
Abstract
Because tumors require a vascular supply for their survival and growth, angiogenesis is considered an important therapeutic target in most human cancers including cancer of the central nervous system. Antiangiogenic therapy has focused on inhibitors of the vascular endothelial growth factor (VEGF) signaling pathway. VEGF pathway-targeted drugs have shown therapeutic efficacy in several CNS tumors and have been tried most frequently in glioblastoma. These therapies, however, have been less effective than anticipated as some patients do not respond to therapy and some receive only modest benefit. Underlying this suboptimal response are multiple mechanisms of drug resistance involving changes in both tumor cells and their microenvironment. In this review, we discuss the multiple proposed mechanisms by which neurological tumors evolve to become resistant to antiangiogenic therapies. A better understanding of these mechanisms, their context, and their interplay will likely facilitate improvements in pharmacological strategies for the targeted treatment of neurological tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Angiogenesis is critical for tumor viability and thus has long been considered an important target for cancer therapy. The focus of antiangiogenic agent development has largely been to inhibit the vascular endothelial growth factor (VEGF) pathway, an essential pathway for angiogenesis, although other antiangiogenic agents have been developed [1]. As evidence emerges that angiogenesis has significant mechanistic complexity, therapeutic resistance and escape have become practical limitations to drug development. Here, we review the mechanisms by which dynamic changes occur in the tumor microenvironment in response to antiangiogenic therapy, leading to drug resistance. These mechanisms include direct selection of clonal cell populations with the capacity to rapidly upregulate alternative proangiogenic pathways, increased invasive capacity, and intrinsic resistance to hypoxia. The implications of normalization of vasculature with subsequently improved vascular function as a result of antiangiogenic therapy are explored, as are the implications of the ability to incorporate and co-opt otherwise normal vasculature. Finally, we consider the extent to which a better understanding of the biology of hypoxia and reoxygenation, as well as the depth and breadth of systems invested in angiogenesis, may enable identification of biomarkers and novel therapeutic targets. Insights gained through this work may offer solutions for personalizing antiangiogenic approaches and improving the outcome of patients with cancer [1]. Antiangiogenic agents have been investigated in the treatment of certain types of brain cancers, particularly glioblastoma, a malignant primary brain tumor with a poor prognosis and a need for novel therapies, as well as vestibular schwannomas and meningiomas arising in the context of type 2 neurofibromatosis [2, 3].
Unfortunately, several studies have suggested that the duration of response to antiangiogenic therapy in cancers, including neurological tumors, is transient [4]. A challenge in interpreting this finding is whether these failures represent biological resistance to the drugs or merely the growth capacity of the tumor eventually exceeding the antitumoral effect of the drug. Clinical and laboratory findings show that neurological as well as other tumors, when challenged with antiangiogenic therapy, either resume growth after a transient period of responsiveness (adaptive resistance) or fail to respond altogether (intrinsic resistance). Extensive laboratory findings in animal models and patient specimens have revealed that a specific profile of molecular changes underpin these two different types of resistance [2]. Interestingly, most, if not all, mechanisms of resistance do not involve disinhibition of VEGF and its signaling; VEGF and its signaling remain inhibited in the resistant tumors [2]. In this review, we discuss the different mechanisms by which brain tumors, glioblastoma in particular, evade antiangiogenic therapy. This information may provide valuable insight in guiding new therapeutic regimens less prone to intrinsic resistance or less likely to promote the evolution of adaptive resistance.
Angiogenesis and its pathways in brain tumors
One of the hallmark necessities of tumors is a vascular supply that, just as for organs of the body, allots exchange of wastes for nutrients and growth factors. One of the most important and established ways in which tumors ensure vascular supply is through angiogenesis, the physiological process by which new vessels are formed from preexisting vessels [5]. In fact, angiogenesis has been shown by a substantial amount of research to be critical in not only tumor growth and survival, but also in tumor progression [6, 7].
The primary mediator of angiogenesis in both physiological and pathophysiological contexts is vascular endothelial growth factor (VEGF) [8]. VEGF is a mitogen that operates via binding to VEGF receptors, resulting in endothelial cell proliferation, migration, and new vessel formation [9]. VEGF is upregulated in a majority of human cancers where its expression is derailed from normal feedback mechanisms (i.e., it remains elevated after establishment of perfusion) [8]. In hypoxic stromal cells and tumor cells high levels of VEGF are expressed to induce branching angiogenesis, and autocrine expression of low levels of VEGF is required for maintenance of mature vessels [10]. Initially, VEGF signaling was thought to occur specifically in endothelial cells of blood vessels. However, it is now known that VEGF has multifaceted activity with multiple cellular targets such as bone marrow-derived myeloid cells, cancer cells, pericytes, etc. [8].
Angiogenesis and VEGF signaling are especially important in glioblastomas as evidenced by their extensive degree of vascularization [11]. In fact, angiogenesis is crucial for glioblastoma development and growth, and the level of angiogenesis correlates with the aggressiveness of glioblastomas [11]. Indeed, glioblastoma is the most lethal brain cancer and the one with the highest degree of endothelial cell proliferation and vascular density [12].
In order to promote angiogenesis, gliomas upregulate VEGF and its downstream pathways in several different ways. VEGF-A, the principal driver of sprouting angiogenesis, is upregulated in glioblastoma and produced by tumor cells as well as tumor-associated stromal and inflammatory cells [13]. There are multiple stimuli and pathways that drive angiogenesis in gliomas such as hypoxia-induced upregulation of HIF, which increases VEGF mRNA levels, HIF-independent VEGF upregulation via EGFR signaling, and many more mechanisms involving upregulation and/or mutation of specific genes [14]. Additionally, increased expression of VEGF receptor 1 (VEGFR-1) and VEGF receptor 2 (VEGFR-2) has been observed in gliomas [15]. There are also a number of non-VEGF signaling pathways that are thought to support angiogenesis in gliomas. Basic fibroblast growth factor (bFGF) is an important proangiogenic growth factor in gliomas, and FGF and FGFR1 (FGF receptor 1) are upregulated in glioblastoma. Additionally, proangiogenic factors such as IL-1α, IL-1β, stem cell factor (SCF), angiopoietin, and IL-8 are upregulated in vitro in the glioma tumor microenvironment [14, 16, 17]. These proangiogenic growth factors will be discussed in further detail in subsequent sections.
Antiangiogenic therapies in brain tumors
Due to its critical role in tumor homeostasis, VEGF and its signaling were proposed as a therapeutic target in cancer over 4 decades ago [1]. Since then, the US Food and Drug Administration (FDA) has, on the basis of phase III clinical trials, approved these agents for treatment of metastatic colorectal cancer, some non-small-cell lung cancers, renal cell cancer, hepatocellular carcinoma, and neuroendocrine tumors [18]. More recently, in 2009, after a series of phase II clinical trials overcame the initial fears of hemorrhage that were associated with using these agents to treat tumors of the central nervous system, bevacizumab, a VEGF neutralizing antibody, was granted accelerated FDA approval for the treatment of recurrent glioblastoma. Antiangiogenic therapies such as bevacizumab may even play a role in the treatment of low grade gliomas [19] and in the treatment of benign brain tumors such as vestibular schwannomas and meningiomas [3].
