
Abstract
TNF-related apoptosis-inducing ligand (TRAIL) is a prominent cytokine capable of inducing apoptosis. It can bind to five different cognate receptors, through which diverse intracellular pathways can be activated. TRAIL’s ability to preferentially kill transformed cells makes it a promising potential weapon for targeted tumor therapy. However, recognition of several resistance mechanisms to TRAIL-induced apoptosis has indicated that a thorough understanding of the details of TRAIL biology is still essential before this weapon can be confidently unleashed. Critical to this aim is revealing the functions and regulation mechanisms of TRAIL’s potent death receptor DR5. Although expression and signaling mechanisms of DR5 have been extensively studied, other aspects, such as its subcellular localization, non-signaling functions, and regulation of its membrane transport, have only recently attracted attention. Here, we discuss different aspects of TRAIL/DR5 biology, with a particular emphasis on the factors that seem to influence the cell surface expression pattern of DR5, along with factors that lead to its nuclear localization. Disturbance of this balance apparently affects the sensitivity of cancer cells to TRAIL-mediated apoptosis, thus constituting an eligible target for potential new therapeutic agents.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
TNF-related apoptosis-inducing ligand (TRAIL, APO2L) is a member of the TNF cytokine superfamily, which was first identified in 1995 through its 23 % C-terminal homology to TNF-α and 28 % homology to Fas ligand, most clearly in the C-terminal ectodomain [1]. The 20-kb human TRAIL gene TNFSF10 is located on chromosome 3q26, comprising five exons and four introns [2]. TRAIL is synthesized as a type II transmembrane protein that can also be cleaved by cysteine proteases and matrix-metalloproteinase-2 (MMP-2) to be released as a 24-kDa soluble form. Although both forms can initiate various intracellular signaling pathways, the transmembrane form is usually defined as a more potent ligand. TRAIL’s ability to induce many different molecular pathways and its other distinguishing features, such as broad range of tissue expression and multiple target receptors, is clearly reflected in its proposed involvement in both the pathogenesis of and/or possible treatment approaches for a variety of diseases including but not limited to cancer, obesity, diabetes, rheumatoid arthritis, and neurodegenerative diseases [3–8].
TRAIL’s having multiple target receptors is a significant aspect that distinguishes it from the other TNF family members. Five such receptors have been identified in humans. TRAIL-R1 (DR4) and TRAIL-R2 (DR5) are able to induce apoptosis through their functional cytoplasmic death domains (DD), when bound by the TRAIL ligand [9–11]. By contrast, TRAIL-R3 (DcR1) and TRAIL-R4 (DcR2) are unable to transmit apoptotic signals due to complete or partial lack of functional death domain, respectively [12, 13]. TRAIL also has a soluble receptor called osteoprotegerin (OPG/TRAIL-R5), which has a weaker affinity for TRAIL, originally defined as a receptor for the receptor activator of nuclear factor kappa-B ligand (RANKL) [14]. Expression levels of transmembrane receptors appear significant for TRAIL’s different actions, and have been correlated with various parameters pertaining to the cancer status or stage [15, 16].
TRAIL’s unique ability to kill many transformed cells through apoptosis while sparing normal cells has naturally generated great hope and enthusiasm, such that many preclinical studies and clinical trials involving recombinant TRAIL or receptor agonists have been implemented [17–19]. In fact, TRAIL’s apoptotic potential is known to be essential for the cells of both the innate and acquired immune systems; both the NK and T cells utilize TRAIL in tumor immunosurveillance, while themselves being resistant to TRAIL-mediated cytotoxicity [20]. TRAIL-induced apoptosis was also shown to switch to necroptosis at acidic extracellular pH, in human H29 colon and HepG2 liver cancer cells [21]. Yet, TRAIL’s potential use in cancer therapeutics currently relies on its apoptotic effect on transformed cells, which is unfortunately hampered by resistance to TRAIL-mediated apoptosis in a considerable number of cancer cell types [22]. Thus, a clear understanding of the resistance mechanisms and unveiling the details of TRAIL/TRAIL-R biology is crucial for designing rational and effective TRAIL therapies. In this review, we aim to summarize the recent updates concerning the biology and regulation of DR5, an essential actor in TRAIL-induced apoptosis. We give a particular emphasis on its differential localization patterns within the cell and related consequences, evaluated in terms of resistance to TRAIL-mediated apoptosis.
Overview of TRAIL signaling and resistance
Soon after TRAIL is synthesized, three molecules form a homotrimer, which is stabilized by binding of a zinc ion through the metal binding site generated by the 230th amino acid of each monomer [23]. Either soluble or membrane-associated, TRAIL induces receptor oligomerization upon ligation [24]. In the case of death receptor binding, this provides a docking site for the Fas-associated protein with death domain (FADD), which interacts with the death domain of either DR4 or DR5, through its own death domain [25]. FADD serves as a bridge between DR4/5 and not single but multiple procaspase-8 molecules, according to recent quantitative mass spectrometry results. Procaspase-8 molecules sequentially bind first to FADD’s death effector domain (DED) and then to each other, through their respective DEDs [26], and establish the death-inducing signaling complex (DISC), also termed the primary signaling complex. In fact, caspase-8 DED chain formation was shown to be essential for the activation of the caspase cascade and apoptotic death. Cleavage and activation of procaspase-8/10 via proper DISC assembly can activate effector caspases 3, 6, and 7, and ultimately lead to apoptosis [27]. TRAIL receptor localization in lipid rafts is closely related to TRAIL sensitivity in cells, and enables the necessary components to initiate DISC formation and the apoptotic process [28]. This process is adequate to induce apoptosis in type 1 cells, while the mitochondrial pathway needs to be involved as well in type 2 cells. In these cells, caspase 8 cleaves the Bid molecule, to form truncated Bid, which, in turn, associates with Bak and Bax proteins to trigger their oligomerization in the mitochondrial membrane, thus promoting disruption and permeabilization. Cleavage of Bid by caspase 8 and later on its association with the outer mitochondrial membrane were recently shown to be two critical processes that lead to Bid activation during TRAIL-mediated apoptosis [29]. Membrane permeabilization results in cytochrome c release, which is an essential component of the apoptosome along with Apaf-1 that recruits and activates procaspase 9. Activated initiator caspase 9 triggers the activation of the effector caspases, thus leading to apoptosis. Recently, TRAIL was also shown to induce caspase-mediated ER stress, and this way to expedite the apoptotic destruction via PERK-eIF2a-ATF4-CHOP pathways activation [30].
