
Abstract
The link between inflammation and cancer is well established. Chronic inflammation promotes cancer initiation and progression. Various studies showed that the underlying mechanisms involve epigenetic alterations. These epigenetic alterations might culminate into an epigenetic switch that transforms premalignant cells into tumor cells or non-invasive into invasive tumor cells, thereby promoting metastasis. Epigenetic switches require an initiating event, which can be inflammation, whereas the resulting phenotype is inherited without the initiating signal. Epigenetic switches are induced and maintained by DNA methylation, histone modifications, polycomb group (PcG)/trithorax group (TrxG) proteins, and feedback loops consisting of transcription factors and microRNAs. Since epigenetic switches are reversible, they might represent an important basis for the design of novel anticancer therapeutics. This review summarizes published evidence of epigenetic switches in cancer development that are induced by inflammation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The link between inflammation and cancer, first perceived in the 19th century by Rudolf Virchow, who observed that tumors are frequently infiltrated with immune cells, is now accepted to play an important role in the initiation and progression of tumors [1–3]. During the last decades, population-based studies have shown that individuals who suffer from chronic inflammatory disorders (caused by chronic infection, dietary factors, obesity, inhaled pollutants, tobacco use, or autoimmunity), have an increased risk for cancer [4]. Among these cancer susceptibility factors, infections represent a major driver of inflammation-induced tumorigenesis and ~20 % of cancer deaths are associated with chronic infection and inflammation [5]. Examples include Helicobacter pylori-induced gastric cancer, human papilloma, hepatitis B or C virus-induced cervical and hepatocellular carcinomas (HCC), and Schistosoma or Bacteroides infections that are associated with bladder and colon cancer, respectively [6, 7]. Chronic inflammation is an aberrantly prolonged form of a beneficial response to tissue injury and pathogenic agents. However, during cancer development, inflammation can be deregulated to promote malignant cell transformation [8]. In other words, inflammation can increase the risk of cancer or promote tumor progression by providing effector molecules produced by infiltrating immune cells in the tumor microenvironment (TME). The TME consists of various, characteristic cellular and physical components. The TME includes cells of the innate immune system, such as macrophages, neutrophils, mast cells, myeloid-derived suppressor cells, dendritic cells, and natural killer cells, as well as adaptive immune cells, such as T and B lymphocytes. Other stromal cells of the TME include fibroblasts, myofibroblasts, adipocytes, neuroendocrine cells, and endothelial cells [9, 10]. In addition, distinct physical features, such as hypoxia and an altered extracellular matrix critically contribute to the TME. Tumor cells interact with the TME via direct cell contact or via secreted cytokines, growth factors, and chemokines that promote proliferation, cell survival and inhibit apoptosis, as well as pro-angiogenic factors and extracellular matrix-modifying enzymes, such as matrix metalloproteinases (MMPs) that promote epithelial–mesenchymal transition (EMT), invasion, and metastasis [11].
Inflammation can exert its pro-tumorigenic properties via induction of epigenetic alterations in tumor cells. Epigenetics defines heritable alterations in gene expression that are not caused by changes in the DNA sequence [12]. For example, DNA methylation, histone modifications, alterations in PcG/TrxG protein expression, and regulatory loops consisting of transcription factors, microRNAs (miRNAs), and long non-coding RNAs can be heritable and are therefore considered epigenetic. However, each of these modifications/factors is not always self-perpetuating or does not change gene expression alone. Therefore, in most cases, the epigenetic inheritance cannot be explained by a single alteration, but by the interplay of different epigenetic mechanisms. Epigenetic switches may be considered as conversions of cellular phenotypes and gene expression patterns from one stable epigenetic state to another without changes in DNA sequence. They require an initiating event (i.e., inflammation), but the resulting cellular state is inherited in the absence of the initiating signal [13]. The new phenotype is often stabilized by feedback loops involving transcription factors and miRNAs, and/or by other epigenetic alterations. Epigenetic switches have been initially described in bacteriophage λ [14], where the switch between lytic and lysogenic state of the virus is regulated by just two DNA-binding proteins. Each state is extremely stable and inherited over many generations, but in response to environmental signals, such as UV light, the switch is induced, which results in a stable change of the state. Later, epigenetic switches were identified in other prokaryotic and eukaryotic organisms, where they are usually more complex, involving various epigenetic modifications and factors, such as DNA methylation, histone modifications, and non-coding RNAs [15–17]. Recent literature suggests that epigenetic switches also play an important role in tumorigenesis, since they can participate in tumor initiation [18, 19] or cancer progression [20, 21]. In this review, we describe the current knowledge about the link between inflammation and epigenetic switches in carcinogenesis.
Inflammation-induced epigenetic reprogramming in cancer
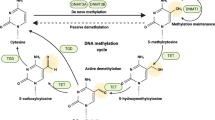
Cancer initiation and progression is often associated with global reorganization of epigenetic modifications, such as DNA methylation or histone modifications [22]. DNA methylation takes place at cytosines that are located within a CpG dinucleotide and is associated with silencing of gene expression (reviewed in [23, 24]). DNA methylation silences transcription by recruitment of methyl-CpG-binding domain proteins, which are capable of recruiting histone-modifying enzymes to facilitate repressive histone modifications [25–27], or to lesser extent by blocking the access of transcription factors that would otherwise activate expression [28, 29]. In mammals, DNA methylation is regulated by three DNA methyltransferases (DNMT1, DNMT3a, and DNMT3b) [23]. Inflammation plays an important role in the modulation of DNA methylation patterns during cancer initiation and progression (Table 1). For example, during Helicobacter pylori-induced gastric carcinogenesis, the pro-inflammatory cytokine interleukin-1 beta (IL-1β) enhances DNMT activity via nitric oxide production, which results in CpG methylation-mediated gene silencing [30], which also leads to down-regulation of the CDH1 gene encoding E-cadherin [31, 32]. Furthermore, transforming growth factor beta (TGF-β) either induces expression and activity of DNMT1, DNMT3A, and DNMT3B resulting in global changes in DNA methylation during EMT in ovarian cancer [33]. Alternatively, TGF-β treatment inhibits the expression of DNMTs in HCC cells and cardiac fibroblasts, resulting in reduced DNA methylation and increased expression of important cancer-related genes, such as CD 133 [34, 35]. Finally, exposure to the pro-inflammatory cytokine interleukin 6 (IL-6) transcriptionally induces and also stabilizes the DNMT1 protein resulting in enhanced methylation, including that of tumor suppressor genes, such as MASPIN [36–38]. Furthermore, acetylation of the IL-6 effector signal transducer and activator of transcription 3 (STAT3) at Lys685 promotes the interaction of STAT3 with DNMT1, resulting in enhanced methylation of tumor suppressor genes [39]. Thus, the pro-tumorigenic functions of cytokine signaling are partially mediated by epigenetic mechanisms. This could be used for therapeutic interventions as has been shown in triple-negative BC where a reduction of acetylated STAT3 with the acetylation inhibitor resveratrol led to demethylation and activation of estrogen receptor α, resulting in sensitization to anti-estrogen therapy [39]. In a mouse model of colitis-associated cancer (CAC), which is based on dextran sulfate sodium (DSS)-induced inflammation, aberrant DNA methylation of FOSB, HOXA5, and KRT7 genes occurred in colon epithelial cells before the development of colon tumors [40]. Exposure of cultured colonic epithelial cells to DSS did not induce DNA methylation suggesting that factors secreted from inflammatory cells were responsible for enhanced methylation. The induction of methylation was also observed in immunodeficient mice that lacked T and B cells, suggesting the infiltrating macrophages might be responsible for the changes in methylation. Finally, IL-1β and tumor necrosis factor alpha (TNF-α) treatment of chondrocytes resulted in global decrease of 5-hydroxy cytosine methylation, showing that inflammation also modulates 5-hydroxymethylation [41], which is another type of DNA methylation recently described to represent an important factor in cancer initiation and progression [42].
