
Abstract
Since their establishment in the early 1970s, the nuclear changes upon apoptosis induction, such as the condensation of chromatin, disassembly of nuclear scaffold proteins and degradation of DNA, were, and still are, considered as the essential steps and hallmarks of apoptosis. These are the characteristics of the execution phase of apoptotic cell death. In addition, accumulating data clearly show that some nuclear events can lead to the induction of apoptosis. In particular, if DNA lesions resulting from deregulation during the cell cycle or DNA damage induced by chemotherapeutic drugs or viral infection cannot be efficiently eliminated, apoptotic mechanisms, which enable cellular transformation to be avoided, are activated in the nucleus. The functional heterogeneity of the nuclear organization allows the tight regulation of these signaling events that involve the movement of various nuclear proteins to other intracellular compartments (and vice versa) to initiate and govern apoptosis. Here, we discuss how these events are coordinated to execute apoptotic cell death.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the seminal paper [1], discrimination of apoptosis from necrosis was based on the changes in the nuclear morphology, namely condensation of chromatin at the nuclear periphery, disassembly of nuclear scaffold proteins, and the formation of apoptotic bodies. As early as 1976, the first biochemical evidence of apoptosis, the degradation of the chromosomal DNA into oligonucleosomal-length fragments after irradiation, was reported [2]. Later on, this degradation was linked to the endonuclease activity [3] and became a routinely used biochemical marker of apoptosis, while apoptosis itself was suggested to be a nucleus-associated process. Still, the exact role of nuclear events in programmed cell death (i.e., whether they initiate apoptotic signaling or are just involved in the resolution of this mode of cell death) remained unknown, and the question about its primacy was raised again only when Nuc-1, a homolog of mammalian DNase II, that is essential for DNA degradation in the nematode Caenorhabditis elegans, was found to act downstream of CED-3 (the homolog of mammalian caspase-3) and CED-4 (the homolog of mammalian Apaf-1) [4], and, in some cases, only after the engulfment of apoptotic cells by phagocytes. Furthermore, experiments with enucleated cells (cytoplasts) showed that, in response to various treatments, the cytoplasts underwent morphological apoptotic changes inhibited by overexpression of the antiapoptotic protein Bcl-2 [5, 6], leading to the conclusion that the presence of the nucleus was required neither for apoptotic cell death nor for the cytoprotective effect of Bcl-2. After that, the involvement of cysteinyl aspartate-specific proteases (caspases) in apoptotic signaling was discovered and, since almost all of them are located and activated in the cytosol [7], apoptosis was suggested to be a cytosolic event. At the same time, other findings pointed out that apoptotic cell death is closely associated to the mitochondria, and ATP produced by the mitochondria was found to be indispensable for the nuclear changes during its course. Finally, multiple lines of evidence indicated that the other cellular organelles also undergo dramatic rearrangements upon apoptosis induction [8], and, at last, apoptosis was established to be a complex process involving all cellular compartments. Now, the extrinsic (plasma membrane death receptors) and intrinsic (mitochondrial) pathways are regarded as the main mechanisms of apoptosis induction, and the others are initiated in the nucleus, endoplasmic reticulum (ER), Golgi apparatus, cytosol, lysosomes or cytoskeleton (for a recent review, see [9]).
A myriad of triggers might initiate apoptosis, and a number of common stimuli, such as DNA damage and oncogene activation, arise from the nucleus, and initially lead to the induction of the nuclear apoptotic machinery. Then, the signal is transduced across the whole cell engaging apoptotic mechanisms outside the nucleus. Yet, various stress stimuli trigger apoptosis in other cellular compartments first. However, again because of the extensive crosstalk between them, a signal, regardless of its origin, ultimately reaches the nucleus, causing distinct morphological changes observed by Kerr et al. [1]. Here, we argue the complexity of nuclear events that can either initiate apoptosis or resolve apoptotic cascades. Starting with the description of how the demolition of the nucleus and its components are executed, we discuss the different aspects of the role of the nucleus and its sub-compartments in the induction of apoptotic cell death.
Caspases: the central machinery of apoptotic cascade
The execution of apoptosis is governed by caspases that can be broadly divided into initiator (caspase-2, -8, -9, -10) and effector (also known as executioner; caspase-3, -6, -7) subgroups. Caspases are expressed as inactive zymogenic precursors (procaspases), which cleavage leads to the formation of active tetrameric enzymes. Cleavage of initiator caspases is usually augmented by assembling of high molecular weight (HMW) protein complexes, serving as special platforms for their activation. Subsequently, initiator caspases cleave, and thereby activate effector caspases that results in the execution of apoptotic cell death followed by the distinct changes in the nuclear morphology [10].
In the extrinsic apoptotic pathway, initially, the ligand binding activates the death domain (DD)-containing tumor necrosis factor (TNF)-superfamily receptors, including CD95/Fas, TNF-R1, TRAIL-R1 (DR4) and TRAIL-R2 (DR5). Then, a group of intracellular adaptor proteins, such as Fas-associated DD (FADD) and/or TNF-R-associated DD (TRADD) proteins, binds to the cytosolic DDs of the death receptors. Subsequently, procaspase-8 and/or -10 are recruited to the corresponding adaptor proteins, leading to the formation of procaspase-8/-10 activation platforms. The most studied of them, death-inducing signaling complex (DISC), is depicted in the Fig. 1 and alternative caspase-8 and -10 activating platforms are described in [11].

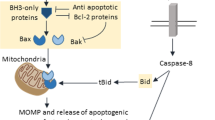
Both extrinsic and intrinsic apoptotic signaling pathways result in the same changes in the nuclear morphology. The extrinsic pathway begins with the stimulation of the death receptors by their cognate ligands that results in the assembly of initiator caspase-8/-10 activation platforms, such as death-inducing signaling complex (DISC). Caspase-8/-10 activation is followed by the cleavage of effector caspase-3, -6 and -7 and, in some cases, processing of Bid: Truncated Bid (tBid) subsequently leads to outer mitochondrial membrane permeabilization (MOMP), thereby, engaging the intrinsic pathway. The major trigger of the intrinsic pathway is DNA damage, following which MOMP is induced via several transcriptional and non-transcriptional mechanisms. MOMP results in the efflux of cytochrome c, which engages the apoptosome formation required for the activation of caspase-9. The mitochondrial proteins Smac/DIABLO and HtrA2/Omi promote apoptosis by neutralizing inhibitors of apoptosis proteins (IAPs), thus reversing their grip on caspase-3, -7, -9. In addition, in response to DNA damage formation of PIDDosome, a platform for caspase-2 activation, might be engaged. Initiator caspase-2 and -9, as well as -8 and -10, propagate the apoptotic signal by direct cleavage of the effector caspases. Effector caspases coordinate the dismantling of the nucleus. Caspase-mediated cleavage of poly(ADP-ribose) polymerase (PARP) prevents DNA repair and facilitates access of nucleases to the chromatin. Then, cleavage of inhibitor of caspase-activated DNase (ICAD) releases CAD, which enters the nucleus and catalyzes DNA degradation. Meanwhile, DNA fragmentation is also promoted in a caspase-independent manner by endonuclease G (EndoG) and apoptosis-inducing factor (AIF), which are released upon MOMP. Caspase-dependent activation of Mst1, PKC-δ and acinus stimulates chromatin condensation. Caspase-mediated cleavage of nuclear lamins contributes to nuclear fragmentation, whereas the proteolysis of the Rho effector ROCK1 results in contraction of the actin cytoskeleton that promotes breakdown of the nuclear envelope, as well as plasma membrane blebbing. Eventually, the cell collapses into apoptotic bodies, which are rapidly engulfed by phagocytic cells. Subsequently, DNA fragments, which were kept within apoptotic bodies, are digested by phagocytic DNase II
The most common trigger of the intrinsic apoptotic pathway is DNA damage that via a sophisticated signaling cascade results in the activation of proapoptotic Bcl-2 family members, such as Bax and Bak, which form channels in the mitochondrial outer membrane (MOM), and hence promote its permeabilization (MOMP) (for a detailed review, see [12]). Apart from this, the activation of caspase-8 or -10 also leads to MOMP through the cleavage of Bid to a truncated form (tBid) [10]. MOMP results in the release of multiple mitochondrial proteins, including cytochrome c, in the cytosol. Cytosolic cytochrome c and Apaf-1, in the presence of dATP, form a large heptameric complex with procaspase-9, called the apoptosome (Fig. 1). Recruitment of caspase-9 occurs via its interaction with Apaf-1, and results in the activation of caspase-9. Notably, the remarkably similar phenotypes of the Apaf-1- and caspase-9-null mice suggest that the Apaf-1-based complex is the only platform for caspase-9 activation [10]. Besides cytochrome c, a number of other proteins such as Smac/DIABLO and HtrA2/Omi are released upon MOMP. When in the cytosol, Smac/DIABLO and HtrA2/Omi abrogate the function of inhibitors of apoptosis proteins (IAPs), which inactivate caspase-9, -3 and -7 (Fig. 1). HtrA2/Omi, after its release from the mitochondria, also accumulates in the nucleus leading to the activation of transcription factor p73 that, in turn, upregulates the expression of several proapoptotic genes, including Bax and Bak [13].
The mechanism of caspase-2 activation, another initiator caspase, is much less understood, but seems to take place upstream of MOMP [14]. It is reported that its activation might occur via homodimerization and subsequent autoproteolysis [15]. Following DNA damage, caspase-2 can be activated in the PIDDosome, a large multimeric complex, comprising RAIDD and C-terminal fragments of PIDD [10] (Fig. 1). However, the significance of this platform in vivo is questionable, since RAIDD- and PIDD-deficient mice do not exhibit significant phenotypes, and do not lack the activity of caspase-2 [10]. In fact, a number of alternative mechanisms of caspase-2 activation have been suggested, and several studies indicate that caspase-2 can also act as an executioner caspase (discussed in [16]).
Once initiator caspases are active, they promote the activation of effector caspases. In addition, other proteases, such as cathepsins, calpains and granzymes, are capable of activating executioner caspases [17] either directly by proteolysis or indirectly by the cleavage of proapoptotic Bcl-2 family proteins (e.g., Bid and Bax). Several studies also reported that the promyelocytic leukemia nuclear bodies (PML NBs) may contribute to the regulation of caspase activity [18–20]. Yet, the implications of these findings have to be further examined.
Effector caspases are known to cleave more than 500 cellular proteins. In particular, caspase-3 and -7 are involved in the processing of the majority of substrates [21], while caspase-6 appears to function in specific physiological contexts [22]. Effector caspases, besides the amplification of the signal by the cleavage of initiator caspases, trigger a cascade of proteolytic events leading to apoptotic destruction of the cell. They orchestrate the degradation of cytoskeletal and nuclear proteins, chromatin condensation, DNA fragmentation and plasma membrane blebbing, finally breaking down the cell into apoptotic/pyknotic bodies, which are rapidly phagocytized. Taken together, both initiator and effector caspases play critical roles in the induction and execution of apoptotic cell death, promoting the fast destruction of cellular content, and its subsequent removal without eliciting an inflammatory response.
Apoptotic changes in the nuclear morphology
Nuclear degradation, one of the hallmarks of apoptosis originally described by Kerr et al. [1], is a multi-step process which has an important function of impeding the release of potentially immunogenic nuclear proteins and nucleic acids into the extracellular space. Since the defects in apoptotic DNA processing predispose to diseases associated with the accumulation of anti-DNA and anti-nucleosomal antibodies [23, 24], the destruction of nuclear content seems to be critical for the prevention of autoimmune disorders development.
Nuclear fragmentation relies on the disintegration of the nuclear lamina and nuclear envelope collapse, and effector caspases participate in both events. In the former one, they directly perform the cleavage of nuclear lamins and lamin-associated proteins. In the latter, executioner caspases activate the Rho effector ROCK1, which drives the contraction of the actin-myosin cytoskeleton and promotes the nuclear envelope breakdown [25] (Fig. 1). In addition, caspases cleave components of the nuclear pore complexes and nuclear transport machinery that also stimulates nuclear envelope permeabilization. Since a number of nuclear envelope proteins, including lamins, lamin-associated polypeptides and nucleoporins, are connected to the chromatin, their cleavage triggers changes in the chromatin conformation allowing access of factors, which modulate chromatin condensation and degradation. Importantly, the disruption of the nuclear envelope, chromatin condensation and DNA fragmentation, all engage either caspase-dependent or caspase-independent mechanisms.
