
Abstract
The intestine harbors enormous numbers of commensal bacteria and is under frequent attack from food-borne pathogens and toxins. A properly regulated immune response is critical for homeostatic maintenance of commensals and for protection against infection and toxins in the intestine. Immunoglobulin A (IgA) isotype antibodies function specifically in mucosal sites such as the intestines to help maintain intestinal health by binding to and regulating commensal microbiota, pathogens and toxins. IgA antibodies are produced by intestinal IgA antibody-secreting plasma cells generated in gut-associated lymphoid tissues from naïve B cells in response to stimulations of the intestinal bacteria and components. Research on generation, migration, and maintenance of IgA-secreting cells is important in our effort to understand the biology of IgA responses and to help better design vaccines against intestinal infections.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As a site for food absorption, the intestines host an enormous number of commensal bacteria constituting the indigenous microbiota and are also a major portal of entry for pathogens. Balanced adaptive immune responses in the intestine are essential for the homeostatic maintenance of the intestinal environment and protection against infection. Immunoglobulin A (IgA) antibodies are a major functional component of the humoral branch of the adaptive immune system, specifically at mucosal sites such as the intestines. The antibodies are predominantly produced by IgA antibody-secreting plasma cells (IgA-ASCs) localized in the intestinal lamina propria (LP) as dimers linked by the joining (J) chain, which contributes to the binding of dimeric IgA to the polymeric immunoglobulin receptor (pIgR) for transportation through intestinal epithelial cells and secretion into the intestinal lumen [1]. Expression of the J chain in IgA-ASCs is preferentially induced in the gut-associated lymphoid tissue (GALT) environments, although underlying mechanisms are not well understood. In the intestinal lumen, secretory IgA (SIgA) antibodies bind to the surface of commensal bacteria to maintain their homeostatic existence with intestinal tissues [2–4]. SIgA antibodies also bind to orally born pathogens and toxins to block them from attaching to and affecting intestinal tissues through a non-inflammatory process commonly known as “immune exclusion” [5–8]. Additionally, SIgA antibodies help facilitate the sampling of intestinal environments by dendritic cells (DCs) in the subepithelial dome region of the Peyer’s patches (PPs). This is accomplished by retro-transporting antigens from the intestinal lumen into the intestinal tissue through the M cells of the follicle-associated epithelium covering the PPs. IgA antibodies within the intestinal epithelial tissues can also intercept invading pathogens and excrete them from the tissues.
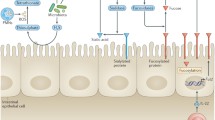
Due to the diverse functions of IgA antibodies in intestinal homeostasis and protection against pathogenic infections, major efforts are underway to understand the generation, distribution and maintenance of IgA-ASCs in intestinal tissues. It has been established that intestinal IgA-ASCs are derived from IgA+ plasmablasts generated from naïve B cells predominantly in GALT. PPs are considered to be the major site of the IgA+ plasmablast cell generation in response to intestinal antigen stimulations (Fig. 1). In addition, IgA+ plasmablasts can be generated in isolated lymphoid follicles (ILFs), small dynamic B cell-rich lymphoid structures scattered abundantly in both the small and large intestines of humans and mice [9]. Unlike PPs that are formed during the embryonic stage, ILFs are predominantly formed postnatally in response to the colonization of commensal bacteria in the intestines, suggesting their involvement in regulating bacterial homeostasis as an additional site of IgA+ plasmablast development [10, 11]. Consistent with this notion, mice lacking PPs displayed the enhanced generation of IgA+ plasmablasts in the ILFs [12, 13]. It was recently reported that the cecal patch (CPs), a lymphoid tissue in the appendix, is also a site for the generation of murine colonic IgA+ secreting cells [14]. IgA+ plasmablasts can also be generated in situ in the intestinal LP of mice, particularly in the absence of PPs and ILFs [15]. Whether the in situ generation of IgA+ cells in intestinal LP occurs in normal physiological conditions is still under debate [16]. At most, the generation of IgA+ cells in the intestinal LP may contribute to a small fraction of the IgA pool under physiological conditions. In addition, there are reports that IgA+ cells could be generated in mesenteric lymph nodes (MLNs) and the peritoneal cavity of mice [17–20]. The importance of these IgA+ cell generation pathways in contribution to the total IgA pool under homeostatic conditions is also under debate [2]. It is likely that different IgA+ cell generation processes operate in concert to ensure proper IgA production in the intestines to maintain intestinal homeostasis and protect against infection.

