
Abstract
The process of embryonic development is highly regulated through the symbiotic control of differentiation and programmed cell death pathways, which together sculpt tissues and organs. The importance of programmed necrotic (RIPK-dependent necroptosis) cell death during development has recently been recognized as important and has largely been characterized using genetically engineered animals. Suppression of necroptosis appears to be essential for murine development and occurs at three distinct checkpoints, E10.5, E16.5, and P1. These distinct time points have helped delineate the molecular pathways and regulation of necroptosis. The embryonic lethality at E10.5 seen in knockouts of caspase-8, FADD, or FLIP (cflar), components of the extrinsic apoptosis pathway, resulted in pallid embryos that did not exhibit the expected cellular expansions. This was the first suggestion that these factors play an important role in the inhibition of necroptotic cell death. The embryonic lethality at E16.5 highlighted the importance of TNF engaging necroptosis in vivo, since elimination of TNFR1 from casp8 −/−, fadd −/−, or cflar −/−, ripk3 −/− embryos delayed embryonic lethality from E10.5 until E16.5. The P1 checkpoint demonstrates the dual role of RIPK1 in both the induction and inhibition of necroptosis, depending on the upstream signal. This review summarizes the role of necroptosis in development and the genetic evidence that helped detail the molecular mechanisms of this novel pathway of programmed cell death.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
The importance of cell death during development has long been recognized. One form of programmed cell death, termed apoptosis, is induced by specific triggers to activate a signaling cascade that ultimately ends in the packaging of the cell by caspase proteases for easy disposal via phagocytosis [1]. There are two prominent apoptotic pathways: intrinsic apoptosis, which is characterized by loss of mitochondrial outer membrane integrity, and extrinsic apoptosis, where ligated death receptors on the cell surface initiate death (Fig. 1) [1]. The role of these apoptotic pathways during development has been characterized using genetically engineered animals. Deletion of pro-death genes involved in intrinsic apoptosis such as apaf1 or bax and bak led to mice with encephalopathy that presented with forebrain outgrowth at E16.5, and the persistence of interdigital webs [2–4]. By comparison, a paradoxical phenotype was observed when components of the extrinsic apoptotic pathway were ablated. Ablation of caspase-8, FADD, or FLIP (cflar) resulted in embryonic lethality at E10.5 with pallid embryos that did not exhibit the expected cellular expansions, suggesting that these factors might play roles in non-apoptotic developmental processes (Fig. 2a) [5–8]. Pharmacological inhibition of caspase-8 in cell culture yielded non-apoptotic death instead of prolonging cell viability under some conditions [9]. These results suggested that caspase-8 was important for opposing an apoptosis-independent form of cell death. This apoptosis-independent, regulated form of cell death, termed necroptosis, has features of necrosis but utilizes a signaling pathway involving Receptor-Interacting Protein (RIP) kinases [10]. This review summarizes the role of necroptosis in development.

Pathways leading to extrinsic apoptosis and necroptosis. Binding of TNF to TNFR1 leads to the recruitment of TRADD, FADD and RIPK1 to form the proinflammatory complex I. TRAF2 binds to TRADD and recruits the E3 ligases cIAP1 and cIAP2 that mediate the ubiquitylation of RIPK1. This leads to the activation of the IKK complex (consisting of NEMO, IKKα and IKKβ) through phosphorylation by TAK1 and the subsequent degradation of IκBα and the release and nuclear translocation of the NF-κB1 complex. In case of RIPK1 deubiquitylation, through cIAP inhibition or ubiquitin removal by the deubiquitylase CYLD, or binding of FasL to CD95, RIPK1 can associate with FADD and recruit procaspase-8 and FLIP in complex IIa. Homodimerization of caspase-8 on FADD can fully activate caspase-8 and leads to apoptosis. However, when FLIP heterodimerizes with caspase-8, the full activation is inhibited and apoptosis is blocked. RIPK1 binds and activates RIPK3, resulting in formation of the necrosome and the activation the pseudokinase MLKL, leading to necroptosis. The caspase-8/FLIP heterodimer also inhibits necroptosis. Upon ligation of TLR3 and 4, necroptosis can be engaged through TRIF-mediated RIPK3 activation. Binding of IFNα/β or IFNγ to their respective receptors also results in activation of necroptosis. Necroptosis induced via either TRIF or IFN receptors can be blocked by RIPK1

Developmental checkpoints of mice deficient for components of extrinsic apoptosis and necroptosis. Lifespan of mice harboring single (a), double (b) or triple (c) deletion of genes involved in extrinsic apoptosis and necroptosis. The embryonic dates listed are for RelA-, HOIP-, and TAK1-deficient mice, rather than animals deficient for other components of these complexes (p50, Sharpin, HOIL1, TAB1, and TAB2)
Mechanisms of necroptosis
Necroptosis describes a novel programmed cell death pathway that phenotypically resembles necrosis, where cells swell and burst, resulting in an immunogenic response [11]. Unlike necrosis caused by excessive damage, necroptosis is mediated through controlled signal transduction pathways (Fig. 1) [12].
Necroptosis is induced after binding of a death ligand to its respective receptor on the cell surface, such as TNFR1, CD95, or the TRAIL receptors (Fig. 1) [13–15]. The molecular details of necroptosis were originally described in the context of TNF-mediated signaling. Upon binding of TNF to TNFR1, pro-survival factors are recruited to the death receptor, to form a pro-inflammatory complex (complex I, C1) [16]. This complex contains numerous proteins involved in NF-κB signaling, as well as TRADD, TRAF2, cIAP1/cIAP2, the linear ubiquitin chain assembly complex (LUBAC), TAK1 and RIPK1. TRADD binds to TNFR through its death domain and is required for the recruitment of the other members of complex 1 [16]. RIPK1 is sequestered in this complex via its ubiquitylation [16].