Chromatin condensation and apoptosis
Histone modifications play an important role in the apoptotic chromatin alterations. Generally, phosphorylation of histones H2A, H2B, H3 and H4, and dephosphorylation of H1 precede chromatin destruction [26]. The most studied histone modification is the phosphorylation of H2B at Ser14 catalyzed by the mitogen-activated protein kinases (MAPKs) in a caspase-independent manner [27], and by Mst1 or PKC-δ kinases in a caspase-dependent manner [28–30] (see Fig. 1). Noteworthy, Mst1 and PKC-δ might also act upstream of caspases promoting apoptotic events upon cleavage [31, 32].
In addition, using an in vitro system, acinus, another nuclear factor that facilitates chromatin condensation was identified [33]. Acinus is able to induce apoptotic chromatin condensation after cleavage by caspase-3 without inducing the fragmentation of DNA (Fig. 1). The stimulation of PKC-δ activation is considered to be connected with acinus isoforms [30]. Although it was suggested that all Mst1, PKC-δ and acinus are involved in apoptotic chromatin condensation [33], later, depletion of acinus was shown not to impede chromatin destruction, but to inhibit oligonucleosomal DNA degradation [34]. This suggests that acinus proteins may also contribute to the activity of apoptotic nucleases or facilitate their access to DNA. Indeed, acinus isoforms were also found to be involved in the pre-mRNA processing and transcriptional regulation [35, 36], therefore, they may modulate the expression of DNA condensation and fragmentation factors. Acinus proteins are highly conserved between mammals and flies, and recent studies on Drosophila have led to a new interesting hypothesis, which therefore could be extended to mammals. Thus, in Drosophila, the loss of acinus function impedes endosomal transport and autophagosome maturation, while the gain of function leads to its stimulation [37]. As acinus proteins are primarily nuclear, the mechanism by which they modulate endocytosis and autophagy is likely to be indirect and seems to be connected with their role in the RNA processing and transcriptional regulation. Hence, acinus may play a crucial role, linking together apoptosis and autophagy. However, there are still many contradictions concerning its function, which require clarification.
DNA fragmentation in apoptotic cells
During apoptosis, DNA is usually degraded in two steps. First, the chromatin is cleaved into 50–300 kb fragments (this step is termed HMW DNA fragmentation), then into 200 bp oligonucleosomal fragments (low molecular weight (LMW) DNA fragmentation).
One of the early events of apoptosis is the cleavage of poly(ADP-ribose) polymerase (PARP), a key enzyme involved in DNA repair (Fig. 1). Importantly, PARP proteolysis not only prevents DNA repair and depletion of NAD+ (a PARP substrate), but also facilitates the access of endonucleases to the chromatin. Accordingly, the maintenance of PARP in an inactive state appears to be crucial for both caspase-dependent and -independent DNA degradation.
Among nucleases activated in a caspase-dependent manner, caspase-activated DNase (CAD or DFF40) is regarded as the major executor of DNA cleavage. In normal cells, it is inactivated by ICAD (DFF45), which acts both as an inhibitor and indispensable chaperone of CAD [38, 39]. Since CAD and ICAD possess nuclear localization signals (NLSs), they were suggested to be mainly nuclear proteins. However, there is also an alternatively spliced isoform of ICAD, the short ICAD-S (or DFF35), that resides in the cytoplasm because of a spliced-out NLS [40]. According to Wang’s group, in non-apoptotic cells, DFF exists in the nucleus as a heterodimer, composed of a 45 kD chaperone and inhibitor subunit (DFF45), and as a 40 kD latent nuclease subunit DFF40 [39]. Apoptotic activation of caspase-3 or -7 is resulting in the cleavage of DFF45 and subsequent release of DFF40 nuclease. DFF40 nuclease activity is further stimulated by specific chromosomal proteins, such as histone H1, HMGB1/2 and topoisomerase II. On the contrary, according to Nagata’s group, CAD is synthesized together with ICAD, which enables the correct folding of CAD [41]. Therefore, CAD exists as an inactive enzyme that forms a complex with ICAD in the cytosol of proliferating cells. When cells are induced to undergo apoptosis, caspases, in particular caspase 3, cleave ICAD, allowing dissociation of the CAD/ICAD complex and CAD translocation to the nucleus, where it cleaves the chromosomal DNA (Fig. 1). ICAD-S may further regulate the CAD DNase by binding to the activated CAD. Caspase-mediated cleavage of ICAD-S is sufficient for CAD dimerization, and facilitates its function as a double-strand-specific DNase that cleaves DNA with formation of oligonucleosomal DNA fragments [42]. Cytosolic localization of CAD/ICAD keeps DNase separate from its target and protects normal cells from the ‘erroneous’ action of this enzyme.
However, knockout of CAD did not affect mouse development and phenotype [43, 44]. Thus, later it was found that DNase II could compensate CAD deficiency in these animals. This nuclease is released from phagocytic lysosomes after the engulfment of apoptotic corpses, and degrades DNA in a caspase-independent manner [44] (Fig. 1); though caspase activity is required for the exposure of phagocytic signals (e.g., phosphatidylserine) on the surface of apoptotic cell. Strikingly, DNase II-null mice appear to die from severe anemia during the perinatal period [45]. The macrophages engulf erythrocytes and apoptotic cells, but cannot degrade DNA, and hence it accumulates in the lysosomes and triggers innate immunity [24, 46]. Therefore, DNase II is required for the degradation of apoptotic DNA by phagocytes. Meanwhile, further studies showed that knockout of CAD also does not completely inhibit autonomous DNA degradation within the cell [47], indicating that there are other nucleases contributing to apoptotic DNA fragmentation. Indeed, candidates, such as cellular endonucleases Nuc70, cyclophilins, DNase I, DNase X, DNase-γ and DNAS1L3, were proposed, but none of them have been shown to fulfill the criteria for apoptotic nuclease in vivo, and their activation manners (caspase-dependent or -independent) have not yet been revealed.
In fact, since caspase inhibition does not fully prevent apoptotic DNA fragmentation inside the cell [48], a significant contribution in this process seems to be made by non-caspase-mediated mechanisms. Accordingly, it was shown that apoptosis-inducing factor (AIF) and endonuclease G (EndoG), which are simultaneously released from the mitochondria following MOMP, translocate to the nucleus, where they contribute to the chromatin condensation and DNA fragmentation [49] (Fig. 1). Importantly, activities of both EndoG and AIF were shown to be caspase-independent [50]. EndoG is a DNase/RNase that triggers chromatin breakdown into HMW DNA fragments, followed by the inter- and intra-nucleosomal DNA cleavages [51]. However, since EndoG-deficient mice do not show any changes in the level of DNA degradation [52, 53], the importance of its contribution to the process is still questionable. As mentioned above, AIF is a flavoprotein that, upon translocation to the nucleus, triggers the condensation of peripheral chromatin and large-scale DNA fragmentation, but does not induce oligonucleosomal DNA fragmentation [54]. Since AIF does not possess endonuclease activity, its nuclear function most likely depends on the cooperation with other proteins. To date, a number of AIF partners, such as calpains, PARP-1, histone H2AX and EndoG [55, 56], have been revealed, but unfortunately, the accumulated data are still insufficient to establish the exact mechanism underlying the function of AIF. Even though the complete deficiency of AIF is embryonically lethal, it does not result from an excess in cell number [57], suggesting that this might be due to the loss of its non-apoptotic function. Indeed, AIF is an essential cellular oxidoreductase [58], and its inhibition, just like the inhibition of EndoG, does not prevent the appearance of HMW DNA fragments [59].
Overall, several enzymes involved in apoptotic DNA degradation have been revealed, but all the attempts to point out at least one enzyme crucial for the process have failed. Nevertheless, further studies still may unravel such a putative key player controlling cellular DNA fragmentation. It also might be that the redundancy of the DNA degradation pathways underlines the significance of protecting surrounding cells from the release of highly immunogenic DNA fragments. Meanwhile, phagocytosis provides a strong backup mechanism for the digestion of DNA released from dying cells during embryonic development; that, however, might not be enough in the case of massive apoptosis. Then, the autonomous cellular DNA degradation may become critical. Hence, the redundancy observed in the DNA degradation pathways inside the cell serves as a reliable defender that prevents the induction of the anti-DNA immune response and cell transformation.
From the end to the beginning: initiation of apoptosis following DNA damage
DNA lesions constantly arise in the cell as a consequence of stochastic errors during DNA replication and generation of reactive oxygen species in the course of metabolism. Moreover, DNA is the main target of genotoxic agents, including ultraviolet light, ionizing radiation, carcinogens, and cytotoxic drugs. The effects of these endogenous and exogenous factors result in various DNA modifications impeding transcription and replication. If not repaired prior to replication, DNA damage might lead to mutations, which accumulation will predispose the cell to malignant transformation. Fortunately, this is normally avoided due to the activation of nuclear apoptotic mechanisms.
Initially, DNA lesions are recognized by the system of DNA damage response (DDR): the highly coordinated network of factors detects the type of injury and delivers the signal to kinases that orchestrate the cellular response. Damage involves either one or both DNA strands. Damage on one strand might engage the activation of base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), or single-strand break repair (SSBR). Lesions of both DNA strands are processed through double-strand break repair (DSBR) that includes three different mechanisms: homologous recombination (HR), non-homologous end joining (NHEJ), and its less known alternative, microhomology-mediated end joining (MMEJ).
Double-strand breaks (DSBs) are the most severe of DNA lesions arising upon cell exposure to genotoxic agents. Due to its mutagenic nature, even one unrepaired DSB can result in cell death [60]. When a DSB occurs during the S and G2 phases of the cell cycle, it is preferentially repaired via the HR pathway (Fig. 2a). DSBs are detected by the MRN complex composed of MRE11, Rad50 and NBS1, and PARP-1 facilitates its recruitment by synthesis of poly(ADP-ribose) chains in the DSB regions [61]. MRN, in turn, promotes binding of ataxia-telangiectasia mutated (ATM) kinase. After binding, ATM is autoactivated and phosphorylates H2AX at Ser139 in the DSB regions. The phosphorylated form of H2AX (γH2AX) serves as a platform for docking of reparation and checkpoint proteins, stimulating signal expansion on the DNA molecule [61]. Subsequent binding of other mediators to γH2AX enables the assembly of multiple DSBR players, including additional ATM [62, 63] and MRN [64] molecules. The balance between the factors recruited at the damage site determines which of the three DSBR mechanisms will be implemented. If a DSB occurs during G1, or the balance is shifted towards NHEJ, this error-prone pathway of DSBR is initiated. The Ku70/80 heterodimer detects and binds DSB ends, driving activation of DNA–protein kinase (DNA-PK), which also amplifies the signal induced by the phosphorylation of H2AX [61] (Fig. 2a).

Transduction of DNA damage signals. a Activation of ataxia-telangiectasia mutated (ATM), Rad3-related (ATR) and DNA–protein kinase (DNA-PK) upon DNA damage. When a double-strand break (DSB) occurs, it is generally repaired via the non-homologous end-joining (NHEJ) or homologous recombination (HR) pathways. NHEJ is engaged upon Ku70/Ku80 binding to DSBs followed by recruitment and activation of DNA-PK. During HR, PARP1 binds to the DSBs and mediates the initial recruitment of the MRE11/Rad50/NBS1 (MRN) complex and subsequent binding and activation of ATM. Single-strand breaks (SSBs) repair is initiated by replication protein A (RPA), which recruits the ATR/ATR-interacting protein (ATRIP) complex and results in ATR activation. Activation of ATM, ATR, and DNA-PK is followed by the phosphorylation of H2AX that amplifies the molecular signal by DNA repair protein recruitment. b ATM, ATR, and DNA-PK can phosphorylate and activate the transcription factor p53 either directly or by means of prior activation of checkpoint kinases Chk1/2. Both ATM and ATR also contribute to the activation of the p38MAPK/MK2 kinase complex. Activation of Chk1/2 and MK2 downstream targets, as well as p53-mediated upregulation of the Cdk2 inhibitor p21, results in transient cell cycle arrest. Unrepaired DNA damage generally leads to permanent cell cycle arrest (senescence) or apoptosis
During the late S- or G2-phases, in addition to ATM, and in some cases, DNA-PK, DSBR activates ATM and Rad3-related (ATR) kinase, the downstream ATM target [65], which is also required for SSBR. In both DSBR and SSBR, the recruitment and activation of ATR are facilitated by the replication protein A (RPA) and ATR-interacting protein (ATRIP). RPA interacts with the single-stranded DNA and stimulates binding of ATRIP, which enables the association of the ATR/ATRIP complex with DNA and recruitment of several ATR mediators [61]. Along with ATM and DNA-PK, ATR leads to the formation of γH2AX (Fig. 2a), as well as other histone modifications implicated in apoptosis.