Other important epigenetic regulators of gene expression are histone modifications. Histones are proteins that associate with DNA and condense it into compact structural units called nucleosomes, which are the building blocks of chromatin. To initiate gene expression, the chromatin has to “open” by a process called chromatin remodeling to allow the binding of transcription factors and RNA polymerase II. Chromatin remodeling is regulated by histone modifications, which includes acetylation, methylation, phosphorylation, and ubiquitination [43]. Histone modifications that are associated with active transcription, also called euchromatin modifications, include histone H3 and H4 acetylation as well as H3K4 and H3K79 trimethylation (H3K4me3, H3K79me3). On the contrary, modifications associated with inactive genes, termed heterochromatin modifications, include the trimethylation of H3K9 and H3K27 (H3K9me3, H3K27me3) [43]. Factors that establish and maintain histone modifications include the PcG and TrxG proteins, which maintain chromatin in the “off” or “on” state, respectively, and thereby mediate repression or activation of gene expression [44, 45]. The PcG and TrxG proteins act antagonistically through DNA elements called PcG/TrxG response elements (PRE/TREs). These are regulatory DNA elements that can preserve the memory of activated or silenced states of their associated genes during several cell generations [45]. PcG and TrxG proteins affect gene expression by integrative regulation of histone modifications, nucleosome composition, and DNA methylation resulting in chromatin structure remodeling [46–48]. For example, the PcG protein Enhancer of zeste homolog 2 (EZH2) is a methyltransferase that converts H3K27 to H3K27me3, which is a hallmark of repressed genes [49]. In contrast, TrxG proteins establish H3K4me3 or H3K79me3 marks, which are characteristic for actively transcribed genes [45]. The modulation of histone modifications has also been associated with inflammation (Table 1). For example, nuclear factor-κB (NF-κB) signaling, which is often induced by inflammation, is mediated by activation of IKKα and IKKβ. IKKβ activates NF-κB by degradation of IκB; however, IKKα lacks this function. Yet, it has been shown that after stimulation with cytokines, IKKα activates NF-κB by translocating to the nucleus and binding to NF-κB-responsive promoters, where it regulates the expression of NF-kB target genes by inducing phosphorylation of specific histone residues [50, 51]. Furthermore, during TGF-β-induced EMT, a genome-wide epigenetic reprogramming was observed [52]. This effect was manifested in a global reduction of the repressive H3K9me2 marks and an increase of the transcriptional activation marks H3K4me3 and H3K36me3. On the other hand, a genome-wide epigenetic reprogramming that facilitates the formation of repressive H3K27me3 modifications is mediated by the long non-coding RNA HOTAIR, which serves as a scaffold for coordination of H3K27-modifying PcG complex [53]. Overexpression of HOTAIR in breast cancer cells resulted in re-targeting of PcG and establishment of new H3K27me3 histone marks in hundreds of genes, including various metastasis suppressors, thereby increasing invasiveness and metastatic potential. Therefore, it comes as no surprise that the expression of HOTAIR is hundreds to nearly two-thousand times elevated in breast cancer metastases when compared to normal breast epithelium [53]. Recently, it has been shown that the expression of HOTAIR is induced by IL-6/STAT3 signaling during malignant transformation of human bronchial epithelial cells caused by cigarette smoke extract [54], suggesting that HOTAIR is another mediator between inflammation and epigenetic reprogramming. Altogether, inflammation can induce various epigenetic alterations that range from modulation of specific genes to genome-wide epigenetic reprogramming, which may culminate in epigenetic switches that promote cancer development.
Inflammation-induced epigenetic switches in tumor initiation
Epigenetic switches can be activated and maintained by feedback loops consisting of transcription factors and miRNAs. This was initially shown by Iliopoulos et al. [18], who used a cell culture model of breast cancer (BC) initiation in which oncogenic transformation of immortalized mammary non-transformed cells was achieved by a transient induction of the SRC oncogene. They showed that induction of SRC for only 5 min is sufficient to drive cells into a transformed state. This short transient signal triggered a switch, which activated a positive feedback loop consisting of NF-κB, IL-6, lin28, and the let-7 miRNA (Fig. 1a). Importantly, even after cessation of ectopic SRC expression, this loop remained active for many generations. Moreover, the cells remained transformed after deactivation of SRC, indicating that the NF-κB/IL-6/lin28/let-7 loop maintains the transformed cell phenotype. Accordingly, repression of any of the loop components resulted in deactivation of the loop and suppression of oncogenic properties. Subsequent studies have extended these findings by identifying the miRNAs miR-21 and miR-181b-1 as direct targets of STAT3, which is a downstream effector of IL-6 [55]. These miRNAs, together with their targets PTEN and CYLD represent active components of the previously identified NF-κB/IL-6/lin28/let-7 loop since they regulate NF-κB activity (Fig. 1a). The inflammatory loops identified by Iliopoulos et al. [18, 55] were applied to a computational model to account for the dynamics of the epigenetic switch [56]. The model suggests that random fluctuations (due to molecular noise or cell-to-cell variability) are able to trigger cell transformation. However, activators (oncogenes) or inhibitors (tumor suppressors) of the feedback loop increase or decrease the robustness of the non-transformed state of the cell toward random fluctuations by modulating the threshold of inflammatory signals needed to promote cell transformation.

Epigenetic switches in cancer initiation. Transient activation of SRC oncogene (a) or co-culture with monocytes (b) triggers the constitutive activation of feedback loops, which induce and maintain transformation of mammary epithelial cells. Modified from Iliopoulos et al. [18] and Rokavec et al. [19]. c Transient exposure to paracrine IL-6 induces constitutive autocrine IL-6 production by cancer cells. d Transient suppression of HNF4A results in permanent activation of an inflammatory feedback loop that induces oncogenic transformation of hepatocytes. Modified from Hatziapostolu et al. [94]. Components indicated in red or green are overexpressed/activated or repressed when feedback loops are active, respectively. Dotted arrows indicate transient stimuli, whereas full arrows represent constitutive activation/repression. See text for details
The components of the loop (IL-6, STAT3, and NF-κB) are also involved in inflammation, indicating that inflammatory signaling is involved in this switch. Importantly, IL-6 was not only a component of the loop, but treatment of immortal mammary epithelial cells with ectopic IL-6 was also sufficient to induce oncogenic transformation, suggesting that the switch can be induced by extrinsic inflammatory signals, which subsequently induce the production of the same inflammatory molecules in the cancer cells. IL-6 is a pro-inflammatory cytokine that is secreted by various components of the TME, but also by cancer cells. IL-6 was found to stimulate the proliferation of premalignant enterocytes in APC min mice as well as in models of CAC and colon cancer [57]. Furthermore, IL-6 produced by myeloid cells functions as a tumor promoter during intestinal carcinogenesis by protecting normal and premalignant intestinal epithelial cells from apoptosis and promoting the proliferation of tumor initiating cells [58, 59]. IL-6 plays an important role in tumor growth by activating its downstream effectors, such as NF-κB and STAT3 [1]. The transcription factors NF-κB and STAT3 promote tumor growth by inhibition of apoptosis [60, 61] or by promoting cell proliferation via up-regulation of the cell cycle regulators cyclin D1, cyclin D2 and cyclin B, as well as the proto-oncogene MYC [8]. Conditional deletion of STAT3 in intestinal epithelial cells prevented the formation of adenomas by controlling the expression of cyclins D1, D2 and B, whereas STAT3 hyperactivation in gp130Y757F mice promoted intestinal tumor growth [59]. Moreover, NF-κB and STAT3 enhance the production of pro-inflammatory cytokines, such as IL-6, IL-11, and TNF-α by myeloid cells, which in turn activate proliferation and survival pathways of epithelial cells via NF-κB and STAT3 activation. Accordingly, blockage of NF-κB activity in myeloid cells reduced the number and size of tumors in a mouse model of CAC/colitis-associated cancer [60].
Another important component of the switches described above are miRNAs, which are short non-protein coding RNAs (20-25 nucleotides) that inhibit the expression of proteins by binding to 3′-untranslated regions (3′-UTR) of their respective mRNAs [62]. They play important roles in cancer development by functioning as tumor suppressors or oncogenes [63, 64]. Growing evidence suggests that miRNAs modulate stress responses (e.g., inflammation) [65]. On one hand, miRNAs mediate the restoration of gene networks after the cessation of the triggering stimuli, such as inflammation, and thus help to confer robustness to biological processes and minimize noise in gene expression [66, 67]. On the other hand, miRNAs can contribute to cell reprogramming by forming feedback loops, such as the NF-κB/IL-6/lin28/let-7 circuitry described above, which confers a stable cell state when the loop is on (transformed) and another state when the loop is off (untransformed). A transient trigger that modulates miRNA expression can therefore induce a stable change in cell phenotype and result in a persistent self-perpetuating response even after the triggering stimulus is removed and thus contribute to epigenetic inheritance [65, 67]. Various miRNAs are regulated by inflammation (Table 1). Moreover, microRNAs encapsuled in exosomes can be transmitted between tumor cells and cells of the TME to modulate tumor properties (reviewed in [68]). Certain miRNAs directly target regulators of epigenetic states, such as DNMT3a, DNMT3b and DNMT1 (miR-29a, miR-29b, miR-29c) [69], HDACs (miR-449a) [70], EZH2 (miR-101) [71], and Bmi-1 (miR-200c) [72], whereas certain miRNA encoding genes are themselves regulated by methylation [73, 74].
Recently, we described another epigenetic switch that converts immortalized human mammary epithelial cells to BC cells [19]. The trigger of the switch was a short co-culture of epithelial cells with monocytes, which are typically found at sites of inflammation. The switch was initiated by monocyte chemotactic protein 1 (MCP-1) secreted by monocytes, which induced the expression of IL-6 in epithelial cells and consequent STAT3-mediated repression of miR-200c. The suppression of miR-200c resulted in the up-regulation of c-Jun N-terminal kinase 2 (JNK2) and NF-κB, which in turn led to constitutive overexpression of IL-6 and the establishment of a feedback loop (Fig. 1b). Interestingly, both, the transient MCP-1-mediated induction of IL-6 that triggers the switch, and the constitutive up-regulation of IL-6 in the loop was mediated by NF-kB, but the transient NF-kB activation was dependent on IKK, whereas the constitutive NF-kB activity was regulated by the PTPRZ1 phosphatase. Besides the direct induction of IL-6 by NF-kB and JNK2, the later also phosphorylated and activated the Heat Shock Factor 1 (HSF1). Binding of HSF1 to IL6 promoter opens its chromatin structure and facilitates the binding of transcription factors, which enhances the expression of IL6 [75]. In our model, HSF1 triggered demethylation of the IL6 promoter, facilitating the binding of the NF-kB and JNK2 effectors p65 and c-Jun, which together drove constitutive expression of IL6. Perhaps, this change in methylation reinforced the loop and made it more resistant to external stimuli. Importantly, the permanent activation of the loop and the stable transformed state was only observed in few mammary epithelial cells, whereas the majority of cells remained untransformed, suggesting that the epigenetic switch occurred only in small number of cells. The IL-6/miR-200c circuit was also manifest in a MMTV-neu mouse model of BC, where deletion of the IL6 gene disabled the loop and impaired mammary tumorigenesis. MMTV-neu mice spontaneously develop mammary carcinomas due to the constitutive expression of the ERBB2 (HER2/neu) receptor tyrosine kinase. This mouse model mimics human BC with HER2 amplification, which accounts for approximately 25 % of all BC cases [76]. The importance of IL-6 in HER2-associated BC has also been described by others. HER2 activation induces IL-6 expression and this is a critical determinant of HER2-mediated oncogenesis, since IL-6 and its effector STAT3 were required for HER2-mediated tumor growth in both, cell line and xenograft-based studies [77]. BC patients with HER2 amplification can be efficiently treated with small molecules or antibodies, such as trastuzumab, that block the HER2 receptor [78]. However, as with every anticancer treatment, resistance represents a problem. Korkaya et al. showed that the activation of an IL-6/STAT3/PTEN/NF-κB inflammatory loop mediates trastuzumab resistance in HER2 positive BC by expanding the cancer stem cell (CSC) population [79]. Accordingly, in xenograft mouse models, inhibition of IL-6 signaling by an IL-6 receptor (IL-6R) blocking antibody overcame acquired and de novo trastuzumab resistance by reducing the CSC population. These results are consistent with observations made by Iliopoulos et al. [80], who showed that IL-6 is sufficient to convert non-CSCs to CSCs in an inducible model of breast carcinogenesis.