For necroptosis to be engaged, RIPK1 requires post-translational modifications, such as deubiquitylation (Fig. 1) [17]. Subsequent to its deubiquitylation, RIPK1 is released from C1 into the cytosol where it binds to FADD and caspase 8 to form complex IIa (C2a), which promotes apoptosis through caspase-8 homo-oligomerization [18]. If caspase-8 activity is diminished, or if FADD levels are low, RIPK1 preferentially binds to RIPK3 via a protein–protein interaction domain known as the RIP Homotypic Interaction Motif (RHIM) domain [19], leading to the subsequent phosphorylation and activation of RIPK3 in complex IIb (C2b), also called the “necrosome” [18]. It is this complex that induces necroptosis [20]. Once activated, RIPK3 recruits, phosphorylates and activates the pseudokinase MLKL, which causes disruption of plasma membrane integrity, resulting in the rapid death of the cell [21]. While the exact mechanism by which MLKL mediates death has not been conclusively elucidated, the current hypothesis predicts that MLKL phosphorylation results in its oligomerization and membrane disruption, resulting in ion influx and osmotic stress, causing a cell death that phenotypically resembles necrosis [22–29].
There is an important molecular interplay between apoptotic pathways and necroptotic pathways which appears to depend on caspase-8 level and activity (Fig. 1) [30]. Caspase-8 deletion or inhibition experiments in vitro and in vivo, suggested that caspase-8 inhibits necroptosis [30]. Subsequent studies suggested that rather than causing apoptosis, an enzymatically active complex of caspase-8, FLIPL, and FADD acted to inhibit necroptosis [31, 32]. Deletion of any of these three genes results in embryonic lethality, suggesting that they may be critical for controlling necroptosis, which was confirmed by experiments involving co-ablation of RIPK3 (Fig. 2b) [31–33]. The exact molecular mechanisms involved in this suppression are unclear but are thought to involve cleavage of RIPK1 and RIPK3, as well as other factors important for formation of C2b, such as the deubiquitylase CYLD [31, 34].
In addition to death receptor-mediated necroptosis, other signals, such as interferons or engagement of TLR signaling through TRIF, can directly activate RIPK3 to promote necroptotic death, bypassing the requirement for RIPK1 (Fig. 1) [35, 36]. In this situation, RIPK1 has an inhibitory role [37]. The different mechanisms for inducing necroptosis have functional consequences for how this pathway is engaged during development.
Developmental checkpoints and necroptosis
In addition to an on-going role in adulthood, suppression of necroptosis appears to be important at three distinct checkpoints during mouse development [20, 38]. These checkpoints were uncovered through the use of necroptosis-prone mouse strains (i.e. casp8 −/−, fadd −/−, or cflar −/− (encoding FLIPL) mice) that were rescued by elimination of RIPK3 (Fig. 2a) [20]. Using these methods, a developmental role for necroptosis was observed at E10.5, E16.5, and P1 [20], discussed in further detail below.
The E10.5 checkpoint: a death receptor-mediated apoptosis trio playing another role?
As discussed above, caspase-8, FADD, and FLIP are necessary to prevent activation of RIPK3-dependent necroptosis in response to TNF signaling (Fig. 1). In this section, we discuss how the genetic evidence resulting from examination of casp8 −/−, fadd −/−, or cflar −/− mice, whose E10.5 lethality was rescued by elimination of RIPK3, supports this conclusion [31–33].
Mice in which caspase-8 has been ablated undergo embryonic lethality at E10.5 (Fig. 2a) [5]. Death was originally attributed to impaired heart muscle development, accumulation of erythrocytes, and neural tube defects. Ex vivo whole embryo culture of E10.5 casp8 −/− embryos, however, showed a delay in lethality and the elimination of the heart and neural tube defects, suggesting that these effects were secondary to the defects in vasculature development in the yolk sac [39]. The formation of the yolk sac vasculature is severely deformed in caspase-8-deficient embryos; in particular the yolk sac primary capillary plexus does not develop into the more mature tree-like hierarchy of vessels seen in WT mice [39].
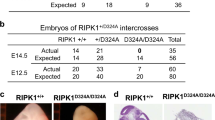
Since the role of caspase-8 in apoptosis was clearly defined, it was important to address whether the embryonic lethality associated with caspase-8 deletion was due to a lack of apoptosis. To answer this, Wallach and colleagues generated transgenic animals using a bacterial artificial chromosome system that expressed caspase-8 with a mutated site in a self-processing site (D387A), rendering it non-cleavable and unable to support apoptosis [40]. When mice expressing this non-apoptotic version of caspase-8 were crossed to casp8 −/− mice, lethality was rescued, demonstrating that the apoptotic function of caspase-8 was not necessary during development [40].
Lethality of casp8 −/− mice was fully rescued when bred with ripk3 −/− mice resulting in a completely normal vascular system, leading to the conclusion that the primary role of caspase-8 during development is to inhibit RIPK3-mediated lethality (Fig. 2b) [31, 33].
Since the activation of caspase-8 depends on the adapter molecule FADD, it was not surprising that the loss of FADD also led to E10.5 lethality with defects in the cardiac area and abdominal hemorrhaging, as in casp8 −/− embryos (Fig. 2a) [6]. Concurrent ablation of RIPK3 from fadd −/− embryos allowed these mice to develop to adulthood, demonstrating that FADD, like caspase-8 is important for RIPK3 inhibition in development (Fig. 2b) [32].
TRADD recruits FADD to C1 via its death domain (Fig. 1) [41]. Interestingly, TRADD-deficient mice (which are resistant to TNF induced toxicity) are morphologically normal and survive to adulthood, suggesting a fundamental difference in the role of FADD and TRADD (Fig. 2a) [42]. Unlike TRADD, FADD contains a death effector domain required for its binding to caspase-8 [43]. Attempts to generate transgenic mice expressing only the DD domain of FADD driven by the vav promoter failed to result in any viable animals, although the authors did not determine at which stage these animals died [44], reiterating the importance of recruitment of caspase-8 to regulate the necrosome. Subsequent work also demonstrated that the death domain of FADD was essential for embryonic development, as transgenic animals expressing FADD with a mutated death domain (R117Q) failed to complement a fadd-deficient background and led to death at E13.5 compared to lethality at E10.5 of fadd-deficient mice alone [45]. While expression of a different mutant in the death domain (V121N) prevented E10.5 lethality, these animals died at E16.5 through apparent engagement of the second checkpoint discussed below [45].