Schematic illustration of major cell types and molecular factors involved in regulation and generation of IgA+ cells and their migration and maintenance in the GALT and intestines. PP Peyer’s patch, ILF isolated lymphoid follicle, CP caecal patch, CSR class switch recombination, SHM somatic hypermutation, DC dendritic cell, ILC2 type 2 innate lymphoid cell, Tfh follicular T help (cell), Tfr follicular regulatory T (cell), RA retinoic acid, PD-1 programmed cell death-1 (molecule)
During differentiation from naïve B cells in GALT, the resultant IgA+ plasmablasts are imprinted to upregulate gut-homing molecules, which direct their migration into the intestinal LP where the IgA+ plasmablasts mature further into IgA-ASCs [21, 22]. In the LP, mature IgA-ASCs can survive for a long period of time, providing a local source of SIgA antibodies [23, 24]. It was suggested that the intestines provide a permissive or supporting environment for the long-term survival of IgA-ASCs. Since the intestinal environment supports long-term maintenance of IgA-ASCs in the absence of obvious antigen stimulation [23, 24], it is possible that intestine-resident IgA+ plasma and memory B cells are critically involved in memory protection against pathogenic infections.
In this article, we will review our current understanding of cellular and molecular mechanisms regulating the generation, migration and distribution of IgA+ cells, including both plasma cells and memory B cells, with special emphasis on the advances made in recent years. Most of those advances are from studies with mouse models unless otherwise indicated.
T cell-dependent differentiation of IgA+ cells
Most IgA+ plasmablasts are generated in the germinal centers of PPs with the help of T cells. Germinal centers are sites in PPs and other secondary lymphoid organs, where antigen-activated B cells proliferate, undergo class switch recombination (CSR) in the immunoglobulin heavy chain (IgH) locus from original IgM to IgA or another isotype, and undergo the somatic hypermutation (SHM) in antigen-recognizing regions of the IgH genes to generate plasmablasts and memory B cells with high-affinity antibodies-producing capacities [25]. T cells play several roles in both the formation of germinal centers and the generation of IgA+ cells. T cells, through the production of cytokines and cell–cell interaction, promote B cell proliferation. In addition, T cells help induction of the expression of activation-induced cytidine deaminase (AID) in B cells, which is the critical enzyme for the execution of CSR and hypermutation. In the absence of T cells, germinal centers failed to form, impairing the generation of IgA cells. Furthermore, IgA+ cells generated in a T cell-independent fashion have restricted antigen specificities with no or low frequencies of somatic hypermutation.
Helper T cells involved in the regulation of IgA+ cell development
Specialized populations of T cells are involved in the promotion of antibody isotype switching. Among them, follicular T help (Tfh) cells are an important helper T cell subset found in the B cell follicles of germinal centers of PPs and other secondary lymphoid organs such as lymph nodes and spleens (reviewed recently in [26]). Tfh cells constitutively express the chemokine receptor CXCR5, important for their localization into the B cell follicles. The expression of CXCR5 has been used to distinguish Tfh cells from other T cell subsets. Tfh cells express cytokines such as IL-21 and IL-4, important in promoting the IgA isotype switch (Fig. 1). Until recently, the developmental origins of Tfh cells and their association with other helper T cell subsets were not fully understood. In addition, how Tfh cells of PPs and other different secondary lymphoid organs are involved in the generation of IgA versus other isotype antibody-producing cells is not clear.
Recently, several reports found that T helper 17 (Th17) cells play an important role in the generation of IgA-producing cells, and one of them found that Th17 cells could give rise to Tfh cells. Th17 cells, through the production of IL-17, are important in maintaining intestinal immune homeostasis. Under homeostatic conditions, Th17 cells are preferentially found in the intestines where their development depends on stimulation from commensal bacteria, particularly segmented filamentous bacteria (SFB), a member of Clostridiales [27, 28]. A recent report found that transferred Th17 cells in recipient mice preferentially home to the intestines and associated lymphoid organs such as PPs, where they acquired a Tfh phenotype, which includes the upregulation of the cell surface molecules CXCR5 and programmed cell death-1 (PD-1), transcription factor BCL6 and the cytokine IL-21 [29]. The acquisition of a Tfh phenotype was associated with the downregulation of Rorγt, IL-17A and other molecules characteristic of Th17 cells [29]. Furthermore, the Th17-derived Tfh cells induce germinal center formation in PPs and promote the generation of IgA+ cells. Mice deficient in Th17 cells were impaired in an antigen-specific intestinal IgA response to immunization of cholera toxin, a model protein antigen commonly used to induce a T cell-dependent IgA response [29]. Consistent with this report, another study found that repletion of mice deficient of T cells with commensal antigen-specific Th17 cells increased the production of intestinal IgA antibodies specific to that antigen [30]. Furthermore, IL-17R−/− mice had lower levels of intestinal IgA antibodies than wild-type mice and displayed systemic anti-microflora antibody responses, likely due to increased infiltration of commensal bacteria into tissues [30]. Interestingly, microbiota-specific Th17 cells are also important for the induction of pIgR expression on intestinal epithelial cells, suggesting Th17 cells are involved in regulating both production and transportation of IgA antibodies for intestinal homeostatic maintenance [30].