The significance of IL-6/STAT3-mediated repression of miR-200c in BC development demonstrated by Rokavec et al. [19] was also observed in other cancer entities. For example, cigarette smoke-induced IL-6 expression leads to suppression of miR-200c and results in transformation and EMT of human bronchial epithelial cells [81]. Furthermore, activation of STAT3 by oncostatin M leads to suppression of miR-200c and let-7 miRNAs, which culminates in the induction of EMT in BC cells [82]. In addition, IL-6 promotes stemness and aggressiveness of glioblastoma tumors by suppressing miR-142-3p [21]. The mechanism of miR-142-3p suppression involves an IL-6 induced hypermethylation of a Sp1-binding site in the miR-142-3p promoter, which decreases the binding of Sp1 and expression of miR-142-3p. In parallel, miR-142-3p suppresses IL-6 expression by targeting the IL6 3′-UTR, thereby forming an IL-6/miR-142-3p feedback loop. Notably, glioblastoma patients with miR-142-3p promoter methylation showed poor survival, whereas orthotopic delivery of miR-142-3p suppressed tumor development in glioblastoma xenotransplanted mice. In contrast, Xiang et al. described a self-inhibitory IL-6/STAT3-induced feedback loop that inhibits IL-6/STAT3 driven carcinogenesis [83]. They showed that STAT3 directly induces the expression of the tumor suppressing miRNA miR-146b. Consequently, miR-146b inhibited NF-κB-dependent expression of IL-6 and subsequent STAT3 activation. However, methylation of miR-146b, which is common in human cancers, prevented the STAT3 induction of miR-146b and therefore STAT3 could exert its oncogenic activities in cancer cells.
Inflammatory loops can also be activated by oncogenes or tumor suppressors that are commonly and aberrantly expressed in tumors. For example, in immortalized mammary epithelial MCF-10a cells, knockdown of p53 and PTEN synergized in the production of IL-6 and activation of IL-6/STAT3/NF-κB signaling, which induced EMT and resulted in metastatic CSCs [84]. The tumorigenicity of these cells was suppressed by blockade of IL-6 or ectopic expression of the STAT3 suppressor SOCS3. Furthermore, in the presence of mutated oncogenic Ras, inflammatory stimuli including cholecystokinin and lipopolysaccharide initiated a positive feedback loop involving NF-kB that further amplified Ras activity to pathological levels [85]. This loop facilitated the expression of inflammatory mediators, such as COX-2, which prolonged Ras signaling and resulted in chronic inflammation and generation of precancerous pancreatic lesions in mice expressing oncogenic K-Ras.
Several studies showed that transient exposure to paracrine IL-6 results in the constitutive expression of IL-6 and establishment of autocrine loops in cancer cells (Fig. 1c). Since IL-6 is frequently produced by tumor stroma, including immune cells [86], tumor infiltration with IL-6-producing immune cells could represent a trigger for constitutive IL-6 expression in cancer cells that facilitates tumorigenesis. IL-6 autocrine loops consist of the NF-κB [18, 19, 87], JNK [19], and ERK [87] signaling pathways, in combination with several miRNAs [18, 19]. Moreover, they are enforced by activation of receptor tyrosine kinases, such as HER2 [77] and IGF-IR [88], that further activate signaling pathways, which may contribute to stabilization of the constitutive expression of IL-6. Methylation of the IL6 promoter also plays an important role in the regulation of IL-6 expression [89]. Since, this methylation is regulated by transcription factors such as p53 [90] and HSF1 [19], targeted demethylation of the IL-6 promoter might be crucial for the establishment of IL-6 autocrine loops. In BC, the estrogen receptor (ER)-negative cells generally express higher levels of IL-6 when compared to ER-positive BC cells [91]. ER-negative BC cells are generally also more mesenchymal, tumorigenic, and invasive and these characteristics depend on autocrine IL-6 and IL-8 expression [77]. Interestingly, prolonged, stem cell enriching mammosphere culture of ER-positive MCF7 cells induced EMT and tumorigenicity by constitutive repression of ERα and induction of IL-6, further indicating that the interplay of IL-6 and estrogen signaling plays an important role in BC aggressiveness [92]. Similar findings were observed in a mouse model of diethylnitrosamine-induced HCC. In this model, the authors observed a gender-biased expression of IL-6, which was higher in male mice. Accordingly, male mice showed increased susceptibility to HCC, while female mice were partially protected from cancer due to inhibition of IL-6 production by estrogen steroid hormones [93].
Hatziapostolou et al. [94] identified an epigenetic switch that regulates hepatocellular oncogenesis. They showed that non-transformed, immortalized hepatocytes undergo oncogenic transformation upon a transient inhibition of hepatocyte nuclear factor 4 alpha (HNF4α). This temporary trigger started the permanent activation of an inflammatory feedback loop consisting of HNF4α, miR-124, IL-6R, STAT3, miR-24, and miR-629 (Fig. 1d). Once this circuit was activated, it maintained the suppression of HNF4α and sustained oncogenesis over multiple generations. Moreover, ectopic expression of any positive factor (IL-6R, STAT3, miR-24, and miR-629) or inhibition of any negative factor (HNF4α and miR-124) also transformed immortalized hepatocytes, indicating that the loop can be induced at any step. Systemic administration of miR-124 and consequent inhibition of the loop prevented and suppressed hepatocellular carcinogenesis in mouse models by inducing tumor-specific apoptosis without toxic side effects. Not only did miR-124 delivery reduce the number and size of induced tumors by 90 % when given throughout the entire course of tumor formation, but it also led to an 80 % decrease in tumor size when treatment was started after the tumors were established. These high efficacies of miRNA-based therapies point to potential therapeutic applications.
In studies by Iliopoulos et al. [18, 55], Rokavec et al. [19] and Hatziapostolou et al. [94], the activation of the inflammatory circuitries was sufficient to induce oncogenic transformation in immortalized, non-transformed cells. However, it was not sufficient to convert primary non-transformed cells, suggesting that preexisting, precancerous genetic or epigenetic alterations are required. During cancer development, it is likely that cells with precancerous mutations develop frequently. Probably, such cells, which represent an intermediate stage, are susceptible for oncogenic transformation induced by epigenetic switches. This hypothesis also supports the multiple-hit theory in which the accumulation of sequential genetic or epigenetic alterations in key growth regulatory genes results in tumor development [95, 96] with inflammation being one of the hits. In the studies by Iliopoulos et al. [18] and Rokavec et al. [19], IL-6 up-regulation was one of the main factors driving tumorigenesis. However, beside tumor promotion, also cancer-suppressing properties have been ascribed to IL-6. For example, oncogene-induced IL-6 expression in primary human fibroblasts resulted in senescence [97]. This induction of senescence was mediated by the up-regulation of the cyclin-dependent kinase inhibitor p15INK4B. The immortalized cell lines used by Iliopoulos et al. [18] and Rokavec et al. [19] harbor a deletion on chromosome 9p21 that includes part of the INK4/ARF locus, which contains the genes that encode the p15INK4B and p16INK4A proteins [98]. Therefore, the pro- or anti-tumorigenic effects of IL-6 on a given cell may depend on the status of its INK4/ARF locus [99].