Embryos lacking cflar die around E10.5 and were originally reported to be phenotypically similar to casp8 or fadd knockout embryos (Fig. 2a) [7]. Unlike cells lacking casp8 −/− or fadd /−, cflar −/− cells were sensitive to apoptosis, presumably because caspase-8 is free to homo-oligomerize [30, 32]. When cflar −/− mice were crossed with ripk3 −/− mice, embryonic lethality was not bypassed and embryos died with the same kinetics and gross phenotype as cflar −/− mice alone (Fig. 2b) [32]. Closer histological examination revealed that tissues in cflar, ripk3 double-deficient embryos underwent apoptotic cell death, reinforcing the importance of FLIP in inhibiting both apoptosis and necroptosis [32]. Subsequently, when fadd was deleted in combination with cflar and ripk3, embryos survived to adulthood (Fig. 2c) [32]. This result suggested that there is a close relationship between both apoptosis and necroptosis during embryonic development and both pathways are likely activated by the same signal, at the same time point.
Tissue specificity of E10.5 death
The shared timing of embryonic lethality and phenotype of death in the casp8 −/−, fadd −/−, and cflar −/− embryos suggested a common role for these proteins in vascular development. This has been empirically tested with the subsequent establishment of conditional genetic models. Consistent with the idea that heart and neural tube defects were secondary to a vascular defect at E10.5, deletion of caspase-8 in the brain or heart had no effect on development [32, 46]. Deletion of caspase-8 in the liver also led to the development of a normal mouse, however, this animal was also protected from lethality resulting from injection with the agonist, anti-CD95 antibody, Jo2 [47]. This suggests that while apoptotic signaling was lost, the liver was not prone to necroptosis in the basal state.
The pallid appearance of the casp8 −/−, fadd −/−, and cflar −/− embryos led to the hypothesis that the lethality of these mice might result from defective blood development. The initial report of the casp8 −/− animal also suggested an inability to recover hematopoietic colony forming units from the embryo [5]. As expected, deletion of caspase-8 from the hematopoietic system using vav-cre was embryonic lethal, although the timing and cause of the death was not reported [48]. The deletion of caspase-8 from differentiated hematological tissues such as T or B cells did not have any effect on the viability of the animals [48–50]. Similarly, loss of caspase-8 from macrophages or dendritic cells resulted in viable animals, although cells from these mice are reported to have an increased ability to stimulate inflammatory cytokine expression through the inflammasome [51].
While there is evidence for a role for necroptosis in development of the hematopoietic system, hematopoietic and endothelial development are closely linked (as hematopoietic stem cells emerge from hemogenic endothelium [52]), making it difficult to determine whether a developmental defect resides in one or both tissues. Endothelial deletion of caspase-8 generates animals that phenocopy the caspase-8 knockout completely [47], but rescue of this lethality with RIPK3 ablation has not been attempted. More detailed analysis of this early stage of development will be required to fully dissect the role of necroptosis at this early checkpoint.
Kinases at E10.5
Molecular experiments have proposed that the necroptotic-signaling pathway involves a kinase cascade, resulting in the phosphorylation and activation of key molecules such as RIPK3. Specifically, RIPK1 activates RIPK3, which in turn phosphorylates and activates MLKL (Fig. 1). To examine this genetically, mice were generated in which RIPK3 lacked its kinase activity (D161N). These mice die at E10.5 from excessive apoptosis as lethality was rescued by the additional ablation of caspase-8 [53]. In contrast, mice expressing other RIPK3 kinase-inactive mutants (D161G, D143N, and K51A) were not embryonic lethal, suggesting that kinase activity and engagement of apoptosis by RIPK3 can be dissociated, and the concomitant addition of RIPK3 inhibitors induced similar levels of apoptosis in cells from each mutant [54]. This symbiotic relationship between apoptotic and necroptotic pathways mimics the phenotype of the cflar knockout animals, where apoptosis occurred in embryos when RIPK3 was eliminated (Fig. 2b) [32].
After TNFR ligation, the NF-κB pathway is engaged by the phosphorylation of IKKα and IKKβ by TAK1 (Fig. 1) [55]. Interestingly, TAK1 inhibition potentiates TNF-induced RIPK1-dependent cell death, suggesting a potential role for TAK1 in limiting necroptosis [56]. TAK1-deficient animals die at E10.5 (Fig. 2a), presumably with the same vascular phenotype as casp8-, fadd-, or cflar-deficient mice, since endothelial specific deletion of TAK1 replicated the germline knockouts [57, 58]. The role of RIPK3 in TAK1 lethality has yet to be reported, although evidence in cell lines shows a delay in death of tak1 −/− cells in the ripk3 −/− background [58].
Ablation of TNFR1 signaling in tak1 −/− animals delays lethality by 2 days, unlike the 6 days delay observed when TNFR1 is ablated in casp8 −/− or hoip −/− (a component of the linear ubiquitin chain assembly complex, LUBAC) mice (discussed below), which could point to additional roles for TAK1 in development (Fig. 2b) [37, 59, 60]. It is conceivable that TGFβ signaling (which activates TAK1), rather than necroptosis, might be responsible for embryonic lethality in the tak1 −/− embryos, as other knockouts in the TGFβ pathway have E10.5 lethality and defects in vascular development [61]. Interestingly, mice deficient in other components of the TNFR/NF-κB signaling pathway such as IKKβ and RelA, die around E14 with defects in the liver [62, 63]; NEMO knockout mice also have a liver pathology but die around E12.5 [64], and RIPK1 knockout mice die at P1 [65] as discussed below (Fig. 2a). Collectively, this suggests that a defect in NF-κB signaling is detrimental to normal development, but may result in a different developmental phenotype compared with animals that die from unregulated RIPK3 activity at E10.5.
E3 ligases at E10.5
Downstream of TNFR ligation with TNF, cIAP1 and 2 are important for the ubiquitylation of components of the TNFR1 receptor C1 (Fig. 1) [66]. Unlike ablation of either cIAP1 or cIAP2 alone, which leads to viable mice, deletion of both genes yields embryonic lethality at E10.5 with pathologies similar to those observed in the casp8 −/− and fadd −/− embryos (Fig. 2a, b) [67]. Deletion of HOIP also leads to embryonic lethality at E10.5 with defects in the endothelium and a similar phenotype (Fig. 2a) [59]. These results implicate cIAP1, cIAP2, and HOIP (and by extension LUBAC) as regulators of necroptosis [68]. Consistent with this hypothesis, endothelial deletion of HOIP results in mice that mimic the endothelial loss of caspase-8 [32, 59]. In the absence of cIAP1/2 or HOIP, it is possible that RIPK1 can no longer be properly ubiquitylated in C1, thus potentially allowing for its disassociation with C1 and association with FADD in C2a or RIPK3 in C2b and the subsequent formation of the necrosome. Conversely, preventing changes in RIPK1 ubiquitylation might lead to inhibition of necroptosis by preventing RIPK1 from leaving C1. Consistent with this hypothesis, mice deficient in A20 (a ubiquitin editor) [69] or CYLD (a deubiquitylase implicated in RIPK1 deubiquitylation) [70] are viable, although a20 −/− mice die early in adulthood from multi-organ inflammation and cyld −/− animals display an increase in inflammatory infiltrates with age [71]. The defects observed in both of these mice likely result from a failure to terminate NF-κB signaling, a predicted consequence of prolonged presence of RIPK1 in C1 [72]. Whether deletion of either A20 or CYLD delays lethality in casp8 −/− , fadd −/− , or cflar −/− mice is currently unknown.
