms of profilin have been discovered to date. The first mammalian profilin gene that was isolated, named PFN1, corresponds to the profilin protein isolated from the thymus, profilin1 [1, 2]. Profilin1 is a small (15,054 Da), highly conserved protein composed of 139 amino acid residues in humans and 140 amino acid residues in rodents. Expression studies in mice have shown that mammalian profilin1 is expressed during all embryonic stages, and additional studies suggest that it is present in nearly all cell and tissue types, including platelets, glia and glioma, and lymphoid cells [1, 2, 5]. In contrast, profilin2 (15,046 Da) is expressed primarily in the developing nervous system and in differentiated neurons, and profilin3 and profilin4 (15,594 and 14,319 Da, respectively) are exclusively expressed in testes [2]. This review focuses on the most prominent isoform, profilin1, due to a plethora of interesting data which have emerged in the last decade linking it to various diseases (Table 1).
In this review, we will discuss profilin1’s biology and its role in actin dynamics in addition to its non-actin-related cellular roles. We will further discuss the cytoskeletal function and dysfunction due to profilin1 dysregulation. Finally, this review discusses the implications of mutant profilin1 in disease with an emphasis on its highly relevant role in amyotrophic lateral sclerosis (ALS).
Profilin1 structure and expression
Human profilin1 tertiary structure was defined using NMR and X-ray crystallography [6] and shows an anti-parallel, 7-stranded β-sheet packed between four α-helices (Fig. 1) [6]. Profilin1 has a complex molecular structure: two of the helices are located at the termini and contribute side chains to a common hydrophobic core involving the β-sheet. Residues on the opposing face of the sheet give rise to a more extensive hydrophobic region juxtaposed by the other two helices [7]. Profilin1’s amino acid sequences are evolutionarily highly conserved across vertebrates and lower organisms [8]. This fact suggests that the profilin1 structure is fundamentally essential to life and must be conserved to maintain its vital actin-regulating function. Profilin1 has an actin-binding domain located on helix 3, which has some contact with helix 4 and strands 4, 5, and 6 in the C-terminal (Fig. 1b). In addition, a poly-l-proline (PLP) binding domain is located at the N-terminal [7, 9].

Profilin1 protein structure and ALS-linked mutations. a Schematic presentation of profilin1, including the five α-helices (tan) and seven β-sheets (green). Eight of the ALS-linked mutations are indicated by black arrows. Three amino acids residues with notable phosphorylation sites on the C-terminal are highlighted (S137 and Y129) and one is at position of T109 that is mutated in ALS. b Three-dimensional structure of human profilin1 (PDB: 1fik) in cartoon representation. The ALS-linked mutations are labeled. Crystal structure data of bovine profilin-1 were downloaded from protein data base (PDB) to show predicted human profilin1 3-D structure: 4X1L and ALS-linked mutation were added using PyMOL (The PyMol Molecular Graphics System, Version 1.5.0.4, Schrodinger, LLC)
Amino acid residues in the actin-binding domain form 2250 Å of combined interfacial surface area in profilin1. This arrangement indicates that the profilin1-actin bond is very strong, hydrophobic, and functionally essential [7, 10]. A surface area of this magnitude is reminiscent of functionally important protein–protein interactions seen in antibody-antigen and phosphatase-substrate interactions [7]. In addition, profilin1 contains two PIP2 binding sites, one of which is located in the PLP binding domain, while the other is located in the actin-binding domain. These dual sites suggest that PIP2 may act as a multi-faceted regulator of profilin1 function as it can prevent both actin and PLP binding [11]. Studies published by Pei et al. [12] and Krishnamoorthy et al. [12, 13] appear to confirm a model in which phosphoinositides bind profilin1 in a complex with actin, forcing the complex to dissociate and release G-actin. Profilin1 domains, amino acid residues, and binding sites dictate its function and interactions with ligands and binding partners. Thus, mutations in these structural components could affect profilin1 functions by radically changing the 3-D structure. These changes have been found to alter profilin1’s binding profile, as discovered by Bosco and colleagues [14], and are discussed later in the ALS section (Fig. 1b).
Studies of profilin1’s spatial expression and tissue-specific abundance show that it comprises 0.2–0.4 % of total protein in non-neuronal tissues, whereas it accounts for only 0.05 % of total protein in the brain. Interestingly, the expression levels of profilin1 and profilin2 seem to inversely complement each other. Profilin2 accounts for 0.01 and 0.02 % of total protein in all tissues sampled except the brain; in this organ, profilin2 content is 15-fold higher [15]. Despite profilin1’s low expression in the brain, it is heavily expressed in activated microglia, likely because these microglia must be highly motile to reach damaged areas of the brain for repair [16]. In accordance with profilin1’s role in cell motility, it is frequently found near the leading edge of the cell membrane, where it aids in lamellipodium formation [15]. It is also found in the nucleus, where it colocalizes with nuclear subcompartments, splicing speckles, and Cajal bodies. This distinct nuclear distribution suggests other cell signaling roles for profilin1 independent of actin polymerization.
Profilin1 protein–protein interactions
Profilin1 interactions with survival of motor neuron protein (SMN) and p42POP (partner of profilin) provide evidence that profilin1 plays a role in RNA processing and neuronal transcription. These putative roles are further supported by the fact that the profilin-actin complex is rapidly removed from the nucleus by exportin-6, but unbound profilin1 is not [2]. A few studies have documented the regulation of profilin1 gene expression, but its ubiquitous presence suggests the possibility of post-translational regulation, which is discussed in the next section. However, although profilin1 expression apparently is not highly dynamic, its level of activation is regulated by the Rho/ROCK pathway and post-translational protein modification. Integrin linked kinase (Ilk)-mediated downregulation of Rho/ROCK leads to a decrease in phosphorylation of profilin1 at the S137 residue, and this event occurs in a Rac1-independent manner despite the initial hypothesis that Rac1 was involved in the process due to its interactions with Rho (Fig. 1a). Upregulation of insulin-like growth factor-1 (IGF-1) has the same effect as that of Ilk, namely, negatively regulating Rho/ROCK levels and consequently raising levels of activated profilin1. The converse is also true. Downregulation of IGF-1 or Ilk leads to an increase in Rho/ROCK, resulting in an increase in phosphorylated profilin1 [17, 18]. Furthermore, profilin1 binds to WAVE [Wiskott–Aldrich syndrome protein (WASP) family verprolin-homologous protein], heat-shock protein (HSP) C300, Nap1, and POP-130, all of which are involved in the Rho/ROCK cascade. The Rho/ROCK cascade is one of the most important regulators of the actin cytoskeleton, further supporting profilin1’s significant contribution to cytoskeletal dynamics. In addition, profilin1 binds to mDia1, mDia2, and mDia3 (mammalian homolog of the Drosophila gene diaphanous), all of which are potent nucleation molecules in actin polymerization [19]. However, the interaction between profilin1 and mDia proteins was found using truncated mDia; therefore, this needs to be replicated using full length mDia proteins [20–22]. Hence, it is likely that an interaction also occurs between profilin1 and the unmodified mDia. Profilin1 also interacts with vascular endothelial growth factor (VEGF), a small protein, and a known neurotropic factor involved in angiogenesis, which was also implicated recently in cell signaling and tumorigenesis [23]. Specifically, VEGF-A binding to VEGF receptor 2 (VEGFR2) triggers phosphorylation of profilin1 Y129, increasing profilin1’s affinity for actin, thereby enhancing actin polymerization at the edge of the cell—the filopodium effect (Fig. 1a). This mechanism has been suggested as a physiological strategy for VEGF-A to initiate cytoskeletal remodeling. However, VEGFR1 can have the same effect as the VEGFR2 activated kinase, so it is likely that some combination of these molecules work together for proper phosphorylation. Interestingly, the VEGFR2-induced phosphorylation of profilin1 Y129 does not appear to be necessary in developmental angiogenesis, as a synthetic Y129F mutation in profilin1 failed to affect normal angiogenesis and arteriogenesis during development, but prevented proper wound healing and arteriogenesis later in life [24].