In terms of angiogenic pathways targeted in brain tumors, the majority of these agents have targeted the VEGF pathway. As mentioned, glioma cells have been shown to secrete VEGF in vivo to support and increase angiogenesis [20], and similar changes have been identified in benign brain tumors such as vestibular schwannomas and meningiomas [21, 22]. The VEGF pathway has been targeted in brain tumors and other cancers using two types of agents (Table 1): agents targeting VEGF directly or receptor tyrosine kinase inhibitors (RTKIs) that typically target multiple receptor tyrosine kinases. Two examples targeting VEGF include VEGF-Trap (afibercept), a soluble VEGF receptor, and bevacizumab, a monoclonal antibody against VEGF-A165 [23]. Examples of RTKIs include sunitib and cediranib (AZD2171) [24].
While the vast majority of antiangiogenic therapies target the VEGF pathway, a few pharmacologic agents have been developed with targets outside this pathway. For example, AMG 386 (trebananib) is thought to inhibit angiogenesis via binding to angiopoietins (Ang 1 and Ang 2), mediators of angiogenesis that will be discussed later [25, 27, 28]. Additionally, cilengitide is a cyclized RGD-containing pentapeptide and a potent antagonist of the αvβ3 and αvβ5 integrins, which are upregulated in several cancers including glioblastoma and whose activation promotes angiogenesis [25, 29].
Clinical observations from use of antiangiogenic therapy for brain cancers
The prototypical VEGF binding agent is bevacizumab (avastin), which is a humanized monoclonal anti-VEGF antibody and was the first anti-VEGF used in patients with glioblastoma [4]. Several mechanisms of action have been proposed to explain the effectiveness of bevacizumab in some patients with glioblastoma, including direct antiglioblastoma effects on VEGFR-expressing glioblastoma cells, direct inhibition of angiogenesis, vascular normalization, and perturbation of the glioma stem cell microvascular niche [4]. Additionally, bevacizumab is thought to have synergistic potential with chemotherapeutic agents due to its ability to promote vascular normalization. In this process, leaky, dysfunctional tumor vessels are replaced with vessels of normal integrity, causing the originally elevated fluid pressure to normalize. This pressure normalization removes the barrier to fluid influx, thereby improving delivery of co-administered chemotherapy [24]. A significant tumor response of glioblastoma to bevacizumab has been observed in multiple studies, and the progression-free survival at 6 months in a recently published article was reported at 42.6 % for monotherapy [25, 30]. Bevacizumab offers a modest (if any) overall survival benefit in patients with glioblastoma because of the tumor’s rapid progression after the brief period of halted growth; this presumably occurs as a result of the tumor’s rapid adaptation to the anti-VEGF therapy [31, 32].
In addition to low- and high-grade gliomas, antiangiogenic therapies such as bevacizumab have been found useful for the treatment of meningiomas and schwannomas, especially those associated with neurofibromatosis type II (NF2). NF2 is a neurocutaneous disorder with autosomal dominant inheritance affecting 1 in 33,000 people worldwide and is characterized by multiple benign neurological tumors. Recently, a retrospective analysis was performed investigating the treatment of meningiomas with bevacizumab in 15 patients with NF2. A volumetric radiographic response was seen in 14 of the patients’ 48 meningiomas (29 %) with a median duration of response of 4 months and median time to progression of 15 months [3]. NF2 is a disease with multiple benign neurological tumors, creating a role for antiangiogenic therapy to avoid the morbidity of multiple surgeries.
In a phase II clinical study evaluating the efficacy of aflibercept (VEGF Trap) in patients with recurrent malignant glioma, 42 patients with glioblastoma and 16 with anaplastic glioma were enrolled after first relapse from concurrent radiation and temozolomide and adjuvant temozolomide [33]. Overall, the progression-free survival rate was 8 % for patients with glioblastoma and 25 % for those with anaplastic glioma; the radiographic response was 18 % for the glioblastoma cohort and 44 % for the anaplastic glioma cohort. The progression-free survival was 12 and 24 weeks for the glioblastoma and anaplastic glioma cohort, respectively. This study supported the hypothesis that agents targeting VEGF directly are less potent as monotherapy and benefit from being part of a combination regimen, while agents targeting the kinase activity of the VEGF receptor have broad-spectrum activity against other receptor tyrosine kinases and are thus often used as monotherapy.
RTKIs are biologically active small molecules that bind the active site of a receptor tyrosine kinase (RTK) to prevent phosphorylation and in doing so modulate signaling [34]. RTKIs vary in which RTKs they effectively inhibit, but most have specificity for multiple RTKs. This makes many postulate that RTKs may be more effective as monotherapy than VEGF-targeted treatments such as bevacizumab, which might be best used in combination regimens. RTKIs are divided into three categories based on the primary RTK inhibited: epidermal growth factor receptor (EGFR), VEGFR, and platelet-derived growth factor receptor (PDGFR) [34].
Erlotinib is an orally available EGFR that binds to the ATP-binding domain to prevent phosphorylation and downstream target activation. First approved for the treatment of lung cancer, erlotinib has been shown to achieve high concentrations in the cerebrospinal fluid, implying satisfactory penetration through the blood-brain barrier [35]. Erlotinib has been evaluated in several single-arm phase II trials. Raizer et al. [36] showed erlotinib monotherapy to have minimal efficacy against recurrent glioblastoma with a progression-free survival (PFS) of 3 % and an overall survival (OS) of 6 months. Yung et al. [37] had better results with a PFS and OS of 20 % and 8.6 months, respectively. Additionally, van de Bent et al. [38] found in a randomized controlled trial that the median survival and 6-month PFS of erlotinib (1.8 months, 11.4 %) were worse than those of the combination of temozolomide and carmustine (2.4 months, 24.1 %). However, the overall survival was not significantly different among the groups with OS of 7.7 months for erlotinib and 7.3 months from temozolomide and carmustine. EGFRvIII mutations correlated with poorer survival in the erlotinib arm, but not in the control arm, suggesting that a subset of patients responds better than others and that blocking downstream EGFR signaling might improve the efficacy of EGFR inhibition in some tumors [34].
Sunitinib, a nonspecific RTKI, was initially approved by the FDA for treatment of renal cell carcinoma and imatinb-resistant gastrointestinal tumors. Sunitib is a small molecular inhibitor of PDGFR, VEGFR, stem cell-like factor receptor (c-KIT), and various other kinases implicated in tumorgenesis [39]. The multiple targets of sunitinib mean that it can cause tumor vascular regression, but also has increased risk for side effects. There are two studies evaluating sunitib for treatment of glioblastoma [40, 41]. The first study demonstrated that in human glioblastoma xenografts implanted in mice, sunitinib has direct antiproliferative effects as demonstrated by decreased MIB-1 staining [40]. The other study was a clinical trial in which 63 patients were stratified into bevacizumab-naïve and bevacizumab-resistant groups and received daily sunitinib treatment [41]. Comparison of the two groups revealed that the bevacizumab-naïve group possessed superior outcomes than the becazimuab-resistant group in terms of radiographic response (10 vs. 0 %), PFS (6 vs. 0 %), and OS (9.4 vs. 4.4 months). While this study showed a failure of sunitinib to offer significant improvement in clinical outcomes, it does highlight the importance of resistance following antiangiogenic therapy.
Classifying resistance mechanisms of brain cancer to antiangiogenic therapy
The enigma posed by antiangiogenic therapy is this: If angiogenesis is so vital to tumor well-being, how then does brain cancer return to growth and progression in the face of potent angiogenesis inhibitors? While this question cannot yet be answered completely, the lens of evolutionary biology can be useful in approaching it.