Inflammation-induced epigenetic switches in tumor progression
There is extensive evidence that inflammation promotes cancer metastasis (reviewed in [100]), which is the most critical aspect of tumor progression since it is responsible for 90 % of cancer mortality [101]. Inflammation can promote different steps of the metastatic process. Metastasizing tumors harbor large numbers of inflammatory cells, especially at their invasive front [102]. These cells secrete pro-inflammatory factors, such as IL-6, TNFα, and IL-1β, that induce, activate, and/or stabilize the EMT transcription factors, Snail, Twist, ZEB1 and ZEB2, which results in EMT and enhanced tumor invasion [103–106]. Furthermore, TNF-α promotes intravasation of cancer cells into blood vessels and the lymphatic system by inducing vascular permeability, cyclooxygenase 2 (COX2)-dependent prostaglandin production, and MMP-mediated tissue remodeling [8, 107]. Moreover, inflammatory mediators enhance the survival of metastatic cells in the circulation and promote the extravasation and colonization of metastatic cells by up-regulating adhesion molecules [1, 8]. As shown in an animal model of BC lung metastasis, a distinct type of monocyte-derived macrophages directly interacts with the extravasating cancer cells, and loss of these macrophages dramatically reduces the number of cancer cells that extravasate from the blood vessels [108]. The number of tumor-associated macrophages (TAMs) correlates with poor prognosis [109] and therapeutic inhibition of macrophage infiltration and/or activity showed promising results in preclinical models and is now being evaluated in clinical studies [110]. For example, inhibition of macrophage infiltration with an antibody against the receptor for colony-stimulating factor 1 (CSF-1), which is a major factor for attracting macrophages, led to a marked reduction of infiltrated TAMs and a significant clinical benefit for patients with diffuse-type giant cell tumors [111]. The recruitment of monocytes is also mediated by the chemokine (C–C motif) ligand 2 (CCL2)—C–C chemokine receptor type 2 (CCR2) axis and blockade of CCL2 suppresses the recruitment of monocytes to the site of metastases and leads to suppression of extravasation and survival of tumor cells [112]. The expression of CCL2 is suppressed by the miR-126/miR-126* pair and hence these miRNAs suppress the recruitment of monocytes and inhibit lung metastasis of BC cells [113]. However, in metastatic BC cells, miR-126 is repressed by DNA methylation, thereby allowing CCL2-mediated monocyte recruitment and promotion of lung metastasis. In addition, exposure of CCR2-positive endothelium to tumor-derived CCL2 leads to an increase in vascular permeability, which promotes extravasation and seeding of cancer cells [114]. Endothelial cells in the TME represent a prime clinical target since they constitute the tumor vasculature that controls passage of nutrients into tissues and maintains blood flow as well as the trafficking of leukocytes [115]. Hypoxia and chronic growth stimulation lead to the dysfunction of endothelial cells which results in structural abnormalities of the vasculature, such as irregular diameters, fragility, and leakiness, thereby enhancing the intravasation and extravasation of cancer cells and contributing to metastasis [114, 116]. Furthermore, activated endothelial cells recruit inflammatory cells via elevated expression of chemokines [117] and cell adhesion molecules, such as E-selectin or vascular cell adhesion molecule 1 (VCAM-1) [118]. Thereby, endothelial cells contribute to tumorigenic inflammation and neo-angiogenesis. Accordingly, experimental activation of the endothelium by administration of cytokines prior to the inoculation of tumor cells resulted in enhanced metastasis [119], while inhibition of endothelial cells attenuated metastasis in several mouse models [120–122].
Metastatic colonization is also supported by cancer-associated fibroblasts (CAF). For example, CAFs were shown to epigenetically repress the expression of miR-200b in gastric cancer cells by increasing the methylation of the miR-200b promoter, resulting in EMT and enhanced migration and invasion [123], Furthermore, in triple-negative breast tumors, CAFs promote the expansion of distinct cancer cell clones by secretion of C-X-C motif chemokine 12 (CXCL12) and insulin-like growth factor 1 [124]. The carcinoma clones selected in this manner are primed for metastasis in the CXCL12-rich microenvironment of the bone marrow, suggesting that signals produced by the TME prime cancer cells for metastasis to specific organs. Complementary, BC cells can convert normal fibroblasts to CAFs by up-regulation of the ADAMTS1 metalloproteinase. This process is mediated by suppression of EZH2 binding to the ADAMTS1 promoter and subsequent decrease of H3K27me3 modification [125]. Finally, MMPs are important molecules that assist tumor cells during all steps of metastasis while inflammatory cells are the major source of these proteases (reviewed in [126]). Expression of MMPs is low in most tissues, but in response to inflammatory cytokines, such as TNF-α, IL-1 and IL-6, the expression of MMPs increases drastically [127, 128]. Moreover, expression of MMPs is regulated by epigenetic mechanisms, such as DNA methylation (reviewed in [129, 130]). The epigenetic regulation of MMPs seem to be connected to inflammation as shown for MMP-3, where IL-1 treatment led to decreased methylation of the MMP3 promoter [131].
Growing evidence suggests that during the metastatic process, cancer cells first switch from epithelial to mesenchymal phenotype (EMT) to acquire mesenchymal characteristics, which allows increased migration and invasion through basal membranes. At distant sites, the circulating tumor cells convert to an epithelial phenotype (MET) to colonize distant organs and form metastases [132, 133]. Such mechanism requires reversible, cellular plasticity. Since epigenetic switches are reversible, they may be extensively involved in the process of metastasis. Scheel el at. [134] utilized immortalized human mammary epithelial cells (HMLE) and treated them with TGF-β, which is a commonly used initiator of EMT. In addition, the authors destabilized adherens junctions with an anti-E-cadherin antibody and activated Wnt signaling by inhibition of the Wnt inhibitor DKK1 and addition of the Wnt ligand Wnt5a. After 14 days of treatment with this EMT-inducing cocktail, the cells had acquired a mesenchymal morphology, which remained stable for more than 10 passages after cessation of the treatment. The transformed cells showed a decrease in epithelial marker proteins, such as E-cadherin and an increase in mesenchymal marker proteins, such as Vimentin, Fibronectin, ZEB1, ZEB2, and Twist. The mesenchymal phenotype was maintained by autocrine loops that consisted of the same factors that initiated the EMT-switch—that is constitutive activation of TGF-β and Wnt signaling. The transformed cells showed increased migration and mammosphere-forming efficiency. When these cells were transformed with a RAS oncogene and implanted into mammary fat pads of mice they gave rise to lung metastases. Whether these cells adopted an epithelial phenotype during metastatic colonization remains to be analyzed.
Several studies implicated the miRNAs of the miR-200 and miR-34 families as important suppressors of EMT [135, 136]: miR-200 targets the EMT transcription factors (EMT-TF) ZEB1 and ZEB2 [137], whereas miR-34 targets the EMT-TF SNAIL [136] and/or ZNF281 [138]. Conversely, ZEB1 and ZEB2 directly repress mir-200 and SNAIL suppresses miR-34, thereby these EMT-TF/miRNA pairs form two double-negative loops that function as bimodal switches to stabilize either the epithelial or the mesenchymal state [139]. Both, miR-34 and miR-200 encoding genes are regulated by p53, suggesting that p53 is a key regulator of cellular plasticity by suppressing EMT and promoting its counterpart, mesenchymal–epithelial transition (MET) [64]. Moreover, ZEB1 was also shown to repress miR-34a expression by binding to the same E-boxes in the miR-34a promoters as SNAIL, thereby further inter-connecting the miR-34/SNAIL and miR-200/ZEB loops [140]. SNAIL and ZEB1 can be activated by TGF-β, which is frequently secreted by immune cells, thereby implying the possibility that inflammation might modulate this circuitry (Fig. 2a). The members of miR-200 and miR-34 families are frequently silenced by DNA methylation in cancer cells [73, 141]. Moreover, a stepwise epigenetic repression of miR-200 promoter, first through the gain of H3K27me3 and then by DNA methylation has been observed in bronchial epithelial cells in response to carcinogen exposure [142].

Epigenetic switches in EMT. a A double-negative feedback loop that maintains either epithelial or mesenchymal cellular state. Modified from Siemens et al. [136]; b IL-6 activated feedback loop that induces EMT and cancer invasion. Components indicated in green maintain epithelial state, whereas components in red induce EMT and maintain mesenchymal state. Modified from Rokavec et al. [143]. See text for details
Recently, we demonstrated that inflammation activates a feedback loop that promotes EMT, invasion, and metastasis. We showed that the pro-inflammatory cytokine IL-6 activates a circuit consisting of IL-6R, STAT3, and miR-34a (Fig. 2b) [143]: IL-6 suppresses the expression of miR-34a via IL-6R-mediated activation of STAT3 and the binding of the later to the first intron of the miR-34a gene. Furthermore, we identified the IL-6R as a direct target of miR-34a, suggesting the existence of an IL-6R/STAT3/miR-34a feedback loop. The loop was confirmed by additional functional analysis and we showed that its activation is involved in EMT, invasion, and metastasis of CRC cell lines. Moreover, the loop was associated with nodal and distant metastasis in CRC patients. In Mir34a-deficient mice, colitis-associated intestinal tumors displayed activation of the loop and in contrast to wild-type animals, progressed to invasive carcinomas. IL-6-treated CRC cells displayed elevated expression of mesenchymal markers, such as SNAIL and ZEB1, indicating that they had undergone EMT. When these cells were injected into tail veins of immune-compromised mice, they developed significantly more lung metastases then non-treated cells. Interestingly, lung metastases that formed from IL-6-treated cells displayed expression levels of mesenchymal markers similar to parental cells before IL-6 treatment, suggesting that these cells underwent MET during the process of metastatic colonization. Furthermore, we showed that miR-34a also targets and represses the soluble IL-6R (s-IL6R) [144]. The s-IL-6R is involved in IL-6 trans-signaling, which induces IL-6/STAT3 signaling also in cells that do not express IL-6R. Instead, s-IL-6R binds together with IL-6 directly to gp130 to induce STAT3 signaling [145]. The importance of the soluble IL-6R has been demonstrated by Becker et al. who showed that blockade of IL-6 receptor (IL-6R) signaling with IL-6R-neutralizing antibody or sgp130Fc, a recombinant protein that blocks IL-6 trans-signaling, reduces tumor growth in a mouse model of colon cancer [146]. Therefore, therapeutic approaches using miR-34a mimetics may inhibit the generation of IL-6R and s-IL-6R, and consequently suppress IL-6 signaling. Such strategies might supplement the IL-6 or IL-6R blocking antibody-based therapeutic approaches that are currently in development for rheumatoid arthritis, cachexia, and cancer [147, 148].