Foxp3+ T cells are another important T cell population in the generation of IgA+ T cells [31, 32]. An earlier study found that a Foxp3-expressing helper T cell subset was able to acquire the Tfh cell phenotype in PPs [31]. When Foxp3+ T cells of spleens or lymph nodes were transferred into T cell-deficient mice, they could induce germinal center formation and increase the generation of IgA+ cells in the PPs and intestinal LP [31]. Correlating with this, in PPs of recipient mice, transferred T cells downregulated Foxp3 and upregulated the expression of CXCR5, ICOS, BCL6 and IL-21, all of which are features of Tfh cells. However, another study found that transferred Foxp3+ T cells could not home to the intestinal LP and associated lymphoid organs, and were unable to differentiate into Tfh cells or support the generation of IgA-producing cells [29]. Therefore, whether Foxp3+ T cell-derived T cells in PPs are bona fide Tfh cells is not clear. The differentiation of Foxp3+ T cells into Tfh-like cells has been also reported in the spleen and other lymphoid organs [33, 34]. Like Foxp3+ T cell-derived Tfh cells in PPs, Foxp3+ T cell-derived Tfh-like cells in the spleen also downregulated the expression of Foxp3 and upregulated expression of molecules associated with Tfh cells, including CXCR5 and PD-1 and BCL6 [33]. However, the Foxp3+ T cell-derived Tfh-like T cells concurrently expressed the gene encoding Blimp-1, which distinguishes them from Tfh cells [33]. Since BCL6 induces expression of CXCR5 on Treg cells, it was concluded that Foxp3+ cell-derived Tfh-like T cells represented a different subset of T cells, named follicular regulatory T (Tfr) cells. Tfr cells suppress T cell proliferation and reduce Tfh and germinal center B cell numbers in spleens. A lack of CXCR5+ Tfr cells leads to greater germinal center reactions in spleens: increased numbers of germinal center B cells, affinity maturation of antibodies, and differentiation of plasma cells [33, 34]. However, it was recently suggested that Foxp3+ Tfr cells function in germinal centers of PPs to promote somatic mutation and affinity maturation of IgA+ cells [2] (Fig. 1). Mechanisms underlying the seemingly opposite functions of Tfr cells in PPs and spleens require further investigation.
Molecular factors mediating T cell regulation of IgA+ cell development
Multiple soluble and cell surface molecules expressed by Tfh cells regulate the generation of IgA+ plasmablast cells in PPs. Among them, PD-1, an immune inhibitory receptor, was recently reported to regulate the generation and selection of IgA-producing cells in the intestine [3] (Fig. 1). PD-1-knockout mice had increased numbers of Tfh cells with altered phenotypes, which in turn resulted in increased numbers of germinal centers and IgA+ cells in PPs. However, PD-1-deficient Tfh cells were impaired in their ability to support the maintenance of IgA-ASCs in the intestinal LP. Furthermore, IgA antibodies generated in PD-1-knockout mice have a reduced capacity to bind commensal bacteria and to maintain their homeostasis in the intestine. PD-1 expressed by Tfh cells in lymphoid organs other than GALT were also found to be important in the selection and survival of long-lived plasma cells secreting different isotype antibodies [35]. In the germinal centers of spleens, PD-1 knockout mice had more cell death and less Tfh cell cytokine production than wild-type mice. However, remaining plasma cells generate antibodies with greater affinity for antigens. What distinguishes the differential effects of PD-1 on the generation of plasma cells secreting antibodies of IgA and other isotypes is not clear.