Cancer can be likened to an ecosystem, composed of heterogeneous cells in metabolic and proliferative coordination and equilibrium [42]. Antiangiogenic therapy is analogous to a selection pressure for the characteristically genetically unstable cancer cells. These cells often have the ability to co-opt support from cells of their microenvironment (e.g., endothelial cells, platelets, fibroblasts, pericytes, and leukocytes), which themselves might also be or become genetically unstable [43]. Thus, the potential mechanisms of resistance to antiangiogenic therapy are indeed adaptive and might be the result of certain subpopulations of tumor and tumor-associated cells becoming selectively advantaged in the face of the selection pressure of antiangiogenic therapy [1].
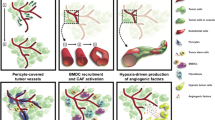
The modes of resistance to antiangiogenic therapy, specifically VEGF inhibitors, have been previously divided into two general categories: adaptive (evasive) resistance and intrinsic (preexisting) non-responsiveness [2]. In the sections to follow the multiple specific hypothesized mechanisms for adaptive resistance (see Fig. 1) will be discussed followed by a broad discussion of intrinsic non-responsiveness.

Six proposed mechanisms by which neurological tumors become resistant to antiangiogenic therapy
Adaptive resistance
As previously mentioned, a subset of patients receiving antiangiogenic therapy such as bevacizumab initially respond to treatment only to later have their tumor’s growth and progression reemerge. This clinical finding correlates with the first of two broad categories of resistance to antiangiogenic therapy—adaptive resistance. The hypothesis of adaptive resistance states that tumors can respond and adapt to the presence of antiangiogenic therapeutics by means that allow them to survive despite continued administration of said therapeutics [2]. Unlike resistance to chemotherapy, most adaptive resistance mechanisms involve transcriptional changes or post-translational protein modifications. These changes are generated more readily and consistently than the DNA gene mutations underpinning resistance to traditional DNA damaging chemotherapy [2]. The implications of these distinct mechanisms are that, when compared to resistance to DNA damaging chemotherapy, resistance to antiangiogenic therapy may occur more frequently, multiple mechanisms may occur simultaneously, and these mechanisms may also be potentially reversible if the agent is switched and reinstituted at a later date.
The impetus for the changes that confer adaptive resistance could be tumor cells sensing decreased VEGF signaling or the effects of VEGF inhibition on the tumor microenvironment, such as hypoxia and consequent hypoxia inducible factors (HIFs), which mediate numerous pathways affecting tumor activity and progression [2, 44]. Generally speaking, tumors adapt to antiangiogenic therapy by modulating their behavior and metabolism such that reinitiating of angiogenesis is no longer essential or by finding new ways to reestablish angiogenesis and/or promote neovascularization [45].
Currently, there are at least six distinct yet possibly interrelated mechanisms of adaptive resistance to antiangiogenic therapies (Fig. 1): first, upregulation and/or activation of alternative proangiogenic signaling pathways; second, recruitment of proangiogenic cells to the tumor to reinitiate angiogenesis; third, increased pericyte coverage of tumor blood vessels, which increases vessel stability and decreases dependence on VEGF signaling; fourth, autophagy as a cytoprotective response to hypoxia; fifth, vasculogenic mimicry; sixth, activation and enhancement of tumor invasion, which provides the tumor access to normal tissue vasculature and relieves dependence on neovascularization.
In the following subsections, the hypothetical mechanisms by which adaptive resistance occurs in cancers of the central nervous system will be supported through discussion of incipient experimental evidence.
Upregulation of alternative proangiogenic signaling pathways
Of the mechanisms for antioangiogenic resistance, upregulation and/or activation of alternative proangiogenic signaling pathways is perhaps the best described and biologically logical. Like other important and complex biological processes, angiogenesis is influenced by multiple signals, although VEGF is considered necessary for angiogenesis in nonpathological contexts [9]. When VEGF signaling is no longer supporting angiogenesis, an alternative pathway may take a more crucial role in the promotion of angiogenesis. Several signaling systems have been found to be upregulated after treatment with antiangiogenic therapy in in vivo and in vitro models, which will be discussed in this section.
Upregulation of proangiogenic factors in relapsing tumors was first noted in mouse models of pancreatic neuroendocrine cancer, Rip1-Tag2 [46]. In these preclinical trials, the genetically engineered Rip1-Tag2 mice were treated with a monoclonal antibody (DC101), and an initial, but transitory response (lasting 10–14 days) was noted with decreased tumor vascularity and halted tumor growth. The relapsing tumor contained significantly higher levels of several proangiogenic factors [fibroblast growth factor 1 (Fgf1) and Fgf2, angiopoiten, ephrin A1, and ephrin A2] when compared to levels in untreated tumors [46]. A similar evasive resistance was noted by Batchelor et al. [47] in their clinical study in which recurrent glioblastoma patients were treated with daily administration of AZD2171 (oral tyrosine kinase inhibitor of VEGF receptors). They observed increased levels of both basic fibroblast growth factor (bFGF) and SDF1α (and viable circulating endothelial cells) in the blood when the tumors escaped treatment after a 28-day response phase [47]. More recent studies have confirmed these initial findings and expanded the number of alternative angiogenic pathways that can compensate for VEGF pathway inhibition. For example, Lucio-Eterovic et al. [16] noted the upregulation of several proangiogenic molecules (e.g., interleukin-1α, transforming growth factor α, etc.) in two glioblastoma cell lines (U87 and NSC23) after bevacizumab treatment. In both cell lines, angiogenin and bFGF were upregulated in response to treatment, with angiogenin being most upregulated [16].
Angiopoietins are a family of molecules that have important roles in angiogenesis in normal and tumor blood vessels. Normally, pericytes express angiopoietin-1 (Ang-1), which promotes blood vessel survival and stabilization by binding Tie2 tyrosine kinase receptor on endothelial cells [48, 49]. However, in glioblastoma there is believed to be increased expression of Ang-1 as well as the Ang-1 context-dependent antagonist/agonist, Ang-2, with Ang-1 being expressed by the tumor cells and Ang-2 by the tumor blood vessels [50]. Current investigations have attempted to address whether Ang-2 might have a proangiogenic function in tumors. Rigamonti et al. [27] reported increased Ang-2 expression in PNET (pancreatic neuroendocrine) tumors as well-enhanced infiltration by TIE2-expressing macrophages in the PNETs upon VEGFR2 inhibition. The proangiogenic function of angiopoietins has yet to be completely delineated in brain cancer.
It is important to note that many of the same molecules (e.g., Ang-2, β8 integrin) implicated in alternative proangiogenic signaling pathways are also thought to operate in other adaptive resistance mechanisms such as vessel co-option and recruitment of proangiogenic cells, which will be discussed in subsequent sections.
Recruitment of proangiogenic marrow-derived cells
In addition to activating alternative angiogenic pathways as discussed in the previous section, the hypoxic tumor conditions resulting from functional loss of vasculature caused by antiangiogenic therapy have been shown to result in recruitment of several different cell types to the tumor to improve its vascularization [2]. Two primary cell types are recruited from the bone marrow: cells that contribute to the process of vasculogenesis and cells that support angiogenesis [45, 51].