The repression of E-cadherin (encoded by the CDH1 gene) represents a hallmark of EMT and is mediated by EMT-TFs, such as SNAIL, that recruit a number of histone-modifying enzymes: e.g., the methyltransferases G9a and SUV39H1, which generate the repressive H3K9me2 and H3K9me3 marks on the CDH1 promoter that mediate CDH1 silencing [149, 150]. Furthermore, SNAIL recruits the EZH2 and Suz12 methyltransferases, which induces H3K27 trimethylation that further silences CDH1 expression [151] (Fig. 3a). During the previously described SRC-induced transformation of mammary epithelial cells and activation of the NF-κB/IL-6/lin28/let-7 loop also the CSC compartment increased [152]. During this process, the miRNA miR-200b was suppressed. The PcG protein Suz12 was identified as a miR-200b target. In CSCs, loss of miR-200b resulted in elevated expression of Suz12, which consequently induced H3K27 trimethylation and repression of CDH1 transcription (Fig. 3a). This stable epigenetic repression of CDH1 was important for maintaining a CSC state [152]. Furthermore, the authors identified additional miRNAs, including miR-15/16, miR-103/107, and miR-145 that are repressed in CSCs and inhibit their growth. These miRNAs share common target genes that encode the Bmi-1 and Suz12 components of PcG complexes, as well as transcription factors Zeb1, Zeb2, and Klf4. Combined, ectopic expression of these miRNAs progressively attenuated the growth of CSCs, suggesting that CSC formation is reinforced by an integrated regulatory circuit of miRNAs, transcription factors, and chromatin-modifying activities that can act as bistable switches to drive cells into either the stem- or the non-stem cell state. This is not the first time that a transient signal was found to induce stem cell formation. It has been shown that transient expression of transcription factors OCT4, SOX2, c-MYC, and KLF4 in fibroblasts results in the generation of induced pluripotent stem cells (iPSCs) [153]. The authors generated iPSCs from human fibroblasts by infection with lentiviral vectors carrying reprogramming factors that can be later excised by Cre-recombinase. After the establishment of the iPSC phenotype, they excised the reprogramming factors and showed that these factor-free iPSCs maintained all characteristics of the pluripotent state. Remarkably, genome-wide expression analyses showed that the factor-free iPSCs more closely resembled human embryonic stem cells than the parental virus-carrying iPSCs. Interestingly, transient treatment with IL-6, which activated the serine/threonine kinase PIM1, replaced c-MYC in iPSC reprogramming [154]. Hence, only transduction of OCT4, SOX2, and KLF4 was required for iPSC reprogramming, further indicating that the reprogramming factors function as triggers that induce a switch, which maintains the pluripotent state.

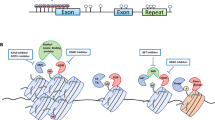
Reorganization of histone modifications at selected promoters during epigenetic switches that occur during non-CSC/CSC and EMT/MET conversions. a The EMT transcription factor SNAIL binds to E-boxes in the CDH1 (E-cadherin) promoter and recruits methyltransferases Suz12, G9a, and SUV39H1, which induce the methylation of repressive marks H3K27 and H3K9, resulting in CDH1 silencing, Furthermore, suppression of miR-200b results in the up-regulation of the miR-200b target Suz12. Enhanced levels of Suz12 lead to silencing of CDH1 and subsequent EMT as well as the formation of CSCs. On the contrary, up-regulation of miR-200b suppresses Suz12 expression and induces the conversion of CSCs to non-CSCs. b The bivalent state of the ZEB1 gene enables the switching between low and high ZEB1 expression. Regulatory regions in the ZEB1 gene contain active H3K4me3 and repressive H3K27me3 marks, which maintain low ZEB1 expression. Upon environmental stimuli, such as TGF-β, the repressive H3K27me3 marks are demethylated and expression of ZEB1 is induced, which leads to EMT and an increase of CSC compartments
Certain genes can be in a bivalent epigenetic state, wherein the chromatin in the regulatory regions contains repressive (i.e., H3K27me3) and active (i.e., H3K4me3) marks simultaneously. Such bivalent domains keep gene expression repressed, but poised for induction [155]. Shi et al. [156] showed that in medulloblastoma cells, certain Sonic hedgehog (Shh) target genes exist in a poised state, containing H3K27me3 and H3K4me3 histone marks. Activation of Shh signaling induced a local epigenetic switch mediated by the Jmjd3 demethylase that removed the H3K27me3 mark at Shh target genes. The switch led to resolution of bivalent domains and activation of Shh target genes, which plays an important role in neuronal development and in medulloblastoma formation. Chafer et al. [20] showed that the ZEB1 gene adopts a bivalent state in BC cells. This bivalent state enables the switching of non-CSCs to CSC. Exposure to certain TME-derived stimuli, such as TGF-β, reduced the presence of repressive H3K27me3 mark at the ZEB1 promoter and enhanced the active H3K4me3 mark (Fig. 3b). This switch resulted in elevated expression of ZEB1 and consequent conversion from non-CSCs to CSCs. Moreover, the cells also underwent EMT. Interestingly, the bivalent state was only observed in basal-, but not in luminal-type BC cells. Consequently, luminal-type cells were unable to switch from non-CSCs to CSCs. Therefore, the ability of non-CSCs to generate CSCs upon signals from the TME may contribute to the aggressive clinical outcome of basal-type BC [157]. This study advances the widely accepted concept that CSCs arise from parental CSCs by symmetric division [158]: Chafer et al. [20] showed that CSCs can also arise from non-CSCs by de-differentiation upon a TME-induced epigenetic switch. Therefore, it is possible that certain non-CSCs located in an inflammatory microenvironment that is rich in EMT-inducing signals (i.e., TGF-β) switch to a CSC state. These findings support a dynamic model of tumor cell plasticity in which inter-conversion between non-CSCs and CSCs occurs frequently, thereby increasing tumorigenic and malignant potential of the tumor. It has been suggested that metastases are formed by CSCs that leave the primary tumor. However, if the CSC/non-CSC plasticity described above occurs in vivo, then also the non-CSCs that have left the primary tumor may be able to form metastases by adopting CSC features. In this case, not only anticancer therapies targeting CSCs should be considered, but also treatment that prevents the non-CSC to CSC conversion. CSCs reside in stem cell niches, which are distinct regions within the TME [159]. The niches consist of non-cancer cells, such as cancer-associated fibroblasts, mesenchymal stem cells, and inflammatory cells, which together secrete factors, such as IL-6 and TGF-β, that maintain stem cell properties of CSCs. Due to cancer cell plasticity, epigenetic switches may be initiated in non-stem cancer cells when they enter a stem cell niche, which may result in de-differentiation and formation of CSCs. This may be prevented by therapies targeting the metastatic niche.
Conclusions and future directions
Epigenetic switches play an important role in cancer development. Interestingly, most of the currently discovered switches are induced by inflammation or involve inflammatory signaling. This is not surprising, since chronic infections and inflammation contribute to about 25 % of all cancers worldwide and also to sporadic cancers, e.g., in primary CRCs, expression signatures of inflammatory signaling have been detected [160]. In many examples, the same inflammatory trigger (e.g., IL-6) that induced the switch is constitutively activated in cancer cells to maintain the new phenotype. This suggests that inflammatory signaling, which initially originates from immune cells is adopted by cancer cells through epigenetic switches. Since epigenetic switches are reversible, they might represent excellent targets for anticancer therapy. This has been demonstrated for the HNF4α/miR-124/IL-6R/STAT3/miR-24/miR-629 switch: systemic administration of miR-124 in a mouse model of HCC resulted in 80 % decrease of tumor mass even when the treatment was started after the tumors were established [94]. However, it is not clear whether the transient treatment with miR-124 permanently reversed the switch, or merely suppressed the HNF4α/miR-124/IL-6R/STAT3/miR-24/miR-629 loop as long as the ectopic miR-124 was present. This is a general concern regarding the majority of described epigenetic switches, which seem to be easily induced and reversed by activating or repressing one of the components of the underlying feedback loop. However, in the in vivo situation, where cells are constantly exposed to extracellular stimuli, the cells might not permanently switch their phenotypes so easily, but only after several conditions are met and the new phenotype is stabilized by additional epigenetic changes. In this case, the permanent therapeutic reversal of an oncogenic switch would probably require the targeting of several components of the epigenetic switch.