Cytokine transforming growth factor β1 (TGF-β1), IL-4, IL-10 and IL-21 are all involved in the generation of IgA-ASCs (Fig. 1). IL-21 produced by Tfh cells is critically required while sources other than T cells are sufficient to generate other cytokines that promote the IgA+ cell generation [36, 37]. IL-21 produced by Tfh cells coordinates with TGF-β1 to promote the generation of IgA+ plasmablast cells. TGF-β1 promotes naive B cell proliferation and differentiation and suppresses IL-21-induced IgG class switching in favor of IgA+ cell generation [36, 37]. Reciprocally, IL-21, through interaction with the IL-21R on B cells, inhibited the TGF-β1-induced IgG2b production to promote the selective generation of IgA+ plasmablasts [37]. However, IL-21 had little effect on the IgA generation by itself and did not alter TGF-β1-induced IgA synthesis, suggesting a synergistic effect of IL-21 and TGF-β1 in promoting the generation of IgA-producing cells.
T cell-independent differentiation of IgA-ASCs
While T cell-dependent differentiation of IgA+ plasmablast cells is essential for the generation of high affinity IgA antibodies, mice deficient of T cells have relatively normal levels of intestinal IgA-ASCs and antibodies, indicating that there exist T cell-independent IgA+ cell differentiation processes [17]. Past studies have found that PPs, ILFs, MLNs and CPs can all support the T cell-independent generation of IgA-ASCs [14, 15, 38]. The intestinal LP was also suggested to support the T cell-independent generation of IgA-ASCs, particularly of B cells derived from the peritoneal cavity in mice [15].
There is increasing evidence that ILFs are an important alternative site for the generation of IgA-ASCs in a T cell-independent fashion (Fig. 1). Unlike PPs, ILFs are generated postnatally in response to the stimulation of commensal bacterial colonization from cryptopatches, which are deposition sites of lymphoid inducer cells. In mice that lack the T cell-dependent generation of IgA-ASCs, there was accelerated maturation of ILFs from cryptopatches and increased numbers of IgA+ cells in ILFs [12, 13]. In addition, we recently found that in the chemokine receptor CCR10 knockout (CCR10−/−) mice in which the migration of CCR10−/− IgA+ plasmablast cells into intestinal LP was impaired, there were compensatory increases in the numbers of ILFs and IgA+ cells in the ILFs [39].
ILFs have also been found abundantly in human intestines [40] and could potentially function in the same fashion as the mouse ILFs in support of the generation of IgA-ASCs. However, since no defined cryptopatches have been found in human intestines, the correlation of human and mouse ILFs has not been fully established. It was argued that human ILFs are derived from similar cryptopatch structures that are generated in the early postnatal stage but that such structures have already matured into ILFs in adult human subjects analyzed for ILFs and cryptopatches. A recent study staining for CCR6+lin−c-kit+ lymphocytes identified clusters of lymphocytes in the human intestine that resemble murine cryptopatches [41]. Furthermore, in humanized mice, the development of human GALT structures, including ILFs, could originate in mouse cryptopatches [42]. Isotype switching to IgA also occurred in these GALT structures, providing strong evidence that human and mouse ILFs share the same developmental processes for the generation of IgA+ cells.
Like the T cell-dependent generation of IgA+ plasmablasts, T cell-independent generation of IgA+ plasmablasts is regulated by a similar set of cytokines, including TGF-β1, IL-4 and IL-10. However, the cellular sources of these cytokines in the absence of T cells are not fully understood. Plasmacytoid dendritic cells (pDCs) were recently identified as a major source of a proliferation-inducing ligand (APRIL), B-cell activating factor (BAFF), IL-6 and IL-10 in promoting T cell-independent IgA CSR and the generation of IgA-ASCs in mice [38] (Fig. 1). In addition, a subset of newly identified innate lymphoid cells (ILC), ILC2 (also called nuocytes or natural helper cells), were recently reported to produce large amounts of cytokines IL-4, IL5, IL6 and IL13 and enhance IgA production in mice [43] (Fig. 1). Furthermore, membrane lymphotoxin b expressed by RORγt+ ILCs was reported to promote inducible nitric oxide synthase (iNOS) expression on CD11c+ DCs, which in turn induces the T cell-independent IgA responses [44].
Regulation of migration of IgA+ plasmablasts into small and large intestines
Major gut-homing molecules involved in the migration of IgA+ plasmablasts
The same process that generates the IgA+ plasmablast cells in GALT also imprints their preferential gut-homing potentials through the induction of a unique combination of chemokine receptors and adhesion molecules. CCR9 and CCR10 are two major mucosa-specific chemokine receptors upregulated on IgA+ plasmablasts during their generation [39, 44–50] (Fig. 1). However, there are also notable differences in the expression patterns of CCR9 and CCR10 on IgA+ cells generated in different GALT and their ligands in the small and large intestines. For example, while IgA+ cells generated in PPs and CPs both express CCR9, IgA+ cells of CPs express significantly higher levels of CCR10 than those of PPs [14]. In addition, CCR10 remains expressed on all IgA-ASCs after their maturation from IgA+ plasmablasts in the intestines, while CCR9 is downregulated on mature IgA-ASCs [45–47, 51]. Furthermore, CCL28, the mucosal ligand of CCR10, is expressed in both small and large intestines while CCL25, the ligand for CCR9, is predominantly expressed in the small intestines [45–47, 51–53].