Profilins1 and 2 also bind to a variety of structural proteins in neurons. These proteins include gephyrin, drebrin, piccolo (was also referred to as aczonin), and delphilin, all of which regulate the actin cytoskeleton implicated in synapse dynamics. Piccolo is a major coordinating factor in the dynamic assembly of F-actin in association with its binding partners, Abp1, profiling, and GITI [25–28]. This association recently was confirmed by knockdown of both Piccolo and profilin2 [29]. These studies provide light on the importance of piccolo and F-actin roles in presynaptic dynamics and regulation of neurotransmitter release. The two profilins bind with roughly equal affinities to all of the structural proteins evaluated to date except for piccolo, which has a higher affinity for profilin2 than for profilin1 [22]. Despite the fact that these interactions between the profilins and structural proteins in neurons have been known for at least 10 years, little is known about their exact functional role. It seems possible that the interactions could provide a molecular mechanism for profilin-regulated remodeling of the structure of the synaptic cleft, with profilin serving as a link to one of the signaling pathways discussed earlier.
Profilin1 post-translational modifications
Profilin1 activity and interactions with other proteins and the corresponding signaling cascades are regulated, in part, by post-translational modifications. Recent studies revealed the biological significance of two of these modifications: phosphorylation of profilin1 at residues S137 and Y129 (Fig. 1a). These phosphorylations increase the affinity of profilin1 for G-actin, which alters actin polymerization dynamics and, thereby, impacts the cell in different ways. For instance, phosphorylation of profilin1 at S137 enhances the migration and invasion of breast cancer cells (MCF7), whereas the phosphorylation at Y129 (which is induced by VEGF-A, as discussed above) at the leading edge of endothelial cells plays a role in endothelial cell migration and angiogenesis [30]. In addition, phosphorylation at Y129 promotes glioblastoma progression by a mechanism that involves accumulation of hypoxia-inducible factor (HIF)-1α in normoxic conditions, which drives aberrant vascularization and glioblastoma progression (Table 1) [31].
Profilin’s role in actin polymerization and depolymerization
The process by which profilin1 increases actin polymerization is complex and it relies on a variety of different factors. One hypothesis is that profilin1 can promote net polymerization by facilitating exchange diffusion. Exchange diffusion describes a process by which ATP-actin or ADP-Pi-actin pairs are added to the filament end, but then dissociate in accordance with random thermal fluctuations (Fig. 2a). Following dissociation, the actin monomers dissociate the ADP or ADP-Pi and exchange it for ATP before binding to F-actin, thus resulting in net polymerization. In this process, profilin1 serves to increase the dissociation rate and accelerate the rate of nucleotide exchange, both of which contribute to an elevated rate of polymerization [4]. Profilin1 may also lower the critical concentration of actin (the necessary amount for polymerization to occur), thus allowing polymerization to begin earlier and last longer [4]. Another current hypothesis is that profilin1 only promotes actin polymerization when either additional actin sequestering molecules or formins are present. The effect of profilin1 on formin-associated strands is concentration-dependent; high profilin1 concentrations inhibit polymerization, whereas lower concentrations of profilin1 in the range 2–5 µM promote faster filament growth. Formins bind to the barbed end of actin filaments and are known to increase polymerization rate independent of profilin1, but the polymerization rate is further increased by the addition of profilin1 [10]. In the presence of other actin sequestering molecules, profilin1 competes for the actin-binding site and serves to shuttle bound actin to the barbed end of the growing polymer [32]. Neither of these proposed mechanisms directly contradict the idea that profilin1 promotes polymerization through enabling faster exchange diffusion, so it is possible that all three mechanisms occur concurrently.

a Diagrammatic scheme depicting profilin1 regulation of actin polymerization. b Diagrammatic scheme depicting the events by which mutations in profilin1 disrupt actin polymerization, thereby potentially causing motor neuron degeneration
Profilin1 in early brain development
Profilin1 is required for normal mouse brain development, which relies on profilin1 regulation of actin. Recent studies by Rust and colleagues [33, 34] showed that profilin1 is required for the radial migration of cerebellar granule neurons (CGN) and for their adhesion with glia cells. Because systemic profilin1 deletion leads to embryonic lethality, the authors chose to develop conditional profilin1 knockout mice. The mice with profilin1 knockout in specific regions of the brain were developed by crossing a conditional profilin1 mouse model with a nestin-cre transgenic line that expresses cre in all brain cells during development. This conditional profilin1 deletion led to a reduced brain size [33]. In addition, the radial migration of CGNs was impaired after profilin1 deletion, as Brdu+-labeled neurons showed shorter mean migration distances in the mutant mice compared to wild type. Due to the actin-binding function of profilin1, it is reasonable to hypothesize that the slowed migration following profilin1 deletion may be explained by impaired CGN binding to glial cells (GCs). Indeed, the authors showed that profilin1 is necessary for normal CGN–GC adhesion. This finding emphasizes the significance of profilin1 in neuronal adhesion, and the importance of the proper cytoskeleton dynamics in the CNS for development and physiological function. Therefore, profilin1 deficiency could have detrimental effects on neuronal development.
A potential mechanism to explain the aberrant radial CGN migration is that impaired glial cell adhesion to CGNs in profilin1 deficient mice severely hampered proper radial migration. Glial cell binding assays using isolated CGNs revealed impaired adhesion, further supporting this explanation [33]. Hence, it is important to explore whether profilin1 mutations are responsible for other developmental neuropathies. Could profilin1 deficiency be directly responsible for the autosomal dominant disorder of Miller–Dieker lissencephaly syndrome and hereditary cerebellar hypoplasia as suggested by recent studies [35, 36]? If profilin1 expression, dysregulation, deficiency, or mutation leads to disease, then the mechanisms and extent of profilin1 involvement in these pathologies should be investigated in detail. The study and discoveries by Kullmann et al. [33, 34, 37] are of historical importance, since they are the first reports to link profilin1 to actin dynamics in neurons, whereas the profilin1 role in actin dynamics/cell motility in endothelial cells is well established [38].
Profilin1 and ALS
The advancement of a new sequencing technology called exome sequencing allows sequencing of all expressed genes (exons). Exome sequencing has resulted in the discovery of a new genetic link between profilin1 and ALS. ALS is a late-onset neurodegenerative disease, which leads to death usually within 3–5 years of diagnosis. Also referred to as Lou Gehrig’s disease, ALS is caused by the progressive death of motor neurons in the brain and spinal cord. The loss of motor neurons impairs motor coordination, which progresses to paralysis and eventually respiratory failure. It is well documented that motor neuron death begins well before the appearance of symptoms or diagnosis of the disease [39]. ALS cases are divided into two primary groups: sporadic (sALS) and familial (fALS). Cases caused by profilin1 mutations fall into the familial category, which accounts for roughly 10 % of total ALS cases [39]. One non-synonymous mutation (R136W) found in sALS category as described below (Chen et al., 2013). Riluzole is the only FDA-approved drug available for ALS treatment, and it confers only a moderate increase in survival [40]. A new drug emerging for the treatment of ALS is Edaravone or RADICUT, which is a free radical scavenger. It is approved as an ALS therapeutic in Japan, and in May 2016, the United States’ FDA approved it as an orphan drug for the treatment of ALS.