References
Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140(6):883–899
Balkwill FR, Mantovani A (2012) Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol 22(1):33–40
Coussens LM, Zitvogel L, Palucka AK (2013) Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science 339(6117):286–291
Aggarwal BB, Vijayalekshmi RV, Sung B (2009) Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res 15(2):425–430
Balkwill F, Charles KA, Mantovani A (2005) Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 7(3):211–217
Karin M (2006) Nuclear factor-kappaB in cancer development and progression. Nature 441(7092):431–436
Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, Housseau F, Pardoll DM, Sears CL (2009) A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 15(9):1016–1022
Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA (2013) Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 13(11):759–771
McAllister SS, Weinberg RA (2014) The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol 16(8):717–727
Chen F, Zhuang X, Lin L, Yu P, Wang Y, Shi Y, Hu G, Sun Y (2015) New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med 13:45
Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA (2014) Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res 2014:149185
Bird A (2007) Perceptions of epigenetics. Nature 447(7143):396–398
Hitchler MJ, Domann FE (2009) Metabolic defects provide a spark for the epigenetic switch in cancer. Free Radic Biol Med 47(2):115–127
Ptashne M, Backman K, Humayun MZ, Jeffrey A, Maurer R, Meyer B, Sauer RT (1976) Autoregulation and function of a repressor in bacteriophage lambda. Science 194(4261):156–161
Herzog VA, Lempradl A, Trupke J, Okulski H, Altmutter C, Ruge F, Boidol B, Kubicek S, Schmauss G, Aumayr K, Ruf M, Pospisilik A, Dimond A, Senergin HB, Vargas ML, Simon JA, Ringrose L (2014) A strand-specific switch in noncoding transcription switches the function of a Polycomb/Trithorax response element. Nat Genet 46(9):973–981
Song J, Angel A, Howard M, Dean C (2012) Vernalization: a cold-induced epigenetic switch. J Cell Sci 125(Pt 16):3723–3731
Sato K, Yamamoto D (2014) An epigenetic switch of the brain sex as a basis of gendered behavior in Drosophila. Adv Genet 86:45–63
Iliopoulos D, Hirsch HA, Struhl K (2009) An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 139(4):693–706
Rokavec M, Wu W, Luo JL (2012) IL6-mediated suppression of miR-200c directs constitutive activation of inflammatory signaling circuit driving transformation and tumorigenesis. Mol Cell 45(6):777–789
Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, D’Alessio AC, Young RA, Weinberg RA (2013) Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 154(1):61–74
Chiou GY, Chien CS, Wang ML, Chen MT, Yang YP, Yu YL, Chien Y, Chang YC, Shen CC, Chio CC, Lu KH, Ma HI, Chen KH, Liu DM, Miller SA, Chen YW, Huang PI, Shih YH, Hung MC, Chiou SH (2013) Epigenetic regulation of the miR142-3p/interleukin-6 circuit in glioblastoma. Mol Cell 52(5):693–706
Tam WL, Weinberg RA (2013) The epigenetics of epithelial–mesenchymal plasticity in cancer. Nat Med 19(11):1438–1449
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16(1):6–21
Jones PA, Baylin SB (2002) The fundamental role of epigenetic events in cancer. Nat Rev Genet 3(6):415–428
Baylin SB, Jones PA (2011) A decade of exploring the cancer epigenome—biological and translational implications. Nat Rev Cancer 11(10):726–734
Easwaran H, Tsai HC, Baylin SB (2014) Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell 54(5):716–727
Wade PA, Gegonne A, Jones PL, Ballestar E, Aubry F, Wolffe AP (1999) Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet 23(1):62–66
Bell AC, Felsenfeld G (2000) Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405(6785):482–485
Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM (2000) CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405(6785):486–489
Hmadcha A, Bedoya FJ, Sobrino F, Pintado E (1999) Methylation-dependent gene silencing induced by interleukin 1beta via nitric oxide production. J Exp Med 190(11):1595–1604
Qian X, Huang C, Cho CH, Hui WM, Rashid A, Chan AO (2008) E-cadherin promoter hypermethylation induced by interleukin-1beta treatment or H. pylori infection in human gastric cancer cell lines. Cancer Lett 263(1):107–113
Huang FY, Chan AO, Rashid A, Wong DK, Cho CH, Yuen MF (2012) Helicobacter pylori induces promoter methylation of E-cadherin via interleukin-1beta activation of nitric oxide production in gastric cancer cells. Cancer 118(20):4969–4980
Cardenas H, Vieth E, Lee J, Segar M, Liu Y, Nephew KP, Matei D (2014) TGF-beta induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics 9(11):1461–1472
You H, Ding W, Rountree CB (2010) Epigenetic regulation of cancer stem cell marker CD133 by transforming growth factor-beta. Hepatology 51(5):1635–1644
Pan X, Chen Z, Huang R, Yao Y, Ma G (2013) Transforming growth factor beta1 induces the expression of collagen type I by DNA methylation in cardiac fibroblasts. PLoS ONE 8(4):e60335
Foran E, Garrity-Park MM, Mureau C, Newell J, Smyrk TC, Limburg PJ, Egan LJ (2010) Upregulation of DNA methyltransferase-mediated gene silencing, anchorage-independent growth, and migration of colon cancer cells by interleukin-6. Mol Cancer Res 8(4):471–481
Hodge DR, Xiao W, Clausen PA, Heidecker G, Szyf M, Farrar WL (2001) Interleukin-6 regulation of the human DNA methyltransferase (HDNMT) gene in human erythroleukemia cells. J Biol Chem 276(43):39508–39511
Liu CC, Lin JH, Hsu TW, Su K, Li AF, Hsu HS, Hung SC (2015) IL-6 enriched lung cancer stem-like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int J Cancer 136(3):547–559
Lee H, Zhang P, Herrmann A, Yang C, Xin H, Wang Z, Hoon DS, Forman SJ, Jove R, Riggs AD, Yu H (2012) Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc Natl Acad Sci U S A 109(20):7765–7769
Katsurano M, Niwa T, Yasui Y, Shigematsu Y, Yamashita S, Takeshima H, Lee MS, Kim YJ, Tanaka T, Ushijima T (2012) Early-stage formation of an epigenetic field defect in a mouse colitis model, and non-essential roles of T- and B-cells in DNA methylation induction. Oncogene 31(3):342–351
Haseeb A, Makki MS, Haqqi TM (2014) Modulation of ten-eleven translocation 1 (TET1), Isocitrate Dehydrogenase (IDH) expression, alpha-Ketoglutarate (alpha-KG), and DNA hydroxymethylation levels by interleukin-1beta in primary human chondrocytes. J Biol Chem 289(10):6877–6885
Kroeze LI, van der Reijden BA (1855) Jansen JH (2015) 5-Hydroxymethylcytosine: an epigenetic mark frequently deregulated in cancer. Biochim Biophys Acta 2:144–154
Li B, Carey M, Workman JL (2007) The role of chromatin during transcription. Cell 128(4):707–719
Mills AA (2010) Throwing the cancer switch: reciprocal roles of polycomb and trithorax proteins. Nat Rev Cancer 10(10):669–682
Steffen PA, Ringrose L (2014) What are memories made of? How Polycomb and Trithorax proteins mediate epigenetic memory. Nat Rev Mol Cell Biol 15(5):340–356
Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G (2007) Genome regulation by polycomb and trithorax proteins. Cell 128(4):735–745
Muller J, Verrijzer P (2009) Biochemical mechanisms of gene regulation by polycomb group protein complexes. Curr Opin Genet Dev 19(2):150–158
Simon JA, Kingston RE (2009) Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 10(10):697–708
Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, Rappsilber J, Lerdrup M, Helin K (2008) A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol 10(11):1291–1300
Saccani S, Pantano S, Natoli G (2002) p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol 3(1):69–75
Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS (2003) A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature 423(6940):659–663
McDonald OG, Wu H, Timp W, Doi A, Feinberg AP (2011) Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol 18(8):867–874
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, Li R, West RB, van de Vijver MJ, Sukumar S, Chang HY (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464(7291):1071–1076
Liu Y, Luo F, Xu Y, Wang B, Zhao Y, Xu W, Shi L, Lu X, Liu Q (2015) Epithelial–mesenchymal transition and cancer stem cells, mediated by a long non-coding RNA, HOTAIR, are involved in cell malignant transformation induced by cigarette smoke extract. Toxicol Appl Pharmacol 282(1):9–19
Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K (2010) STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell 39(4):493–506
Gerard C, Gonze D, Lemaigre F, Novak B (2014) A model for the epigenetic switch linking inflammation to cell transformation: deterministic and stochastic approaches. PLoS Comput Biol 10(1):e1003455
Fenton JI, Hursting SD, Perkins SN, Hord NG (2006) Interleukin-6 production induced by leptin treatment promotes cell proliferation in an Apc (Min/+) colon epithelial cell line. Carcinogenesis 27(7):1507–1515
Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M (2009) IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15(2):103–113
Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, Nebelsiek T, Lundgren-May T, Canli O, Schwitalla S, Matthews V, Schmid RM, Kirchner T, Arkan MC, Ernst M, Greten FR (2009) gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 15(2):91–102
Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M (2004) IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118(3):285–296
Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M (2006) The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell 124(3):601–613