Consistent with their expression patterns, CCR9 and its ligand CCL25 play an important role in the homing of IgA+ plasmablasts into the small intestine [46, 47]. The functional importance of CCR10 and its ligand CCL28 in the intestinal IgA response was not clear until recently. In an earlier study, anti-CCL28 antibody blockage impaired the intestinal IgA response to oral immunization of cholera toxin (CT) [49]. However, anti-CCL28 antibody treatment did not affect intestinal IgA response to rotavirus infection [54], and the homeostatic IgA antibody levels in the intestines of CCR10-knockout mice were normal [55], suggesting that CCR10 and CCL28 are not required for intestinal IgA production in response to stimulations from commensal bacteria or pathogens. This might be partially due to redundant roles of CCR10 and CCR9 [54], at least in the small intestine. However, considering the unique expression patterns of CCR10 and CCL28, CCR10 likely has other functions that are distinct from those of CCR9.
Using a novel strain of CCR10-knockout/EGFP-knockin mice generated in our laboratory [56], we recently discovered several critical roles of CCR10 in intestinal IgA response and homeostasis [39]. We found that in CCR10-knockout/EGFP-knockin mice, enhanced generation of IgA+ cells in ILFs compensated for the defective migration of CCR10-deficient IgA+ cells into the intestine [39]. However, the compensatorily generated IgA-ASCs are qualitatively different from IgA-ASCs of wild-type mice, likely because they are generated in a T cell-independent process [13, 39]. Correlating with this, CCR10-knockout/EGFP-knockin mice have increased amounts of commensal bacteria in the large intestine [39]. In addition, these mice have a profoundly defective intestinal IgA memory response to pathogen infection due to the impaired maintenance of long-lived IgA-ASCs and IgA+ memory B cells in the intestines [39].
In addition to the two chemokine receptors, the adhesion molecule α4β7 integrin is expressed on intestine-homing IgA+ cells. Through the interaction with its ligand mucosal addressin cell adhesion molecule-1 (MAdCAM-1) expressed on postcapillary venules of the small and large intestines, α4β7 helps the homing of IgA+ plasmablasts to the intestines [57] (Fig. 1).
Regulation of gut-homing molecules on IgA+ cells
DCs and T cells of the GALT imprint intestine-homing properties on newly generated IgA+ plasmablasts by secreting specific factors [58]. Among those factors, retinoic acid (RA), a metabolite of vitamin A, is capable of inducing the expression of α4β7 and CCR9 on IgA+ plasmablasts [58] (Fig. 1). Consistent with this, RA, when included with antigens in the skin immunization, can trigger the generation of gut-homing IgA+ plasmablasts in the skin-draining lymph nodes and enhance levels of antigen-specific IgA antibodies in the intestinal lumen of immunized mice [59]. RA is generated from vitamin A by retinal dehydrogenases, which are highly expressed by DCs of PPs and MLNs but not some other lymphoid organs such as spleens or inguinal peripheral lymphoid nodes [58, 60–62].
DCs of GALT were also involved in promoting CCR10 expression on IgA+ plasmablasts. In an in vitro study of human B cells, RA increased the percentage of CCR10+ IgA+ cells generated from naïve B cells in the presence of IL-21 [36]. In addition, 1,25-Dihydroxyvitamin D3, the active metabolite of Vitamin D, dramatically increased the proportion of CD19+IgD−CD38+ cells expressing high levels of CCR10 that were generated from naïve human B cells [63]. It was suggested that 1,25-dihydroxyvitamin D3 activates vitamin D receptors, which then bind to a vitamin D response element in the promoter region of the human CCR10 gene for inducible expression of CCR10. Interestingly, DCs of the CPs induce a higher CCR10 expression on IgA+ cells than DCs of the PPs do in mice while they induced similar CCR9 expression [14]. Whether the differential capacities of DCs of CPs and PPs in the induction of CCR10 expression are due to their differential expression of RA and 1,25-dihydroxyvitamin D3 is not known. In addition, vitamin D did not induce the CCR10 expression on mouse IgA+ cells in vitro [63, 64]. While an explanation for this is that the promoter region of the mouse CCR10 gene does not contain a vitamin D response element [63, 64], whether vitamin D plays a role in the expression of CCR10 in vivo has yet to be elucidated.