Exome sequencing has revealed eight mutations in the profilin1 gene in familial and sporadic ALS cases in the United States, Europe, and China; (A20T, C71G, G118V, M114T, E117G, T109M, R136W, Q139L) (Fig. 1b) [41–44]. A study of 10 fALS and 540 sALS patients in China reported one non-synonymous mutation (R136W) in one early onset sporadic ALS patient [44]. They also found a synonymous mutation (L88L) in a late-onset sALS patient, which would not have caused a mutation of the profilin1 protein, but may affected its expression since its RNA sequence changed by one nucleotide base. Although research teams outside of the United States and Europe have searched for profilin1 mutations in their local fALS patients, these studies did not identify any abnormalities, suggesting that profilin1 mutations may not be uniformly distributed across ALS patient populations in different geographical areas [41, 43–51, 54].
It is crucial to note that some mutations in profilin1 associated with fALS are in residues composing the profilin1 actin-binding domain (Fig. 3). This important observation has been a guide to subsequent research as investigators hypothesized that profilin1 mutations cause ALS as a result of altered actin binding (Fig. 2b). There are other fALS-linked profilin1 mutations (e.g., T109M and Q139L) that are located on PLP domain (Fig. 3) [41, 52, 53]. Therefore, the hypothesis that an actin-binding impairment resulting from profilin1 mutations causes toxicity in ALS may not be the only mechanism of neurodegeneration. Elegant studies by two independent research teams provide support for the concept that profilin1 mutations contribute to ALS pathogenesis by diverse mechanisms [52–54]. These recent studies provide new evidence that the mechanism of profilin1 toxicity is more complex than first thought and may involve the PLP binding domain as well as the actin-binding domain. The finding of mutations in the PLP domain of profilin1 does not dispute the toxicity caused by actin-binding domain mutations, and hence, actin dynamics and cytoskeletal dysfunction cannot be ruled out but may be part of a bigger picture of neuronal dysfunction. The mechanism(s) by which mutations in profilin1 induce ALS is a hot topic that is being intensively pursued to search for novel mechanisms involved in the pathogenesis of ALS and to identify new therapeutic targets to slow its progression. The next section will detail the seminal papers attempting to understand these elusive mechanisms.

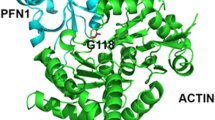
a Schematic demonstration of profilin1 actin binding. Predicted structure of human profilin1 PDB: 4X1L (yellow) and β-actin PDB: 2BTF (green) complex based on a bovine structure (2BTF) represented in cartoon. The mutated profilin1 residues involved in ALS are colored and shown in space fill. All these images were generated using PyMOL. Residues in profilin1 that makes contact with beta actin [7] are shown in cyan in dots on the top of actin (shown in green). b Crystal structure of human profilin-1 showing ALS-linked mutations. Three-dimensional structure of profilin1 was downloaded from PDB: 4X1L and mutated using PyMOL. Actin-binding domain is shown in cyan in dots
Profilin1 mutations cause ALS by inducing protein aggregation
Neuronal cultures (Neuro-2A cells) transfected with mutant profilin1 variants C71G, M114T, and G118V contain protein aggregates as evidenced by the presence of profilin1 in the insoluble fractions of cell lysates [43]. This finding indicates that mutant profilin1 accumulates in neurons, possibly in the form of inclusions and aggregates. Therefore, the authors probed for expression patterns of wild type (WT) and mutant profilin1 (C71G, G118V, and M114T) in N2A cells and primary motor neurons transfected with the profilin1 variants. They found that WT profilin1 is diffuse across the cytoplasm, whereas the mutant profilin1 variants assembled in cytoplasmic ubiquitinated aggregates. These findings are significant as the formation of aggregates is one of the hallmarks of ALS, and proving that mutant profilin1 forms aggregates in neurons further strengthens its causal link to ALS. These findings also offer a window into the mechanism of how mutations in profilin1 may lead to ALS. It is worth noting that the E117G variant was found in both controls and ALS patients; it did not form aggregates in vitro, unlike the other mutants. Therefore, E117G behaves similarly to WT, indicating that this profilin1 mutation may be a benign mutation or could be a risk factor.
A recent study by Boopathy et al. [14] showed that the mutant profilin1 proteins C71G, M114T, and G118V were destabilized in vitro under conditions, in which the WT and E117G forms were unaffected. The authors measured shifts in the fluorescence signal of the proteins when adding an increasing concentration of urea. Chemical and thermal denaturation studies provided evidence for severe destabilization of mutant profilin1. In addition, all three of the stated mutants that were susceptible to destabilization had lower melting temperatures than the WT. Both of these findings indicate that the C71G, M114T, and G118V mutants are unstable compared to WT. This instability conferred by the mutations is a probable factor in their ALS-causing mechanism. The authors then analyzed crystal structures of the WT, E117G, and M114T [14]. The structures of the WT and E117G were similar as they had a small cleft on their surfaces putatively associated with protein instability, further confirming that the E117G is merely a risk factor for ALS. Interestingly, the M114T mutation caused this cleft to expand and appear as a large cavity near the protein core. Computer analyses also predicted that the C71G form would have a cavity larger than that observed in WT. It is possible that these cavities contribute to the instability of the ALS-mutant profilin1 proteins. These cavities lead to destabilization of profilin1, which may encourage profilin1 aggregation. The aggregation of profilin1, in turn, may lead to a fibril-like transition intermediate and act as a “seed” for progressive aggregation. If proven, the aggregates formed by profilin1 may accumulate in motor neurons and lead to neurodegeneration, suggesting a possible mechanism for ALS caused by profilin1 mutations, neurotoxicity, and progressive neuronal death. At least four of the profilin1 mutations are in the actin-binding domain, and two are in the PLP domain of the protein. One may hypothesize that these mutations may destabilize profilin1 and trigger aggregation, thereby leading to a toxic “gain of function,” “loss of function” or other pathogenic disruptions in the homeostasis of actin dynamics, ultimately interrupting critical cellular functions associated with profilin1. This scenario would explain the impact of mutant profilin1 on primary motor neurons [43] and now confirmed in transgenic mice overexpressing the G118V mutation (Kiaei et al., SfN abstract 2015). Collectively, growing evidence supports the hypothesis that mutations in profilin1 contribute to ALS via a cytoskeletal destabilization mechanism. However, the discovery of other mechanisms is expected, since profilin1 interacts with a large pool of ligands and binding partners that may also be disrupted by mutations of profilin1. Following this logic, exploring the following questions becomes critical to defining the impact of profilin1 on neuronal function and designing interventions to restore homeostasis. Is it possible to restore the native profilin1 3-D structure? Could novel molecules that enhance the stability of profilin1 by correcting its folding (such as chaperones or small molecular stabilizers) be effective therapies? If so, will restoration of profilin1’s normal structure prevent the pathogenesis of ALS and/or stop its progression? If it is confirmed that “loss of function” of profilin1 is a major contributing factor to ALS progression, then gene therapy using a neuron-specific promoter to drive expression of WT profilin1 may hold some promise. Since all profilin1 mutations are autosomal dominant, and all the patients already have one wild-type copy of the PFN1 gene, therefore, loss of function may manifest itself at later stages of life when there is higher demand for profilin1 function.