In response to the hypoxic stimulus, bone marrow-derived precursors of pericytes and endothelial cells are recruited; these cells are thought to improve tumor vascularization through the process of vasculogenesis [52–54]. Instead of new vessels being formed from pre-existing vessels (angiogenesis), vasculogenesis is a process by which endothelial precursor cells called angioblasts migrate and then undergo differentiation in response to a variety of signals (e.g., growth factors) in their local environment to form new blood vessels [55]. To promote angiogenesis, myeloid BMDCs are also recruited to the tumor in hypoxic conditions where they secrete various proangiogenic molecules such as matrix metalloproteinase (MMP)-9, Bv8, and G-CSF [56].
More specifically, several studies have identified the mechanism by which antiangiogenic therapy/hypoxia induce glioblastoma tumor cells to secrete factors in order to recruit BDMCs, which promote both vasculogenesis and angiogenesis, to the site of the tumor. For example, Du et al. [54] demonstrated the importance of HIF1α, which is upregulated in response to tumor hypoxia, in the recruitment of BMDCs. By transplanting bone marrow cells from β-actin-EGFP mice into HIF1α-proficient and HIF1α-deficient mice, they found that tumors in HIF1α-proficient mice contained about three times the amount of CD45+ monocytic cells, seven times more PDGFRβ+/Sca-1+ pericyte precursor cells, and nearly four times the number of endothelial precursor cells as HIF1α-deficient (knockout) mice [54]. Aghi et al. [57] showed that CD45+ myeloid cells are attracted to and retained within glioblastoma tumors by stromal-derived factor 1α (SDF1α/CXCL12). Du et al. [54] added to this finding by showing one important way in which HIF1α attracts BMDCs to the tumor is through induction of CXCL12. These findings have been supported by subsequent studies; for example, Guo et al. [58] demonstrated that endothelial precursor cells (identified based on gene expression profiling and cell markers) could be isolated from malignant gliomas. However, the original source of these cells was not investigated. Furthermore, in addition to HIF1α, other molecules such as PDGF-BB have been implicated in hypoxia-induced recruitment of BMDCs [51]. PDGF has been shown to upregulate CXC12 expression in human endothelial cells, further emphasizing the importance of the CXC12-CXCR4 pathway in pericyte recruitment [59]. Interestingly, as discussed in the next section, PDGF may also modify interactions between endothelial cells and pericytes in hypoxic gliomas.
Most of the BMDCs are myeloid CD45+ cells, which, after leaving the bone marrow as mature monocytes, migrate to and enter into the tumor where they mature into macrophages [60]. There are in fact two types of tumor-associated macrophages in human gliomas: M1 macrophages mediate antitumor immunity, and M2 macrophages support angiogenesis [61]. M2 macrophages produce substantial amounts of various growth factors (e.g., VEGF, IL-8, bFGF) and metalloproteases (MMP-1,2,9) [60, 62, 63]. These secreted molecules work together in a coordinated fashion to promote neovascularization; for example, the extracellular matrix degradation promoted by metalloproteinases is an important first step in angiogenesis [64]. Additionally, the actions of the metalloproteases might also be important in facilitating the infiltration of BMDCs leading to a positive feedback cycle that promotes continued angiogenesis [65].
In summary, in response to hypoxia, tumor cells secrete various factors such as HIF and PDGF to recruit cells from the bone marrow in order to promote neovascularization. The bone marrow-derived endothelial cell and pericyte precursors support vasculogenesis, whereas the bone marrow-derived CD45+ cells become M2 macrophages that secrete a variety of factors to support angiogenesis.
Increased pericyte coverage
Pericytes, one type of vascular mural cell, have a multifaceted and important role in vessel stabilization and formation and, more generally, in the tumor microenvironment. Currently, aberrations in endothelial cells, pericytes, and their interactions are thought to contribute to the abnormalities in tumor vasculature (e.g., disorganization, increased permeability) [66]. In response to environmental stress, pericytes are recruited to vascular endothelial cells (via PDGF-B signaling), where they are thought to provide trophic support to endothelial cells via secretion of molecules such as VEGF [66–68]. Pericytes also play important roles in hypoxic remodeling of vessels; they, along with the basement membrane, are thought to provide a scaffold that facilitates vascular regrowth after administration of antiangiogenic therapies [69].
Pericytes’ role in resistance to antiangiogenic therapy was initially supported by two important observations: first, differences exist in pericyte coverage before and after administration of antiangiogenic therapy; second, tumor vessels with inadequate pericyte coverage are more susceptible to anti-VEGF agents [69–74]. In response to antiangiogenic therapy, pericytes become more numerous and more tightly associated with the endothelial cells than they were in pretreatment tumor vasculature [73]. These observations suggest that in response to hypoxic stress or absence of survival signals (e.g., VEGF), endothelial cells can recruit pericytes to protect themselves from death due to insufficient survival signals that occur via VEGF signaling [2].
The finding that perciytes possess stabilizing effects on endothelial cells in response to antiangiogenic therapy makes dual targeting of endothelial cells and pericytes a reasonable therapeutic strategy. In fact, it has been shown in mouse models that inhibiting PDGF-mediated pericyte recruitment increases the efficacy of antiangiogenic therapy as evidenced by increased vessel regression and tumor hypoxia [70, 75]. Furthermore, di Tomaso et al. [76] detected the presence of PDGF-C (an isoform of platelet-derived growth factor) in the U87MG human glioblastoma cell line. The tumors overexpressing PDGF-C had smaller vessel diameters and decreased vascular permeability than the parental and siRNA-transfected tumors. Importantly, PDGF-C overexpressing tumors possessed more extensive coverage with perivascular cells and thicker basement membranes. Finally, parental tumors, but not PDGF-C overexpressing tumors, had decreased vessel density upon application of a VEGFR2 antibody (DC101) [76]. Taken together, these findings suggest PDGF-C may allow for vessel stabilization to escape the vascular normalization associated antiangiogenic therapy in human gliomas. Many current therapies (e.g., sunitinib, pazopanib, sorafenib) are potent inhibitors of VEGF and PDGF receptors; thus, some of their efficacy may be attributable to impairment of pericyte recruitment. In a more recent study, pericyte depletion was observed in response to sunitinib therapy in vivo in two different metastatic breast cancer cell lines [77]. However, in response to pericyte depletion, these tumors became more metastatic, demonstrating how pericytes can influence a variety of tumor properties. This being said, few studies have specifically investigated pericytes' role in brain cancer vasculature stabilization as a means of adaptive resistance to antiangiogenic therapy.
Upregulating hypoxia survival mechanisms
Autophagy, a lysosomal degradation pathway, is an interesting and distinct mechanism by which glioblastoma has been hypothesized to resist antiangiogenic therapy. Autophagy occurs when an isolation membrane forms by enclosing cellular structures targeted for destruction to create an autophagosome; the autophagosome then fuses with the lysosome so that enzymatic degradation of autophagosome contents can occur [78]. While formation of autophagosomes is typically associated with cell death (e.g., in apoptosis), several studies suggest that autophagy allows cells to cope with stressors both intrinsic and extrinsic (e.g., chemotherapy, radiotherapy) by destroying damaged proteins and organelles before these damaged contents trigger apoptotic cell death as a survival strategy [79–82].