If SSBs are not repaired efficiently, they might terminate gene transcription and generate toxic DNA DSBs during replication. To prevent the formation of DSBs, SSB repair must be completed before DNA replication. To accomplish this, cells should be able to detect unrepaired SSBs, and delay the cell cycle progression to allow more time for repair; however, to date there is no evidence supporting the coordination of SSB repair and replication in the human cells. Recently, it has been shown that ATM can restrict the replication of SSB-containing DNA, and thus prevent the formation of DSB [66] (Fig. 2a).
Once activated, ATM, ATR and DNA-PK also phosphorylate the checkpoint kinases Chk1 and/or Chk2, two other central DDR kinases that are the major downstream targets of ATR and ATM, respectively. Chk1/2, along with the later identified complex consisting of p38MAPK and MK2, which is also activated upon DNA damage in the ATM- and ATR-dependent manner, controls the G1/S, intra-S and G2/M checkpoints, and arrests the cell cycle when DDR is initiated (reviewed in [67]) (Fig. 2b).
When the repair is accomplished successfully, the cell re-enters the cell cycle. In the case of its failure, DDR proteins are recruited to the break sites again with the subsequent activation of p53 [68] and/or other transcription factors, mainly p63 and p73 [69]. Their targets include the genes involved in the DDR, cell cycle arrest, autophagy, cellular senescence and apoptosis. Initially, their expression contributes to the delay of the cell cycle (mainly via p21) and favors DNA repair. However, if homeostasis is not re-established, the accumulation of the p53-inducible proapoptotic products occurs, and can lead to apoptosis. Alternatively, depending on the degree and nature of stress, as well as on the cell-type and cellular context, other mechanisms of cell death or cellular senescence might be initiated (Fig. 2b).
Notably, mutations in both ATM and DNA-PK lead to embryonic lethality in mice, suggesting that these kinases have additional functions essential for the development [70]. This indicates that DNA-PK also plays an important role in checkpoint signaling, which is evident only in the absence of ATM [71]. As for the p38MAPK/MK2 complex, it is crucial for the survival of p53-deficient cells affected by genotoxic agents [72].
p53: the key mediator in apoptosis induction
The tumor suppressor p53 has been implicated in many important cellular processes including the DDR, cell cycle arrest, and apoptotic cell death. Since the impairment of p53 signaling protects cancer cells, detailed understanding of its role in apoptosis will be extremely valuable. Indeed, though p53-null mice are viable, they develop tumors with virtually 100 % frequency [73]. Moreover, p53 is inactivated in over 50 % of human cancers, while the deregulation of other elements of its network accounts for almost all remaining cases, making reactivation of the p53 pathway in tumors a prospective strategy for anti-cancer therapy.
In the absence of stress stimuli, p53 has low activity and a short half-life. Two critical players for its suppression have been identified: the E3 ubiquitin ligase MDM2, and its close homolog MDMX (MDM4). When MDM2 or MDMX genes are knocked out in mice, the animals die in utero due to aberrant p53-induced apoptosis, which is completely rescued by the loss of p53 gene [74]. MDM2 binds to the p53 transcription-activating domain preventing it from the interaction with other factors, and mediates p53 ubiquitination and subsequent proteasomal degradation (as represented by the blue arrows in Fig. 3). Moreover, MDM2 directly interacts with the mRNA encoding p53 and suppresses its translation [75]. MDMX also binds to p53 preventing its activation, and promotes p53 ubiquitination presumably forming a heterodimer with MDM2 [76]. In addition to MDM2, other E3 ubiquitin ligases for p53 (e.g., COP1, PIRH2, and TRIM24) are described [77]; however, there is no evidence to date that any of them are critical for the regulation of p53.

Different routes of p53-mediated apoptotic cell death. In response to extensive DNA damage, the interactions between p53, MDM2 and MDMX are disrupted by posttranslational modifications (only some phosphorylation events are shown), preventing inhibition and proteasomal degradation of p53 (p53 is normally kept at low levels by MDM2 and MDMX, as represented by the blue arrows). Activated p53 upregulates proapoptotic genes (as highlighted in the box), and represses antiapoptotic genes (not shown). As a result, p53 predominantly engages the intrinsic apoptotic pathway. In addition, p53 transactivates MDM2 and Wip1 phosphatase, providing the negative feedback loops that help to restrain p53 at the end of a stress response. Non-transcriptional activities of p53, as shown on the right, include translocation of its monoubiquitinated form to the mitochondria, where, after being deubiquitinated by HAUSP, it promotes Bak/Bax oligomerization through direct physical interaction with Bak/Bax or by binding of antiapoptotic proteins Bcl-2, Bcl-XL and Mcl-1. On the other hand, cytosolic p53 interacts with the sarco-ER Ca2+-ATPase (SERCA) pumps at the endoplasmic reticulum (ER) and mitochondrial-associated membranes (MAMs), potentiating Ca2+ influx followed by an enhanced transfer to the mitochondria. These transcription-independent functions of p53, along with the transcription-mediated events, prompt MOMP leading to the release of cytochrome c and subsequent apoptosis
In response to DNA damage and other stress factors, p53 is modified by various posttranslational modifications (PTMs), which disrupt its interaction with MDM2 and MDMX. This prevents p53 degradation and facilitates its accumulation in the nucleus. PTMs implicated in p53 regulation include phosphorylation, acetylation and ubiquitination. Generally, phosphorylation plays the key role in the modulation of the p53 network, and is carried out by more than 30 kinases (ATM, ATR, DNA-PK, and Chk1/2 being the most studied). Acetylation seems to be downstream of phosphorylation in p53 signaling network upon DNA damage. At least, the most important Ser15 phosphorylation event is required for the association of p53 with p300 and CBP acetyltransferases [78]. Their recruitment results in local histone acetylation [79], and subsequent chromatin relaxation [80], which permits the induction of transcription. In addition, Tip60 acetyltransferase, which is also implicated in DSBR, was shown to be essential for acetylation of p53 at Lys120, a modification crucial for p53-mediated apoptosis [81]. Other various aspects of the acetylation network are highlighted in a recent review [82].
The complexity of p53 regulation by PTMs is further enhanced by the existence of reverse mechanisms. Thus, dephosphorylation of MDM2 and MDMX is performed by the protein phosphatases 1 (PP1), PP2A and Wip1. The latter is itself a transcription target of p53 and the p53/Wip1 feedback loop leading to ATM inactivation appears to contribute to the oscillation of p53 level in response to DNA damage (Fig. 3). Notably, Wip1 also reverses the activity of ATM, Chk2 and p38MAPK, and can directly dephosphorylate γH2AX [83]. The deacetylation network includes histone deacetylases (HDACs) that remove acetyl groups from p53, generally suppressing its activity. Human HDACs comprise 11 HDAC and 7 SIRT proteins [84]. Deubiquitination also plays a central role in the regulation of p53 activity. Among at least 91 deubiquitinating enzymes (DUBs), some proteins contribute to p53 activation, while others suppress it. One of DUBs, HAUSP (or USP7), was shown to be indispensable for the development. Normally, it functions by deubiquitinating MDM2 and MDMX, and consequently, its knockout results in mouse embryonic lethality accompanied by high p53 levels [85]. Another DUB, USP10, directly deubiquitinates p53 in the cytoplasm. Upon genotoxic stress, USP10 is phosphorylated by ATM and translocates to the nucleus, where it stabilizes p53 by deubiquitination [86]. Accordingly, a vast multitude of p53 PTMs determines its ability to respond to DNA damage, and the subsequent outcome.
Upon initial activation by PTMs, p53 forms tetrameric complexes, which have a much higher affinity for the DNA binding and upregulate the transcription of numerous genes, including MDM2, COP1, PIRH2, TRIM24 and Wip1 genes encoding p53 negative regulators. Some p53 targets, such as the most well-known Cdk inhibitor p21, are involved in the initial cell cycle delay in response to DNA damage. However, p21 transactivation is only transient, since extensive DNA damage might result in apoptosis accompanied by a low level of p21 expression [87]. Moreover, p21-knockout animals show increased levels of apoptosis [88, 89], and p21 disruption tips the balance from senescence to apoptosis in the cells treated with the topoisomerase inhibitors [90], suggesting an inverse correlation between apoptosis and p21 levels.
p53 apoptotic targets include members of both the extrinsic and intrinsic apoptotic pathways [91] (Fig. 3). The p53-mediated intrinsic apoptosis occurs upon induction of such proapoptotic genes as Bax, Puma, Noxa, Bid and Apaf-1. The p53-induced extrinsic pathway involves upregulation of CD95/Fas, CD95/Fas ligand and TRAIL-R2. Caspase-2 activation can be driven by p53 through the induction of PIDD gene [91] (Fig. 3). In addition, p53 regulates the expression of a number of microRNAs (miRNAs), some of which exhibit proapoptotic functions and contribute to the execution of apoptosis. Indeed, the comparison of miRNA expression profiles in the wild-type and p53-null MEFs revealed that p53 affects the expression of 145 miRNAs [92].
Generally, p53 affinity for binding sites is modulated by PTMs of its DNA-binding domain, which correlates with the fact that over 80 % of cancer-derived p53 mutations are located within this domain [93]. However, other aspects, such as cofactor recruitment to p53 and its target sites, are also critical for the regulation of p53 activity and, along with p53 PTMs, they have a strong impact on apoptosis. Hence, an open question remains: how exactly p53 chooses its target genes and this has to be addressed in the future studies.
In addition to the transcriptional activity of p53, its transcription-independent functions also contribute to apoptosis (Fig. 3). For instance, in response to stress stimuli, monoubiquitinated p53 translocates to MOM, where, after activation by HAUSP-mediated deubiquitination [94], it physically interacts with the pro- (Bak, Bax) and antiapoptotic (Bcl-2, Bcl-XL and Mcl-1) members of the Bcl-2 protein family, thereby, triggering MOMP [95]. Remarkably, cytosolic p53 has also been shown to modulate the Ca2+ transfer from the ER to mitochondria, which is a critical step in the induction of apoptosis. Accordingly, stress-activated p53 accumulates at the ER and mitochondria-associated membranes (MAMs), and directly binds to the sarco-ER Ca2+-ATPase (SERCA) pumps, promoting Ca2+ overload in the mitochondria that drives the loss of membrane potential, and culminates in apoptotic cell death [96, 97]. Importantly, induction of Ca2+-dependent apoptosis critically depends on p53, and hence cells lacking it fail to trigger Ca2+-dependent apoptotic mechanisms upon chemotherapy treatment [97] indicating the importance of restoring the p53/Ca2+ signaling for re-establishing tumor sensitivity to anti-cancer therapies.
p53 activation upon perturbation of ribosome biogenesis
The nucleoli seem to function as stress sensors that monitor the strength of cellular stress and modulate the activity of p53, determining cell fate by regulating the translocation of nucleolar contents to the nucleoplasm. In unstressed cells, rRNAs and a number of proteins, such as nucleostemin (NS), nucleophosmin (NPM), nucleolin (NCL), ARF (p14ARF in human), MYBBP1A and ribosomal proteins (RPs), are confined to the nucleoli, and p53 level is kept low. Following DNA damage, oncogenic stress, serum starvation and other stimuli, ribosome biogenesis is halted, nucleolar integrity is disrupted, and subsequently ribosomal (nucleolar) stress occurs. Consequently, nucleolar rRNAs and proteins are released leading to p53 stabilization and activation by several non-exclusive mechanisms (see Fig. 4). First, when in the nucleoplasm, NS, NPM, NCL, ARF and RPs (e.g., RPL5, RPL11, RPS7, RPS25, RPL37, RPS15, RPS20, RPL26 and RPS27A) associate with MDM2 inhibiting its E3 ubiquitin ligase activity toward p53. Second, RPL11, RPS7, RPS25, RPL37, RPS15, and RPS20 downregulate MDMX levels, though in distinct ways, thereby, also promoting p53 activation [98, 99]. Third, upon export in the cytoplasm, RPL26 and RPS27A bind to the 5′-UTR of p53 mRNA increasing its translation level. At the same time, RPL26 and RPS27A have been reported to be ubiquitinated by MDM2 for proteosomal degradation under non-stressed conditions, hence, providing an additional MDM2/p53 regulatory loop [100]. Fourth, RPS26 and MYBBP1A promote the acetylation of p53 and its subsequent transcriptional activity [101, 102]. While RPS26 has been shown to act through forming a complex with p53 and p300, MYBBP1A has been found to serve as a platform for p53 tetramerization, a prerequisite for the efficient activity of p53 that, in turn, provides appropriate binding sites for p300 and promotes the acetylation of p53.