A plethora of pathways?
The complex regulation of RIPK3-mediated necroptosis is not yet fully understood. Numerous other genetically modified animals die around E10.5 with phenotypes that mimic those seen in mice prone to RIPK3-dependent embryonic lethality. It is tempting to speculate that these mice provide evidence for novel signaling pathways that mediate necroptosis. FOXO1-deficient mice die at E11 with vascular effects that are phenocopied by the conditional loss of FOXO1 in endothelia [73, 74]. Perhaps FOXO1 could be an important transcription factor for the components that control the necroptosis pathway. Similar phenotypes are seen in animals deficient for Notch signaling (such as Jagged 1 germline knockouts [75] and Snail1 endothelial specific knockouts [76]), suggesting a possible role for these signals in limiting necroptosis. Mice with deletions in genes that affect different biological functions such as methylation (Jmjd5 knockout [77]), altered signal transduction (RapGEF2 knockout [78]), or nucleic acid sensing (Npm1 knockout [79]) also show early embryonic lethality with vascular defects. Investigating the role of these pathways in regulating necroptosis, formally tested by eliminating RIPK3 or MLKL, would resolve these possibilities.
A novel developmental checkpoint: E16.5
The molecular details of how TNF activates necroptosis have been well described in vitro. What is the genetic evidence that supports the role for TNF engaging necroptosis in vivo? Recent work has demonstrated that elimination of TNFR1 from casp8 −/−, fadd −/−, or cflar −/− embryos delays embryonic lethality from 10.5 until E16.5 (Fig. 2b) [37]. In each of these cases, the yolk sac vasculature appears intact, and examination of these embryos did not lead to an obvious cause of death. Similarly, removal of TNFR1 from hoip-deficient animals also delayed lethality to this later E16.5 checkpoint, with complete recovery of vascular development (Fig. 2b) [59]. In contrast to hoip −/− ,tnfr1 −/− embryos, hoip −/− ,tnf −/− embryos have vascular defects, leading to lethality at E12.5 [59]. These results suggest that in addition to TNF, lymphotoxin, which also engages TNFR1, might also play a role in death signaling at E10.5, a hypothesis that has not been directly examined in either the hoip −/− or casp8 −/− contexts [37, 59]. While casp8 −/− embryos are rescued to adulthood by ablation of ripk3 −/−, formal demonstration that hoip −/− animals die from unregulated RIPK3 or necroptosis remains elusive, since no hoip −/− , ripk3 −/− or hoip −/− , mlkl −/− mice have been reported [31, 33, 59].
Since both HOIP and cIAPs are proposed to ubiquitylate RIPK1, it seems strange that there is a partial rescue of lethality in both casp8 −/− and hoip −/− embryos to E16.5 by TNFR1 ablation, but cIAP1,cIAP2 double-deficient mice crossed to the tnfr1 −/− background are rescued to birth (Fig. 2b, c) [67]. Even more surprising is that the loss of ripk1 −/− or ripk3 −/− bypasses the E10.5 lethality of cIAP1,cIAP2 double-deficient mice leading to E12.5 and E16.5 lethality, respectively (Fig. 2c) [67]. This suggests that a combination of necroptosis and apoptosis mediates lethality in many of these genetic mouse models, thus further confounding the analysis of necroptosis signaling in development.
The cause of death in animals that die at the E16.5 checkpoint remains unclear. Hoip −/−, tnfr1 −/− mice display severe heart defects with chamber dilation and blood vessel dilation throughout the heart and other organs, yet similar defects were not seen in the casp8 −/−, tnfr1 −/− mice [37, 59]. Some insight can be gained by histological examination of cflar −/−, ripk3 −/−, tnfr1 −/− animals, as histological analysis revealed that liver and spleen from these mice undergo apoptotic rather than necroptotic death (Dillon, Green unpublished).
To date, it remains unclear which signals might drive activation of necroptosis at the E16.5 checkpoint. However, it is tempting to speculate that this signaling may occur through another TNFR superfamily member. While signaling through both CD95 and TRAIL has been implicated in necroptotic cell death in vitro, neither has yet been shown to be responsible for the embryonic death observed at E16.5 [13, 80]. Additional experiments will be required to delineate the developmental signals inducing necroptosis at this point.
P1 checkpoint: dying at birth
RIPK1 is key in inducing cell death downstream of TNFR1 signaling through its inclusion in either C2a or C2b [16]. However, in response to direct activation of RIPK3 via innate immune signals such as TRIF, RIPK1 appears to inhibit rather than drive cell death (Fig. 1). Genetic experiments described below provided support for this initially counter-intuitive hypothesis.
Unlike the casp8 −/−, fadd −/−, or cflar −/− mice that die at E10.5 or the ripk3- and mlkl-deficient animals that reach adulthood without a phenotype, the deletion of RIPK1 leads to a surprising and novel phenotype, death at birth (P1) [65]. Examination of ripk1 −/− mice showed an increase in cell death in both the thymus and adipose tissues as well as widespread edema [65]. Given that RIPK1 plays an important role in TNF-induced NF-κB signaling, it was assumed that this phenotype was largely attributable to these pro-survival functions of RIPK1 [65]. However, to address the possibility that cell death plays a role in this postnatal lethality, three research groups eliminated both apoptosis and necroptosis from ripk1 −/− embryos by deleting both caspase-8 (or FADD) and RIPK3 (Fig. 2c) [37, 81, 82]. Elimination of both of these pathways led to an animal that survived into adulthood and was grossly normal. However, ripk1 −/−, casp8 −/− and ripk1 −/−, ripk3 −/− pups failed to reach weaning, with the former dying at birth with the same kinetics as the ripk1 −/− mouse, and the latter succumbing slowly over the first week of life (Fig. 2b) [37, 81, 82]. From these data, it was clear that postnatally RIPK1 inhibits both apoptotic and necroptotic lethality triggered through both caspase-8 and RIPK3, respectively.