Intriguingly, non-canonical signaling pathways for TRAIL death receptors have also been identified. In addition to the cell death pathway, DR4 and DR5 were shown to also activate proliferative and survival pathways. Co-immunoprecipitation experiments have proved that both DR4 and DR5 can be part of a secondary signaling complex, which also contains receptor-interacting protein 1 (RIP1) and TNF receptor-associated factor 2 (TRAF2) [31]. This complex, independent of the DISC, associates with different components to activate various molecules related to pathways of survival, proliferation, angiogenesis, and migration/invasion, such as NF-κB, JNKs, p38, ERKs, PKC, PI3 K/Akt, and Src (reviewed in [32]).
TRAIL-induced apoptotic signaling pathways are subject to inhibition at multiple levels, the majority of which are extensively reported and reviewed in the related literature. These include, among many others, overexpression of caspase 8 homologue cellular FLICE-like inhibitory protein (c-FLIP) preventing DISC formation; competition of DcR1 and DcR2 decoy receptors with DR4/5 for TRAIL binding, or activation of intracellular molecules with antiapoptotic potential, such as NF-κB [33–35]. Many additional means of resistance to TRAIL-mediated apoptosis have been reported, which have been thoroughly reviewed in a recent report [36]. Recently, DcRs originating from stromal cells have also been shown to affect TRAIL sensitivity of tumors through transcellular regulation at the supracellular level [37]. Furthermore, the nuclear localization of DR5 has started to attract attention as a potent mechanism of resistance to TRAIL-induced apoptosis in cancer cells, accordingly constituting a potential therapeutic target.
Essentials of DR5 biology and regulation
DR5, encoded by TNFRSF10B, is quite conserved among both close and distant lineages, including humans, mice, and rats. Although the single TRAIL death receptor identified in mice is named mDR5, it has 60 % homology in sequence to both human DR4 and DR5, which also exhibit 58 % homology to each other [1, 38]. Though TRAIL can generate an apoptotic signal by binding to both DR4 and DR5, these two receptors differ in various aspects. Apoptosis induction capacity of these receptors is unequal; there are studies favoring either DR4 or DR5 in different cell types and conditions for more effective induction of apoptosis upon TRAIL binding. However, DR5 is generally accepted to be a more potent apoptotic trigger compared with DR4 [39, 40]. In a study investigating TRAIL binding affinity at 37 °C, DR5 was found to be the strongest among all TRAIL receptors [41].
DR4 and DR5 transcripts compared: 3′-UTR makes the difference
In addition to physiological differences, some major structural distinctions also exist between DR4 and DR5 at both DNA and RNA levels. Human DR4 gene is located on chromosome 8p21 and spans 33.711 base pairs. On the other hand, the gene coding for DR5 resides on 8p22–p21, and comprises 49.055 base pairs. This difference becomes more pronounced when these genes are transcribed; DR4 transcript is 1764 nucleotide long, whereas the longest DR5 isoform is 4154 nucleotides, more than twice as long (Fig. 1). The two transcripts differ greatly in length mainly in the 3′-UTR regions. Whereas the 3′-UTR region of the DR4 transcript is only 251 nt long, corresponding to one-seventh of the whole mRNA, the 3′-UTR region of the DR5 mRNA spans 2538 nucleotides. This length corresponds to more than half of the entire transcript, even longer than the total length of DR4 mRNA.

DR4 and DR5 transcripts compared. CDS coding sequence, UTR untranslated region, miRDB microRNA Database
Transcriptional modulation of DR5: a long list of regulators
To date, a vast number of studies have investigated the transcriptional regulation of TNFRSF10B by a variety of factors including proteins or other organic molecules. Not surprisingly, p53 occurs early in the list [42]. Second most prominent of such transcription factors is the C/EBP-homologous protein (CHOP), which has an important role in ER stress response and has a binding site on the TNFRSF10B promoter [43–45]. Promoter deletion and mutation studies have identified Elk1, which is activated by the ERK pathway, as a partner of CHOP in transcriptional induction [46]. NF-κB is another major transcriptional regulator of DR5 [47]. Accordingly, DR5 possesses an NF-κB consensus binding site in its first intronic region [48]. The JNK pathway, on the other hand, activates transcription factors c-Jun and c-Fos, which, in turn, form a dimeric AP-1 complex. AP-1 upregulates DR5 expression in response to JNK activation [49]. JNK/Sp1-dependent upregulation of DR5 expression by bile acids was also shown [50]. Interestingly, FOXO transcription factors have been reported to induce DR5 transcription [51], although there is no direct evidence of these transcription factors binding to the DR5 promoter. Yet, not all transcription factors affecting DR5 mRNA levels are transcription inducers. Yin Yang 1 (YY1) was shown to transcriptionally repress DR5 expression in certain cancer cell lines [52]. A recent study also identified Bcl-2 19 kDa interacting protein (BNIP3) as a transcriptional repressor of DR5 in glioblastoma cell lines [53]. Detailed information on specific transcription factors, their exact binding positions on TNFRSF10B promoter, and binding scores can be obtained from the UCSC Genome Browser.
Recently, a comprehensive list of 161 transcription factors has been investigated in a number of cell types using chromatin immunoprecipitation technique within the context of The Encyclopedia of DNA Elements (ENCODE) Project [54]. Not included in this list is the androgen receptor (AR). We have previously shown by immunohistochemistry that DR5 expression is increased in prostate cancer patients who received androgen ablation therapy (AAT) [55], which may indicate AR involvement in transcriptional regulation of DR5. Consistently, the TNFRSF10B promoter appears in the list of AR binding sites, determined by ChIP analysis (Supplementary data of [56]). Taken together, our results and ChIP data imply that AR binding might inhibit DR5 transcription. Nevertheless, elucidating AR’s role in death receptor regulation requires further research.
A good number of organic molecules have also been reported to influence DR5 transcription. One of them is ibuprofen, a widely used non-steroidal anti-inflammatory drug (NSAID) [57]. Ibuprofen increases DR5 transcription in HCT116 colon cancer cell line with no observable effect on DR4 expression. Baicalein, a natural flavonoid, also induces transcription of DR5 and triggers apoptosis in TRAIL resistant cells. In SW480 colon cancer cells, CHOP expression is upregulated by baicalein, followed by induction of DR5 expression and restoration of sensitivity to TRAIL-induced apoptosis. However, in T-cell leukemia Jurkat cells and prostate cancer cell lines PC3 and DU145, baicalein increases DR5 transcription in a reactive oxygen species (ROS)-dependent manner [58]. A recently reviewed extensive list of nutraceuticals upregulating DR4/DR5 expression can be found elsewhere [59].