Increased aggregation propensity was examined with purified pathogenic mutants (C71G, M114T, G118V, T109M, and A20T) using biophysical techniques [51, 54]. A significant increase in aggregation propensity was found in mutant profilin1 as compared to WT profilin1 and the less pathogenic mutant forms of profilin1 (E117G and Q139L) [53, 54]. Analysis of T109M mutants indicated a conformational stability similar to WT profilin1, despite the fact that this mutant exhibited the highest aggregation propensity compared to other forms. This suggests that the increased aggregation in the mutant forms is independent of the changes in conformational stability [51, 53, 54].
Structural studies of the intrinsic fluorescence and far-UV CD spectra showed differences between the pathogenic mutant forms and WT profilin1 which correlated with the differences in aggregation propensity. These differences in tertiary structures (i.e., folding) are significant, because the differences correlate with the aggregation propensity profiles of mutant profilin1 proteins. This supports the hypothesis that increased aggregation propensity of the ALS-linked profilin1 mutants is promoted by changes in protein folding, i.e., secondary and tertiary structures.
This strengthens the theory that ALS-causing mutations in profilin1 induced pathogenicity by causing increased aggregation of mutant profilin1.
These authors also conducted circular dichroism (CD) analyses, which revealed a transient, partially folded, transition state between the folded and unfolded states of profilin1. Kinetic analyses suggest that this state is thermodynamically comparable to the fully unfolded state, i.e., it is unstable, indicating that there is an intermediate state for WT profilin1 during folding, but it does not accumulate as it is highly transient. It can be speculated that mutations in profilin1 could transform the transitional intermediate to an atypical intermediate state and consequently slow its degradation, which may result in its aggregation. If confirmed, this concept would be a significant finding as it could offer more insight into the mechanism by which mutant profilin1 contributes to neurotoxicity during ALS. Notably, the hypothesis that profilin1 mutations lead to ALS by promoting the formation of protein aggregates is gaining support based on functional studies in primary motor neurons and yeast cells [43, 55]. In other words, these findings support the hypothesis that mutations in profilin1 disrupt the de novo conformation of profilin1 in a manner that leads to the accumulation of an intermediate form (fibril-like), which becomes toxic for neurons leading to ALS.
Profilin 1 with ALS-linked mutations causes TDP-43 aggregation
TAR-DNA binding protein-43 (TDP-43) has been shown to play a pivotal role in the pathogenesis of neurodegenerative diseases, such as ALS and frontotemporal lobe degenerations (FTLD). TDP-43 accumulates in ubiquitin-positive inclusions in affected regions of the postmortem spinal cord and brain sections of ALS and FTLD patients [56, 57]. Recent studies show that ALS-causing mutant profilin1 forms cytoplasmic aggregates (as discussed earlier), which are positive for ubiquitin and p62 in human neuroblastoma cells; these aggregates sequester endogenous TDP-43 [58]. The transient overexpression of C71G mutant profilin1 protein formed more aggregates than overexpressed WT. E117G and WT transient overexpression in cell cultures exhibited similar aggregation profiles confirming the suggestion that the E117G mutation is a risk factor rather than a cause of ALS [58]. As discussed above, mutations in profilin1 render it prone to aggregation, which may trigger a series of events that promote toxicity in motor neurons. Postmortem tissue analysis from mutant profilin1 cases of ALS show that TDP-43 is immunoreactive with skein-like neuronal cytoplasmic inclusions in cranial nerve XII [42]. These tissues exhibited p62 and TDP-43 positive inclusions in glial cells but not in spinal cord neurons. These findings raise the possibility that mutant profilin1 may lead to a conformational change in TDP-43, which, in turn, may lead to aggregation. Thus, profilin1 with ALS-linked mutations may act as a “seed” to induce accumulation of TDP-43 as aggregates [58].
In recent studies, TDP-43 and mutant profilin1 were found to coaggregate in vitro [58]. The pathways involved in recruiting TDP-43 to aggregates are unknown and whether a conformational change in TDP-43 following its interaction with mutant profilin1 could be a basis for a “gain-of-toxic function” is yet to be determined. Interestingly, Woerner et al. [59] showed that cytoplasmic aggregates enhanced toxicity by sequestration and mislocalization of proteins containing disordered and low-complexity sequences in HEK293T cells [59]. Although this study was conducted in a single cell line, elegant approaches (using artificial beta sheet proteins) were used to show that the location of aggregates (whether in the cytoplasm or the nucleus) is important as the toxicity of the aggregates depends on their intracellular location. This finding further implies that cytoplasmic TDP-43 aggregates in ALS are likely toxic and may constitute a mechanism of neuronal death. Therefore, it is of great interest to confirm whether TDP-43 and profilin1 coaggregate in vivo, since aggregation of profilin1 is implicated in motor neuron degeneration. Indeed, if profilin1 and TDP-43 coaggregate in vivo, it is possible that the aggregation of mutant profilin1 serves as a “seed” for TDP-43 recruitment in aggregate inclusions.
Mutant profilin1 and stress granule dynamics
In addition to destabilizing profilin1 and inducing aggregate formation, how else do proflin1 mutations impact the cell? A recent study by Gitler and colleagues [55] investigated the effects of mutant profilin1 on stress granules using a yeast-based system and primary neurons. Stress granules, also called ribonucleoprotein granules, are small RNA–protein assemblies which form in the cytoplasm in response to cellular stress. Stress granules were investigated as potential factors in ALS pathogenesis [60]. The authors used a yeast stress-granule reporter cell line to show that WT profilin1 localizes to stress granules following arsenite treatment; whereas the three ALS-linked mutations (C71G, G118V, and M114T) showed impaired targeting to stress granules under the same conditions [55]. Thus, ALS-linked mutations alter the ability of profilin1 to be recruited to stress granules, raising the possibility that the subsequent accumulation of mutant profilin1 is a mechanism of neuronal degeneration in ALS. If confirmed in other models, this hypothesis is of great importance, since it links mutant profilin1 and stress granules to ALS pathogenesis. In addition, cytoplasmic aggregates distinct from stress granules were observed in yeast cells transfected with C71G, and to a lesser extent, the M114T mutation. Taken together, these observations provide evidence for a possible novel model of ALS pathogenesis in which profilin1 mutations alter stress granule dynamics, although further research is necessary to shed more light on the topic.
Profilin1 mutations in ALS and structural instability
Profilin1 mutations are believed to cause ALS by impairing the ability of profilin1 to bind actin, but the specifics of this dysregulation are unknown. Many of ALS-linked profilin1 mutations are located in the actin-binding domain of profilin1, supporting the hypothesis that changes in the affinity of profilin1 for actin may be responsible for ALS. Furthermore, tissue culture models expressing the C71G, M114T, and G118V mutations have lower levels of bound actin when compared to WT. Mutants expressing the E117G mutation did not demonstrate a significantly decreased affinity for actin [43, 51, 53].
A more recent study in ALS patients by Smith et al. [42] discovered the ALS-linked mutations: A20T and Q139L. Structural modeling revealed that the A20T mutation creates internal mismatches at several residues and theoretically alters the conformation of the beta sheet. Additional studies are needed to determine the effects of the Q139L mutation [42]. Another missense mutation, T109M, has also been implicated in ALS patients (Fig. 1b). This mutation occurs at a phosphorylation site, and it remains to be determined whether any functional changes related to ALS pathogenesis may result from this mutation, or if it will affect only the phosphorylation status [41]. A recent study showed that a profilin1 mutation in the position T109M does not affect actin binding as the location of this mutation is outside the actin-binding domain [52]. This mutation indicated that the PLP domain, as well as the actin-binding domain, actin polymerization, and other functions of profilin1 may play important roles in the pathogenesis of ALS. Interestingly, the authors also showed that the T109M mutant-induced stress granules were at a level comparable to other ALS-linked mutations C71G, M114T, and G118V [52].