Schematic representation of the ribosomal (nucleolar) stress response. Under normal conditions, ribosome biogenesis occurs normally and p53 activity is kept low (indicated by the blue arrows). Following ribosomal stress, ribosome biogenesis is perturbed, and nucleolar proteins, including rRNAs and ribosomal proteins (RPs), are released into the nucleoplasm leading to the activation of p53. 5S RNP, consisting of RPL5, RPL11 and 5S rRNA, seems to be the main player in eliciting p53 response upon impairment of ribosomal biogenesis; it acts via direct binding and inhibition of MDM2, and is essential for the sequestration of MDM2 by ARF. Along with 5S RNP and ARF, free rRNAs and RPs, and other nucleolar proteins [e.g., nucleostemin (NS), nucleophosmin (NPM) and nucleolin (NCL)] also contribute to MDM2 inhibition by binding, while a number of RPs act through the inhibition of MDMX or upregulation of p53 mRNA translation (RPL26 and RPS27A). In addition, RPS26 and MYBBP1A promote p53-mediated transcription through the facilitation of p53 acetylation by p300. The role of PICT1 remains controversial, but since, for instance, it facilitates the formation of 5S RNP and can directly increase p53 activity by binding, it appears to be an important mediator of the ribosomal stress response
Notably, PICT1 (or GLTSCR2) has been identified as another member of the family of nucleolar proteins that regulate cellular p53 levels in response to ribosomal stress. PICT1 was found to retain RPL11 in the nucleoli, thereby, inhibiting the interaction between RPL11 and MDM2 that, following ribosomal stress, would normally take place in the nucleoplasm and promote the activation of p53 [103]. In contrast, later nucleoplasmic redistribution of PICT1 was shown to allow for the direct interaction between PICT1 and p53, which stabilizes the latter [104] (Fig. 4). Then, PICT1 was reported to bind 5S rRNA and facilitate the formation of 5S RNP (a complex that includes RPL11 and RPL5) and its integration into the ribosome [105, 106]. In fact, 5S RNP appears to be the predominant player in eliciting p53 response through the sequestration of MDM2, and its formation may be the explanation of the mechanisms underlying earlier observations by [103]. Noteworthy, Sloan et al. [106] have also shown that the formation of 5S RNP is regulated by PICT1 and is essential for the activation of p53 by ARF, another protein released upon ribosomal stress, which can be also activated by oncogene overexpression. Other functions of PICT1 were also reported. For example, a recent study described PICT1 as a negative regulator of NPM. In particular, PICT1 has been found to enhance the proteasomal degradation of NPM and decrease the transforming activity induced by NPM overexpression [107]. Overall, PICT1 is an important regulator of the ribosome biogenesis and ribosomal stress, and consequently, of p53 levels. Yet, no definite conclusion could be drawn about PICT1 functions, since most of obtained by now results contradict to each other.
Importantly, imbalances in the production of RPs can also result in ribosomal stress and apoptosis. In fact, RPs, which genes are widely dispersed in the human genome, may work as sensors for genomic instability (e.g., aneuploidy) [108]. Thus, once DNA copy number changes, an imbalance in RP expression will lead to the upregulation of p53, in turn, resulting in the induction of apoptotic cell death. Intriguingly, zebrafish lacking RPL11, even display embryonic lethality due to extensive p53-mediated apoptosis, which is rescued by simultaneous knockdown of the p53 gene [109]. However, in some cases RP imbalances can down-modulate p53 levels. Thus, when the expression of RPL5 and RPL11 is reduced, it dramatically impairs ribosomal stress and p53-dependent apoptosis in multiple tumor cells [98, 100]. In fact, RPs possess a diverse range of ribosome-independent functions, and are involved in various physiological and pathological processes. Indeed, RPs play important role in tumorigenesis, immune signaling and development that are independent of the translational machinery. In this way, some ribosome-free RPs possess oncogenic activity (e.g., RPS13), and some act as oncoprotein inhibitors (e.g., RPL5, RPL11 and RPS14 that were shown to suppress c-Myc activity and regulate its mRNA turnover) (for further details see [99]). Some oncogenes itself (e.g., c-Myc) are associated with enhanced RP expression. Indeed, rapidly and actively proliferating cancer cells need more ribosome machineries compared with the normal cells. Thus, c-Myc-promoted tumor growth is dramatically abolished when the enhanced ribosome biogenesis in tumor is reduced to normal [110]. Importantly, this makes cancer cells more sensitive to ribosomal stress than normal cells, and, hence, RPs, as well as other proteins involved in ribosome biogenesis, could be potential targets for the development of novel anti-cancer drugs.
Promyelocytic leukemia nuclear bodies as scaffolds for p53-mediated apoptosis
The nucleus is an incredibly complex organelle, which, in addition to the discussed above nucleoli, contains a number of other spatially and functionally differently organized compartments. On the one hand, the presence of such distinct microenvironments provides necessary regulation of cellular signaling; on the other hand, as nuclear substructures share and exchange different components, an extensive crosstalk exists. An important role in this interplay belongs to PML NBs, dynamic subnuclear structures of 0.2–1.0 micrometer in diameter, which are implicated in the regulation of a broad range of cellular processes, including stress response, apoptosis, cellular senescence and antiviral defense. In unstressed or low stress conditions, PML NBs, for instance, participate in the DDR and DNA repair, while, upon DNA damage, they might increase in number and size and contribute to apoptosis induction [111]. Accordingly, mice and cells deficient in PML, the key organizer of PML NBs, are resistant to diverse apoptotic stimuli, including genotoxic stress [112].
PML NBs, as well as the nucleoli, contribute to both p53-dependent (Fig. 5) and -independent apoptotic pathways (discussed below and shown in Fig. 6), serving as dynamic scaffolds for PTMs of several transcription factors, including p53 family members, and for the accumulation of diverse cofactors that regulate activity of these transcriptional factors. Thus, PML directly interacts with p53 in vivo and facilitates its PTMs; in addition, PML itself may inhibit MDM2 by sequestering it in the nucleoli. At the same time, PML NB stability and scaffold function also seem to be modulated by PTMs of PML and PML NB components [113]. Interestingly, the SUMOylation, a complicated but highly controlled PTM, seems to be critical for its regulation. Indeed, PML NBs are enriched with SUMOylated proteins, including enzymes of the SUMOylation machinery. PML is able to recruit unmodified proteins as well as SUMOylated, and might itself act as an E3 SUMO ligase [114]. However, other PTMs also appear to be important for the function of PML NBs, such as phosphorylation, ubiquitination, acetylation and cleavage as discussed below.

Regulation of p53 activity in the promyelocytic leukemia nuclear bodies (PML NBs). In response to DNA damage, several kinases, including Chk2 and CK1, as well as HIPK2 in lethally damaged cells, are activated. Under unstressed or mildly stressed conditions, HIPK2 is targeted for proteasomal degradation (the blue arrows on the top). MDM2-mediated ubiquitination of p53 in unstressed cells is promoted by DAXX and HAUSP, as indicated by the blue arrows in the middle. Following DNA damage, phosphorylation of p53 by Chk2, CK1 and HIPK2 in PML NBs disrupts p53/MDM2 interaction. In addition, PML inhibits MDM2 by sequestering it in the nucleoli. Moreover, upon severe genotoxic stress, Axin, DAXX or Tip60 (not shown) might form complexes with HIPK2 and p53, enhancing the function of HIPK2 against p53. In turn, the phosphorylation of p53 drives its acetylation. In PML NBs, acetylation is mainly performed by CBP, while Pin1 promotes this event. Activated p53 induces the expression of its target genes, including PML

p63, p73, FLASH and Nur77 as initiators of p53-independent apoptosis. In response to DNA damage, c-Abl kinase is activated and accumulated in the nucleus. Subsequently, p63 and p73 are phosphorylated, and their targeting for proteasomal degradation by ITCH (blue arrows) is prevented. Moreover, p73 is recruited to PML NBs, where it is acetylated by p300 (and to the lesser extent by pCAF), while YAP and Pin1 promote the modification. In addition, such RPs as RPL5, RPL11 and RPS14 bind to p73 enhancing its transcriptional activity. Activated p63 and p73 induce the expression of several proapoptotic genes engaging the intrinsic and extrinsic apoptotic pathways. At the same time, p73 might move to the cytoplasm and promote MOMP independently of its transcriptional activity (not shown). In unstressed cells, FLASH colocalizes with Sp100 in PML NBs. Following CD95/Fas activation, FLASH relocates to the cytosol and accumulates at the mitochondria, where it binds caspase-8, and promotes its activation followed by Bid cleavage. Nur77, when in the nucleus, acts as a transcription factor that promotes the expression of prosurvival factors. In response to stress stimuli, Nur77 translocates to the cytoplasm and reverses antiapoptotic function of Bcl-2 promoting the formation of Bak/Bax channels that results in MOMP. Translocation of nuclear PML to the cytoplasm may lead to its accumulation at the ER and MAMs and subsequent upregulation of the Ca2+ transfer to the mitochondria followed by the loss of membrane potential and apoptosis
The most extensively studied transcription factor recruited in PML NBs is p53. Notably, PML itself is a p53 target gene, providing a positive feedback loop that potentiates p53-dependent apoptosis [115] (Fig. 5). Phosphorylation of p53 in PML NBs can be mediated by Chk2, CK1 and HIPK2, whereas CK1 additionally phosphorylates MDM2 [116], and PML can promote autophosphorylation of Chk2, thereby, enhancing its activity [117]. Intriguingly, HIPK2 appears to function in a stress dose-dependent manner. In normal and sublethal conditions, HIPK2 is targeted by Siah-1, MDM2 and other E3 ubiquitin ligases for the proteolytic degradation [118]. In lethally damaged cells, the level of E3 ubiquitin ligases activity is critically reduced (e.g., due to phosphorylation of Siah-1 by ATM and ATR), and HIPK2 remains stable. Moreover, the phosphorylation of p53 by HIPK2 severely depends on the presence of PML, as well as Axin and DAXX, which also interact with PML. Upon severe DNA damage, these proteins, together with Tip60, form the Axin/PML/HIPK2/p53, DAXX/Axin/HIPK2/p53 and Axin/Tip60/HIPK2/p53 complexes that enhance HIPK2 activity [119–121]. In addition, under lethal genotoxic stress, HIPK2 is processed in a caspase-dependent manner, with the formation of a hyperactive kinase, which potentiates the phosphorylation of p53 [118]. Caspase-6, an established p53-target gene, was found to be required for this processing, thereby, p53 can drive a feed-forward loop to potentiate activity of HIPK2. These mechanisms stimulate HIPK2-mediated phosphorylation of p53 that rapidly promotes its acetylation and subsequent apoptosis. Meanwhile, in response to p53 phosphorylation, the prolyl-isomerase Pin1 that also resides in PML NBs, induces conformational changes in p53, additionally potentiating its acetylation, which is generally carried out by CBP, p300 and Tip60 [122].