Pasquinelli AE (2012) MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet 13(4):271–282
Di Leva G, Briskin D, Croce CM (2012) MicroRNA in cancer: new hopes for antineoplastic chemotherapy. Ups J Med Sci 117(2):202–216
Rokavec M, Li H, Jiang L, Hermeking H (2014) The p53/microRNA connection in gastrointestinal cancer. Clin Exp Gastroenterol 7:395–413
Leung AK, Sharp PA (2010) MicroRNA functions in stress responses. Mol Cell 40(2):205–215
Ebert MS, Sharp PA (2012) Roles for microRNAs in conferring robustness to biological processes. Cell 149(3):515–524
Gurtan AM, Sharp PA (2013) The role of miRNAs in regulating gene expression networks. J Mol Biol 425(19):3582–3600
Neviani P, Fabbri M (2015) Exosomic microRNAs in the Tumor Microenvironment. Front Med (Lausanne) 2:47
Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, Volinia S, Guler G, Morrison CD, Chan KK, Marcucci G, Calin GA, Huebner K, Croce CM (2007) MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA 104(40):15805–15810
Noonan EJ, Place RF, Pookot D, Basak S, Whitson JM, Hirata H, Giardina C, Dahiya R (2009) miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene 28(14):1714–1724
Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, Brenner JC, Yu J, Kim JH, Han B, Tan P, Kumar-Sinha C, Lonigro RJ, Palanisamy N, Maher CA, Chinnaiyan AM (2008) Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 322(5908):1695–1699
Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, Dirbas FM, Somlo G, Pera RA, Lao K, Clarke MF (2009) Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 138(3):592–603
Lodygin D, Tarasov V, Epanchintsev A, Berking C, Knyazeva T, Korner H, Knyazev P, Diebold J, Hermeking H (2008) Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle 7(16):2591–2600
Hermeking H (2012) MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer 12(9):613–626
Inouye S, Fujimoto M, Nakamura T, Takaki E, Hayashida N, Hai T, Nakai A (2007) Heat shock transcription factor 1 opens chromatin structure of interleukin-6 promoter to facilitate binding of an activator or a repressor. J Biol Chem 282(45):33210–33217
Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A et al (1989) Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 244(4905):707–712
Hartman ZC, Yang XY, Glass O, Lei G, Osada T, Dave SS, Morse MA, Clay TM, Lyerly HK (2011) HER2 overexpression elicits a proinflammatory IL-6 autocrine signaling loop that is critical for tumorigenesis. Cancer Res 71(13):4380–4391
Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr, Davidson NE, Tan-Chiu E, Martino S, Paik S, Kaufman PA, Swain SM, Pisansky TM, Fehrenbacher L, Kutteh LA, Vogel VG, Visscher DW, Yothers G, Jenkins RB, Brown AM, Dakhil SR, Mamounas EP, Lingle WL, Klein PM, Ingle JN, Wolmark N (2005) Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 353(16):1673–1684
Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, Quraishi AA, Tawakkol N, D’Angelo R, Paulson AK, Chung S, Luther T, Paholak HJ, Liu S, Hassan KA, Zen Q, Clouthier SG, Wicha MS (2012) Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell 47(4):570–584
Iliopoulos D, Hirsch HA, Wang G, Struhl K (2011) Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci USA 108(4):1397–1402
Zhao Y, Xu Y, Li Y, Xu W, Luo F, Wang B, Pang Y, Xiang Q, Zhou J, Wang X, Liu Q (2013) NF-kappaB-mediated inflammation leading to EMT via miR-200c is involved in cell transformation induced by cigarette smoke extract. Toxicol Sci 135(2):265–276
Guo L, Chen C, Shi M, Wang F, Chen X, Diao D, Hu M, Yu M, Qian L, Guo N (2013) Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial–mesenchymal transition. Oncogene 32(45):5272–5282
Xiang M, Birkbak NJ, Vafaizadeh V, Walker SR, Yeh JE, Liu S, Kroll Y, Boldin M, Taganov K, Groner B, Richardson AL, Frank DA (2014) STAT3 induction of miR-146b forms a feedback loop to inhibit the NF-kappaB to IL-6 signaling axis and STAT3-driven cancer phenotypes. Sci Signal 7 (310):ra11
Kim G, Ouzounova M, Quraishi AA, Davis A, Tawakkol N, Clouthier SG, Malik F, Paulson AK, D’Angelo RC, Korkaya S, Baker TL, Esen ES, Prat A, Liu S, Kleer CG, Thomas DG, Wicha MS, Korkaya H (2015) SOCS3-mediated regulation of inflammatory cytokines in PTEN and p53 inactivated triple negative breast cancer model. Oncogene 34(6):671–680
Daniluk J, Liu Y, Deng D, Chu J, Huang H, Gaiser S, Cruz-Monserrate Z, Wang H, Ji B, Logsdon CD (2012) An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest 122(4):1519–1528
Quante M, Varga J, Wang TC, Greten FR (2013) The gastrointestinal tumor microenvironment. Gastroenterology 145(1):63–78
Che Q, Liu BY, Wang FY, He YY, Lu W, Liao Y, Gu W, Wan XP (2014) Interleukin 6 promotes endometrial cancer growth through an autocrine feedback loop involving ERK-NF-kappaB signaling pathway. Biochem Biophys Res Commun 446(1):167–172
Rojas A, Liu G, Coleman I, Nelson PS, Zhang M, Dash R, Fisher PB, Plymate SR, Wu JD (2011) IL-6 promotes prostate tumorigenesis and progression through autocrine cross-activation of IGF-IR. Oncogene 30(20):2345–2355
Nile CJ, Read RC, Akil M, Duff GW, Wilson AG (2008) Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum 58(9):2686–2693
D’Anello L, Sansone P, Storci G, Mitrugno V, D’Uva G, Chieco P, Bonafe M (2010) Epigenetic control of the basal-like gene expression profile via Interleukin-6 in breast cancer cells. Mol Cancer 9:300
Chiu JJ, Sgagias MK, Cowan KH (1996) Interleukin 6 acts as a paracrine growth factor in human mammary carcinoma cell lines. Clin Cancer Res 2(1):215–221
Guttilla IK, Phoenix KN, Hong X, Tirnauer JS, Claffey KP, White BA (2012) Prolonged mammosphere culture of MCF-7 cells induces an EMT and repression of the estrogen receptor by microRNAs. Breast Cancer Res Treat 132(1):75–85
Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M (2007) Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 317(5834):121–124
Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, Jaeger SA, Ogata H, Karin M, Struhl K, Hadzopoulou-Cladaras M, Iliopoulos D (2011) An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 147(6):1233–1247
Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61(5):759–767
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133(6):1019–1031
Cowell JK, LaDuca J, Rossi MR, Burkhardt T, Nowak NJ, Matsui S (2005) Molecular characterization of the t(3;9) associated with immortalization in the MCF10A cell line. Cancer Genet Cytogenet 163(1):23–29
Drost J, Agami R (2009) Transformation locked in a loop. Cell 139(4):654–656
Kitamura T, Qian BZ, Pollard JW (2015) Immune cell promotion of metastasis. Nat Rev Immunol 15(2):73–86
Mehlen P, Puisieux A (2006) Metastasis: a question of life or death. Nat Rev Cancer 6(6):449–458
Pollard JW (2008) Macrophages define the invasive microenvironment in breast cancer. J Leukoc Biol 84(3):623–630
Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, Oberyszyn TM, Hall BM (2009) Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 28(33):2940–2947
Yu H, Kortylewski M, Pardoll D (2007) Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 7(1):41–51
Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP (2009) Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 15(5):416–428
Yadav A, Kumar B, Datta J, Teknos TN, Kumar P (2011) IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res 9(12):1658–1667
Nguyen DX, Bos PD, Massague J (2009) Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 9(4):274–284
Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, Lang RA, Pollard JW (2009) A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE 4(8):e6562
Murdoch C, Muthana M, Coffelt SB, Lewis CE (2008) The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 8(8):618–631
Ruffell B, Coussens LM (2015) Macrophages and Therapeutic Resistance in Cancer. Cancer Cell 27(4):462–472
Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I, Jones T, Jucknischke U, Scheiblich S, Kaluza K, Gorr IH, Walz A, Abiraj K, Cassier PA, Sica A, Gomez-Roca C, de Visser KE, Italiano A, Le Tourneau C, Delord JP, Levitsky H, Blay JY, Ruttinger D (2014) Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 25(6):846–859
Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW (2011) CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475(7355):222–225
Zhang Y, Yang P, Sun T, Li D, Xu X, Rui Y, Li C, Chong M, Ibrahim T, Mercatali L, Amadori D, Lu X, Xie D, Li QJ, Wang XF (2013) miR-126 and miR-126* repress recruitment of mesenchymal stem cells and inflammatory monocytes to inhibit breast cancer metastasis. Nat Cell Biol 15(3):284–294
Wolf MJ, Hoos A, Bauer J, Boettcher S, Knust M, Weber A, Simonavicius N, Schneider C, Lang M, Sturzl M, Croner RS, Konrad A, Manz MG, Moch H, Aguzzi A, van Loo G, Pasparakis M, Prinz M, Borsig L, Heikenwalder M (2012) Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell 22(1):91–105
Dudley AC (2012) Tumor endothelial cells. Cold Spring Harb Perspect Med 2(3):a006536
De Bock K, Mazzone M, Carmeliet P (2011) Antiangiogenic therapy, hypoxia, and metastasis: risky liaisons, or not? Nat Rev Clin Oncol 8(7):393–404
Laubli H, Spanaus KS, Borsig L (2009) Selectin-mediated activation of endothelial cells induces expression of CCL5 and promotes metastasis through recruitment of monocytes. Blood 114(20):4583–4591
Pober JS, Sessa WC (2007) Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 7(10):803–815
Okahara H, Yagita H, Miyake K, Okumura K (1994) Involvement of very late activation antigen 4 (VLA-4) and vascular cell adhesion molecule 1 (VCAM-1) in tumor necrosis factor alpha enhancement of experimental metastasis. Cancer Res 54(12):3233–3236