It has been hypothesized that the devascularization that results from antiangiogenic therapy induces hypoxia, and in response to this hypoxia, autophagy occurs, alloting resistance to antiangiogenic therapy in glioblastoma [83]. Autophagy was first reported as a novel resistance mechanism to anti-VEGF therapy in glioblastoma by Hu et al. [83]. This study included multiple pieces of evidence for the hypothesis that anti-VEGF therapy promotes autophagy because of its hypoxic effects. Hypoxia-induced autophagy was found to be dependent on signaling through the hypoxia-inducible factor 1α (HIF-1α)/AMPK pathway, and treatment of hypoxic cells with autophagy inhibitors caused a shift from autophagic to apoptotic cell death in vitro. Additionally, in glioblastomas clinically resistant to the VEGF-neutralizing antibody bevacizumab, increased regions of hypoxia and higher levels of autophagy-mediating BNIP3 were found when compared with pretreatment specimens from the same patients. When treated with bevacizumab alone, human glioblastoma xenografts showed increased BNIP3 expression and hypoxia-associated growth, which could be prevented by addition of the autophagy inhibitor chloroquine. Finally, in vivo targeting of the essential autophagy gene ATG7 also disrupted tumor growth when combined with bevacizumab treatment [83]. This initial study’s findings also have been supported by findings from other groups. Shen et al. [84] found that ZD6474 (a small-molecule inhibitor of VEGFR, EGFR, and RET tyrosine kinases) induced autophagy in U251 glioblastoma cells. Furthermore, in a xenograft mouse model, chloroquine, a pharmacological inhibitor of autophagy, and ZD6474 both individually inhibited U251 tumor growth; a combination of these agents increased the apoptotic cell number compared with application of either agent alone [84].
Together, these findings elucidate a unique mechanism of resistance to antiangiogenic therapy in which hypoxia-mediated autophagy promotes tumor cell survival. One strong implication of these findings is that autophagy inhibitors may help prevent resistance to antiangiogenic therapy used in the clinic. In the context of other adaptive resistance mechanisms, this mechanism may be a part of a larger strategy in that it could allow the brain cancer cells to manage the initial hypoxic insult and then go on to recruit another mechanism to increase oxygenation/angiogenesis and subsequent proliferation. The cytoprotective autophagic response is thus one plausible adaptive response mechanism to antiangiogenic therapy.
Vasculogenic mimicry
Vasculogenic mimicry is a unique hypothesized way by which gliomas are thought to promote vascularization following antiangiogenic therapy [24]. In vasculogenic mimicry (VM), glioblastoma tumor stem-like cells differentiate into endothelial cells or pericytes, which are then organized into vessel-like structures that are perfused via connections with preexisting vessels in the tumor microenvironment [85–88]. One recent experiment investigated the hypothesis that VEGF receptor 2 (Flk-1) is an important mediator of VM in glioblastoma [89]. Treatment of two glioblastoma cell lines (U87 and GSDC) with Flk-1 gene knockdown followed by implantation or treatment of implanted wild-type tumors with the Flk-1 kinase inhibitor SU1498 impaired vascular structure and function, with significant decreases in smooth muscle α-actin expression and tube formation observed [89]. Unlike treatment with SU1498, bevacizumab administration failed to reduce expression of smooth muscle α-actin and, relatedly, did not induce dysfunction of vascular formation in either cell line [89]. Furthermore, VEGF administration failed to improve the impaired capability of tube formation in Flk-1shRNA-treated cells [89]. Together these findings suggest that Flk-1 is essential for VM in glioblastoma and signaling through Flk-1 can occur in the absence of VEGF. Wang et al. [87] found that different signaling cascades govern different steps in the maturation of multipotent glioblastoma tumor stem cells to endothelial cells. NOTCH1 silencing or γ-secretase inhibition impairs the differentiation of CD133+ cells into endothelial progenitors, whereas VEGFR2 inhibition interferes with maturation of tumor endothelial progenitors in endothelium [87].
Hypoxia also appears to be important in the regulation of glioblastoma stem cell differentiation into endothelial cells, as glioblastoma stem cell-derived endothelial cells are often localized to deeper, hypoxic areas of the tumor and are less likely to be found at the tumor surface [90]. Thus, vasculogenic mimicry is a hypothesized way by which brain tumors ensure vascularization in a VEGF-independent fashion. Hypoxia-driven Notch signaling and/or molecules such as Flk-1 might be important in mediating vasculogenic mimicry and possibly do so in a sequential fashion.
Increased perivascular invasiveness
Enhancement of invasiveness is a problematic, clinically documented mechanism of adaptive resistance that occurs in glioblastoma in response to antiangiogenic therapy [91]. Invasiveness is a process that allows for glioblastoma to co-opt vessels, thereby abrogating the tumor’s necessity for neovascularization [2]. Vessel co-option is the recruitment of local blood vessels by a tumor occurring as the tumor invades the surrounding tissue [92].
The antiangiogenic therapy-induced adaptive phenotype of increased invasiveness was first demonstrated in mouse models of orthotopic glioblastoma. In these models, neovascularization was inhibited via pharmacological blockade of VEGF with SU5416 (semaxanib) or inhibited via genetic deletion of angiogenic factors such as VEGF, HIF1α, and matrix metalloproteinase 9 [93]. These glioblastoma cells were observed to become more invasive, continue to grow (although at a slower rate), and also co-opt blood vessels (referred to as a perivascular tumor invasion phenotype) [2, 93]. Interestingly, other studies showed that glioblastoma cells treated with antiangiogenic agents invade normal brain tissue in a different manner than untreated glioblastoma cells with treated cells migrating as multicellular layers along normal blood vessels rather than as single cells migrating along basement membranes of leptomeninges, blood vessels, and ventricles [2]. The difference in invasive patterns lends additional support to the idea that antiangiogenic therapy causes increased invasiveness.
Since the first model, multiple mouse models have demonstrated increased invasive properties upon genetic and pharmacologic inhibition of angiogenesis in glioblastoma with both hypoxia-dependent and -independent mechanisms being proposed [93–96]. c-Met is a hepatocyte growth factor receptor (HGFR) tyrosine kinase that activates endothelial cells and also affects multiple properties of cancer cells, promoting proliferation, invasion, survival, etc. [97]. Eckerich et al. [98] reported that c-Met transcription and protein levels were elevated in half of glioblastoma cell lines and primary cultures and HIF1α levels were increased with hypoxia. Transfection of siRNA against HIF-1α abrogated the hypoxic induction of c-Met, suggesting that c-Met expression is upregulated by a HIF-1α-dependent mechanism [98]. Furthermore, multiple studies have shown that increased c-Met expression/activity in glioblastoma correlates with increased tumor invasiveness in response to hypoxia and antiangiogenic treatment and also correlates with poorer survival and increased invasion [76, 96, 98].
Additionally, hypoxia-independent invasive resistance mechanisms exist. Lu et al. [96] demonstrated that VEGF is a direct, negative regulator of invasiveness through use of different mouse astrocytoma cell lines that differed only in their VEGF expression. It was found that intratumoral VEGF levels inversely correlate with the extent of MET phosphorylation and invasiveness of the glioblastoma tumor cells [96]. Furthermore, interactions between the HGF and VEGF pathways were elucidated; HGF-dependent MET phosphorylation was inhibited in a dose-dependent fashion by VEGF-mediated enhanced recruitment of the non-receptor protein tyrosine phosphatase 1B (PTP1B) to a MET/VEGFR2 heterocomplex [96]. These findings suggest coadministration of agents targeting both the VEGF pathway and c-Met in glioma tumors might be a viable treatment modality for glioblastoma [45].