HuR in the scene: DR5 transcript stabilized at the cost of supressed translation
Translational modulation of DR5 also appears complicated. The large 3′-UTR region of the DR5 gene implies extensive post-transcriptional regulation. Many microRNAs (miRNAs) are claimed to be involved in this process. According to a recently updated bioinformatics tool (miRDB) [60], DR5 3′-UTR region is predicted to be targeted by 49 miRNAs, whereas only 6 miRNAs are anticipated to bind to the DR4 3′-UTR region. Numerous RNA binding proteins (RBPs) are known to be involved in post-transcriptional regulation of gene expression (reviewed in [61]). These proteins bind mRNAs in the nucleus and dictate their cytoplasmic fate by directly functioning in or influencing diverse processes, such as transcription, pre-mRNA splicing and polyadenylation, RNA modification, transport, localization, translation, and turnover [62].
According to CLIPdb, a database generated based on crosslinking immunoprecipitation-sequencing (CLIP-Seq) data sets, 32 different RBPs bind to the DR5 transcript [63]. The embryonic lethal abnormal vision-like RNA binding protein 1 (ELAVL1) or Human antigen R (HuR) is one such RBP, which not only binds to the DR5 mRNA but also to numerous other transcripts, such as p21 [64], beta-adrenergic receptor [65], VEGF [66], and androgen receptor [67]. HuR has two N-terminal RNA recognition motifs (RRMs) which have a high affinity for AU-rich sequences, a nucleoplasmic shuttling sequence, and a C-terminal RRM, which can recognize poly(A) tails [68, 69]. When bound to the AU-rich elements in the 3′-UTR region of a target mRNA, HuR is reported to increase the stability of the target transcript and generally inhibit its translation [69–71]. Similarly, HuR binds to the 3′-UTR sequence of the DR5 transcript under stress conditions, leading to its stabilization [72] (Fig. 1). This increased mRNA stability by HuR at the cost of suppressed translation of DR5 may naturally lead to little or no correlation between its mRNA and protein levels. As shown in another study, binding of HuR in response to various cancer-associated stressors in pancreatic ductal adenocarcinomas represses DR5 translation, inferred from increased DR5 protein levels following HuR silencing [73]. A recent study carried out with non-small cell lung cancer (NSCLC) patients has identified four important TNFRSF10B gene polymorphisms associated with high risk of death [74]. One of these four SNPs is located in the 3′-UTR sequence, further supporting the significance of this region.
Nuclear localization of DR5 resists to apoptosis: importin-β1 as the culprit
Subcellular localization of TRAIL death receptors is recently recognized as a phenomenon influencing the outcome of TRAIL signaling and treatment approaches. In a 2012 study carried out with 231 early stage colorectal carcinoma (CRC) patients, membrane DR4 staining was detected in 71 % of samples. In contrast, positive DR5 staining on the membrane was observed in a mere 16 % of cases, the remaining 84 % being exclusively cytoplasmic or negative [75]. Jin and coworkers have elegantly demonstrated that TRAIL-resistant colon cancer cells (SW480), negatively selected by repetitive treatment with soluble TRAIL, were deficient in transporting DR4 to the cell membrane [76]. In addition to disrupted cell surface expression of DR4, they observed a lack of resistance to Fas and paclitaxel, and no change in the expression levels of various apoptosis mediators including DR4. This clearly indicates that failure in death receptor transport alone is sufficient to confer resistance to apoptosis.
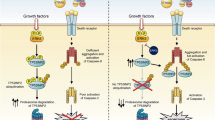
Although the fact that DR5 functions as a plasma membrane-bound receptor might imply exclusive transmembrane expression, we and several other groups have reported cytoplasmic and nuclear expression of both death-inducing TRAIL receptors [77–79] (cellular compartments containing TRAIL receptors are reviewed elsewhere [80]). We already know examples of cell surface receptors that undergo proteolytic cleavage or internalization upon ligand binding and subsequently translocate into the nucleus to affect various cellular events [81–83]. On the other hand, a death receptor’s presence in the nucleus may also reflect a mere mislocalization due to dysregulation of cellular processes, a well-known characteristic of cancer cells. Under careful scrutiny, however, DR5 was recently shown to bear two nuclear localization signals (NLS) [84], suggesting that nuclear translocation does not occur accidentally but is a regulated event. The same study demonstrated physical association between DR5 and importin β1, suggesting the presence of a nuclear import pathway mediated by importin β1 (Fig. 2). In fact, knockdown of importin β1 by an siRNA approach inhibited nuclear transport of DR5 and upregulated its expression on the cell surface, providing increased sensitivity to TRAIL-induced apoptosis in HeLA and HepG2 cells.

Factors affecting intracellular fate of DR5. Two different routes for the DR5 protein to follow within the cell are: plasma membrane localization via mechanisms still not fully elucidated, and nuclear localization through binding of importin β1. When HuR binds to DR5 mRNA under stress conditions, its translation is suppressed
Suspecting that DR5 might translocate to nucleus for a specific function, Haselmann and colleagues investigated whether it associated with any nuclear proteins in human pancreatic ductal adenocarcinoma cells (PDAC) [85]. DR5 was found to interact with Drosha and DGCR8, the core components of the microRNA processing complex, and the related regulatory proteins p68, hnRNPA1, NF45, and NF90. This interaction inhibits maturation of let-7 miRNA family members, resulting in impaired suppression of particularly the let-7 target genes LIN28B and HMGA2, thereby providing a growth advantage to tumor cells. The above-mentioned data clearly indicate that DR5 has dual but evidently opposite functions; it is capable of inducing apoptotic cell death when bound by TRAIL on the cell surface, but facilitates cell survival and/or proliferation once inside the nucleus. Thus, further investigation of spatial receptor regulation and elucidation of factors mediating receptor trafficking is crucial. A first step toward achieving this goal was taken in 2004 [86]. In the quest for factors differentially affecting regulation of death receptors DR4 and DR5, the authors inhibited the functions of around 550 genes by various siRNAs. Intriguingly, silencing genes responsible for the signal recognition particle (SRP) pathway, a well-known canonical mechanism for sorting membrane and secreted proteins, resulted in suppression of DR4 surface expression and DR4-induced apoptosis. There was no change in the total protein level of DR4, and no significant effect on DR5 surface expression or DR5-mediated apoptosis. Thus, to be expressed on the cell surface, DR5 seems to utilize a non-canonical mechanism, the details of which still remain elusive.