Several cytokines derived from Tfh cells were identified to induce the expression of intestine-homing molecules on IgA+ plasmablasts. In vitro studies found that TGF-β1 and IL-21, two cytokines important in the generation of IgA+ plasmablasts, also downregulated CXCR5 and upregulated CCR10 on human IgA+ plasmablasts, suggesting their role in enabling exit of IgA+ plasmablasts from germinal centers and migration into the intestinal mucosa [36]. Whether the ability to induce gut-homing properties of IgA+ plasmablasts is unique to Tfh cells of GALT is not clear.
Differential regulation of migration and responses of IgA+ plasmablasts in the small and large intestines
Considering the differential expression of chemokines by the small and large intestines, regulation of the migration and localization of IgA+ plasmablasts into the small and large intestines is likely different. Our analysis of mice found that a significant percentage of IgA+ cells of the small intestines co-expressed CCR9 and CCR10 while IgA+ cells of the large intestines expressed only CCR10 (unpublished observations) (Fig. 1). In addition, compared to wild-type mice, CCR10-knockout mice had more severely impaired migration of IgA+ cells into the large intestines than into the small intestines [39]. Appendectomy reduced IgA+ cells in the large but not the small intestines, likely because IgA+ cells generated in the CPs of the appendix predominantly contribute to the large intestine [14].
Types of antigens and their routes of stimulations are also important factors in regulating IgA responses in the small and large intestines. It was recently reported that while the inoculation of germ-free (GF) mice with Bacteroides acidifaciens or Lactobacillus johnsonii induced same levels of IgA production in the small intestine, the Bacteroides acidifaciens-associated mice had significantly higher levels of IgA production in the large intestine [65]. In another study, SFB induced a lower frequency of IgA+ cells but they stimulated development of ILFs more efficiently than nonpathogenic Escherichia coli [66]. These studies indicate that different microorganisms use different pathways to induce intestinal IgA responses. In addition, IgA+ plasma cells generated from different immunization routes could migrate into the small and large intestines using different homing molecules. Intra-rectal immunization with protein antigens induced generation of IgA+ plasmablasts capable of homing into both the small and large intestines [67]. However, migration into the small intestines was found to be independent of CCR9/CCR10 and instead dependent on α4β7. In contrast, IgA+ plasmablasts induced by intra-nasal immunization expressed low levels of α4β7 and were usually excluded from the gut. However, intra-nasal immunization increased Ag-specific IgA+ cells in the small intestine of β7-knockout mice, demonstrating that intestinal homing of IgA+ plasmablasts is a competitive process and that α4β7 determines not only the intestinal localization of IgA+ plasmablasts generated in GALT but also the intestinal exclusion of lymphocytes primed in other inductive sites [67]. Further research is required to fully understand the molecular mechanisms underlying the regulation of differential expression of the small and large intestine homing molecules.
Maintenance of IgA-ASCs and establishment of IgA+ memory
Considering that immune reactivity at mucosal sites is critical for local control of pathogens and prevention of their spreading, understanding how IgA-ASCs and memory B cells are maintained in the intestine has significant implications in the development of vaccines against many medically important pathogens that infect through the intestines. Recent studies have revealed that the maintenance of intestinal IgA-ASCs is uniquely different from that of plasma cells producing other isotypes of antibodies [23, 68], and have begun to shed light on mechanisms of long-term intestinal IgA-ASCs maintenance and IgA memory.
IgA-ASCs are long-lived in the intestinal environment
Recent studies in both mice and humans found that IgA-ASCs could survive in intestinal tissues for extended periods, even in the absence of antigen stimulations. In one study, the temporary colonization of germ-free mice with a strain of E. coli that were unable to replicate resulted in the generation of bacterium-specific IgA-ASCs that last for months without sign of receding after bacteria clearance. This suggests that IgA-ASCs generated against the bacteria are long-lived cells [23]. However, when immunized mice were re-colonized with different strains of bacteria, newly generated IgA-ASCs gradually became dominant, indicating that the maintenance of the IgA-ASCs repertoire in the intestines is modulated by the current status of commensal bacteria. Therefore, while IgA-ASCs are capable of long-term survival, their presence in the intestine would be restricted from the competition of newly formed IgA-ASCs in response to the current commensal stimulation. In an ex vivo culture study of human small intestines, IgA-secreting CD27+CD138+ plasma cells and their production of IgA antibodies could be sustained for more than 4 weeks [24]. One recent sequence analysis study of IgA variable heavy chain genes from human ileal biopsies found the occurrence of many clonally related sequences, suggesting a local expansion of IgA precursor cells, most likely IgA+ plasmablasts [69]. A similar murine IgA sequence study also found evidence for expanded clones of IgA-ASCs [70]. In addition, the IgA repertoire of an individual mouse was stable and could be recalled after a temporary depletion of IgA-ASCs from the intestine [70], indicative of functional IgA memory. However, the origin of this memory is not clear. Whether IgA memory towards pathogenic infection is regulated in the same way as the memory to commensal bacteria is unclear as well.