Witz IP (2008) The selectin-selectin ligand axis in tumor progression. Cancer Metastasis Rev 27(1):19–30
Matsuo Y, Amano S, Furuya M, Namiki K, Sakurai K, Nishiyama M, Sudo T, Tatsumi K, Kuriyama T, Kimura S, Kasuya Y (2006) Involvement of p38alpha mitogen-activated protein kinase in lung metastasis of tumor cells. J Biol Chem 281(48):36767–36775
Kobayashi K, Matsumoto S, Morishima T, Kawabe T, Okamoto T (2000) Cimetidine inhibits cancer cell adhesion to endothelial cells and prevents metastasis by blocking E-selectin expression. Cancer Res 60(14):3978–3984
Kurashige J, Mima K, Sawada G, Takahashi Y, Eguchi H, Sugimachi K, Mori M, Yanagihara K, Yashiro M, Hirakawa K, Baba H, Mimori K (2015) Epigenetic modulation and repression of miR-200b by cancer-associated fibroblasts contribute to cancer invasion and peritoneal dissemination in gastric cancer. Carcinogenesis 36(1):133–141
Zhang XH, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, Smid M, Foekens JA, Massague J (2013) Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 154(5):1060–1073
Tyan SW, Hsu CH, Peng KL, Chen CC, Kuo WH, Lee EY, Shew JY, Chang KJ, Juan LJ, Lee WH (2012) Breast cancer cells induce stromal fibroblasts to secrete ADAMTS1 for cancer invasion through an epigenetic change. PLoS ONE 7(4):e35128
Deryugina EI, Quigley JP (2006) Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev 25(1):9–34
Hojilla CV, Mohammed FF, Khokha R (2003) Matrix metalloproteinases and their tissue inhibitors direct cell fate during cancer development. Br J Cancer 89(10):1817–1821
Cairns RA, Khokha R, Hill RP (2003) Molecular mechanisms of tumor invasion and metastasis: an integrated view. Curr Mol Med 3(7):659–671
Labrie M, St-Pierre Y (2013) Epigenetic regulation of mmp-9 gene expression. Cell Mol Life Sci 70(17):3109–3124
Clark IM, Swingler TE, Sampieri CL, Edwards DR (2008) The regulation of matrix metalloproteinases and their inhibitors. Int J Biochem Cell Biol 40(6–7):1362–1378
Couillard J, Esteve PO, Pradhan S, St-Pierre Y (2011) 5-Aza-2′-deoxycytidine and interleukin-1 cooperate to regulate matrix metalloproteinase-3 gene expression. Int J Cancer 129(9):2083–2092
Tsai JH, Yang J (2013) Epithelial–mesenchymal plasticity in carcinoma metastasis. Genes Dev 27(20):2192–2206
Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A, Nieto MA (2012) Metastatic colonization requires the repression of the epithelial–mesenchymal transition inducer Prrx1. Cancer Cell 22(6):709–724
Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, Weinberg RA (2011) Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145(6):926–940
Pa Gregory, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Ma Vadas, Khew-Goodall Y, Goodall GJ (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10(5):593–601
Siemens H, Jackstadt R, Hunten S, Kaller M, Menssen A, Gotz U, Hermeking H (2011) miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 10(24):4256–4271
Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T (2008) A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 9(6):582–589
Hahn S, Jackstadt R, Siemens H, Hunten S, Hermeking H (2013) SNAIL and miR-34a feed-forward regulation of ZNF281/ZBP99 promotes epithelial-mesenchymal transition. EMBO J 32(23):3079–3095
Rokavec M, Li H, Jiang L, Hermeking H (2014) The p53/miR-34 axis in development and disease. J Mol Cell Biol 6(3):214–230
Ahn YH, Gibbons DL, Chakravarti D, Creighton CJ, Rizvi ZH, Adams HP, Pertsemlidis A, Gregory PA, Wright JA, Goodall GJ, Flores ER, Kurie JM (2012) ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J Clin Invest 122(9):3170–3183
Vrba L, Jensen TJ, Garbe JC, Heimark RL, Cress AE, Dickinson S, Stampfer MR, Futscher BW (2010) Role for DNA methylation in the regulation of miR-200c and miR-141 expression in normal and cancer cells. PLoS ONE 5(1):e8697
Tellez CS, Juri DE, Do K, Bernauer AM, Thomas CL, Damiani LA, Tessema M, Leng S, Belinsky SA (2011) EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells. Cancer Res 71(8):3087–3097
Rokavec M, Oner MG, Li H, Jackstadt R, Jiang L, Lodygin D, Kaller M, Horst D, Ziegler PK, Schwitalla S, Slotta-Huspenina J, Bader FG, Greten FR, Hermeking H (2014) IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest 124(4):1853–1867
Li H, Rokavec M, Hermeking H (2015) Soluble IL6R represents a miR-34a target: potential implications for the recently identified IL-6R/STAT3/miR-34a feed-back loop. Oncotarget 6(17):14026–14032
Chalaris A, Garbers C, Rabe B, Rose-John S, Scheller J (2011) The soluble Interleukin 6 receptor: generation and role in inflammation and cancer. Eur J Cell Biol 90(6–7):484–494
Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, Burg J, Strand S, Kiesslich R, Huber S, Ito H, Nishimoto N, Yoshizaki K, Kishimoto T, Galle PR, Blessing M, Rose-John S, Neurath MF (2004) TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 21(4):491–501
Weidle UH, Klostermann S, Eggle D, Kruger A (2010) Interleukin 6/interleukin 6 receptor interaction and its role as a therapeutic target for treatment of cachexia and cancer. Cancer Genomics Proteomics 7(6):287–302
Rossi JF, Lu ZY, Jourdan M, Klein B (2015) Interleukin-6 as a Therapeutic Target. Clin Cancer Res 21(6):1248–1257
Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, Evers BM, Zhou BP (2012) G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J Clin Invest 122(4):1469–1486
Dong C, Wu Y, Wang Y, Wang C, Kang T, Rychahou PG, Chi YI, Evers BM, Zhou BP (2013) Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene 32(11):1351–1362
Herranz N, Pasini D, Diaz VM, Franci C, Gutierrez A, Dave N, Escriva M, Hernandez-Munoz I, Di Croce L, Helin K, Garcia de Herreros A, Peiro S (2008) Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol Cell Biol 28(15):4772–4781
Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K (2010) Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell 39(5):761–772
Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, Cook EG, Hargus G, Blak A, Cooper O, Mitalipova M, Isacson O, Jaenisch R (2009) Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136(5):964–977
Brady JJ, Li M, Suthram S, Jiang H, Wong WH, Blau HM (2013) Early role for IL-6 signalling during generation of induced pluripotent stem cells revealed by heterokaryon RNA-Seq. Nat Cell Biol 15(10):1244–1252
Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125(2):315–326
Shi X, Zhang Z, Zhan X, Cao M, Satoh T, Akira S, Shpargel K, Magnuson T, Li Q, Wang R, Wang C, Ge K, Wu J (2014) An epigenetic switch induced by Shh signalling regulates gene activation during development and medulloblastoma growth. Nat Commun 5:5425
Leidy J, Khan A, Kandil D (2014) Basal-like breast cancer: update on clinicopathologic, immunohistochemical, and molecular features. Arch Pathol Lab Med 138(1):37–43
O’Brien CA, Kreso A, Jamieson CH (2010) Cancer stem cells and self-renewal. Clin Cancer Res 16(12):3113–3120
Plaks V, Kong N, Werb Z (2015) The Cancer Stem Cell Niche: how Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell 16(3):225–238
Hussain SP, Harris CC (2007) Inflammation and cancer: an ancient link with novel potentials. Int J Cancer 121(11):2373–2380
Niwa T, Tsukamoto T, Toyoda T, Mori A, Tanaka H, Maekita T, Ichinose M, Tatematsu M, Ushijima T (2010) Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res 70(4):1430–1440
Kominsky DJ, Keely S, MacManus CF, Glover LE, Scully M, Collins CB, Bowers BE, Campbell EL, Colgan SP (2011) An endogenously anti-inflammatory role for methylation in mucosal inflammation identified through metabolite profiling. J Immunol 186(11):6505–6514
Yang CH, Yue J, Fan M, Pfeffer LM (2010) IFN induces miR-21 through a signal transducer and activator of transcription 3-dependent pathway as a suppressive negative feedback on IFN-induced apoptosis. Cancer Res 70(20):8108–8116
Loffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermuller J, Kretzschmar AK, Burger R, Gramatzki M, Blumert C, Bauer K, Cvijic H, Ullmann AK, Stadler PF, Horn F (2007) Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 110(4):1330–1333
Kong D, Piao YS, Yamashita S, Oshima H, Oguma K, Fushida S, Fujimura T, Minamoto T, Seno H, Yamada Y, Satou K, Ushijima T, Ishikawa TO, Oshima M (2012) Inflammation-induced repression of tumor suppressor miR-7 in gastric tumor cells. Oncogene 31(35):3949–3960
Patel SA, Bhambra U, Charalambous MP, David RM, Edwards RJ, Lightfoot T, Boobis AR, Gooderham NJ (2014) Interleukin-6 mediated upregulation of CYP1B1 and CYP2E1 in colorectal cancer involves DNA methylation, miR27b and STAT3. Br J Cancer 111(12):2287–2296
Olaru AV, Selaru FM, Mori Y, Vazquez C, David S, Paun B, Cheng Y, Jin Z, Yang J, Agarwal R, Abraham JM, Dassopoulos T, Harris M, Bayless TM, Kwon J, Harpaz N, Livak F, Meltzer SJ (2011) Dynamic changes in the expression of MicroRNA-31 during inflammatory bowel disease-associated neoplastic transformation. Inflamm Bowel Dis 17(1):221–231
Yang P, Li Q-J, Feng Y, Zhang Y, Markowitz GJ, Ning S, Deng Y, Zhao J, Jiang S, Yuan Y, Wang H-Y, Cheng S-Q, Xie D, Wang X-F (2012) TGF-β-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell 22(3):291–303
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rokavec, M., Öner, M.G. & Hermeking, H. lnflammation-induced epigenetic switches in cancer. Cell. Mol. Life Sci. 73, 23–39 (2016). https://doi.org/10.1007/s00018-015-2045-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-2045-5