Membrane trafficking of DR5 still unsolved
The importance of spatial distribution of death receptors and particularly membrane trafficking issues is rapidly gaining ground, as reflected in several recent reviews [87, 88]. TRAIL sensitive breast cancer cells (MDA-MB-231) developed resistance to TRAIL upon prolonged exposure to subtoxic doses of soluble TRAIL molecule [89]. This acquired resistance was found to result from diminished surface expression, with unaltered total amounts of DR4 and DR5, as well as upregulated c-FLIP and Stat5 expressions. As stated above, unlike DR4, the routes controlling DR5 localization at the cell membrane and the related regulatory mechanisms are still unrevealed (Fig. 2). In a study published in 2006, the authors demonstrated that apigenin, a widely found dietary flavonoid in nature, sensitized malignant cells to TRAIL treatment [90]. This effect of apigenin was shown to be due to increased protein stability of DR5, rather than increased transcriptional activity on its promoter. Interestingly, increased DR5 levels appeared in the membrane fractions, but not in the cytosolic fractions. Researchers also reported that no change was evident in DR4 levels.
Accumulation of autophagosomes can confer TRAIL resistance via DR4 and DR5 sequestration within the cytoplasm, resulting in downregulation of their surface expressions. Inhibition of autophagy leads to increased TRAIL responsiveness via restoration of receptor surface expressions, without altering the total protein levels [91]. Liu et al. showed that detachment of esophageal carcinoma cells (EC9706) did not affect total DR5 protein levels in cells but provided relocalization of DR5 on the cell surface [92]. DR5 oligomerization also occured, rendering cells sensitive to anoikis, a special form of apoptosis triggered by detachment of anchorage-dependent cells. These findings may have especially important implications, considering that in order for metastasis to occur, epithelial cells must first detach from their original tissue. Supporting observations have come from skin carcinoma cells, detachment of which increased sensitivity to TRAIL treatment [93]; as well as MCF-7 cells, anchorage of which suppressed TRAIL sensitivity [94]; and ovarian cancer cell lines, which acquired increased sensitivity to TRAIL upon detachment [95]. On the contrary, a number of studies have described strong association between TRAIL signaling and metastasis [96–98]. However, triple-negative metastatic breast cancer cells injected into mammary glands of female mice exhibited decreased metastatic potential upon DR5-agonistic mAb (lexatumumab) treatment [99].
In human tumor samples, high DR5 expression has been correlated with pancreatic ductal adenocarcinoma cell invasion into lymph vessels and diminished metastasis-free survival of patients with KRAS-mutated colorectal cancer [100]. Taken together with Liu’s findings [92], these data may indicate that increased surface expression of DR5 in detaching cells occurs as a defense mechanism against metastasis, rather than being a cause of the metastatic phenotype. However, more research is required to test this hypothesis before jumping to any conclusion.
Excessive internalization disrupts sensitivity: also valid for DR5/TRAIL relationship
In addition to disturbance of membrane trafficking, excessive internalization of death receptors can also influence the outcome of TRAIL treatment. On its cytoplasmic domain, DR5 contains ESEHLL and YTML sequences which are clathrin-dependent endocytosis motifs ([D/E]XXXL[L/I], YXXØ) [101, 102]. In contrast to Fas and TNF receptors, DR4/DR5 internalization upon ligand binding is cell type-dependent and is not obligatory for induction of apoptosis [103]. Investigation of a variety of breast cancer cell lines revealed that constitutive endocytosis of DR4 and DR5 causes deficient cell surface expression of these receptors regardless of mRNA and total protein levels, resulting in TRAIL resistance [104]. The same group also showed in MDA-MB-231 breast cancer cells that the reduced cell surface expressions of TRAIL death receptors was evident following rapid internalization of TRAIL with DR4 and likely DR5 via endocytosis [105]. Another study had also reported clathrin-mediated DR5 endocytosis upon TRAIL binding in several different cell lines [106]. In this setting, DISC activation was shown to lead to cleavage of proximal components of the endocytic machinery, which, in turn, resulted in inhibition of endocytosis, augmentation of the caspase cascade, and apoptosis. This suggested existence of a feedback loop.
Ubiquitination as a regulatory mechanism of DR5 as well as DR4 actions: another uncompleted story
Ubiquitin-mediated degradation is a prevalent mechanism utilized by the cell to get rid of misfolded, malfunctioning or aged proteins and is vital for maintaining cellular homeostasis. An array of specific proteins and enzymes participate in this process. Ubiquitin, named for being ubiquitously expressed in almost all eukaryotic cells, is covalently attached to target proteins. The position (such as K48, K29, K63, and others) and number (monoubiquitination/polyubiquitination) of bonds determine the nature of the signal and dictate the outcome. Multiple membrane proteins including TRAIL receptors are regulated by ubiquitin-mediated degradation. The E3 ubiquitin ligase c-Cbl has been shown to bind to both DR4 and DR5, enabling regulation of these receptors via proteosomal and lysosomal pathways [107]. The differential action of c-Cbl in this setting is thought to be determined by its phosphorylation status at the Tyr-731 residue. c-Cbl was shown to catalyze monoubiquitination of TRAIL receptors and take part in both steady-state turnover of receptors and development of early phase of acquired resistance to TRAIL.
Preferential ubiquitination of DR4 over DR5, to be downregulated from the cell surface leading to TRAIL resistance, was also reported via exogenous overrepresentation of the membrane-associated RING-CH (MARCH) proteins, such as MARCH-1 and -8 [108]. Endogenous MARCH-8 was directly correlated with steady-state ubiquitination of DR4, and the conserved membrane-proximal lysine 273 was reported as one of the potential acceptor sites. Yet, there is still a large lack of information on ubiquitin-mediated regulation of both death receptors, regarding the degree of specificity, location, the particular type, and outcome of the ubiquination reactions.
The use of various proteasome inhibitors were shown to affect TRAIL sensitivity in different cell types. Bortezomib (VELCADE) was shown to increase both total and surface expressions of DR4 and DR5 in NSCLC cell lines [109]. ALLN, on the other hand, upregulated DR5 total protein expression while having no effect on DR4 levels in LNCaP cells [110]. Similarly, proteasome inhibitors PSI-1 and MG132 were found to upregulate DR5 mRNA and protein levels and enhance TRAIL-mediated apoptosis in human lung cancer cells, without altering DR4 or TRAIL expressions [48]. This effect was at the transcriptional level with the involvement of NF-κB and ROS-dependent p53 activation. A parallel outcome, albeit through a different mechanism, was referred to above [72]. According to the results of the mentioned report, proteasomal inhibition by bortezomib resulted in a sixfold increase in mRNA levels after 24-h treatment, while protein levels increased threefold following 18 h of treatment. The authors report an eightfold rise in HuR bound to DR5 mRNA following bortezomib treatment in LNCaP cells. These data indicate that the ubiquitin–proteasome system (UPS) plays an important role in regulation of DR5 expression, and is capable of rendering cells resistant to TRAIL-mediated apoptosis. The UPS system contributes to TRAIL resistance not only by its effect on DR5, but also on DR4, TRAIL, and numerous downstream participants of TRAIL signaling. A review of UPS’s role in TRAIL/TRAIL-R system has been recently published [111].