Inhibition of necroptosis by RIPK1 was unexpected since RIPK1 appears to be necessary for necroptotic death at the E10.5 and E16.5 checkpoints, as demonstrated by the delay of lethality of casp8 −/− mice to birth when crossed onto the ripk1 −/− background [37, 81, 82]. This observation is consistent with earlier reports that RIPK1 deficiency allowed fadd −/− animals to reach birth [83]. In contrast to ripk1 −/− mice, mice expressing a kinase inactive version of RIPK1 survive to adulthood, suggesting that kinase activity of RIPK1 is dispensable for development, [53, 84]. The kinase activity of RIPK1 is important for necroptosis induced through TNFR1 [85], and therefore casp8 −/− or fadd −/− mice would be predicted to survive until birth when crossed to mice expressing a kinase inactive version of RIPK1, but this cross has yet to be reported.
At the P1 death checkpoint, ripk1 −/− ripk3 −/− animals appear to have increased apoptotic cell death in the intestines [37, 82]. Mice with intestine-specific ablation of caspase-8, FADD, or RIPK1, survive to birth, but they succumb within the first month and display gross abnormalities of the gut epithelia, providing evidence that both necroptosis and apoptosis are important mediators of cell death in the gut [86–89]. Necroptosis may also play a role in maintenance of the skin. Ripk1 −/−, tnfr1 −/− mice displayed skin pathology and died on average by P14 [37]. RIPK3 can be activated in the absence of RIPK1 via TRIF and interferons [37]. To test whether engagement of TRIF and interferon signaling causes lethality in the ripk1 −/−, tnfr1 −/− mice, ripk1 −/−, tnfr1 −/−, trif −/− and ripk1 −/−, tnfr1 −/−, ifnar −/− animals were examined [37]. Despite a increase in median survival to reach weaning, the skin was significantly affected in ripk1 −/−, tnfr1 −/−, trif −/− and ripk1 −/−, tnfr1 −/−, ifnar −/− animals and these animals displayed a loss of hair consistent with the phenotype seen in skin specific deletion of caspase-8, FADD, or RIPK1 [86, 90–92].
It is formally possible that inflammation rather than cell death per se causes the early lethality of ripk1 −/− animals. Ripk1 −/− mice display systematic multi-organ inflammation and intestinal damage immediately at birth, which appears to depend on MyD88 and TNFR signaling, respectively [65, 82]. Ablation of neither MyD88 nor TNFR is capable of extending the lifespan of ripk1 −/− mice past weaning at P21, despite a reduction of inflammation in ripk1 −/−, myd88 −/− mice and intestinal cell death in ripk1 −/−, tnfr1 −/− animals [82]. Furthermore, the original source of MyD88 or TNFR signals driving this pathology is also unknown, and while it is tempting to speculate that colonization by bacteria activates these signaling pathways, raising ripk1 −/− mice in abiotic conditions had no effect on lifespan [82].
The genetic data reviewed here revealed an unexpected role for RIPK1 in suppressing early postnatal lethality caused by Caspase-8/FADD-mediated apoptosis and RIPK3-mediated necroptosis, supporting a model in which RIPK1 may alternatively activate or inhibit necroptosis depending on different upstream activation signals.
Necroptosis in other organisms
The developmental biology of necroptosis has been well described in mice, but whether the pathway is conserved in other organisms besides humans is currently under study [93]. Zebrafish have been used as a tractable alternative genetic model to study development, including hematopoiesis [94]. Zebrafish appear to have an intact death receptor-mediated apoptosis pathway, but knockdown of FADD via morpholino oligomer did not alter development [95]. Zebrafish also have multiple caspase-8 homologues, which has complicated attempts to determine whether an intact necroptosis pathway exists in this species [95].
While the necroptosis pathway has been well studied in human cells, there are differences in the mechanisms of necroptosis in humans versus mice. For instance, human MLKL (hMLKL) is inhibited by the drug NSA, while mMLKL is not [96], suggesting the molecular details of MLKL activation differ between the species. Despite these potential differences, it is possible that defects in regulation of necroptosis affects human development, as one family of presumably necroptosis prone humans with caspase-8 mutations has previously been identified [97]. These patients show a T cell immunodeficiency that mimics what is seen in mice with T cell-specific caspase-8 loss [97]. These patients might survive development because of either maintenance of residual caspase-8 activity or redundancy with other caspases, allowing for the inhibition of RIPK3. Consistent with this hypothesis, humans have a caspase-8 homologue, caspase-10, whose role in necroptosis to date has not been well defined [98].
Future perspectives
In the last several years, genetic investigations have led to important insights into the molecular mechanisms of necroptosis. From the initial observation that casp8−/− mice are rescued by loss of RIPK3, a detailed understanding of the pathway has emerged. It has been become increasingly apparent that all of the major components of this pathway are involved in other important pathways within the cell such as responding to DNA damage and inflammatory cytokine production. Future work will require dissecting these distinct roles. Analysis of the developmental biology and physiology of animals with mutations in the necroptosis pathway and its regulation will continue to contribute to our evolving understanding.