That being said, c-Met signaling, while well established, is not the only hypothesized mediator of increased invasiveness in response to antiangiogenic therapy. For example, integrins such as β8 integrin have also been linked to invasive growth properties of glioblastoma as well as increased angiogenesis. Interestingly Tchaicha et al. [99] found that poorly invasive tumors were highly angiogenic and had low β8 integrin expression, whereas high invasiveness was associated with decreased neovascularization and elevated β8 integrin expression in U87 glioblastoma cells. These associations were also observed after genetic manipulation of β8 integrin expression through the use of shRNA and transfection. This study lends support to the idea of heterogeneity among adaptive resistance strategies depending on the tumor type and/or its microenvironmental conditions [99].
Intrinsic resistance
As previously mentioned, a substantial subset of patients with high-grade gliomas fail to respond in any capacity to antiangiogenic treatment [100]. This is most likely due to intrinsic resistance of the tumor to antiangiogenic therapy acquired as a consequence of tumor development and/or the hypoxic selection pressures that occur during this time period [2]. Intrinsic or pre-existing resistance in a tumor occurs when antiangiogenic therapy fails to produce any beneficial effect (i.e., no tumor shrinkage, growth cessation, or decreased growth rate) [2].
While it may be difficult to definitively distinguish rapidly developed adaptive resistance from intrinsic resistance [2], clinical reports suggest that anti-VEGF agents such as bevacizumab, sorafenib, and sunitinib fail to show even transitory radiographic response or clinical benefits in some patients with tumors being of increased size at the time of their first monitoring after initiation of therapy [2, 47, 101]. Intrinsic resistance can be attributed to the preexistence of one or more of the aforementioned adaptive mechanisms. The identification of resistance markers might hopefully allow identification of intrinsic resistance by screening patients before treatment, thereby saving these patients the cost and morbidity of treatments that were never going to be effective.
Future perspectives
There are several areas from which advancements in our understanding of resistance to antiangiogenic therapy and therapeutic strategies might arise. Brain tumors, especially glioblastoma, are regarded as highly heterogeneous, which may explain the discrepancies in tumor response to antiangiogenic therapy [45, 102]. The fact that VEGF signaling remains inhibited makes combination therapy a viable option for treatment of glioblastoma. Therapies that take into account factors such as biomarkers and radiographic features to predict which patients will respond to antiangiogenic therapy will be beneficial for improving patient outcomes [34, 45, 103]. Furthermore, new delivery vehicles such as mesenchymal stem cells and nanoparticles are being developed, which might increase the efficacy of existing therapies [104, 105].
Our understanding of the mechanisms that underlie resistance to antiangiogenic therapy and knowledge of the specific pathways that mediate this resistance continue to improve. Over the past few years, new adaptive resistance mechanisms and new insights into how different pathways might interact have emerged. While dichotomized into separate mechanisms in this review, reality is likely more nuanced with individual mechanisms possibly occurring simultaneously and in a coordinated fashion as a part of a larger and more complex strategy of tumor resistance. These discoveries have the potential to improve the efficacy of current therapeutics for antiangiogenic treatment of human brain cancer either by identifying new targets or via administration of multiple pathway inhibitors.
References
Bottsford-Miller JN, Coleman RL, Sood AK (2012) Resistance and escape from antiangiogenesis therapy: clinical implications and future strategies. J Clin Oncol Off J Am Soc Clin Oncol 30:4026–4034. doi:10.1200/JCO.2012.41.9242
Bergers G, Hanahan D (2008) Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 8:592–603. doi:10.1038/nrc2442
Nunes FP, Merker VL, Jennings D et al (2013) Bevacizumab treatment for meningiomas in NF2: a retrospective analysis of 15 patients. PLoS One 8:e59941. doi:10.1371/journal.pone.0059941
Chamberlain MC (2011) Bevacizumab for the treatment of recurrent glioblastoma. Clin Med Insights Oncol 5:117–129. doi:10.4137/CMO.S7232
Adams RH, Alitalo K (2007) Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 8:464–478. doi:10.1038/nrm2183
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Baeriswyl V, Christofori G (2009) The angiogenic switch in carcinogenesis. Semin Cancer Biol 19:329–337. doi:10.1016/j.semcancer.2009.05.003
Moens S, Goveia J, Stapor PC et al (2014) The multifaceted activity of VEGF in angiogenesis—implications for therapy responses. Cytokine Growth Factor Rev 25:473–482. doi:10.1016/j.cytogfr.2014.07.009
Hoeben A, Landuyt B, Highley MS et al (2004) Vascular endothelial growth factor and angiogenesis. Pharmacol Rev 56:549–580. doi:10.1124/pr.56.4.3
Lee S, Chen TT, Barber CL et al (2007) Autocrine VEGF signaling is required for vascular homeostasis. Cell 130:691–703. doi:10.1016/j.cell.2007.06.054
Bello L, Giussani C, Carrabba G et al (2004) Angiogenesis and invasion in gliomas. Cancer Treat Res 117:263–284
Kobayashi N, Allen N, Clendenon NR, Ko LW (1980) An improved rat brain-tumor model. J Neurosurg 53:808–815. doi:10.3171/jns.1980.53.6.0808
Hanahan D, Folkman J (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86:353–364
Cea V, Sala C, Verpelli C (2012) Antiangiogenic therapy for glioma. J Signal Transduct 2012:e483040. doi:10.1155/2012/483040
Plate KH, Breier G, Weich HA, Risau W (1992) Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 359:845–848. doi:10.1038/359845a0
Lucio-Eterovic AK, Piao Y, de Groot JF (2009) Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin Cancer Res Off J Am Assoc Cancer Res 15:4589–4599. doi:10.1158/1078-0432.CCR-09-0575
Karcher S, Steiner H-H, Ahmadi R et al (2006) Different angiogenic phenotypes in primary and secondary glioblastomas. Int J Cancer J Int Cancer 118:2182–2189. doi:10.1002/ijc.21648
Cook KM, Figg WD (2010) Angiogenesis inhibitors: current strategies and future prospects. CA Cancer J Clin 60:222–243. doi:10.3322/caac.20075
Lakka SS, Rao JS (2008) Antiangiogenic therapy in brain tumors. Expert Rev Neurother 8:1457–1473. doi:10.1586/14737175.8.10.1457
Valter MM, Wiestler OD, Pietsche T, Pietsch T (1999) Differential control of VEGF synthesis and secretion in human glioma cells by IL-1 and EGF. Int J Dev Neurosci Off J Int Soc Dev Neurosci 17:565–577
Berkman RA, Merrill MJ, Reinhold WC et al (1993) Expression of the vascular permeability factor/vascular endothelial growth factor gene in central nervous system neoplasms. J Clin Invest 91:153–159. doi:10.1172/JCI116165