Altogether, the impact on the tertiary structure, phosphorylation, loss or gain of new function, stress granules, and aggregation due to profilin1 mutations could contribute to the demise of motor neurons in ALS (Fig. 4).

Diseases caused by dysregulation or mutations of profilin1
Profilin1 in neuronal migration
Profilin1’s interactions with a variety of cytoskeletal and cellular signaling molecules (actin and ligands, such as N-WASP, Mena, VASP, dynamin1, Arp2/3 complex) provide a link between actin polymerization and other cellular processes. Profilin1 is essential to early embryogenesis [5]. Embryos from profilin1-null mice die as early as the two cell stage [5]. This finding implies a role for profilin1 in early cell cleavage [15]. Profilin1 is also involved in cellular motility and migration [15, 61]. Profilin1’s interaction with actin provides a necessary mechanism for lamellipodium formation in migrating cells by recruiting actin to the leading edge and accelerating actin polymerization at the lamellipodium [62]. Recent studies have suggested that profilin1 plays an additional actin-independent role in cell migration by blocking PIP2, which regulates recruitment of other motility-enhancing complexes to the leading edge of the cell membrane. While this process takes place in all migrating cells, profilin1 plays a particularly important role in the migration of cerebellar granule neurons (CGNs) as described in the previous section [33].
Profilin1 is vital for proper myelination of the peripheral nervous system, and its expression is indispensable for proper Schwann cell (SC) development. SCs produced in the absence of profilin1 are either underdeveloped or unable to migrate properly, which results in axon hypomyelination [63]. Profilin1 knockdown has also been shown to lead to defects in the Purkinje cell layer (PCL), specifically resulting in large gaps caused by aberrant purkinje cell (PC) migration as described above [33]. These abnormalities, however, originate from mechanisms other than cell-autonomous defects, as PC-specific knockdown of profilin1 resulted in no damage to the PCL. This finding may indicate that the impaired migration of PCs is linked to that of CGNs when profilin1 is removed. Despite the compromised PCL, the function of the parallel fiber–Purkinje cell (PF–PC) synapses was preserved after profilin1 knockdown, suggesting that profilin1 is important for synapse dynamics. Despite proper PF–PC synapse formation, profilin1 knockout mice demonstrate compromised motor coordination, as would be expected given the role of the PCL in muscular control [33].
Profilin1 and synaptic plasticity
Profilin1 is an important regulator of synaptic plasticity that may relate to its contribution to actin dynamics and cytoskeletal integrity. The necessity of profilin1 in synaptogenesis is evident in the neuronal disorder fragile X syndrome [64]. In this developmental neurological disorder, profilin1 levels were implicated, since it is the only profilin isoform required for development. In contrast, profilin2 is required for spine stability and plasticity in mature neurons [65, 66]. Fragile X syndrome presents primarily with impaired spine maturation. Therefore, these findings establish a reasonable link between profilin1 and spine formation, i.e., neurodevelopment. Mutations in the gene fragile X mental retardation 1 (fmr1) lead to fragile X syndrome. The fmr1 KO mouse, a model for fragile X syndrome, exhibits immature dendritic spines, a phenotype of fragile X syndrome. Interestingly, this phenotype is rescued in neurons of the fmr1 KO model when profilin1 is overexpressed indicating the significance of profilin1’s role in synaptogenesis (Fig. 4) [64].
Profilin1 in vascular hypotension
Given profilin1’s many roles, it is anticipated that dysregulation of mutant profilin1 could result in a wide variety of diseases and medical conditions (Table 1). One such condition is vascular inflammation. Profilin1 is proposed to be a player in hypertrophic signaling pathway activation via the actin cytoskeleton, inducing stress via excessive remodeling of the vasculature via vasoconstriction. The Rho/ROCK pathway has been implicated in the process, as well as the mitogen-activated protein (MAP) kinase cascade, which translates signals from growth factors and mechanical strain into regulation and synthesis of proteins. In murine mesenteric arteries overexpressing profilin1, increased numbers of stress fibers were observed, leading to an increase in α-1 and β-1 integrins, which are implicated in arterial stiffness [67]. The exact link between profilin1 and arterial stiffness is unknown, but it seems likely that overly high profilin1 levels cause extensive remodeling of the vasculature and the appearance of stress fibers, triggering signaling cascades that release additional integrins, which, in turn, lead to arterial rigidity. The model used in the study by Hassona et al. [67] overexpressed profilin1 in the mesenteric arteries, which was associated with an accelerated onset of hypertension, increased stress fiber formation, and activation of both Rho/ROCK and hypertrophic signaling pathways. Increased levels of endothelial nitric oxide synthase were also reported [67].
Profilin gene locus deletion and Miller–Dieker syndrome (MDS)
Miller–Dieker syndrome is a rare condition characterized by classical type 1 lissencephaly and facial abnormalities. Most cases of MDS are thought to be caused by a cytogenetically detectable deletion of chromosome 17p, involving substrand 13.3. However, some cases differ by having no abnormality detected using cytogenetic analysis. Profilin1 has been localized to the 17p13.3 chromosome, and whether any DNA sequences of PFN1 gene deleted must be verified [36]. If proven, then this will be the only recognized functional gene to be deleted in this syndrome. Patients in whom the MDS deletion does not encompass the profilin1 gene still display the disease phenotype, however, indicating that the profilin1 deletion does not play a major role in its primary presentation. Further studies will be necessary to define the exact contribution of profilin1 deletion to MDS development [36].
Profilin1 in breast cancer and metastasis
Choi et al. [68] showed in 2014 that profilin1 interacts with C-terminus of Hsc-70 interacting protein (CHIP), a molecule involved in metastasis. Profilin1 downregulation also correlates with breast cancer metastasis (Table 1; Fig. 4). The two molecules interact, resulting in ubiquitination and subsequent degradation of profilin1. In this case, CHIP serves as a regulator of profilin1 and is likely the cause for its observed downregulation in breast cancer cells. Interestingly, despite profilin1’s involvement in lamellipodium formation and cell motility in healthy cells, CHIP-mediated knockdown of profilin1 in breast cancer cells resulted in a promigratory phenotype characterized by abundance of F-actin structure. This event contributed to the ability of the cancer to metastasize in response to lowering of profilin1 levels [68]. Profilin1 also interacts with estrogen receptor alpha (ERα), which is known to mediate apoptosis in certain cell lines, in addition to contributing to metastasis. In these cells, tamoxifen (Tam) has been shown to upregulate profilin1, which subsequently colocalizes in the nucleus with ERα. Profilin1 was observed to dramatically decrease ERα-mediated transactivation of the estrogen response element (ERE) promoter in the presence of both 17α-estradiol (E2) and Tam, but had no effect without the other two molecules. These findings suggest that all three (profilin1-Tam-E2) form a series of corepressors for ERα. Thus, the profilin1-Tam-E2 set of corepressors inhibits ERα-mediated metastasis. In addition, profilin1’s inhibition of ERα activity leads to an increased expression of cleaved caspase 9 and caspase 3, both of which are associated with caspase-dependent apoptosis. Therefore, profilin1 upregulation in cancer cells provides a potential pathway for destruction of cells expressing the tumorigenic phenotype [69].