References
Green DR, Llambi F (2015) Cell Death Signaling. Cold Spring Harb Perspect Biol 7(12). doi:10.1101/cshperspect.a006080
Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P (1998) Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 94(6):727–737
Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW (1998) Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 94(6):739–750
Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, Thompson CB (2000) The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell 6(6):1389–1399
Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D (1998) Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9(2):267–276
Yeh WC, de la Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, El-Deiry WS, Lowe SW, Goeddel DV, Mak TW (1998) FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279(5358):1954–1958
Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, Wakeham A, Mirtsos C, Suzuki N, Bonnard M, Goeddel DV, Mak TW (2000) Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 12(6):633–642
Zhang J, Winoto A (1996) A mouse Fas-associated protein with homology to the human Mort1/FADD protein is essential for Fas-induced apoptosis. Mol Cell Biol 16(6):2756–2763
Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P (1998) Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 187(9):1477–1485
Green DR, Oberst A, Dillon CP, Weinlich R, Salvesen GS (2011) RIPK-dependent necrosis and its regulation by caspases: a mystery in five acts. Mol Cell 44(1):9–16. doi:10.1016/j.molcel.2011.09.003
Weinlich R, Dillon CP, Green DR (2011) Ripped to death. Trends Cell Biol 21(11):630–637. doi:10.1016/j.tcb.2011.09.002
Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P (2014) Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15(2):135–147. doi:10.1038/nrm3737
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1(6):489–495. doi:10.1038/82732
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325(5938):332–336. doi:10.1126/science.1172308
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK (2009) Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137(6):1112–1123. doi:10.1016/j.cell.2009.05.037
Brenner D, Blaser H, Mak TW (2015) Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 15(6):362–374. doi:10.1038/nri3834
Justus SJ, Ting AT (2015) Cloaked in ubiquitin, a killer hides in plain sight: the molecular regulation of RIPK1. Immunol Rev 266(1):145–160. doi:10.1111/imr.12304
Linkermann A, Green DR (2014) Necroptosis. N Engl J Med 370(5):455–465. doi:10.1056/NEJMra1310050
Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM (2002) Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem 277(11):9505–9511. doi:10.1074/jbc.M109488200
Weinlich R, Green DR (2014) The two faces of receptor interacting protein kinase-1. Mol Cell 56(4):469–480. doi:10.1016/j.molcel.2014.11.001
Silke J, Rickard JA, Gerlic M (2015) The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol 16(7):689–697. doi:10.1038/ni.3206
Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, Bertin J, Gough PJ, Savvides S, Martinou JC, Bertrand MJ, Vandenabeele P (2014) MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 7(4):971–981. doi:10.1016/j.celrep.2014.04.026
Galluzzi L, Kepp O, Kroemer G (2014) MLKL regulates necrotic plasma membrane permeabilization. Cell Res 24(2):139–140. doi:10.1038/cr.2014.8
Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, Pierotti C, Garnier JM, Dobson RC, Webb AI, Tripaydonis A, Babon JJ, Mulcair MD, Scanlon MJ, Alexander WS, Wilks AF, Czabotar PE, Lessene G, Murphy JM, Silke J (2014) Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci USA 111(42):15072–15077. doi:10.1073/pnas.1408987111
Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RC, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J, Alexander WS (2013) The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39(3):443–453. doi:10.1016/j.immuni.2013.06.018
Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, Oberst A, Quarato G, Low J, Cripps JG, Chen T, Green DR (2015) Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. doi:10.1038/cdd.2015.70
Shin YK, Kim J, Yang Y (2014) Switch for the necroptotic permeation pore. Structure 22(10):1374–1376. doi:10.1016/j.str.2014.09.002
Su L, Quade B, Wang H, Sun L, Wang X, Rizo J (2014) A plug release mechanism for membrane permeation by MLKL. Structure 22(10):1489–1500. doi:10.1016/j.str.2014.07.014
Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, Wakefield R, Frase S, Moldoveanu T, Green DR (2016) Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol Cell. doi:10.1016/j.molcel.2016.01.011
Oberst A, Green DR (2011) It cuts both ways: reconciling the dual roles of caspase 8 in cell death and survival. Nat Rev Mol Cell Biol 12(11):757–763. doi:10.1038/nrm3214
Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR (2011) Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471(7338):363–367. doi:10.1038/nature09852
Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, Mak TW, Wallach D, Green DR (2012) Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep 1(5):401–407. doi:10.1016/j.celrep.2012.03.010
Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES (2011) RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471(7338):368–372. doi:10.1038/nature09857
O’Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, Green DR, Ting AT (2011) Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol 13(12):1437–1442. doi:10.1038/ncb2362
Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, Rall GF, Degterev A, Balachandran S (2013) Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci USA 110(33):E3109–E3118. doi:10.1073/pnas.1301218110
Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES (2013) Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 288(43):31268–31279. doi:10.1074/jbc.M113.462341
Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, Janke LJ, Kelliher MA, Kanneganti TD, Green DR (2014) RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157(5):1189–1202. doi:10.1016/j.cell.2014.04.018
Weinlich R, Oberst A, Dillon CP, Janke LJ, Milasta S, Lukens JR, Rodriguez DA, Gurung P, Savage C, Kanneganti TD, Green DR (2013) Protective roles for caspase-8 and cFLIP in adult homeostasis. Cell Rep 5(2):340–348. doi:10.1016/j.celrep.2013.08.045