Koutsimpelas D, Stripf T, Heinrich UR et al (2007) Expression of vascular endothelial growth factor and basic fibroblast growth factor in sporadic vestibular schwannomas correlates to growth characteristics. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol 28:1094–1099. doi:10.1097/MAO.0b013e31814b2787
Rosen LS (2002) Clinical experience with angiogenesis signaling inhibitors: focus on vascular endothelial growth factor (VEGF) blockers. Cancer Control J Moffitt Cancer Cent 9:36–44
Vasudev NS, Reynolds AR (2014) Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis 17:471–494. doi:10.1007/s10456-014-9420-y
Reardon DA, Turner S, Peters KB et al (2011) A review of VEGF/VEGFR-targeted therapeutics for recurrent glioblastoma. J Natl Compr Cancer Netw JNCCN 9:414–427
Zustovich F, Landi L, Lombardi G et al (2013) Sorafenib plus daily low-dose temozolomide for relapsed glioblastoma: a phase II study. Anticancer Res 33:3487–3494
Rigamonti N, Kadioglu E, Keklikoglou I et al (2014) Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell Rep 8:696–706. doi:10.1016/j.celrep.2014.06.059
Herbst RS, Hong D, Chap L et al (2009) Safety, pharmacokinetics, and antitumor activity of AMG 386, a selective angiopoietin inhibitor, in adult patients with advanced solid tumors. J Clin Oncol Off J Am Soc Clin Oncol 27:3557–3565. doi:10.1200/JCO.2008.19.6683
Desgrosellier JS, Cheresh DA (2010) Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 10:9–22. doi:10.1038/nrc2748
Reardon DA (2014) Update on the use of angiogenesis inhibitors in adult patients with brain tumors. Clin Adv Hematol Oncol HO 12:293–303
Norden AD, Drappatz J, Muzikansky A et al (2009) An exploratory survival analysis of anti-angiogenic therapy for recurrent malignant glioma. J Neurooncol 92:149–155. doi:10.1007/s11060-008-9745-8
Xu T, Chen J, Lu Y, Wolff JE (2010) Effects of bevacizumab plus irinotecan on response and survival in patients with recurrent malignant glioma: a systematic review and survival-gain analysis. BMC Cancer 10:252. doi:10.1186/1471-2407-10-252
De Groot JF, Lamborn KR, Chang SM et al (2011) Phase II study of aflibercept in recurrent malignant glioma: a North American Brain Tumor Consortium study. J Clin Oncol Off J Am Soc Clin Oncol 29:2689–2695. doi:10.1200/JCO.2010.34.1636
Lau D, Magill ST, Aghi MK (2014) Molecularly targeted therapies for recurrent glioblastoma: current and future targets. Neurosurg Focus 37:E15. doi:10.3171/2014.9.FOCUS14519
Deng Y, Feng W, Wu J et al (2014) The concentration of erlotinib in the cerebrospinal fluid of patients with brain metastasis from non-small-cell lung cancer. Mol Clin Oncol 2:116–120. doi:10.3892/mco.2013.190
Raizer JJ, Abrey LE, Lassman AB et al (2010) A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol 12:95–103. doi:10.1093/neuonc/nop015
Yung WKA, Vredenburgh JJ, Cloughesy TF et al (2010) Safety and efficacy of erlotinib in first-relapse glioblastoma: a phase II open-label study. Neuro-Oncol 12:1061–1070. doi:10.1093/neuonc/noq072
Van den Bent MJ, Brandes AA, Rampling R et al (2009) Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol Off J Am Soc Clin Oncol 27:1268–1274. doi:10.1200/JCO.2008.17.5984
Chow LQM, Eckhardt SG (2007) Sunitinib: from rational design to clinical efficacy. J Clin Oncol Off J Am Soc Clin Oncol 25:884–896. doi:10.1200/JCO.2006.06.3602
De Boüard S, Herlin P, Christensen JG et al (2007) Antiangiogenic and anti-invasive effects of sunitinib on experimental human glioblastoma. Neuro-Oncol 9:412–423. doi:10.1215/15228517-2007-024
Kreisl TN, Smith P, Sul J et al (2013) Continuous daily sunitinib for recurrent glioblastoma. J Neurooncol 111:41–48. doi:10.1007/s11060-012-0988-z
Merlo LMF, Pepper JW, Reid BJ, Maley CC (2006) Cancer as an evolutionary and ecological process. Nat Rev Cancer 6:924–935. doi:10.1038/nrc2013
Akino T, Hida K, Hida Y et al (2009) Cytogenetic abnormalities of tumor-associated endothelial cells in human malignant tumors. Am J Pathol 175:2657–2667. doi:10.2353/ajpath.2009.090202
Kaur B, Khwaja FW, Severson EA et al (2005) Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro-Oncol 7:134–153. doi:10.1215/S1152851704001115
Lu KV, Bergers G (2013) Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. CNS Oncol 2:49–65. doi:10.2217/cns.12.36
Casanovas O, Hicklin DJ, Bergers G, Hanahan D (2005) Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 8:299–309. doi:10.1016/j.ccr.2005.09.005
Batchelor TT, Sorensen AG, di Tomaso E et al (2007) AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 11:83–95. doi:10.1016/j.ccr.2006.11.021
Eklund L, Olsen BR (2006) Tie receptors and their angiopoietin ligands are context-dependent regulators of vascular remodeling. Exp Cell Res 312:630–641. doi:10.1016/j.yexcr.2005.09.002
Bergers G, Song S (2005) The role of pericytes in blood-vessel formation and maintenance. Neuro-Oncol 7:452–464. doi:10.1215/S1152851705000232
Zagzag D, Amirnovin R, Greco MA et al (2000) Vascular apoptosis and involution in gliomas precede neovascularization: a novel concept for glioma growth and angiogenesis. Lab Investig J Tech Methods Pathol 80:837–849
Boer JC, Walenkamp AME, den Dunnen WFA (2014) Recruitment of bone marrow derived cells during anti-angiogenic therapy in GBM: the potential of combination strategies. Crit Rev Oncol Hematol 92:38–48. doi:10.1016/j.critrevonc.2014.05.001
Asahara T, Murohara T, Sullivan A et al (1997) Isolation of putative progenitor endothelial cells for angiogenesis. Science 275:964–967
Song S, Ewald AJ, Stallcup W et al (2005) PDGFRbeta+ perivascular progenitor cells in tumours regulate pericyte differentiation and vascular survival. Nat Cell Biol 7:870–879. doi:10.1038/ncb1288
Du R, Lu KV, Petritsch C et al (2008) HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 13:206–220. doi:10.1016/j.ccr.2008.01.034
Patan S (2004) Vasculogenesis and angiogenesis. Cancer Treat Res 117:3–32
Shojaei F, Wu X, Malik AK et al (2007) Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+ Gr1+ myeloid cells. Nat Biotechnol 25:911–920. doi:10.1038/nbt1323
Aghi M, Cohen KS, Klein RJ et al (2006) Tumor stromal-derived factor-1 recruits vascular progenitors to mitotic neovasculature, where microenvironment influences their differentiated phenotypes. Cancer Res 66:9054–9064. doi:10.1158/0008-5472.CAN-05-3759
Guo K-T, Juerchott K, Fu P et al (2012) Isolation and characterization of bone marrow-derived progenitor cells from malignant gliomas. Anticancer Res 32:4971–4982
Song N, Huang Y, Shi H et al (2009) Overexpression of platelet-derived growth factor-BB increases tumor pericyte content via stromal-derived factor-1alpha/CXCR4 axis. Cancer Res 69:6057–6064. doi:10.1158/0008-5472.CAN-08-2007
Mantovani A, Sozzani S, Locati M et al (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23:549–555
Mantovani A, Allavena P (2004) Sica A () Tumour-associated macrophages as a prototypic type II polarised phagocyte population: role in tumour progression. Eur J Cancer Oxf Engl 40:1660–1667. doi:10.1016/j.ejca.2004.03.016
Heusinkveld M, van der Burg SH (2011) Identification and manipulation of tumor associated macrophages in human cancers. J Transl Med 9:216. doi:10.1186/1479-5876-9-216