DAXX, mentioned as a binding partner of HIPK2, regulates p53 function at multiple levels. In normal conditions, DAXX binds to MDM2 and the deubiquitinase HAUSP, and promotes the effect of HAUSP on MDM2, thereby enhancing stability of MDM2. DNA damage leads to the dissociation of the complex, and hence to the degradation of MDM2 and subsequent stabilization of p53. Released HAUSP also contributes to p53-mediated apoptosis by deubiquitinating p53 [122]. As discussed in the next section, DAXX is an important player in the p53-independent pathway. Another protein that colocalizes to PML NBs, TRADD, may promote apoptosis in a p53-dependent manner upon certain conditions [123], but further work is necessary to determine the mechanisms of its regulation by PML.
Overall, PML NBs seem to be important regulators of p53 activity that constantly recruit and release a variety of p53 regulators depending on the cellular conditions. However, so far, the majority of studies in the field were performed on PML IV, the isoform that is expressed at very low levels in most cell lines [124]. Thereby the relevance of obtained results remains obscure, and, importantly, the fundamental issue of isoform-specific PML functions still needs further clarifications.
p53-independent apoptosis
Given the fact that p53-mutated cells do not lose the ability to delay the cell cycle progression and to undergo apoptosis, a p53 backup system exists. Like the p53 network, this system is complex and uses several strategies. The most studied players in non-p53-mediated apoptosis are close p53 homologs, p63 and p73. Since p63 and p73 are mutated in less than 1 % of human cancers, they do not seem to be as effective at inducing apoptosis as p53, and yet they are essential in certain physiological conditions. While p63 is crucial in craniofacial, limb and skin development [125] and, for its function in oocytes, is regarded as the ‘guardian of the female germline’ [126], p73 is important during neurogenesis [127]. Moreover, p63 and p73 are implicated in the DDR, and their loss severely impairs the induction of p53-dependent apoptosis [126].
In unstressed conditions, the levels of p63 and p73, similar to p53, are kept low by E3 ubiquitin ligases; however, not by MDM2, but mostly by ITCH ligase [128, 129]. After activation in response to stress stimuli, p63 and p73 can transactivate a number of proapoptotic genes (Fig. 6), many of which are p53-responsive [130]. Thus, p63 has been shown to share with p53 such targets as Bax, Apaf-1, CD95/Fas and TRAIL-R1/2, while p73 upregulates Bax, Puma, Noxa and CD95/Fas [127, 131]. In addition, p63 and p73 have their own transcriptional targets (e.g., TNF-R1 for p63 [130]; Bim and GRAMD4 for p73 [127, 132]). A number of microRNAs have also been shown to function as transcriptional targets of p63 and p73. They appear to aid p63 and p73 in promoting growth arrest and apoptosis [133].
While p63-dependent apoptosis is primarily modulated by p63 transcriptional activity, p73 also engages several transcription-independent mechanisms (reviewed in [127]). Accordingly, cytosolic p73 also can move to the mitochondria and promote MOMP. Thereby, p73 substitutes not only transcriptional, but also transcription-independent functions of p53 in cancer cells in which p53 is mutated or inactive.
Activation of both p63 and p73 upon DNA damage depends on phosphorylation. The c-Abl kinase is the most known of the upstream factors responsible for p63 and p73 phosphorylation (Fig. 6). Although multiple kinases, including ATM, Chk2 and a downstream effector of c-Abl, p38MAPK, show the ability to phosphorylate p63 and p73 [134, 135]. p73 activity also increases upon phosphorylation by c-Jun amino-terminal kinase (JNK), another kinase activated by c-Abl [136], and acetylation by p300 and pCAF [137].
While c-Abl does not directly phosphorylate p53 [138], it does increase p53 activity through the phosphorylation of MDM2 and MDMX [139, 140] and via function of its downstream kinase JNK [141]. On the other hand, c-Abl seems to be necessary for the full activation of ATM and ATR-related pathways that, in turn, triggers p53 activity [142]. In addition, recently, phosphorylation and stabilization of HIPK2, a kinase that primes p53-mediated apoptosis, have been shown to be promoted by c-Abl in response to γ- and ultraviolet radiation [143]. Thus, the activation of c-Abl, along with p53-independent pathway, stimulates p53-dependent apoptotic mechanisms.
Interestingly, in the same way as p53-mediated apoptosis, p73-dependent pathway is regulated by RPs (Fig. 6). Accordingly, RPL5, RPL11 and RPS14, which have never been shown to bind to p53, can directly bind to p73 and circumvent its inhibition by MDM2. MDM2 was shown to bind p73 and impede its transcriptional activity, although without targeting it for degradation. Subsequently, p73 is released and p73-mediated apoptosis might be engaged [144].
Moreover, among the p53 family, PML modulates functions not only of p53, but also of p63 and p73 [145]. In addition, PML, being a p53-responsive gene, is a direct transcriptional target of p73 [146] (Fig. 6). PML was shown to bind p73 in PML NBs and promote its acetylation by p300, meanwhile, Pin1 was found to stimulate this PTM, and is essential for the upregulation of several proapoptotic genes (e.g., Bax) by p73 [137]. YAP is another critical mediator of p73 function, which localizes in PML NBs. Following DNA damage, it binds to p73 and prevents it from degradation by ITCH, and, at the same time, promotes the p300-mediated acetylation of p73. In unstressed conditions, the interaction between YAP and p73 is negatively regulated by the Akt (PKB)-dependent phosphorylation of YAP that results in the translocation of YAP to the cytosol [137, 146].
Noteworthy, besides the discussed above transcriptionally active (TA) isoforms of p63 and p73, their genes also encode the ΔNp63 and ΔNp73 isoforms lacking the N-terminal transcriptional domain. Generally, ΔNp63 and ΔNp73 block activities of p53, the full-length p63 and p73, and, as a consequence, inhibit apoptosis. In response to DNA damage, and probably other stimuli, they appear to be degraded. In their function, ΔNp63 and ΔNp73 resemble p53 mutants. Indeed, it may seem that the partial depletion of p53 cannot be worse than its complete loss, but it can, since p53 mutants aggregate with normal p53, p63 and p73 and impede their functions [147]. In addition, p53 mutants were shown to bind cellular proteins, as well as PML isoform IV [148]. However, while in normal cells, p53 associates with PML only upon stress signals, in cancer cells the interaction between mutant p53 and PML appears to be constitutive. As a scaffold function of PML NBs strictly depends on the ability of PML to recruit transcription factors and its regulators that might prevent proper activation of apoptosis even under severe treatment. On the other hand, the complete absence of PML further reduces survival of p53 mutant mice [149]. Indeed, a reduction or a loss of PML expression is frequently observed in tumors [150]. Therefore, combined treatment of patients harboring a p53 mutation together with drugs enhancing PML function could be beneficial. Meanwhile, ΔNp63α isoform also was reported to interact with PML [151], as well as with YAP, activator of p73 [152]. Truly, even in the absence of mutant variants, final outcomes of cellular events depend rather on the ratio between isoforms of the p53 family proteins, than on p53, p63 and p73 activities alone, implying the importance of gene expression evaluation, while studying cell signaling.
Other proteins implicated in p53-independent apoptosis include DAXX. Although DAXX was mentioned in the section above as a PML NB compound, initially, it was discovered as a cytoplasmic protein that interacts with the CD95/Fas death receptor and engages JNK-mediated apoptosis [153]. Later, however, DAXX was shown to promote CD95/Fas-induced apoptosis not directly by binding to DISC, but from the nucleus [154]. As DAXX can suppress the expression of several antiapoptotic genes (e.g., of the prosurvival nuclear factor-κB (NF-κB) target genes) [155], in this manner it might modulate the induction of CD95/Fas-dependent apoptosis signaling from PML NBs. Nevertheless, several subsequent studies also reported the translocation of DAXX to the cytoplasm in response to different stress stimuli [156]. Though, there was another work, which again contradicted their results showing that endogenous DAXX has nuclear localization both in non-stressed and stressed conditions [157]. But as new studies describing cytoplasmic relocation of DAXX and suggesting mechanisms of this movement are still emerging [158]. It is still unclear whether DAXX translocates to the cytoplasm in physiological conditions or not, and hence the distribution of DAXX should be assessed more carefully. On the other hand, not only pro-, but also antiapoptotic roles of DAXX have been reported, and, strikingly, either partial [159] or complete [160] DAXX knockout in mice results in embryonic lethality accompanied by extensive apoptosis. Yet, whether it is only due to the disruption of the DAXX-mediated stimulation of MDM2 activity against p53 in normal conditions, or because of other antiapoptotic functions of DAXX remains to be established. Nevertheless, despite many unanswered questions, nuclear DAXX appears to be important for both p53-dependent and -independent apoptotic pathways, as well as for other cellular processes, such as chromatin remodeling and regulation [161].
Furthermore, PML NBs were reported to contribute to the CD95/Fas-induced apoptosis due to the interaction of Sp100, its constitutive component, with FLASH (or CASP8AP2). Initially, FLASH was proposed to bind procaspase-8 within DISC upon the engagement of CD95/Fas [162]. Later, however, FLASH was shown to reside in PML NBs in unstressed conditions, and to relocate to the cytoplasm in response to CD95/Fas stimulation. Upon its translocation to the cytoplasm, FLASH was reported to accumulate at the mitochondria, where it facilitates processing of caspase-8 that promotes the intrinsic apoptotic pathway via the cleavage of Bid [163] (Fig. 6). A potential mechanism for the nucleocytoplasmic shuttling of FLASH was also suggested [158], though it involves DAXX, which cytosolic translocation is still a matter of debate. Later, antiapoptotic functions of FLASH have also been proposed [164], and other reports revealed multiple roles of FLASH. Indeed, FLASH knockout in mice is lethal early in embryogenesis, as FLASH seems to be an essential regulator of histone transcription and cell cycle during embryogenesis [165]. Moreover, the impact of FLASH in the apoptotic cell death is still questioned. In the cell, FLASH is primarily located in histone locus bodies (HLBs), whereas only a fraction of FLASH is detected in PML NBs. Interestingly, a recent study suggested that FLASH may be recruited to PML NBs predominantly for its degradation [166].
An orphan nuclear receptor Nur77 was also shown to localize in PML NBs. Precisely, PML interacts with the DNA-binding domain of Nur77, and thereby represses its transactivation activity and the expression of survival molecules [167]. On the other hand, in response to some apoptotic stimuli, Nur77 translocates to the mitochondria [168], where it converts Bcl-2 function from an antiapoptotic to a proapoptotic, and hence induces the release of cytochrome c and caspase cascade activation [169] (Fig. 6). Most of the apoptotic stimuli that lead to Nur77-dependent apoptosis act through its PTMs. One of the possible modes of Nur77 activation includes its phosphorylation by JNK followed by the nuclear export of Nur77, and p38MAPK-mediated induction of Nur77 interaction with Bcl-2 [170]. Interestingly, a recently performed screening of a library of Nur77-targeting compounds revealed that Nur77 also may cross into the mitochondrial inner membrane, cause the dissipation of the mitochondrial membrane potential, and induce autophagy [171], that might provide an alternative way of cell death induction in cancer therapy.
Intriguingly, PML may modulate apoptotic response independently of its scaffold function in PML NBs. Thus, similar to p53, although earlier, PML was shown to translocate to MAMs and direct the regulation of Ca2+ influx from the ER to mitochondria depending on the stress condition (Fig. 6). Accordingly, PML-null cells are protected from cell death induced by stimuli that rely on the changes in Ca2+ signaling [172]. In fact, this is likely to be just one of many cytosolic functions of PML, however, only a few are suggested, while the rest remains to be unknown.
Taken together, p53-independent apoptosis, in a similar manner to p53-mediated apoptotic pathway, appears to be under the tight regulation of PML NBs. Thereby, PML NBs are crucial cellular mediators that integrate a myriad of stimuli and transfer the signal to a range of factors, among which p53 family proteins play the central role.
Concluding remarks
Morphological and biochemical changes, including chromatin condensation and DNA fragmentation, for many years were, and still are, the hallmarks of apoptosis. Since their establishment in 1972, more than 20 years of extensive studies were required to identify endonucleases that can cleave chromatin during apoptosis; and their activity was found to be modulated by different cellular signals that underline their important role in both physiological and pathological processes. Work from Wang [39] and Nagata’s [41] laboratories established a missing link between apoptotic caspases and nuclear events and described a mechanism for disassembly of the most complex of all biological molecules, chromatin, during apoptosis. Moreover, it became clear that some elements of this mechanism are highly conserved between species, as one of the major functions of apoptotic endonucleases is to eliminate unwanted or damaged cells from the tissues to maintain genomic stability. Thereby, the strategy of maintaining metazoan life involves a security system in a fantastically designed, highly efficient and tightly controlled removal kit that is capable, not only of killing unwanted cells on cue, but of burying the evidence, and fast.