Conclusion and discussion
Since its discovery, profilin1 has been implicated in a number of diseases. This fact is not surprising as profilin1 plays a crucial role in regulating the cytoskeleton in addition to other cellular functions. In this review, we focused on profilin1’s involvement in fALS due to a significant interest by the scientific community in the link between profilin1 variants and fALS. As presented in this review, select mutations in profilin1 are found in a subpopulation of fALS patients. Whereas the link between mutant profilin1 and ALS is widely accepted, details of the mechanisms by which mutant profilin1 leads to ALS are unclear. This review assembles recent findings that point toward possible mechanisms by which mutant profilin1 may contribute to the pathogenesis of ALS. Reports show that profilin1 containing ALS-linked mutations forms aggregates in neuronal cultures, primary motor neurons, and yeast cells. Further structural studies by Bosco and colleagues [14] show that ALS-linked mutations lead to the formation of cavities in the profilin1 protein core. These cavities likely cause instability of mutant profilin1 and lead to conformational changes that contribute to abnormal function. These unstable mutants may be misfolded and targeted for degradation. These findings raise the question of how profilin1 instability affects motor neuron status in ALS, a complex issue that is unresolved.
Another important finding is the link between stress granules and profilin1. Wild-type profilin1 localizes to stress granules in yeast cells and in mammalian cells including primary mouse cortical neurons. ALS-linked profilin1 mutants alter the ability of profilin1 to be recruited to or cleared from stress granules, i.e., the mutations in profilin1 alter cellular stress granule dynamics. This discovery may offer additional insight into mechanisms of ALS as the connection between stress granules and ALS has not been fully described.
Another interesting finding is the involvement of profilin1 in the neurodevelopmental disorder fragile X syndrome (FXS). This link implies that profilin1 is crucial for synaptogenesis. Therefore, it is possible that profilin1 mutations initiate events that predispose to ALS as early as the embryonic stage. If confirmed, this finding would pose a paradigm shift in how we view the mechanisms of ALS, since it infers that ALS pathology may be initiated decades earlier than the onset of symptoms.
It is interesting that there appears to be no compensatory mechanism to overcome the loss of profilin1, for instance, in FXS, which argues for a “loss-of-function” toxicity. In MDS cases, microdeletions of distal 17p region, where the profilin1 gene is located, are linked to some cases of MDS [36]. On the other hand, the toxicity of mutant profilin1 may be due to a “toxic gain of function,” which can be inferred from its role as a “seed” for TDP-43 aggregation. In addition, the fact that the profilin1 mutation is autosomal dominant argues against the “loss-of-function” hypothesis, although reduction of F-actin may represent a conceptual loss of function for mutant profilin1.
In our effort to understand the toxicity of profilin1 in ALS in vivo, we generated a novel mouse model that expresses the G118V mutant form of profilin1. This new mouse model appears to recapitulate the phenotypes and pathogenic cellular abnormalities of ALS, and thus serves as a much-needed, novel animal model for this disease. Additional studies are needed to determine the exact pathway(s) by which the mutations contribute to the axonal and cytoskeletal disruptions that are characteristic of the disease.
References
Carlsson L, Nystrom LE, Sundkvist I, Markey F, Lindberg U (1977) Actin polymerizability is influenced by profilin, a low molecular weight protein in non-muscle cells. J Mol Biol 115(3):465–483
Birbach A (2008) Profilin, a multi-modal regulator of neuronal plasticity. BioEssays 30(10):994–1002
Kang F, Purich DL, Southwick FS (1999) Profilin promotes barbed-end actin filament assembly without lowering the critical concentration. J Biol Chem 274(52):36963–36972
Yarmola EG, Bubb MR (2009) How depolymerization can promote polymerization: the case of actin and profilin. BioEssays 31(11):1150–1160
Witke W, Sutherland JD, Sharpe A, Arai M, Kwiatkowski DJ (2001) Profilin I is essential for cell survival and cell division in early mouse development. Proc Natl Acad Sci USA 98(7):3832–3836
Metzler WJ, Farmer BT 2nd, Constantine KL, Friedrichs MS, Lavoie T, Mueller L (1995) Refined solution structure of human profilin I. Protein Sci 4(3):450–459
Schutt CE, Myslik JC, Rozycki MD, Goonesekere NC, Lindberg U (1993) The structure of crystalline profilin-beta-actin. Nature 365(6449):810–816
Bhargav SP, Vahokoski J, Kallio JP, Torda AE, Kursula P, Kursula I (2015) Two independently folding units of Plasmodium profilin suggest evolution via gene fusion. Cell Mol Life Sci 72(21):4193–4203
Metzler WJ, Bell AJ, Ernst E, Lavoie TB, Mueller L (1994) Identification of the poly-l-proline-binding site on human profilin. J Biol Chem 269(6):4620–4625
Kovar DR, Harris ES, Mahaffy R, Higgs HN, Pollard TD (2006) Control of the assembly of ATP- and ADP-actin by formins and profilin. Cell 124(2):423–435
Lambrechts A, Jonckheere V, Peleman C, Polet D, De Vos W, Vandekerckhove J, Ampe C (2006) Profilin-I-ligand interactions influence various aspects of neuronal differentiation. J Cell Sci 119(Pt 8):1570–1578
Pei W, Du F, Zhang Y, He T, Ren H (2012) Control of the actin cytoskeleton in root hair development. Plant Sci 187:10–18
Krishnamoorthy P, Sanchez-Rodriguez C, Heilmann I, Persson S (2014) Regulatory roles of phosphoinositides in membrane trafficking and their potential impact on cell-wall synthesis and re-modelling. Ann Bot 114(6):1049–1057
Boopathy S, Silvas TV, Tischbein M, Jansen S, Shandilya SM, Zitzewitz JA, Landers JE, Goode BL, Schiffer CA, Bosco DA (2015) Structural basis for mutation-induced destabilization of profilin 1 in ALS. Proc Natl Acad Sci USA 112(26):7984–7989
Ding Z, Bae YH, Roy P (2012) Molecular insights on context-specific role of profilin-1 in cell migration. Cell Adhes Migr 6(5):442–449
Dong JH, Ying GX, Zhou CF (2004) Entorhinal deafferentation induces the expression of profilin mRNA in the reactive microglial cells in the hippocampus. Glia 47(1):102–108
Montani L, Petrinovic MM (2014) Targeting axonal regeneration: the growth cone takes the lead. J Neurosci 34(13):4443–4444
Nolle A, Zeug A, van Bergeijk J, Tonges L, Gerhard R, Brinkmann H, Al Rayes S, Hensel N, Schill Y, Apkhazava D et al (2011) The spinal muscular atrophy disease protein SMN is linked to the Rho-kinase pathway via profilin. Hum Mol Genet 20(24):4865–4878