Other aspects of DR5 regulation
Post-translational modifications are well known to be crucial in regulation of cellular processes. Membrane-associated or secreted proteins are especially highly subject to these modifications. Death receptor O-glycosylation was found to control TRAIL sensitivity in tumor cells, according to a comprehensive study carried out with 119 human cancer cell lines [112]. O-Glycosylation of DR5, which bears four o-glycosylation sites, promotes receptor clustering and increases TRAIL responsiveness. Knockdown of syndecan-1, the dominant form of syndecans in multiple myeloma, via a siRNA approach provided increased TRAIL-induced apoptosis through elevated expression of TRAIL receptor o-glycosylation enzyme [113].
Analysis of the genome-wide mRNA expression data of the NCI60 panel of human cancer cell lines revealed consistently upregulated expression of H-ras in TRAIL resistant cancer cell lines [114]. Accordingly, suppression of H-ras sensitized these cells to anti-DR5 antibody-induced apoptosis via increased translocation of DR5 to the cell surface, with unaltered total DR5 protein levels. However, the same group also reported that expression of a constitutively active H-ras mutant (H-RasV12) elevated DR5 but not DR4 surface expression in OSCC3 oral squamous cancer cell lines, with little or no effect on the mRNA or protein levels of either receptor [115]. This treatment restored TRAIL sensitivity in these cells. Whether Ras can be used as a biomarker for TRAIL sensitivity in cancer cells, however, is an issue that requires further analysis.
Clustering of receptors in specific membrane domains is another important factor in determining TRAIL sensitivity. Quercetin, a ubiquitous flavonoid and a potential anti-cancer molecule, was demonstrated to increase TRAIL sensitivity of colon cancer cells by simply mediating redistribution of DR4 and DR5 into lipid rafts at the plasma membrane with no effect on mRNA or protein levels [116]. Toad venom bufalin also enhanced TRAIL-induced apoptosis in breast cancer cells via a similar effect on death receptor distribution [117].
Concluding remarks and future perspectives
TRAIL is an important molecule with crucial roles in maintaining vertebrate homeostasis, tumor surveillance, immune system, and metabolism. Discovery of its ability to selectively kill tumor cells and its relatively low cytotoxicity to normal cells has put a focus on potential use of TRAIL in cancer treatment. Nevertheless, diverse resistance mechanisms identified in various cancer cells constitute a challenging obstacle for its efficient use in targeted therapy approaches. Although such mechanisms have been intensively studied and frequently reviewed, intracellular localization of DR5 and related regulatory mechanisms seem to have been underestimated so far as an important means of resistance to TRAIL-mediated apoptosis. Yet unraveling the fact that DR5 contains NLS sequences and performs specific oncogenic functions in the nucleus has obliged scientific community to revise the understanding of TRAIL biology. Current data suggest that there is still an unknown mechanism which determines whether DR5 will translocate into the nucleus or head for the plasma membrane. Disruption of this balance in favor of nuclear transport seems to greatly contribute to TRAIL resistance. Elucidation of the molecular mechanisms controlling the membrane trafficking of DR5 is necessary for the development of novel approaches to tip the balance toward the opposite direction. Surprisingly, for over 10 years, since the 2004 publication excluding SRP-mediated pathway involvement in DR5 membrane localization [86], this mechanism has remained elusive.
In 2010, translocation of the beta-adrenergic receptor to the cell membrane was shown to be mediated by the HuR protein, which binds to the β-AR mRNA and accompanies it to the cell’s periphery [118]. Recently, HuR was demonstrated to mediate membrane localization of several proteins by binding to the 3′-UTR sequences of various transcripts including TNFRSF13C, where a new model of 3′-UTR-dependent protein localization (UDPL) was proposed [119]. Although HuR suppresses the translation of DR5 under stress conditions by binding to its 3′UTR sequence, it may also assist its membrane transport under other conditions. This assumption requires a thorough investigation for further evaluation.
Metastasis is one of the major obstacles on the way to effective cancer therapy. DR5 seems apparently to be linked to it, however, with contrasting evidence supporting either a suppressive [120] or promoting [121] effect. We favor the former possibility and think that the membrane trafficking process of DR5 may be related to its association with metastasis, again an area requiring thorough investigation.