IgA+ memory B cells in intestinal IgA response
In addition to long-lived IgA-ASCs, IgA+ memory B cells are important in memory maintenance and response. Whether IgA memory B cells represent a major component of intestinal memory during pathogen infection remains a debated topic. In mouse studies using oral immunization of cholera toxin to induce intestinal IgA responses, it was found that the IgA memory response to cholera toxin re-challenge was mediated by memory B cells but not by long-lived IgA-ASCs [71–73]. A similar argument has been proposed to describe the IgA memory response to pathogen infections [39, 74]. Memory B cells also contribute significantly to IgA memory responses in humans immunized with an oral cholera vaccine [75]. However, it was recently reported that repeated immunization with the same strain of commensal bacteria only had an additive effect on the generation of IgA-ASCs, which argues against IgA memory response from these types of infections [23]. There are several possible explanations to account for this difference. First, immunization with commensal bacteria is different from that of protein antigens or pathogens. Second, the presence of previously formed IgA-ASCs and antibodies specific to the commensal bacteria is sufficient to suppress their invasion so that no strong memory IgA response is evoked. This would not be the case if a pathogenic infection is reintroduced. Third, it could be argued that a T cell-independent IgA response might be predominantly promoted by immunization with the bacteria, while oral immunization with T cell-dependent antigens would induce more efficient memory B cell development [39, 76].
Molecular factors involved in the maintenance of intestinal IgA memory
Considering that IgA-ASCs and memory B cells have distinct properties compared with other types of plasma and memory B cells, molecular factors involved in their intestinal maintenance and regulation are likely different. Besides molecules commonly involved in the survival and proliferation of plasma and memory cells, there are also several unique requirements for efficient maintenance of IgA+ ASCs and memory B cells in the intestines. Among the common survival factors, a proliferation-inducing ligand (APRIL) and its receptor B cell maturation antigen (BCMA) are involved in the maintenance of IgA-ASCs and plasma cells of other isotypes [24, 77–79]. In ex vivo culture studies of human intestinal sections, IgA antibody production and plasma cell survival were also found to depend on IL-6 [24]. However, these factors do not explain unique intestinal maintenance requirements of IgA-ASCs or IgA+ memory B cells.
Our laboratory recently identified CCR10 as an important regulator of the intestinal maintenance of IgA-ASCs and memory B cells [39]. In CCR10-knockout mice infected with the intestinal bacterial pathogen Citrobacter rodentium, pathogen-specific long-lived IgA-producing plasma cells and IgA+ memory B cells were not properly maintained in the intestines, and the IgA memory response to pathogen reinfection was severely impaired. Additionally, we found that while IgA+ memory B cells of spleens did not express CCR10, IgA+ memory B cells isolated from the intestine did, suggesting that CCR10, through interaction with its mucosal ligand CCL28, may be directly involved in their maintenance in the intestine. Relating to this, earlier studies of murine rotavirus infection found that rotavirus-specific memory B cells expressed α4β7, a molecule important for the efficient migration of memory B cells into the intestines [74]. IgA-ASCs were recently reported to express a functional membrane B cell receptor (BCR) capable of inducing calcium mobilization and phosphorylation of extracellular signal-regulated kinase 1/2 and serine/threonine kinase AKT important for the survival of IgA-ASCs, suggesting that the maintenance of IgA memory is also modulated by specific antigens [80].