Sakamaki K, Inoue T, Asano M, Sudo K, Kazama H, Sakagami J, Sakata S, Ozaki M, Nakamura S, Toyokuni S, Osumi N, Iwakura Y, Yonehara S (2002) Ex vivo whole-embryo culture of caspase-8-deficient embryos normalize their aberrant phenotypes in the developing neural tube and heart. Cell Death Differ 9(11):1196–1206. doi:10.1038/sj.cdd.4401090
Kang TB, Oh GS, Scandella E, Bolinger B, Ludewig B, Kovalenko A, Wallach D (2008) Mutation of a self-processing site in caspase-8 compromises its apoptotic but not its nonapoptotic functions in bacterial artificial chromosome-transgenic mice. J Immunol 181(4):2522–2532
Hsu H, Shu HB, Pan MG, Goeddel DV (1996) TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84(2):299–308
Pobezinskaya YL, Kim YS, Choksi S, Morgan MJ, Li T, Liu C, Liu Z (2008) The function of TRADD in signaling through tumor necrosis factor receptor 1 and TRIF-dependent Toll-like receptors. Nat Immunol 9(9):1047–1054. doi:10.1038/ni.1639
Park HH, Lo YC, Lin SC, Wang L, Yang JK, Wu H (2007) The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol 25:561–586. doi:10.1146/annurev.immunol.25.022106.141656
Pellegrini M, Bath S, Marsden VS, Huang DC, Metcalf D, Harris AW, Strasser A (2005) FADD and caspase-8 are required for cytokine-induced proliferation of hemopoietic progenitor cells. Blood 106(5):1581–1589. doi:10.1182/blood-2005-01-0284
Imtiyaz HZ, Zhou X, Zhang H, Chen D, Hu T, Zhang J (2009) The death domain of FADD is essential for embryogenesis, lymphocyte development, and proliferation. J Biol Chem 284(15):9917–9926. doi:10.1074/jbc.M900249200
Krajewska M, You Z, Rong J, Kress C, Huang X, Yang J, Kyoda T, Leyva R, Banares S, Hu Y, Sze CH, Whalen MJ, Salmena L, Hakem R, Head BP, Reed JC, Krajewski S (2011) Neuronal deletion of caspase 8 protects against brain injury in mouse models of controlled cortical impact and kainic acid-induced excitotoxicity. PLoS One 6(9):e24341. doi:10.1371/journal.pone.0024341
Kang TB, Ben-Moshe T, Varfolomeev EE, Pewzner-Jung Y, Yogev N, Jurewicz A, Waisman A, Brenner O, Haffner R, Gustafsson E, Ramakrishnan P, Lapidot T, Wallach D (2004) Caspase-8 serves both apoptotic and nonapoptotic roles. J Immunol 173(5):2976–2984
Lemmers B, Salmena L, Bidere N, Su H, Matysiak-Zablocki E, Murakami K, Ohashi PS, Jurisicova A, Lenardo M, Hakem R, Hakem A (2007) Essential role for caspase-8 in Toll-like receptors and NFkappaB signaling. J Biol Chem 282(10):7416–7423. doi:10.1074/jbc.M606721200
Beisner DR, Ch’en IL, Kolla RV, Hoffmann A, Hedrick SM (2005) Cutting edge: innate immunity conferred by B cells is regulated by caspase-8. J Immunol 175(6):3469–3473
Salmena L, Hakem R (2005) Caspase-8 deficiency in T cells leads to a lethal lymphoinfiltrative immune disorder. J Exp Med 202(6):727–732. doi:10.1084/jem.20050683
Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D (2013) Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38(1):27–40. doi:10.1016/j.immuni.2012.09.015
Hirschi KK (2012) Hemogenic endothelium during development and beyond. Blood 119(21):4823–4827. doi:10.1182/blood-2011-12-353466
Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, Roose-Girma M, Warming S, Dixit VM (2014) Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343(6177):1357–1360. doi:10.1126/science.1249361
Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, Ouellette M, King BW, Wisnoski D, Lakdawala AS, DeMartino MP, Casillas LN, Haile PA, Sehon CA, Marquis RW, Upton J, Daley-Bauer LP, Roback L, Ramia N, Dovey CM, Carette JE, Chan FK, Bertin J, Gough PJ, Mocarski ES, Kaiser WJ (2014) RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell 56(4):481–495. doi:10.1016/j.molcel.2014.10.021
Mihaly SR, Ninomiya-Tsuji J, Morioka S (2014) TAK1 control of cell death. Cell Death Differ 21(11):1667–1676. doi:10.1038/cdd.2014.123
Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, Vandenabeele P, Bertrand MJ (2013) RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ 20(10):1381–1392. doi:10.1038/cdd.2013.94
Shim JH, Xiao C, Paschal AE, Bailey ST, Rao P, Hayden MS, Lee KY, Bussey C, Steckel M, Tanaka N, Yamada G, Akira S, Matsumoto K, Ghosh S (2005) TAK1, but not TAB 1 or TAB 2, plays an essential role in multiple signaling pathways in vivo. Genes Dev 19(22):2668–2681. doi:10.1101/gad.1360605
Morioka S, Broglie P, Omori E, Ikeda Y, Takaesu G, Matsumoto K, Ninomiya-Tsuji J (2014) TAK1 kinase switches cell fate from apoptosis to necrosis following TNF stimulation. J Cell Biol 204(4):607–623. doi:10.1083/jcb.201305070
Peltzer N, Rieser E, Taraborrelli L, Draber P, Darding M, Pernaute B, Shimizu Y, Sarr A, Draberova H, Montinaro A, Martinez-Barbera JP, Silke J, Rodriguez TA, Walczak H (2014) HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep 9(1):153–165. doi:10.1016/j.celrep.2014.08.066
Xiao Y, Li H, Zhang J, Volk A, Zhang S, Wei W, Zhang S, Breslin P, Zhang J (2011) TNF-alpha/Fas-RIP-1-induced cell death signaling separates murine hematopoietic stem cells/progenitors into 2 distinct populations. Blood 118(23):6057–6067. doi:10.1182/blood-2011-06-359448
Chang H, Huylebroeck D, Verschueren K, Guo Q, Matzuk MM, Zwijsen A (1999) Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development 126(8):1631–1642
Beg AA, Baltimore D (1996) An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science 274(5288):782–784
Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM (1999) Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science 284(5412):321–325
Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J, Elia AJ, Mak TW (2000) Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes Dev 14(7):854–862
Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P (1998) The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 8(3):297–303
Silke J, Vucic D (2014) IAP family of cell death and signaling regulators. Methods Enzymol 545:35–65. doi:10.1016/B978-0-12-801430-1.00002-0
Moulin M, Anderton H, Voss AK, Thomas T, Wong WW, Bankovacki A, Feltham R, Chau D, Cook WD, Silke J, Vaux DL (2012) IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J 31(7):1679–1691. doi:10.1038/emboj.2012.18
de Almagro MC, Goncharov T, Newton K, Vucic D (2015) Cellular IAP proteins and LUBAC differentially regulate necrosome-associated RIP1 ubiquitination. Cell Death Dis 6:e1800. doi:10.1038/cddis.2015.158
Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A (2000) Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 289(5488):2350–2354
Reiley WW, Zhang M, Jin W, Losiewicz M, Donohue KB, Norbury CC, Sun SC (2006) Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat Immunol 7(4):411–417. doi:10.1038/ni1315