Still, this sequence of nuclear events characterizes an executional step of apoptotic cell death. Meanwhile, accumulating data clearly show that some nuclear events can have a signaling function in apoptosis. Hence, a variety of different, potentially lethal stimuli, such as DNA damage as a result of inappropriate cell division, viral infection or exposure to ionizing radiation or chemotherapeutic drugs, are all recognized by a set of transcription factors and signaling molecules that initiate such adaptive reactions as the cell cycle arrest, protein synthesis shutoff or survival mechanisms of autophagy. However, if in any of these cases damage cannot be properly eliminated by efficient repair systems, activation of endogenously controlled mechanisms will lead to cell death. As discussed above, this can be initiated within the nucleus in a p53-dependent or -independent manner. The mechanisms involve a set of proteins located in the nucleus, and some of them are translocated to other intracellular compartments to expand the apoptotic response.
Recently, unforeseen complexities in this crosstalk have started to emerge. Three research groups have independently found that, depending on the severity of stimuli, proteins released from the mitochondria not only can initiate apoptosis from the cytosol or upon translocation to the nucleus, but also might cause DNA damage leading to the genomic instability and carcinogenesis [173–175]. These unexpected events result from the limited MOMP accompanied by sublethal activation of caspase-3. Although all groups have reached the same conclusion, mechanistic details leading to this effect were different. Liu et al. presented the evidence that caspase-3-mediated DNA damage requires EndoG activity, and its attenuation significantly reduced radiation-induced DNA damage and prevented oncogenic transformation [174], suggesting EndoG as a downstream effector of caspase-3 in this unanticipated sequence of events. On the other hand, it was demonstrated that simultaneous caspase-3-mediated activation of CAD and inactivation of the key DDR pathways, which required mild proteolysis of PARP, ATM and KAP1 [173]. Importantly, this unforeseen complexity was not limited to one cell-type, tissue or apoptotic inducer, but rather was observed for different types of cells, tissues and inducers. However, it is still unclear whether differences leading to the same final step (carcinogenesis) are cell-, tissue- or treatment-specific. Thus, although the nucleus plays an important role in both signaling and execution of apoptosis, as well as in the ‘reversible’ apoptotic response leading to cellular transformation, it is clear that not only nuclear events, but rather the tight communication between all intracellular compartments is essential for proper apoptotic response to damage.
References
Kerr JFR, Wyllie AH, Currie ARD (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26:239–257
Skalka M, Matyasova J, Cejkova M (1976) DNA in chromatin of irradiated lymphoid tissues degrades in vivo into regular fragments. FEBS Lett 72:271–275
Wyllie AH (1980) Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature 284:555–556
Ellis HM, Horvitz HR (1986) Genetic control of programmed cell death in the nematode C. elegans. Cell 44:817–829. doi:10.1016/0092-8674(86)90004-8
Jacobson MD, Burne JF, Raff MC (1994) Programmed cell death and Bcl-2 protection in the absence of a nucleus. EMBO J 13:1899–1910
Schulze-Osthoff K, Walczak H, Dröge W, Krammer PH (1994) Cell nucleus and DNA fragmentation are not required for apoptosis. J Cell Biol 127:15–20. doi:10.1083/jcb.127.1.15
Zhivotovsky B, Samali A, Gahm A, Orrenius S (1999) Caspases : their intracellular localization and translocation during apoptosis. Cell Death Differ 6:644–651
Ferri KF, Kroemer G (2001) Organelle-specific initiation of cell death pathways. Nat Cell Biol 3:E255–E263. doi:10.1038/ncb1101-e255
Galluzzi L, Bravo-San Pedro JM, Kroemer G (2014) Organelle-specific initiation of cell death. Nat Cell Biol 16:728–736. doi:10.1038/ncb3005
Parrish AB, Freel CD, Kornbluth S (2013) Cellular mechanisms controlling caspase activation and function. Cold Spring Harb Perspect Biol. doi:10.1101/cshperspect.a008672
Zamaraev AV, Kopeina GS, Zhivotovsky B, Lavrik IN (2015) Cell death controlling complexes and their potential therapeutic role. Cell Mol Life Sci 72:505–517. doi:10.1007/s00018-014-1757-2
Gillies LA, Kuwana T (2014) Apoptosis regulation at the mitochondrial outer membrane. J Cell Biochem 115:632–640. doi:10.1002/jcb.24709
Marabese M, Mazzoletti M, Vikhanskaya F, Broggini M (2008) HtrA2 enhances the apoptotic functions of p73 on bax. Cell Death Differ 15:849–858. doi:10.1038/cdd.2008.7
Bouchier-Hayes L, Oberst A, McStay GP et al (2009) Characterization of cytoplasmic caspase-2 activation by induced proximity. Mol Cell 35:830–840. doi:10.1016/j.molcel.2009.07.023
Baliga BC, Read SH, Kumar S (2004) The biochemical mechanism of caspase-2 activation. Cell Death Differ 11:1234–1241. doi:10.1038/sj.cdd.4401492
Aksenova VI, Bylino OV, Zhivotovsky BD, Lavrik IN (2013) Caspase-2: what do we know today? Mol Biol 47:165–180. doi:10.1134/S0026893313010020
Vandenabeele P, Orrenius S, Zhivotovsky B (2005) Serine proteases and calpains fulfill important supporting roles in the apoptotic tragedy of the cellular opera. Cell Death Differ 12:1219–1224. doi:10.1038/sj.cdd.4401719
Tang J, Xie W, Yang X (2005) Association of caspase-2 with the promyelocytic leukemia protein nuclear bodies. Cancer Biol Ther 4:645–649. doi:10.4161/cbt.4.6.1729
Tan JAT, Sun Y, Song J et al (2008) SUMO conjugation to the matrix attachment region-binding protein, special AT-rich sequence-binding protein-1 (SATB1), targets SATB1 to promyelocytic nuclear bodies where it undergoes caspase cleavage. J Biol Chem 283:18124–18134. doi:10.1074/jbc.M800512200
Hayashi N, Shirakura H, Uehara T, Nomura Y (2006) Relationship between SUMO-1 modification of caspase-7 and its nuclear localization in human neuronal cells. Neurosci Lett 397:5–9. doi:10.1016/j.neulet.2005.11.057
Houde C, Banks KG, Coulombe N et al (2004) Caspase-7 expanded function and intrinsic expression level underlies strain-specific brain phenotype of caspase-3-null mice. J Neurosci 24:9977–9984. doi:10.1523/JNEUROSCI.3356-04.2004
Slee EA, Adrain C, Martin SJ (2001) Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem 276:7320–7326. doi:10.1074/jbc.M008363200
Napirei M, Karsunky H, Zevnik B et al (2000) Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet 25:177–181. doi:10.1038/76032
Kawane K, Fukuyama H, Yoshida H et al (2003) Impaired thymic development in mouse embryos deficient in apoptotic DNA degradation. Nat Immunol 4:138–144. doi:10.1038/ni881
Croft DR, Coleman ML, Li S et al (2005) Actin-myosin-based contraction is responsible for apoptotic nuclear disintegration. J Cell Biol 168:245–255. doi:10.1083/jcb.200409049
Martelli AM, Zweyer M, Ochs RL et al (2001) Nuclear apoptotic changes : an overview. J Cell Biochem 82:634–646
Lu CR, Shi Y, Luo Y et al (2010) MAPKs and Mst1/Caspase-3 pathways contribute to H2B phosphorylation during UVB-induced apoptosis. Sci China Life Sci 53:663–668. doi:10.1007/s11427-010-4015-3
Cheung WL, Ajiro K, Samejima K et al (2003) Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell 113:507–517. doi:10.1016/S0092-8674(03)00355-6
Graves JD, Draves KE, Gotoh Y et al (2001) Both phosphorylation and caspase-mediated cleavage contribute to regulation of the Ste20-like protein kinase Mst1 during CD95/Fas-induced apoptosis. J Biol Chem 276:14909–14915. doi:10.1074/jbc.M010905200
Hu Y, Liu Z, Yang S-J, Ye K (2007) Acinus-provoked protein kinase C delta isoform activation is essential for apoptotic chromatin condensation. Cell Death Differ 14:2035–2046. doi:10.1038/sj.cdd.4402214
Basu A, Akkaraju GR (1999) Regulation of caspase activation and cis-diamminedichloroplatinum(II)-induced cell death by protein kinase C. Biochemistry 38:4245–4251. doi:10.1021/bi982854q
Lee KK, Ohyama T, Yajima N et al (2001) MST, a physiological caspase substrate, highly sensitizes apoptosis both upstream and downstream of caspase activation. J Biol Chem 276:19276–19285. doi:10.1074/jbc.M005109200
Sahara S, Aoto M, Eguchi Y et al (1999) Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nature 401:168–173. doi:10.1038/43678
Joselin AP, Schulze-Osthoff K, Schwerk C (2006) Loss of Acinus inhibits oligonucleosomal DNA fragmentation but not chromatin condensation during apoptosis. J Biol Chem 281:12475–12484. doi:10.1074/jbc.M509859200
Schwerk C, Prasad J, Degenhardt K et al (2003) ASAP, a novel protein complex involved in RNA processing and apoptosis. Mol Cell Biol 23:2981–2990. doi:10.1128/MCB.23.8.2981
Vucetic Z, Zhang Z, Zhao J et al (2008) Acinus-S’ represses retinoic acid receptor (RAR)-regulated gene expression through interaction with the B domains of RARs. Mol Cell Biol 28:2549–2558. doi:10.1128/MCB.01199-07
Haberman AS, Akbar MA, Ray S, Krämer H (2010) Drosophila acinus encodes a novel regulator of endocytic and autophagic trafficking. Development 137:2157–2166. doi:10.1242/dev.044230
Liu X, Li P, Widlak P et al (1998) The 40-kDa subunit of DNA fragmentation factor induces DNA fragmentation and chromatin condensation during apoptosis. Proc Natl Acad Sci USA 95:8461–8466. doi:10.1073/pnas.95.15.8461
Liu X, Zou H, Slaughter C, Wang X (1997) DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 89:175–184. doi:10.1016/S0092-8674(00)80197-X
Widlak P, Lanuszewska J, Cary RB, Garrard WT (2003) Subunit structures and stoichiometries of human DNA fragmentation factor proteins before and after induction of apoptosis. J Biol Chem 278:26915–26922. doi:10.1074/jbc.M303807200
Enari M, Sakahira H, Yokoyama H et al (1998) A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391:43–50. doi:10.1038/34112
Widlak P, Li LY, Wang X, Garrard WT (2001) Action of recombinant human apoptotic endonuclease G on naked DNA and chromatin substrates: cooperation with exonuclease and DNase I. J Biol Chem 276:48404–48409. doi:10.1074/jbc.M108461200
Zhang J, Liu X, Scherer DC et al (1998) Resistance to DNA fragmentation and chromatin condensation in mice lacking the DNA fragmentation factor 45. Proc Natl Acad Sci USA 95:12480–12485. doi:10.1073/pnas.95.21.12480
McIlroy D, Tanaka M, Sakahira H et al (2000) An auxiliary mode of apoptotic DNA fragmentation provided by phagocytes. Genes Dev 14:549–558. doi:10.1101/gad.14.5.549
Krieser RJ, MacLea KS, Longnecker DS et al (2002) Deoxyribonuclease IIalpha is required during the phagocytic phase of apoptosis and its loss causes perinatal lethality. Cell Death Differ 9:956–962. doi:10.1038/sj.cdd.4401056
Kawane K, Ohtani M, Miwa K et al (2006) Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 443:998–1002. doi:10.2492/inflammregen.29.204