Abbreviations
- AR:
-
Androgen receptor
- BNIP3:
-
Bcl-2 19 kDa interacting protein
- c-FLIP:
-
Cellular FLICE-like protein
- DD:
-
Death domain
- DED:
-
Death effector domain
- ELAVL1:
-
Embryonic lethal abnormal vision-like RNA-binding protein 1
- FADD:
-
FAD-associated death domain protein
- FLICE:
-
FADD-like interleukin-1 beta-converting enzyme
- HuR:
-
Human antigen R
- NLS:
-
Nuclear localization signal
- PDAC:
-
Pancreatic ductal adenocarcinoma cells
- RIP1:
-
Receptor-interacting protein 1
- SRP:
-
Signal recognition particle
- TRAF2:
-
TNF receptor-associated factor 2
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- TRAIL-R:
-
TRAIL receptors
- UPS:
-
Ubiquitin-proteasome system
- UDPL:
-
3′-UTR-dependent protein localization
- YY1:
-
Yin Yang 1
References
Wiley SR et al (1995) Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 3(6):673–682
Gong B, Almasan A (2000) Genomic organization and transcriptional regulation of human Apo2/TRAIL gene. Biochem Biophys Res Commun 278(3):747–752
Harith HH, Morris MJ, Kavurma MM (2013) On the TRAIL of obesity and diabetes. Trends Endocrinol Metab 24(11):578–587
Audo R et al (2015) Osteoprotegerin and tumor necrosis factor-related apoptosis-inducing ligand as prognostic factors in rheumatoid arthritis: results from the ESPOIR cohort. Arthritis Res Ther 17:193
de Miguel D et al (2016) Onto better TRAILs for cancer treatment. Cell Death Differ 23(5):733–747
Di Bartolo BA et al (2011) TNF-related apoptosis-inducing ligand (TRAIL) protects against diabetes and atherosclerosis in Apoe/mice. Diabetologia 54(12):3157–3167
Huang Y et al (2005) The role of TNF related apoptosis-inducing ligand in neurodegenerative diseases. Cell Mol Immunol 2(2):113–122
Hilliard B et al (2001) Roles of TNF-related apoptosis-inducing ligand in experimental autoimmune encephalomyelitis. J Immunol 166(2):1314–1319
Pan G et al (1997) The receptor for the cytotoxic ligand TRAIL. Science 276(5309):111–113
Walczak H et al (1997) TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J 16(17):5386–5397
Falschlehner C et al (2007) TRAIL signalling: decisions between life and death. Int J Biochem Cell Biol 39(7–8):1462–1475
Degli-Esposti MA et al (1997) Cloning and characterization of TRAIL-R3, a novel member of the emerging TRAIL receptor family. J Exp Med 186(7):1165–1170
Degli-Esposti MA et al (1997) The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 7(6):813–820
Emery JG et al (1998) Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem 273(23):14363–14367
Sanlioglu AD et al (2009) High TRAIL death receptor 4 and decoy receptor 2 expression correlates with significant cell death in pancreatic ductal adenocarcinoma patients. Pancreas 38(2):154–160
Koksal IT et al (2008) Tumor necrosis factor-related apoptosis inducing ligand-R4 decoy receptor expression is correlated with high Gleason scores, prostate-specific antigen recurrence, and decreased survival in patients with prostate carcinoma. Urol Oncol 26(2):158–165
Walczak H et al (1999) Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med 5(2):157–163
Ashkenazi A et al (1999) Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Investig 104(2):155–162
Bellail AC et al (2009) TRAIL agonists on clinical trials for cancer therapy: the promises and the challenges. Rev Recent Clin Trials 4(1):34–41
Mirandola P et al (2004) Activated human NK and CD8+ T cells express both TNF-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors but are resistant to TRAIL-mediated cytotoxicity. Blood 104(8):2418–2424
Meurette O et al (2005) TRAIL (TNF-related apoptosis-inducing ligand) induces necrosis-like cell death in tumor cells at acidic extracellular pH. Ann N Y Acad Sci 1056:379–387
Zhang L, Fang B (2005) Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther 12(3):228–237
Hymowitz SG et al (2000) A unique zinc-binding site revealed by a high-resolution X-ray structure of homotrimeric Apo2L/TRAIL. Biochemistry 39(4):633–640
Walczak H, Haas TL (2008) Biochemical analysis of the native TRAIL death-inducing signaling complex. Methods Mol Biol 414:221–239
Bodmer JL et al (2000) TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat Cell Biol 2(4):241–243
Dickens LS et al (2012) A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol Cell 47(2):291–305
Pennarun B et al (2010) Playing the DISC: turning on TRAIL death receptor-mediated apoptosis in cancer. Biochim Biophys Acta 1805(2):123–140
Song JH et al (2007) Lipid rafts and nonrafts mediate tumor necrosis factor related apoptosis-inducing ligand induced apoptotic and nonapoptotic signals in non small cell lung carcinoma cells. Cancer Res 67(14):6946–6955
Huang K et al (2016) Cleavage by caspase 8 and mitochondrial membrane association activate bid during TRAIL-induced apoptosis. J Biol Chem 291(22):11843–11851
Lee DH et al (2016) TRAIL-induced caspase activation is a prerequisite for activation of the endoplasmic reticulum stress-induced signal transduction pathways. J Cell Biochem 117(5):1078–1091
Varfolomeev E et al (2005) Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J Biol Chem 280(49):40599–40608
Azijli K et al (2013) Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: discord in the death receptor family. Cell Death Differ 20(7):858–868
Krueger A et al (2001) FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol 21(24):8247–8254
Aydin C et al (2007) Decoy receptor-2 small interfering RNA (siRNA) strategy employing three different siRNA constructs in combination defeats adenovirus-transferred tumor necrosis factor-related apoptosis-inducing ligand resistance in lung cancer cells. Hum Gene Ther 18(1):39–50
Sanlioglu AD et al (2006) Adenovirus-mediated IKKbetaKA expression sensitizes prostate carcinoma cells to TRAIL-induced apoptosis. Cancer Gene Ther 13(1):21–31
Trivedi R, Mishra DP (2015) Trailing TRAIL resistance: novel targets for TRAIL sensitization in cancer cells. Front Oncol 5:69
O’Leary L et al (2016) Decoy receptors block TRAIL sensitivity at a supracellular level: the role of stromal cells in controlling tumour TRAIL sensitivity. Oncogene 35(10):1261–1270
Chaudhary PM et al (1997) Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity 7(6):821–830
Kelley RF et al (2005) Receptor-selective mutants of apoptosis-inducing ligand 2/tumor necrosis factor-related apoptosis-inducing ligand reveal a greater contribution of death receptor (DR) 5 than DR4 to apoptosis signaling. J Biol Chem 280(3):2205–2212
LeBlanc HN, Ashkenazi A (2003) Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ 10(1):66–75
Truneh A et al (2000) Temperature-sensitive differential affinity of TRAIL for its receptors. DR5 is the highest affinity receptor. J Biol Chem 275(30):23319–23325