Reiley WW, Jin W, Lee AJ, Wright A, Wu X, Tewalt EF, Leonard TO, Norbury CC, Fitzpatrick L, Zhang M, Sun SC (2007) Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med 204(6):1475–1485. doi:10.1084/jem.20062694
Sun SC (2008) Deubiquitylation and regulation of the immune response. Nat Rev Immunol 8(7):501–511. doi:10.1038/nri2337
Dharaneeswaran H, Abid MR, Yuan L, Dupuis D, Beeler D, Spokes KC, Janes L, Sciuto T, Kang PM, Jaminet SC, Dvorak A, Grant MA, Regan ER, Aird WC (2014) FOXO1-mediated activation of Akt plays a critical role in vascular homeostasis. Circ Res 115(2):238–251. doi:10.1161/CIRCRESAHA.115.303227
Furuyama T, Kitayama K, Shimoda Y, Ogawa M, Sone K, Yoshida-Araki K, Hisatsune H, Nishikawa S, Nakayama K, Nakayama K, Ikeda K, Motoyama N, Mori N (2004) Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J Biol Chem 279(33):34741–34749. doi:10.1074/jbc.M314214200
Xue Y, Gao X, Lindsell CE, Norton CR, Chang B, Hicks C, Gendron-Maguire M, Rand EB, Weinmaster G, Gridley T (1999) Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet 8(5):723–730
Wu ZQ, Rowe RG, Lim KC, Lin Y, Willis A, Tang Y, Li XY, Nor JE, Maillard I, Weiss SJ (2014) A Snail1/Notch1 signalling axis controls embryonic vascular development. Nat Commun 5:3998. doi:10.1038/ncomms4998
Oh S, Janknecht R (2012) Histone demethylase JMJD5 is essential for embryonic development. Biochem Biophys Res Commun 420(1):61–65. doi:10.1016/j.bbrc.2012.02.115
Satyanarayana A, Gudmundsson KO, Chen X, Coppola V, Tessarollo L, Keller JR, Hou SX (2010) RapGEF2 is essential for embryonic hematopoiesis but dispensable for adult hematopoiesis. Blood 116(16):2921–2931. doi:10.1182/blood-2010-01-262964
Grisendi S, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K, Pandolfi PP (2005) Role of nucleophosmin in embryonic development and tumorigenesis. Nature 437(7055):147–153. doi:10.1038/nature03915
Zhang M, Harashima N, Moritani T, Huang W, Harada M (2015) The roles of ROS and caspases in TRAIL-induced apoptosis and necroptosis in human pancreatic cancer cells. PLoS One 10(5):e0127386. doi:10.1371/journal.pone.0127386
Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, Huang C, Sundararajan A, Guo H, Roback L, Speck SH, Bertin J, Gough PJ, Balachandran S, Mocarski ES (2014) RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci USA 111(21):7753–7758. doi:10.1073/pnas.1401857111
Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, Vince JE, Lawlor KE, Ninnis RL, Anderton H, Hall C, Spall SK, Phesse TJ, Abud HE, Cengia LH, Corbin J, Mifsud S, Di Rago L, Metcalf D, Ernst M, Dewson G, Roberts AW, Alexander WS, Murphy JM, Ekert PG, Masters SL, Vaux DL, Croker BA, Gerlic M, Silke J (2014) RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157(5):1175–1188. doi:10.1016/j.cell.2014.04.019
Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J (2011) Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 471(7338):373–376. doi:10.1038/nature09878
Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Cook M, Finger J, Hughes-Earle A, Harris PA, Kaiser WJ, Mocarski ES, Bertin J, Gough PJ (2014) Cutting Edge: rIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol 192(12):5476–5480. doi:10.4049/jimmunol.1400499
Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, Lenardo MJ (2003) A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem 278(51):51613–51621. doi:10.1074/jbc.M305633200
Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, Eftychi C, Lin J, Corona T, Hermance N, Zelic M, Kirsch P, Basic M, Bleich A, Kelliher M, Pasparakis M (2014) RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513(7516):90–94. doi:10.1038/nature13608
Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, Becker C (2011) Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 477(7364):335–339. doi:10.1038/nature10400
Takahashi N, Vereecke L, Bertrand MJ, Duprez L, Berger SB, Divert T, Goncalves A, Sze M, Gilbert B, Kourula S, Goossens V, Lefebvre S, Gunther C, Becker C, Bertin J, Gough PJ, Declercq W, van Loo G, Vandenabeele P (2014) RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature 513(7516):95–99. doi:10.1038/nature13706
Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, Kirsch P, Sterner-Kock A, van Loo G, Pasparakis M (2011) FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477(7364):330–334. doi:10.1038/nature10273
Lee P, Lee DJ, Chan C, Chen SW, Ch’en I, Jamora C (2009) Dynamic expression of epidermal caspase 8 simulates a wound healing response. Nature 458(7237):519–523. doi:10.1038/nature07687
Kovalenko A, Kim JC, Kang TB, Rajput A, Bogdanov K, Dittrich-Breiholz O, Kracht M, Brenner O, Wallach D (2009) Caspase-8 deficiency in epidermal keratinocytes triggers an inflammatory skin disease. J Exp Med 206(10):2161–2177. doi:10.1084/jem.20090616
Bonnet MC, Preukschat D, Welz PS, van Loo G, Ermolaeva MA, Bloch W, Haase I, Pasparakis M (2011) The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity 35(4):572–582. doi:10.1016/j.immuni.2011.08.014
Tanzer MC, Matti I, Hildebrand JM, Young SN, Wardak A, Tripaydonis A, Petrie EJ, Mildenhall AL, Vaux DL, Vince JE, Czabotar PE, Silke J, Murphy JM (2016) Evolutionary divergence of the necroptosis effector MLKL. Cell Death Differ. doi:10.1038/cdd.2015.169
Clements WK, Traver D (2013) Signalling pathways that control vertebrate haematopoietic stem cell specification. Nat Rev Immunol 13(5):336–348. doi:10.1038/nri3443
Eimon PM, Kratz E, Varfolomeev E, Hymowitz SG, Stern H, Zha J, Ashkenazi A (2006) Delineation of the cell-extrinsic apoptosis pathway in the zebrafish. Cell Death Differ 13(10):1619–1630. doi:10.1038/sj.cdd.4402015
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148(1–2):213–227. doi:10.1016/j.cell.2011.11.031
Su H, Bidere N, Zheng L, Cubre A, Sakai K, Dale J, Salmena L, Hakem R, Straus S, Lenardo M (2005) Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science 307(5714):1465–1468. doi:10.1126/science.1104765
Wang J, Chun HJ, Wong W, Spencer DM, Lenardo MJ (2001) Caspase-10 is an initiator caspase in death receptor signaling. Proc Natl Acad Sci USA 98(24):13884–13888. doi:10.1073/pnas.241358198
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Dillon, C.P., Tummers, B., Baran, K. et al. Developmental checkpoints guarded by regulated necrosis. Cell. Mol. Life Sci. 73, 2125–2136 (2016). https://doi.org/10.1007/s00018-016-2188-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2188-z