Li LY, Luo X, Wang X (2001) Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412:95–99. doi:10.1038/35083620
Jayaraj R, Gupta N, Rao PVL (2009) Multiple signal transduction pathways in okadaic acid induced apoptosis in HeLa cells. Toxicology 256:118–127. doi:10.1016/j.tox.2008.11.013
Saelens X, Festjens N, Vande Walle L et al (2004) Toxic proteins released from mitochondria in cell death. Oncogene 23:2861–2874. doi:10.1038/sj.onc.1207523
Arnoult D, Gaume B, Karbowski M et al (2003) Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J 22:4385–4399. doi:10.1093/emboj/cdg423
Kalinowska M, Garncarz W, Pietrowska M et al (2005) Regulation of the human apoptotic DNase/RNase endonuclease G: involvement of Hsp70 and ATP. Apoptosis 10:821–830. doi:10.1007/s10495-005-0410-9
Irvine RA, Adachi N, Shibata DK et al (2005) Generation and characterization of endonuclease G null mice. Mol Cell Biol 25:294–302. doi:10.1128/MCB.25.1.294-302.2005
David KK, Sasaki M, Yu S-W et al (2006) EndoG is dispensable in embryogenesis and apoptosis. Cell Death Differ 13:1147–1155. doi:10.1038/sj.cdd.4401787
Susin SA, Lorenzo HK, Zamzami N et al (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397:441–446. doi:10.1038/17135
Baritaud M, Boujrad H, Lorenzo HK et al (2010) Histone H2AX: the missing link in AIF-mediated caspase-independent programmed necrosis. Cell Cycle 9:3166–3173. doi:10.4161/cc.9.16.12552
Liu KC, Huang YAT, Wu PP et al (2011) The roles of AIF and Endo G in the apoptotic effects of benzyl isothiocyanate on DU 145 human prostate cancer cells via the mitochondrial signaling pathway. Int J Oncol 38:787–796. doi:10.3892/ijo.2010.894
Joza N, Susin SA, Daugas E et al (2001) Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 410:549–554. doi:10.1038/35069004
Miramar MD, Costantini P, Ravagnan L et al (2001) NADH oxidase activity of mitochondrial apoptosis-inducing factor. J Biol Chem 276:16391–16398. doi:10.1074/jbc.M010498200
Yuste VJ, Sánchez-López I, Solé C et al (2005) The contribution of apoptosis-inducing factor, caspase-activated DNase, and inhibitor of caspase-activated DNase to the nuclear phenotype and DNA degradation during apoptosis. J Biol Chem 280:35670–35683. doi:10.1074/jbc.M504015200
Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461:1071–1078. doi:10.1038/nature08467
Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40:179–204. doi:10.1016/j.molcel.2010.09.019
Lou Z, Minter-Dykhouse K, Franco S et al (2006) MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell 21:187–200. doi:10.1016/j.molcel.2005.11.025
Dimitrova N, De Lange T (2006) MDC1 accelerates nonhomologous end-joining of dysfunctional telomeres. Genes Dev 20:3238–3243. doi:10.1101/gad.1496606
Goldberg M, Stucki M, Falck J et al (2003) MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature 421:952–956. doi:10.1038/nature01445
Jazayeri A, Falck J, Lukas C et al (2006) ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 8:37–45. doi:10.1038/ncb1337
Khoronenkova SV, Dianov GL (2015) ATM prevents DSB formation by coordinating SSB repair and cell cycle progression. Proc Natl Acad Sci USA 112:3997–4002. doi:10.1073/pnas.1416031112
Reinhardt HC, Yaffe MB (2009) Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol 21:245–255. doi:10.1016/j.ceb.2009.01.018
Batchelor E, Mock CS, Bhan I et al (2008) Recurrent initiation: a mechanism for triggering p53 pulses in response to DNA damage. Mol Cell 30:277–289. doi:10.1016/j.molcel.2008.03.016
Allocati N, Di Ilio C, De Laurenzi V (2012) P63/p73 in the control of cell cycle and cell death. Exp Cell Res 318:1285–1290. doi:10.1016/j.yexcr.2012.01.023
Gurley KE, Kemp CJ (2001) Synthetic lethality between mutation in Atm and DNA-PKcs during murine embryogenesis. Curr Biol 11:191–194. doi:10.1016/S0960-9822(01)00048-3
Stiff T, O’Driscoll M, Rief N et al (2004) ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res 64:2390–2396. doi:10.1158/0008-5472.CAN-03-3207
Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB (2007) p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 11:175–189. doi:10.1016/j.ccr.2006.11.024
Donehower LA, Harvey M, Slagle BL et al (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356:215–221. doi:10.1038/356215a0
Murray-Zmijewski F, Slee EA, Lu X (2008) A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol 9:702–712. doi:10.1038/nrm2451
Candeias MM, Malbert-Colas L, Powell DJ et al (2008) P53 mRNA controls p53 activity by managing Mdm2 functions. Nat Cell Biol 10:1098–1105. doi:10.1038/ncb1770
Huang L, Yan Z, Liao X et al (2011) The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc Natl Acad Sci USA 108:12001–12006. doi:10.1073/pnas.1102309108
Hock AK, Vousden KH (2014) The role of ubiquitin modification in the regulation of p53. Biochim Biophys Acta 1843:137–149. doi:10.1016/j.bbamcr.2013.05.022
Lambert PF, Kashanchi F, Radonovich MF et al (1998) Phosphorylation of p53 serine 15 increases interaction with CBP. J Biol Chem 273:33048–33053. doi:10.1074/jbc.273.49.33048
Hu W, Feng Z, Levine AJ (2012) The regulation of multiple p53 stress responses is mediated through MDM2. Genes Cancer 3:199–208. doi:10.1177/1947601912454734
Espinosa JM (2008) Mechanisms of regulatory diversity within the p53 transcriptional network. Oncogene 27:4013–4023. doi:10.1038/onc.2008.37
Tang Y, Luo J, Zhang W, Gu W (2006) Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell 24:827–839. doi:10.1016/j.molcel.2006.11.021
Reed SM, Quelle DE (2015) p53 acetylation: regulation and consequences. Cancers (Basel) 7:30–69. doi:10.3390/cancers7010030
Le Guezennec X, Bulavin DV (2010) WIP1 phosphatase at the crossroads of cancer and aging. Trends Biochem Sci 35:109–114. doi:10.1016/j.tibs.2009.09.005
Ling H, Peng L, Seto E, Fukasawa K (2012) Suppression of centrosome duplication and amplifcation by deacetylases. Cell Cycle 11:3779–3791. doi:10.4161/cc.21985
Kon N, Kobayashi Y, Li M et al (2010) Inactivation of HAUSP in vivo modulates p53 function. Oncogene 29:1270–1279. doi:10.1038/onc.2009.427
Bhattacharya S, Ghosh MK (2014) Cell death and deubiquitinases: perspectives in cancer. Biomed Res Int 2014:435197. doi:10.1155/2014/435197
Zhang Y, Gao Y, Zhang G et al (2011) DNMT3a plays a role in switches between doxorubicin-induced senescence and apoptosis of colorectal cancer cells. Int J Cancer 128:551–561. doi:10.1002/ijc.25365
Muñoz-Espín D, Cañamero M, Maraver A et al (2013) Programmed cell senescence during mammalian embryonic development. Cell 155:1104–1118. doi:10.1016/j.cell.2013.10.019
Storer M, Mas A, Robert-Moreno A et al (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. doi:10.1016/j.cell.2013.10.041
Hayward RL, Macpherson JS, Cummings J et al (2003) Antisense Bcl-xl down-regulation switches the response to topoisomerase I inhibition from senescence to apoptosis in colorectal cancer cells, enhancing global cytotoxicity. Clin Cancer Res 9:2856–2865
Fridman JS, Lowe SW (2003) Control of apoptosis by p53. Oncogene 22:9030–9040. doi:10.1038/sj.onc.1207116
He L, He X, Lim LP et al (2007) A microRNA component of the p53 tumour suppressor network. Nature 447:1130–1134. doi:10.1038/nature05939
Olivier M, Eeles R, Hollstein M et al (2002) The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat 19:607–614. doi:10.1002/humu.10081
Marchenko ND, Wolff S, Erster S et al (2007) Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J 26:923–934. doi:10.1038/sj.emboj.7601560
Wolff S, Erster S, Palacios G, Moll UM (2008) p53’s mitochondrial translocation and MOMP action is independent of Puma and Bax and severely disrupts mitochondrial membrane integrity. Cell Res 18:733–744. doi:10.1038/cr.2008.62
Giorgi C, Bonora M, Missiroli S et al (2015) Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget 6:1435–1445
Giorgi C, Bonora M, Sorrentino G et al (2015) p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci 112:1779–1784. doi:10.1073/pnas.1410723112
Zhang Y, Lu H (2009) Signaling to p53: ribosomal proteins find their way. Cancer Cell 16:369–377. doi:10.1016/j.ccr.2009.09.024
Zhou X, Liao W, Liao J et al (2015) Ribosomal proteins : functions beyond the ribosome. J Mol Cell Biol 7:92–104. doi:10.1093/jmcb/mjv014
Zhou X, Liao J-M, Liao W-J, Lu H (2012) Scission of the p53-MDM2 loop by ribosomal proteins. Genes Cancer 3:298–310. doi:10.1177/1947601912455200
Cui D, Li L, Lou H et al (2014) The ribosomal protein S26 regulates p53 activity in response to DNA damage. Oncogene 33:2225–2235. doi:10.1038/onc.2013.170
Ono W, Hayashi Y, Yokoyama W et al (2014) The nucleolar protein Myb-binding protein 1A (MYBBP1A) enhances p53 tetramerization and acetylation in response to nucleolar disruption. J Biol Chem 289:4928–4940. doi:10.1074/jbc.M113.474049
Sasaki M, Kawahara K, Nishio M et al (2011) Regulation of the MDM2-P53 pathway and tumor growth by PICT1 via nucleolar RPL11. Nat Med 17:944–951. doi:10.1038/nm.2392
Lee S, Kim J-Y, Kim Y-J et al (2012) Nucleolar protein GLTSCR2 stabilizes p53 in response to ribosomal stresses. Cell Death Differ 19:1613–1622. doi:10.1038/cdd.2012.40
Donati G, Peddigari S, Mercer CA, Thomas G (2013) 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep 4:87–98. doi:10.1016/j.celrep.2013.05.045
Sloan KE, Bohnsack MT, Watkins NJ (2013) The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep 5:237–247. doi:10.1016/j.celrep.2013.08.049
Kim J-Y, Cho Y-E, An Y-M et al (2015) GLTSCR2 is an upstream negative regulator of nucleophosmin in cervical cancer. J Cell Mol Med 19:1245–1252. doi:10.1111/jcmm.12474
Kim T, Leslie P, Zhang Y (2014) Ribosomal proteins as unrevealed caretakers for cellular stress and genomic instability. Oncotarget 5:860–871
Chakraborty A, Uechi T, Higa S et al (2009) Loss of ribosomal protein L11 affects zebrafish embryonic development through a p53-dependent apoptotic response. PLoS ONE 4(1):e4152. doi:10.1371/journal.pone.0004152
Barna M, Pusic A, Zollo O et al (2008) Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 456:971–975. doi:10.1038/nature07449
Bernardi R, Pandolfi PP (2007) Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 8:1006–1016. doi:10.1038/nrm2277
Wang ZG, Ruggero D, Ronchetti S et al (1998) PML is essential for multiple apoptotic pathways. Nat Genet 20:266–272. doi:10.1038/3073
Lallemand-Breitenbach V, de Thé H (2010) PML nuclear bodies. Cold Spring Harb Perspect Biol. doi:10.1101/cshperspect.a000661
Van Damme E, Laukens K, Dang TH, van Ostade X (2010) A manually curated network of the pml nuclear body interactome reveals an important role for PML-NBs in SUMOylation dynamics. Int J Biol Sci 6:51–67. doi:10.7150/ijbs.6.51
De Stanchina E, Querido E, Narita M et al (2004) PML is a direct p53 target that modulates p53 effector functions. Mol Cell 13:523–535. doi:10.1016/S1097-2765(04)00062-0
Knippschild U, Gocht A, Wolff S et al (2005) The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal 17:675–689. doi:10.1016/j.cellsig.2004.12.011