Wallar BJ, Alberts AS (2003) The formins: active scaffolds that remodel the cytoskeleton. Trends Cell Biol 13(8):435–446
Moseley JB, Sagot I, Manning AL, Xu Y, Eck MJ, Pellman D, Goode BL (2004) A conserved mechanism for Bni1- and mDia1-induced actin assembly and dual regulation of Bni1 by Bud6 and profilin. Mol Biol Cell 15(2):896–907
Peng J, Wallar BJ, Flanders A, Swiatek PJ, Alberts AS (2003) Disruption of the diaphanous-related formin Drf1 gene encoding mDia1 reveals a role for Drf3 as an effector for Cdc42. Curr Biol 13(7):534–545
Witke W (2004) The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol 14(8):461–469
Pronto-Laborinho AC, Pinto S, de Carvalho M (2014) Roles of vascular endothelial growth factor in amyotrophic lateral sclerosis. Biomed Res Int 2014:947513
Simons M, Schwartz MA (2012) Profilin phosphorylation as a VEGFR effector in angiogenesis. Nat Cell Biol 14(10):985–987
Wang X, Kibschull M, Laue MM, Lichte B, Petrasch-Parwez E, Kilimann MW (1999) Aczonin, a 550-kD putative scaffolding protein of presynaptic active zones, shares homology regions with Rim and Bassoon and binds profilin. J Cell Biol 147(1):151–162
Fenster SD, Kessels MM, Qualmann B, Chung WJ, Nash J, Gundelfinger ED, Garner CC (2003) Interactions between Piccolo and the actin/dynamin-binding protein Abp1 link vesicle endocytosis to presynaptic active zones. J Biol Chem 278(22):20268–20277
Fenster SD, Chung WJ, Zhai R, Cases-Langhoff C, Voss B, Garner AM, Kaempf U, Kindler S, Gundelfinger ED, Garner CC (2000) Piccolo, a presynaptic zinc finger protein structurally related to bassoon. Neuron 25(1):203–214
Kim S, Ko J, Shin H, Lee JR, Lim C, Han JH, Altrock WD, Garner CC, Gundelfinger ED, Premont RT et al (2003) The GIT family of proteins forms multimers and associates with the presynaptic cytomatrix protein Piccolo. J Biol Chem 278(8):6291–6300
Waites CL, Leal-Ortiz SA, Andlauer TF, Sigrist SJ, Garner CC (2011) Piccolo regulates the dynamic assembly of presynaptic F-actin. J Neurosci 31(40):14250–14263
Fan T, Zhai H, Shi W, Wang J, Jia H, Xiang Y, An L (2012) Overexpression of profilin 3 affects cell elongation and F-actin organization in Arabidopsis thaliana. Plant Cell Rep 71:684–697
Fan Y, Arif A, Gong Y, Jia J, Eswarappa SM, Willard B, Horowitz A, Graham LM, Penn MS, Fox PL (2012) Stimulus-dependent phosphorylation of profilin-1 in angiogenesis. Nat Cell Biol 14(10):1046–1056
Mahoney NM, Janmey PA, Almo SC (1997) Structure of the profilin-poly-l-proline complex involved in morphogenesis and cytoskeletal regulation. Nat Struct Biol 4(11):953–960
Kullmann JA, Neumeyer A, Wickertsheim I, Bottcher RT, Costell M, Deitmer JW, Witke W, Friauf E, Rust MB (2012) Purkinje cell loss and motor coordination defects in profilin1 mutant mice. Neuroscience 223:355–364
Kullmann JS, Grigoleit JS, Lichte P, Kobbe P, Rosenberger C, Banner C, Wolf OT, Engler H, Oberbeck R, Elsenbruch S et al (2013) Neural response to emotional stimuli during experimental human endotoxemia. Hum Brain Mapp 34(9):2217–2227
Turkmen S, Demirhan O, Hoffmann K, Diers A, Zimmer C, Sperling K, Mundlos S (2006) Cerebellar hypoplasia and quadrupedal locomotion in humans as a recessive trait mapping to chromosome 17p. J Med Genet 43(5):461–464
Kwiatkowski DJ, Aklog L, Ledbetter DH, Morton CC (1990) Identification of the functional profilin gene, its localization to chromosome subband 17p13.3, and demonstration of its deletion in some patients with Miller–Dieker syndrome. Am J Hum Genet 46(3):559–567
Rust MB, Kullmann JA, Witke W (2012) Role of the actin-binding protein profilin1 in radial migration and glial cell adhesion of granule neurons in the cerebellum. Cell Adhes Migr 6(1):13–17
Ding Z, Lambrechts A, Parepally M, Roy P (2006) Silencing profilin-1 inhibits endothelial cell proliferation, migration and cord morphogenesis. J Cell Sci 119(Pt 19):4127–4137
Majoor-Krakauer D, Willems PJ, Hofman A (2003) Genetic epidemiology of amyotrophic lateral sclerosis. Clin Genet 63(2):83–101
Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O (2003) An outcome study of riluzole in amyotrophic lateral sclerosis–a population-based study in Ireland, 1996–2000. J Neurol 250(4):473–479
Ingre C, Landers JE, Rizik N, Volk AE, Akimoto C, Birve A, Hubers A, Keagle PJ, Piotrowska K, Press R et al (2013) A novel phosphorylation site mutation in profilin 1 revealed in a large screen of US, Nordic, and German amyotrophic lateral sclerosis/frontotemporal dementia cohorts. Neurobiol Aging 34(6):1708e1–6
Smith BN, Vance C, Scotter EL, Troakes C, Wong CH, Topp S, Maekawa S, King A, Mitchell JC, Lund K et al (2015) Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol Aging 36(3):1602.e17–27
Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, Lowe P, Koppers M, McKenna-Yasek D, Baron DM et al (2012) Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488(7412):499–503
Chen Y, Zheng ZZ, Huang R, Chen K, Song W, Zhao B, Chen X, Yang Y, Yuan L, Shang HF (2013) PFN1 mutations are rare in Han Chinese populations with amyotrophic lateral sclerosis. Neurobiol Aging 34(7):1922.e21–25
Yang S, Fifita JA, Williams KL, Warraich ST, Pamphlett R, Nicholson GA, Blair IP (2013) Mutation analysis and immunopathological studies of PFN1 in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging 34(9):2235.e7–10
Smith BN, Vance C, Scotter EL, Troakes C, Wong CH, Topp S, Maekawa S, King A, Mitchell JC, Lund K et al (2014) Novel mutations support a role for profilin 1 in the pathogenesis of ALS. Neurobiol Aging 36:e17–e27
Tiloca C, Ticozzi N, Pensato V, Corrado L, Del Bo R, Bertolin C, Fenoglio C, Gagliardi S, Calini D, Lauria G et al (2013) Screening of the PFN1 gene in sporadic amyotrophic lateral sclerosis and in frontotemporal dementia. Neurobiol Aging. 34(5):1517.e9–10
Daoud H, Dobrzeniecka S, Camu W, Meininger V, Dupre N, Dion PA, Rouleau GA (2013) Mutation analysis of PFN1 in familial amyotrophic lateral sclerosis patients. Neurobiol Aging 34:1311.e1–2
Lattante S, Le Ber I, Camuzat A, Brice A, Kabashi E (2013) Mutations in the PFN1 gene are not a common cause in patients with amyotrophic lateral sclerosis and frontotemporal lobar degeneration in France. Neurobiol Aging 34(6):1709.e1–2
van Blitterswijk M, Baker MC, Bieniek KF, Knopman DS, Josephs KA, Boeve B, Caselli R, Wszolek ZK, Petersen R, Graff-Radford NR et al (2013) Profilin-1 mutations are rare in patients with amyotrophic lateral sclerosis and frontotemporal dementia. Amyotroph Lateral Scler Frontotemporal Degener 14(5–6):463–469
Del Poggetto E, Chiti F, Bemporad F (2015) The Folding process of Human Profilin-1, a novel protein associated with familial amyotrophic lateral sclerosis. Sci Rep 5:12332