Wu GS et al (1997) KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet 17(2):141–143
Yamaguchi H, Wang HG (2004) CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem 279(44):45495–45502
Yoshida T et al (2005) Proteasome inhibitor MG132 induces death receptor 5 through CCAAT/enhancer-binding protein homologous protein. Cancer Res 65(13):5662–5667
Kouhara J et al (2007) Fenretinide up-regulates DR5/TRAIL-R2 expression via the induction of the transcription factor CHOP and combined treatment with fenretinide and TRAIL induces synergistic apoptosis in colon cancer cell lines. Int J Oncol 30(3):679–687
Oh YT et al (2010) ERK/ribosomal S6 kinase (RSK) signaling positively regulates death receptor 5 expression through co-activation of CHOP and Elk1. J Biol Chem 285(53):41310–41319
Eckhardt I, Roesler S, Fulda S (2013) Identification of DR5 as a critical, NF-kappaB-regulated mediator of Smac-induced apoptosis. Cell Death Dis 4:e936
Chen JJ et al (2008) Proteasome inhibitors enhance TRAIL-induced apoptosis through the intronic regulation of DR5: involvement of NF-kappa B and reactive oxygen species-mediated p53 activation. J Immunol 180(12):8030–8039
Zou W et al (2004) c-Jun NH2-terminal kinase-mediated up-regulation of death receptor 5 contributes to induction of apoptosis by the novel synthetic triterpenoid methyl-2-cyano-3,12-dioxooleana-1, 9-dien-28-oate in human lung cancer cells. Cancer Res 64(20):7570–7578
Higuchi H et al (2004) Bile acids up-regulate death receptor 5/TRAIL-receptor 2 expression via a c-Jun N-terminal kinase-dependent pathway involving Sp1. J Biol Chem 279(1):51–60
Chen Q et al (2010) Resveratrol induces growth arrest and apoptosis through activation of FOXO transcription factors in prostate cancer cells. PLoS One 5(12):e15288
Baritaki S et al (2007) Regulation of tumor cell sensitivity to TRAIL-induced apoptosis by the metastatic suppressor Raf kinase inhibitor protein via Yin Yang 1 inhibition and death receptor 5 up-regulation. J Immunol 179(8):5441–5453
Burton TR et al (2013) BNIP3 acts as transcriptional repressor of death receptor-5 expression and prevents TRAIL-induced cell death in gliomas. Cell Death Dis 4:e587
Gerstein MB et al (2012) Architecture of the human regulatory network derived from ENCODE data. Nature 489(7414):91–100
Koksal IT et al (2010) Effects of androgen ablation therapy in TRAIL death ligand and its receptors expression in advanced prostate cancer. Urol Int 84(4):445–451
Massie CE et al (2007) New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep 8(9):871–878
Todo M et al (2013) Ibuprofen enhances TRAIL-induced apoptosis through DR5 upregulation. Oncol Rep 30(5):2379–2384
Taniguchi H et al (2008) Baicalein overcomes tumor necrosis factor-related apoptosis-inducing ligand resistance via two different cell-specific pathways in cancer cells but not in normal cells. Cancer Res 68(21):8918–8927
Prasad S et al (2014) Targeting death receptors for TRAIL by agents designed by Mother Nature. Trends Pharmacol Sci 35(10):520–536
Wong N, Wang X (2015) miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res 43(Database issue):D146–D152
Gerstberger S, Hafner M, Tuschl T (2014) A census of human RNA-binding proteins. Nat Rev Genet 15(12):829–845
Glisovic T et al (2008) RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett 582(14):1977–1986
Yang YC et al (2015) CLIPdb: a CLIP-seq database for protein-RNA interactions. BMC Genom 16:51
Wang W et al (2000) HuR regulates p21 mRNA stabilization by UV light. Mol Cell Biol 20(3):760–769
Blaxall BC et al (2000) Purification and characterization of beta-adrenergic receptor mRNA-binding proteins. J Biol Chem 275(6):4290–4297
Goldberg-Cohen I, Furneauxb H, Levy AP (2002) A 40-bp RNA element that mediates stabilization of vascular endothelial growth factor mRNA by HuR. J Biol Chem 277(16):13635–13640
Yeap BB et al (2002) Novel binding of HuR and poly(C)-binding protein to a conserved UC-rich motif within the 3′-untranslated region of the androgen receptor messenger RNA. J Biol Chem 277(30):27183–27192
Fan XC, Steitz JA (1998) HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proc Natl Acad Sci USA 95(26):15293–15298
Fan XC, Steitz JA (1998) Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J 17(12):3448–3460
Brennan CM, Steitz JA (2001) HuR and mRNA stability. Cell Mol Life Sci 58(2):266–277
Takeuchi O (2015) HuR keeps interferon-beta mRNA stable. Eur J Immunol 45(5):1296–1299
Kandasamy K, Kraft AS (2008) Proteasome inhibitor PS-341 (VELCADE) induces stabilization of the TRAIL receptor DR5 mRNA through the 3′-untranslated region. Mol Cancer Ther 7(5):1091–1100
Pineda DM et al (2012) HuR’s post-transcriptional regulation of Death Receptor 5 in pancreatic cancer cells. Cancer Biol Ther 13(10):946–955
Schabath MB et al (2013) TNFRSF10B polymorphisms and haplotypes associated with increased risk of death in non-small cell lung cancer. Carcinogenesis 34(11):2525–2530
Kriegl L et al (2012) Microsatellite instability, KRAS mutations and cellular distribution of TRAIL-receptors in early stage colorectal cancer. PLoS One 7(12):e51654
Jin Z et al (2004) Deficient tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor transport to the cell surface in human colon cancer cells selected for resistance to TRAIL-induced apoptosis. J Biol Chem 279(34):35829–35839
Spierings DC et al (2003) Expression of TRAIL and TRAIL death receptors in stage III non-small cell lung cancer tumors. Clin Cancer Res 9(9):3397–3405
Sanlioglu AD et al (2007) Differential expression of TRAIL and its receptors in benign and malignant prostate tissues. J Urol 177(1):359–364
Ganten TM et al (2009) Prognostic significance of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor expression in patients with breast cancer. J Mol Med (Berl) 87(10):995–1007
Bertsch U et al (2014) Compartmentalization of TNF-related apoptosis-inducing ligand (TRAIL) death receptor functions: emerging role of nuclear TRAIL-R2. Cell Death Dis 5:e1390
Carpenter G, Liao HJ (2009) Trafficking of receptor tyrosine kinases to the nucleus. Exp Cell Res 315(9):1556–1566
Bryant DM, Stow JL (2005) Nuclear translocation of cell-surface receptors: lessons from fibroblast growth factor. Traffic 6(10):947–954
Kasuga K et al (2007) Generation of intracellular domain of insulin receptor tyrosine kinase by gamma-secretase. Biochem Biophys Res Commun 360(1):90–96
Kojima Y et al (2011) Importin beta1 protein-mediated nuclear localization of death receptor 5 (DR5) limits DR5/tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-induced cell death of human tumor cells. J Biol Chem 286(50):43383–43393