Bernardi R, Papa A, Pandolfi PP (2008) Regulation of apoptosis by PML and the PML-NBs. Oncogene 27:6299–6312. doi:10.1038/onc.2008.305
Sombroek D, Hofmann TG (2009) How cells switch HIPK2 on and off. Cell Death Differ 16:187–194. doi:10.1038/cdd.2008.154
Li Q, He Y, Wei L et al (2011) AXIN is an essential co-activator for the promyelocytic leukemia protein in p53 activation. Oncogene 30:1194–1204. doi:10.1038/onc.2010.499
Li Q, Wang X, Wu X, Rui Y, Liu W, Wang J et al (2007) Daxx cooperates with the axin/HIPK2/p53 complex to induce cell death. Cancer Res 67:66–74. doi:10.1158/0008-5472.CAN-06-1671
Li Q, Lin S, Wang X et al (2009) Axin determines cell fate by controlling the p53 activation threshold after DNA damage. Nat Cell Biol 11:1128–1134. doi:10.1038/ncb1927
Krieghoff-Henning E, Hofmann TG (2008) Role of nuclear bodies in apoptosis signalling. Biochim Biophys Acta 1783:2185–2194. doi:10.1016/j.bbamcr.2008.07.002
Morgan M, Thorburn J, Pandolfi PP, Thorburn A (2002) Nuclear and cytoplasmic shuttling of TRADD induces apoptosis via different mechanisms. J Cell Biol 157:975–984. doi:10.1083/jcb.200204039
Condemine W, Takahashi Y, Zhu J et al (2006) Characterization of endogenous human promyelocytic leukemia isoforms. Cancer Res 66:6192–6198. doi:10.1158/0008-5472.CAN-05-3792
Yang A, Schweitzer R, Sun D et al (1999) p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398:714–718. doi:10.1038/19539
Levine AJ, Tomasini R, McKeon FD et al (2011) The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol 12:259–265. doi:10.1038/nrm3086
Yoon M, Ha J, Lee M (2015) Structure and apoptotic function of p73. BMB Rep 48:81–90. doi:10.5483/BMBRep.48.2.255
Rossi M, De Laurenzi V, Munarriz E et al (2005) The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J 24:836–848. doi:10.1038/sj.emboj.7600444
Rossi M, Aqeilan RI, Neale M et al (2006) The E3 ubiquitin ligase Itch controls the protein stability of p63. Proc Natl Acad Sci USA 103:12753–12758. doi:10.1073/pnas.0603449103
Vilgelm A, El-Rifai W, Zaika A (2008) Therapeutic prospects for p73 and p63: rising from the shadow of p53. Drug Resist Updat 11:152–163. doi:10.1016/j.drup.2008.08.001
Gressner O, Schilling T, Lorenz K et al (2005) TAp63alpha induces apoptosis by activating signaling via death receptors and mitochondria. EMBO J 24:2458–2471. doi:10.1038/sj.emboj.7600708
Busuttil V, Droin N, McCormick L et al (2010) NF-kappaB inhibits T-cell activation-induced, p73-dependent cell death by induction of MDM2. Proc Natl Acad Sci USA 107:18061–18066. doi:10.1073/pnas.1006163107
Boominathan L (2010) The guardians of the genome (p53, TA-p73, and TA-p63) are regulators of tumor suppressor miRNAs network. Cancer Metastasis Rev 29:613–639. doi:10.1007/s10555-010-9257-9
Amelio I, Grespi F, Annicchiarico-Petruzzelli M, Melino G (2012) p63 the guardian of human reproduction. Cell Cycle 11:4545–4551. doi:10.4161/cc.22819
Bolcun-Filas E, Rinaldi VD, White ME, Schimenti JC (2014) Reversal of female infertility by Chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science 343:533–536. doi:10.1126/science.1247671
Jones EV, Dickman MJ, Whitmarsh AJ (2007) Regulation of p73-mediated apoptosis by c-Jun N-terminal kinase. Biochem J 405:617–623. doi:10.1042/BJ20061778
Conforti F, Sayan AE, Sreekumar R, Sayan BS (2012) Regulation of p73 activity by post-translational modifications. Cell Death Dis 3:e285. doi:10.1038/cddis.2012.27
Ben-Yehoyada M, Ben-Dor I, Shaul Y (2003) c-Abl tyrosine kinase selectively regulates p73 nuclear matrix association. J Biol Chem 278:34475–34482. doi:10.1074/jbc.M301051200
Goldberg Z, Sionov RV, Berger M et al (2002) Tyrosine phosphorylation of Mdm2 by c-Abl: implications for p53 regulation. EMBO J 21:3715–3727. doi:10.1093/emboj/cdf384
Zuckerman V, Lenos K, Popowicz GM et al (2009) c-Abl phosphorylates Hdmx and regulates its interaction with p53. J Biol Chem 284:4031–4039. doi:10.1074/jbc.M809211200
Dhanasekaran DN, Reddy EP (2008) JNK signaling in apoptosis. Oncogene 27:6245–6251. doi:10.1038/onc.2008.301
Wang X, Zeng L, Wang J et al (2011) A positive role for c-Abl in Atm and Atr activation in DNA damage response. Cell Death Differ 18:5–15. doi:10.1038/cdd.2010.106
Reuven N, Adler J, Porat Z et al (2015) The tyrosine kinase c-Abl promotes homeodomain-interacting protein kinase 2 (HIPK2) accumulation and activation in response to DNA damage. J Biol Chem 290:16478–16488. doi:10.1074/jbc.M114.628982
Zhou X, Hao Q, Zhang Q et al (2014) Ribosomal proteins L11 and L5 activate TAp73 by overcoming MDM2 inhibition. Cell Death Differ 22:755–766. doi:10.1038/cdd.2014.167
Salomoni P, Dvorkina M, Michod D (2012) Role of the promyelocytic leukaemia protein in cell death regulation. Cell Death Dis 3:e247-6. doi:10.1038/cddis.2011.122
Lapi E, Di Agostino S, Donzelli S et al (2008) PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol Cell 32:803–814. doi:10.1016/j.molcel.2008.11.019
Joerger AC, Rajagopalan S, Natan E et al (2009) Structural evolution of p53, p63, and p73: implication for heterotetramer formation. Proc Natl Acad Sci USA 106:17705–17710. doi:10.1073/pnas.0905867106
Haupt S, Di Agostino S, Mizrahi I et al (2009) Promyelocytic leukemia protein is required for gain of function by mutant p53. Cancer Res 69:4818–4826. doi:10.1158/0008-5472.CAN-08-4010
Haupt S, Mitchell C, Corneille V et al (2013) Loss of PML cooperates with mutant p53 to drive more aggressive cancers in a gender-dependent manner. Cell Cycle 12:1722–1731. doi:10.4161/cc.24805
Gurrieri C, Capodieci P, Bernardi R et al (2004) Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J Natl Cancer Inst 96:269–279. doi:10.1093/jnci/djh043
Bernassola F, Oberst A, Melino G, Pandolfi PP (2005) The promyelocytic leukaemia protein tumour suppressor functions as a transcriptional regulator of p63. Oncogene 24:6982–6986. doi:10.1038/sj.onc.1208843
Tomlinson V, Gudmundsdottir K, Luong P et al (2010) JNK phosphorylates Yes-associated protein (YAP) to regulate apoptosis. Cell Death Dis 1:e29. doi:10.1038/cddis.2010.7
Yang X, Khosravi-Far R, Chang HY, Baltimore D (1997) Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell 89:1067–1076. doi:10.1016/S0092-8674(00)80294-9
Torii S, Egan DA, Evans RA, Reed JC (1999) Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs). EMBO J 18:6037–6049. doi:10.1093/emboj/18.21.6037
Croxton R, Puto LA, De Belle I et al (2006) Daxx represses expression of a subset of antiapoptotic genes regulated by nuclear factor-kappaB. Cancer Res 66:9026–9035. doi:10.1158/0008-5472.CAN-06-1047
Lindsay CR, Morozov VM, Ishov AM (2008) PML NBs (ND10) and Daxx: from nuclear structure to protein function. Front Biosci 13:7132–7142. doi:10.2741/3216
Lindsay CR, Giovinazzi S, Ishov AM (2009) Daxx is a predominately nuclear protein that does not translocate to the cytoplasm in response to cell stress. Cell Cycle 8:1544–1551. doi:10.4161/cc.8.10.8379
Tanaka M, Kamitani T (2010) Cytoplasmic relocation of Daxx induced by Ro52 and FLASH. Histochem Cell Biol 134:297–306. doi:10.1007/s00418-010-0734-6
Michaelson JS, Bader D, Frank K et al (1999) Loss of Daxx, a promiscuously interacting protein, results in extensive apoptosis in early mouse development. Genes Dev 13:1918–1923. doi:10.1101/gad.13.15.1918
Ishov AM, Vladimirova OV, Maul GG (2004) Heterochromatin and ND10 are cell-cycle regulated and phosphorylation-dependent alternate nuclear sites of the transcription repressor Daxx and SWI/SNF protein ATRX. J Cell Sci 117:3807–3820. doi:10.1242/jcs.01230
Salomoni P (2013) The PML-interacting protein DAXX: histone loading gets into the picture. Front Oncol 3:152. doi:10.3389/fonc.2013.00152
Imai Y, Kimura T, Murakami A et al (1999) The CED-4-homologous protein FLASH is involved in Fas-mediated activation of caspase-8 during apoptosis. Nature 398:777–785. doi:10.1038/19709
Milovic-Holm K, Krieghoff E, Jensen K et al (2007) FLASH links the CD95 signaling pathway to the cell nucleus and nuclear bodies. EMBO J 26:391–401. doi:10.1038/sj.emboj.7601504
Chen S, Evans HG, Evans DR (2012) FLASH knockdown sensitizes cells to Fas-mediated apoptosis via down-regulation of the anti-apoptotic proteins, MCL-1 and cflip short. PLoS ONE 7(3):e32971. doi:10.1371/journal.pone.0032971
De Cola A, Bongiorno-Borbone L, Bianchi E et al (2011) FLASH is essential during early embryogenesis and cooperates with p73 to regulate histone gene transcription. Oncogene. doi:10.1038/onc.2011.274
Vennemann A, Hofmann TG (2013) SUMO regulates proteasome-dependent degradation of FLASH/Casp8AP2. Cell Cycle 12:1914–1921. doi:10.4161/24943
Wu W-S, Xu Z-X, Ran R et al (2002) Promyelocytic leukemia protein PML inhibits Nur77-mediated transcription through specific functional interactions. Oncogene 21:3925–3933. doi:10.1038/sj.onc.1205491
Li H, Kolluri SK, Gu J et al (2000) Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science 289:1159–1164. doi:10.1126/science.289.5482.1159
Kolluri SK, Zhu X, Zhou X et al (2008) A short Nur77-derived peptide converts Bcl-2 from a protector to a killer. Cancer Cell 14:285–298. doi:10.1016/j.ccr.2008.09.002
Zhou Y, Zhao W, Xie G et al (2014) Induction of Nur77-dependent apoptotic pathway by a coumarin derivative through activation of JNK and p38 MAPK. Carcinogenesis 35:2660–2669. doi:10.1093/carcin/bgu186
Wang W, Wang Y, Chen H et al (2014) Orphan nuclear receptor TR3 acts in autophagic cell death via mitochondrial signaling pathway. Nat Chem Biol 10:133–140. doi:10.1038/nchembio.1406
Giorgi C, Ito K, Lin H-K et al (2010) PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 330:1247–1251. doi:10.1126/science.1189157
Ichim G, Lopez J, Ahmed SU et al (2015) Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell 57:860–872. doi:10.1016/j.molcel.2015.01.018
Liu X, He Y, Li F et al (2015) Caspase-3 promotes genetic instability and carcinogenesis. Mol Cell 58:284–296. doi:10.1016/j.molcel.2015.03.003
Tang HL, Tang HM, Mak KH et al (2012) Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell 23:2240–2252. doi:10.1091/mbc.E11-11-0926
Acknowledgments
This work was supported by Grant from the Russian Science Foundation (14-25-0056). The work in the authors’ laboratories is also supported by Grants from the Russian Foundation for Basic Research, Russian President Fund, Dynasty Foundation, as well as the Stockholm and Swedish Cancer Societies, the Swedish Childhood Cancer Foundation, and the Swedish Research Council. We apologize to those authors whose primary works could not be cited owing to space limitations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Prokhorova, E.A., Zamaraev, A.V., Kopeina, G.S. et al. Role of the nucleus in apoptosis: signaling and execution. Cell. Mol. Life Sci. 72, 4593–4612 (2015). https://doi.org/10.1007/s00018-015-2031-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-2031-y