Solinas G, Germano G, Mantovani A, Allavena P (2009) Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol 86:1065–1073. doi:10.1189/jlb.0609385
Rundhaug JE (2005) Matrix metalloproteinases and angiogenesis. J Cell Mol Med 9:267–285
Pollard JW (2004) Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4:71–78. doi:10.1038/nrc1256
Barlow KD, Sanders AM, Soker S et al (2012) Pericytes on the tumor vasculature: Jekyll or Hyde? Cancer Microenviron 6:1–17. doi:10.1007/s12307-012-0102-2
Abramsson A, Lindblom P, Betsholtz C (2003) Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest 112:1142–1151. doi:10.1172/JCI18549
Jain RK, Booth MF (2003) What brings pericytes to tumor vessels? J Clin Invest 112:1134–1136. doi:10.1172/JCI200320087
Mancuso MR, Davis R, Norberg SM et al (2006) Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest 116:2610–2621. doi:10.1172/JCI24612
Bergers G, Song S, Meyer-Morse N et al (2003) Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest 111:1287–1295. doi:10.1172/JCI17929
Jain RK (2005) Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307:58–62. doi:10.1126/science.1104819
Kamba T, McDonald DM (2007) Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br J Cancer 96:1788–1795. doi:10.1038/sj.bjc.6603813
Baluk P, Hashizume H, McDonald DM (2005) Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev 15:102–111. doi:10.1016/j.gde.2004.12.005
Benjamin LE, Hemo I, Keshet E (1998) A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Dev Camb Engl 125:1591–1598
Erber R, Thurnher A, Katsen AD et al (2004) Combined inhibition of VEGF and PDGF signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. FASEB J Off Publ Fed Am Soc Exp Biol 18:338–340. doi:10.1096/fj.03-0271fje
Di Tomaso E, London N, Fuja D et al (2009) PDGF-C induces maturation of blood vessels in a model of glioblastoma and attenuates the response to anti-VEGF treatment. PLoS One 4:e5123. doi:10.1371/journal.pone.0005123
Welti JC, Powles T, Foo S et al (2012) Contrasting effects of sunitinib within in vivo models of metastasis. Angiogenesis 15:623–641. doi:10.1007/s10456-012-9291-z
Xie Z, Klionsky DJ (2007) Autophagosome formation: core machinery and adaptations. Nat Cell Biol 9:1102–1109. doi:10.1038/ncb1007-1102
Boya P, González-Polo R-A, Casares N et al (2005) Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 25:1025–1040. doi:10.1128/MCB.25.3.1025-1040.2005
Lum JJ, Bauer DE, Kong M et al (2005) Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120:237–248. doi:10.1016/j.cell.2004.11.046
Sato K, Tsuchihara K, Fujii S et al (2007) Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res 67:9677–9684. doi:10.1158/0008-5472.CAN-07-1462
Hu Y-L, Jahangiri A, DeLay M, Aghi MK (2012) Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. Cancer Res 72:4294–4299. doi:10.1158/0008-5472.CAN-12-1076
Hu Y-L, DeLay M, Jahangiri A et al (2012) Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res 72:1773–1783. doi:10.1158/0008-5472.CAN-11-3831
Shen J, Zheng H, Ruan J et al (2013) Autophagy inhibition induces enhanced proapoptotic effects of ZD6474 in glioblastoma. Br J Cancer 109:164–171. doi:10.1038/bjc.2013.306
Folberg R, Maniotis AJ (2004) Vasculogenic mimicry. APMIS Acta Pathol Microbiol Immunol Scand 112:508–525. doi:10.1111/j.1600-0463.2004.apm11207-0810.x
Cheng L, Huang Z, Zhou W et al (2013) Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153:139–152. doi:10.1016/j.cell.2013.02.021
Wang R, Chadalavada K, Wilshire J et al (2010) Glioblastoma stem-like cells give rise to tumour endothelium. Nature 468:829–833. doi:10.1038/nature09624
Ricci-Vitiani L, Pallini R, Biffoni M et al (2010) Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 468:824–828. doi:10.1038/nature09557
Francescone R, Scully S, Bentley B et al (2012) Glioblastoma-derived tumor cells induce vasculogenic mimicry through Flk-1 protein activation. J Biol Chem 287:24821–24831. doi:10.1074/jbc.M111.334540
Soda Y, Marumoto T, Friedmann-Morvinski D et al (2011) Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci USA 108:4274–4280. doi:10.1073/pnas.1016030108
Soda Y, Myskiw C, Rommel A, Verma IM (2013) Mechanisms of neovascularization and resistance to anti-angiogenic therapies in glioblastoma multiforme. J Mol Med Berl Ger 91:439–448. doi:10.1007/s00109-013-1019-z
Holash J, Maisonpierre PC, Compton D et al (1999) Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 284:1994–1998
Rubenstein JL, Kim J, Ozawa T et al (2000) Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia N Y N 2:306–314
De Groot JF, Fuller G, Kumar AJ et al (2010) Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro-Oncol 12:233–242. doi:10.1093/neuonc/nop027
Pennacchietti S, Michieli P, Galluzzo M et al (2003) Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 3:347–361
Lu KV, Chang JP, Parachoniak CA et al (2012) VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 22:21–35. doi:10.1016/j.ccr.2012.05.037
Li Y, Li A, Glas M et al (2011) c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc Natl Acad Sci USA 108:9951–9956. doi:10.1073/pnas.1016912108
Eckerich C, Zapf S, Fillbrandt R et al (2007) Hypoxia can induce c-Met expression in glioma cells and enhance SF/HGF-induced cell migration. Int J Cancer J Int Cancer 121:276–283. doi:10.1002/ijc.22679
Tchaicha JH, Reyes SB, Shin J et al (2011) Glioblastoma angiogenesis and tumor cell invasiveness are differentially regulated by β8 integrin. Cancer Res 71:6371–6381. doi:10.1158/0008-5472.CAN-11-0991
Saltz LB, Lenz H-J, Kindler HL et al (2007) Randomized phase II trial of cetuximab, bevacizumab, and irinotecan compared with cetuximab and bevacizumab alone in irinotecan-refractory colorectal cancer: the BOND-2 study. J Clin Oncol Off J Am Soc Clin Oncol 25:4557–4561. doi:10.1200/JCO.2007.12.0949
Galanis E, Anderson SK, Lafky JM et al (2013) Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): a north central cancer treatment group trial. Clin Cancer Res Off J Am Assoc Cancer Res 19:4816–4823. doi:10.1158/1078-0432.CCR-13-0708
Snuderl M, Fazlollahi L, Le LP et al (2011) Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 20:810–817. doi:10.1016/j.ccr.2011.11.005
Jahangiri A, Aghi MK (2012) Biomarkers predicting tumor response and evasion to anti-angiogenic therapy. Biochim Biophys Acta 1825:86–100. doi:10.1016/j.bbcan.2011.10.004
Gu G, Hu Q, Feng X et al (2014) PEG-PLA nanoparticles modified with APTEDB peptide for enhanced anti-angiogenic and anti-glioma therapy. Biomaterials 35:8215–8226. doi:10.1016/j.biomaterials.2014.06.022
Feng Y, Zhu M, Dangelmajer S et al (2014) Hypoxia-cultured human adipose-derived mesenchymal stem cells are non-oncogenic and have enhanced viability, motility, and tropism to brain cancer. Cell Death Dis 5:e1567. doi:10.1038/cddis.2014.521
Acknowledgments
This work was supported by funding to M.K.A.’s laboratory from the NIH (1 R01 NS079697).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Flanigan, P.M., Aghi, M.K. Adaptation to antiangiogenic therapy in neurological tumors. Cell. Mol. Life Sci. 72, 3069–3082 (2015). https://doi.org/10.1007/s00018-015-1916-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-1916-0