Haselmann V et al (2014) Nuclear death receptor TRAIL-R2 inhibits maturation of let-7 and promotes proliferation of pancreatic and other tumor cells. Gastroenterology 146(1):278–290
Ren YG et al (2004) Differential regulation of the TRAIL death receptors DR4 and DR5 by the signal recognition particle. Mol Biol Cell 15(11):5064–5074
Twomey JD et al (2015) Spatial dynamics of TRAIL death receptors in cancer cells. Drug Resist Updat 19:13–21
Schneider-Brachert W, Heigl U, Ehrenschwender M (2013) Membrane trafficking of death receptors: implications on signalling. Int J Mol Sci 14(7):14475–14503
Yoshida T et al (2009) Repeated treatment with subtoxic doses of TRAIL induces resistance to apoptosis through its death receptors in MDA-MB-231 breast cancer cells. Mol Cancer Res 7(11):1835–1844
Horinaka M et al (2006) The dietary flavonoid apigenin sensitizes malignant tumor cells to tumor necrosis factor-related apoptosis-inducing ligand. Mol Cancer Ther 5(4):945–951
Di X et al (2013) Accumulation of autophagosomes in breast cancer cells induces TRAIL resistance through downregulation of surface expression of death receptors 4 and 5. Oncotarget 4(9):1349–1364
Liu GC et al (2009) Detachment of esophageal carcinoma cells from extracellular matrix causes relocalization of death receptor 5 and apoptosis. World J Gastroenterol 15(7):836–844
Grosse-Wilde A et al (2008) TRAIL-R deficiency in mice enhances lymph node metastasis without affecting primary tumor development. J Clin Investig 118(1):100–110
Goldberg GS et al (2001) Global effects of anchorage on gene expression during mammary carcinoma cell growth reveal role of tumor necrosis factor-related apoptosis-inducing ligand in anoikis. Cancer Res 61(4):1334–1337
Lane D et al (2008) Cell detachment modulates TRAIL resistance in ovarian cancer cells by downregulating the phosphatidylinositol 3-kinase/Akt pathway. Int J Gynecol Cancer 18(4):670–676
Trauzold A et al (2006) TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene 25(56):7434–7439
Ishimura N et al (2006) Trail induces cell migration and invasion in apoptosis-resistant cholangiocarcinoma cells. Am J Physiol Gastrointest Liver Physiol 290(1):G129–G136
Hoogwater FJ et al (2010) Oncogenic K-Ras turns death receptors into metastasis-promoting receptors in human and mouse colorectal cancer cells. Gastroenterology 138(7):2357–2367
Malin D et al (2011) Enhanced metastasis suppression by targeting TRAIL receptor 2 in a murine model of triple-negative breast cancer. Clin Cancer Res 17(15):5005–5015
von Karstedt S et al (2015) Cancer cell-autonomous TRAIL-R signaling promotes KRAS-driven cancer progression, invasion, and metastasis. Cancer Cell 27(4):561–573
Kozik P et al (2010) A screen for endocytic motifs. Traffic 11(6):843–855
Akazawa Y et al (2009) Death receptor 5 internalization is required for lysosomal permeabilization by TRAIL in malignant liver cell lines. Gastroenterology 136(7):2365–2376 e1–e7
Kohlhaas SL et al (2007) Receptor-mediated endocytosis is not required for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. J Biol Chem 282(17):12831–12841
Zhang Y, Zhang B (2008) TRAIL resistance of breast cancer cells is associated with constitutive endocytosis of death receptors 4 and 5. Mol Cancer Res 6(12):1861–1871
Zhang Y, Yoshida T, Zhang B (2009) TRAIL induces endocytosis of its death receptors in MDA-MB-231 breast cancer cells. Cancer Biol Ther 8(10):917–922
Austin CD et al (2006) Death-receptor activation halts clathrin-dependent endocytosis. Proc Natl Acad Sci USA 103(27):10283–10288
Song JJ et al (2010) c-Cbl-mediated degradation of TRAIL receptors is responsible for the development of the early phase of TRAIL resistance. Cell Signal 22(3):553–563
van de Kooij B et al (2013) Ubiquitination by the membrane-associated RING-CH-8 (MARCH-8) ligase controls steady-state cell surface expression of tumor necrosis factor-related apoptosis inducing ligand (TRAIL) receptor 1. J Biol Chem 288(9):6617–6628
Liu X et al (2007) The proteasome inhibitor PS-341 (bortezomib) up-regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL-induced apoptosis despite up-regulation of c-FLIP and survivin expression in human NSCLC cells. Cancer Res 67(10):4981–4988
Lee YJ et al (2011) Inhibition of the ubiquitin-proteasome system sensitizes TRAIL-resistant prostate cancer cells by up-regulation of death receptor 5. Mol Med Rep 4(6):1255–1259
Sarhan D, D’Arcy P, Lundqvist A (2014) Regulation of TRAIL-receptor expression by the ubiquitin-proteasome system. Int J Mol Sci 15(10):18557–18573
Wagner KW et al (2007) Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med 13(9):1070–1077
Wu YH et al (2012) Removal of syndecan-1 promotes TRAIL-induced apoptosis in myeloma cells. J Immunol 188(6):2914–2921
Chen JJ et al (2014) H-Ras regulation of TRAIL death receptor mediated apoptosis. Oncotarget 5(13):5125–5137
Chen JJ et al (2013) TRAIL induces apoptosis in oral squamous carcinoma cells—a crosstalk with oncogenic Ras regulated cell surface expression of death receptor 5. Oncotarget 4(2):206–217
Psahoulia FH et al (2007) Quercetin enhances TRAIL-mediated apoptosis in colon cancer cells by inducing the accumulation of death receptors in lipid rafts. Mol Cancer Ther 6(9):2591–2599
Yan S et al (2014) Bufalin enhances TRAIL-induced apoptosis by redistributing death receptors in lipid rafts in breast cancer cells. Anticancer Drugs 25(6):683–689
Tholanikunnel BG et al (2010) Novel mechanisms in the regulation of G protein-coupled receptor trafficking to the plasma membrane. J Biol Chem 285(44):33816–33825
Berkovits BD, Mayr C (2015) Alternative 3′ UTRs act as scaffolds to regulate membrane protein localization. Nature 522(7556):363–367
Grosse-Wilde A, Kemp CJ (2008) Metastasis suppressor function of tumor necrosis factor-related apoptosis-inducing ligand-R in mice: implications for TRAIL-based therapy in humans? Cancer Res 68(15):6035–6037
Fritsche H et al (2015) TRAIL-R2 promotes skeletal metastasis in a breast cancer xenograft mouse model. Oncotarget 6(11):9502–9516
Acknowledgments
This work was supported by the Research Fund of Akdeniz University, Project Number TDK-2015-942.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interests.
Rights and permissions
About this article

Cite this article
Mert, U., Sanlioglu, A.D. Intracellular localization of DR5 and related regulatory pathways as a mechanism of resistance to TRAIL in cancer. Cell. Mol. Life Sci. 74, 245–255 (2017). https://doi.org/10.1007/s00018-016-2321-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2321-z