Freischmidt A, Schopflin M, Feiler MS, Fleck AK, Ludolph AC, Weishaupt JH (2015) Profilin 1 with the amyotrophic lateral sclerosis associated mutation T109M displays unaltered actin binding and does not affect the actin cytoskeleton. BMC Neurosci 16:77
Del Poggetto E, Bemporad F, Tatini F, Chiti F (2015) Mutations of profilin-1 associated with amyotrophic lateral sclerosis promote aggregation due to structural changes of its native state. ACS Chem Biol 10(11):2553–2563
Del Poggetto E, Gori L, Chiti F (2016) Biophysical analysis of three novel profilin-1 variants associated with amyotrophic lateral sclerosis indicates a correlation between their aggregation propensity and the structural features of their globular state. Biol Chem 397(9):927–937
Figley MD, Bieri G, Kolaitis RM, Taylor JP, Gitler AD (2014) Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J Neurosci 34(24):8083–8097
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351(3):602–611
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(5796):130–133
Tanaka Y, Nonaka T, Suzuki G, Kametani F, Hasegawa M (2016) Gain-of-function profilin 1 mutations linked to familial amyotrophic lateral sclerosis cause seed-dependent intracellular TDP-43 aggregation. Hum Mol Genet 25(7):1420–1433
Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, Tatzelt J, Mann M, Winklhofer KF, Hartl FU et al (2016) Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 351(6269):173–176
Kent L, Vizard TN, Smith BN, Topp SD, Vance C, Gkazi A, Miller J, Shaw CE, Talbot K (2014) Autosomal dominant inheritance of rapidly progressive amyotrophic lateral sclerosis due to a truncation mutation in the fused in sarcoma (FUS) gene. Amyotroph Lateral Scler Frontotemporal Degener 15:557–562
Bottcher RT, Wiesner S, Braun A, Wimmer R, Berna A, Elad N, Medalia O, Pfeifer A, Aszodi A, Costell M et al (2009) Profilin 1 is required for abscission during late cytokinesis of chondrocytes. EMBO J 28(8):1157–1169
Small JV, Stradal T, Vignal E, Rottner K (2002) The lamellipodium: where motility begins. Trends Cell Biol 12(3):112–120
Montani L, Buerki-Thurnherr T, de Faria JP, Pereira JA, Dias NG, Fernandes R, Goncalves AF, Braun A, Benninger Y, Bottcher RT et al (2014) Profilin 1 is required for peripheral nervous system myelination. Development 141(7):1553–1561
Michaelsen-Preusse K, Zessin S, Grigoryan G, Scharkowski F, Feuge J, Remus A, Korte M (2016) Neuronal profilins in health and disease: relevance for spine plasticity and Fragile X syndrome. Proc Natl Acad Sci USA 113(12):3365–3370
Gorlich A, Zimmermann AM, Schober D, Bottcher RT, Sassoe-Pognetto M, Friauf E, Witke W, Rust MB (2012) Preserved morphology and physiology of excitatory synapses in profilin1-deficient mice. PLoS One 7(1):e30068
Pilo Boyl P, Di Nardo A, Mulle C, Sassoe-Pognetto M, Panzanelli P, Mele A, Kneussel M, Costantini V, Perlas E, Massimi M et al (2007) Profilin2 contributes to synaptic vesicle exocytosis, neuronal excitability, and novelty-seeking behavior. EMBO J 26(12):2991–3002
Hassona MD, Abouelnaga ZA, Elnakish MT, Awad MM, Alhaj M, Goldschmidt-Clermont PJ, Hassanain H (2010) Vascular hypertrophy-associated hypertension of profilin1 transgenic mouse model leads to functional remodeling of peripheral arteries. Am J Physiol Heart Circ Physiol 298(6):H2112–H2120
Choi YN, Lee SK, Seo TW, Lee JS, Yoo SJ (2014) C-Terminus of Hsc70-interacting protein regulates profilin1 and breast cancer cell migration. Biochem Biophys Res Commun 446(4):1060–1066
Kanaujiya JK, Lochab S, Kapoor I, Pal P, Datta D, Bhatt ML, Sanyal S, Behre G, Trivedi AK (2013) Proteomic identification of Profilin1 as a corepressor of estrogen receptor alpha in MCF7 breast cancer cells. Proteomics 13(14):2100–2112
Fan Y, Potdar AA, Gong Y, Eswarappa SM, Donnola S, Lathia JD, Hambardzumyan D, Rich JN, Fox PL (2014) Profilin-1 phosphorylation directs angiocrine expression and glioblastoma progression through HIF-1alpha accumulation. Nat Cell Biol 16(5):445–456
Zou L, Jaramillo M, Whaley D, Wells A, Panchapakesa V, Das T, Roy P (2007) Profilin-1 is a negative regulator of mammary carcinoma aggressiveness. Br J Cancer 97(10):1361–1371
Bae YH, Ding Z, Zou L, Wells A, Gertler F, Roy P (2009) Loss of profilin-1 expression enhances breast cancer cell motility by Ena/VASP proteins. J Cell Physiol 219(2):354–364
Karamchandani JR, Gabril MY, Ibrahim R, Scorilas A, Filter E, Finelli A, Lee JY, Ordon M, Pasic M, Romaschin AD et al (2015) Profilin-1 expression is associated with high grade and stage and decreased disease-free survival in renal cell carcinoma. Hum Pathol 46(5):673–680
Zoidakis J, Makridakis M, Zerefos PG, Bitsika V, Esteban S, Frantzi M, Stravodimos K, Anagnou NP, Roubelakis MG, Sanchez-Carbayo M et al (2012) Profilin 1 is a potential biomarker for bladder cancer aggressiveness. Mol Cell Proteomics. 11(4):M111.009449
Yao W, Ji S, Qin Y, Yang J, Xu J, Zhang B, Xu W, Liu J, Shi S, Liu L et al (2014) Profilin-1 suppresses tumorigenicity in pancreatic cancer through regulation of the SIRT3-HIF1alpha axis. Mol Cancer 13:187
Li Z, Zhong Q, Yang T, Xie X, Chen M (2013) The role of profilin-1 in endothelial cell injury induced by advanced glycation end products (AGEs). Cardiovasc Diabetol 12:141
Moustafa-Bayoumi M, Alhaj MA, El-Sayed O, Wisel S, Chotani MA, Abouelnaga ZA, Hassona MD, Rigatto K, Morris M, Nuovo G et al (2007) Vascular hypertrophy and hypertension caused by transgenic overexpression of profilin 1. J Biol Chem 282(52):37632–37639
Wang X, Zhang N, Ning X, Li T, Wu P, Peng S, Fan Y, Bu D, Gong K (2014) Higher prevalence of novel mutations in VHL gene in Chinese Von Hippel–Lindau disease patients. Urology 83(3):675.e1–5
Pae M, Romeo GR (2014) The multifaceted role of profilin-1 in adipose tissue inflammation and glucose homeostasis. Adipocyte 3(1):69–74
Romeo GR, Moulton KS, Kazlauskas A (2007) Attenuated expression of profilin-1 confers protection from atherosclerosis in the LDL receptor null mouse. Circ Res 101(4):357–367
Acknowledgments
The authors are grateful to Dr. Nancy Rusch for helpful discussions and comments on the manuscript. Dr. Ziad Ghneim is acknowledged for his edits and constructive discussions. This work was supported by funds from a UAMS startup fund, the UAMS Center for Translational Neurosciences, NIGMS IDeA Program Award P30 GM110702, and NINDS R21 (NS088653). E.Z.F. contributed to this study at UAMS as a Metcalf intern from University of Chicago.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors have no conflict of interest to declare.
Rights and permissions
About this article

Cite this article
Alkam, D., Feldman, E.Z., Singh, A. et al. Profilin1 biology and its mutation, actin(g) in disease. Cell. Mol. Life Sci. 74, 967–981 (2017). https://doi.org/10.1007/s00018-016-2372-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2372-1