Future perspective
Studies in recent years have made significant progress towards understanding the roles of IgA antibodies in the regulation of intestinal homeostasis of commensals and the immune system. In addition, cellular and molecular factors involved in the differentiation and localization of IgA-producing cells in the intestinal mucosa are also better elucidated. However, many important questions remain to be addressed. Much of the current understanding of regulation of IgA responses focuses on their generation and maintenance under homeostatic conditions in relationship with stimulations of commensal bacteria. Whether IgA responses against intestinal pathogen infection under inflammatory conditions are regulated in a similar fashion is not well understood. In addition, while the intestinal mucosa were found to be able to support the long-term survival of IgA-ASCs, how the continuous stimulation from commensal bacteria and food-born antigens affects the presence of long-term maintenance of IgA-ASCs against intestinal pathogens needs to be investigated further. Are there any anatomic niches in the intestine and associated lymphoid organs for long-lived IgA-ASCs and memory B cells? What are cellular and molecular mechanisms for the establishment and maintenance of IgA memory in the intestine against pathogen infections. Considering the important roles of IgA antibodies in the prevention of mucosal infection, understanding these questions will help to design better vaccination strategies against pathogen infections.
Abbreviations
- AID:
-
Activation-induced cytidine deaminase
- APRIL:
-
A proliferation-inducing ligand
- BAFF:
-
B cell activating factor
- BCMA:
-
B cell maturation antigen
- BCR:
-
B cell receptor
- CP:
-
Caecal patch
- CSR:
-
Class switch recombination
- CT:
-
Cholera toxin
- DC:
-
Dendritic cell
- GALT:
-
Gut-associated lymphoid tissue
- GF:
-
Germ-free
- IgA:
-
Immunoglobulin A
- IgA-ASC:
-
IgA antibody-secreting plasma cell
- IgH:
-
Immunoglobulin heavy chain
- ILC:
-
Innate lymphoid cell
- ILF:
-
Isolated lymphoid follicle
- LP:
-
Lamina propria
- LTi:
-
Lymphoid tissue inducer
- MLN:
-
Mesenteric lymph node
- PD-1:
-
Programmed cell death-1
- pDC:
-
Plasmacytoid dendritic cell
- pIgR:
-
Polymeric immunoglobulin receptor
- PP:
-
Peyer’s patch
- RA:
-
Retinoic acid
- RAG:
-
Recombinase-activating gene
- SFB:
-
Segmented filamentous bacteria
- SHM:
-
Somatic hypermutation
- SIgA:
-
Secretory IgA
- TCR:
-
T cell receptor
- Tfh:
-
Follicular T help
- Tfr:
-
Follicular regulatory T
- TGF-β1:
-
Transforming growth factor β1
- TH17:
-
T helper 17
- Treg:
-
Regulatory T
References
Brandtzaeg P, Prydz H (1984) Direct evidence for an integrated function of J chain and secretory component in epithelial transport of immunoglobulins. Nature 311(5981):71–73
Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y, Tsutsui Y, Qin H, Honda K, Okada T et al (2014) Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 41(1):152–165
Kawamoto S, Tran TH, Maruya M, Suzuki K, Doi Y, Tsutsui Y, Kato LM, Fagarasan S (2012) The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science 336(6080):485–489
Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W et al (2014) Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158(5):1000–1010
Corthesy B (2007) Roundtrip ticket for secretory IgA: role in mucosal homeostasis? J Immunol 178(1):27–32
Cerutti A, Rescigno M (2008) The biology of intestinal immunoglobulin A responses. Immunity 28(6):740–750
Mantis NJ, Rol N, Corthesy B (2011) Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol 4(6):603–611
Boullier S, Tanguy M, Kadaoui KA, Caubet C, Sansonetti P, Corthesy B, Phalipon A (2009) Secretory IgA-mediated neutralization of Shigella flexneri prevents intestinal tissue destruction by down-regulating inflammatory circuits. J Immunol 183(9):5879–5885
Hamada H, Hiroi T, Nishiyama Y, Takahashi H, Masunaga Y, Hachimura S, Kaminogawa S, Takahashi-Iwanaga H, Iwanaga T, Kiyono H et al (2002) Identification of multiple isolated lymphoid follicles on the antimesenteric wall of the mouse small intestine. J Immunol 168(1):57–64
Lorenz RG, Chaplin DD, McDonald KG, McDonough JS, Newberry RD (2003) Isolated lymphoid follicle formation is inducible and dependent upon lymphotoxin-sufficient B lymphocytes, lymphotoxin beta receptor, and TNF receptor I function. J Immunol 170(11):5475–5482
Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG, Eberl G (2008) Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456(7221):507–510
Lorenz RG, Newberry RD (2004) Isolated lymphoid follicles can function as sites for induction of mucosal immune responses. Ann N Y Acad Sci 1029:44–57
Tsuji M, Suzuki K, Kitamura H, Maruya M, Kinoshita K, Ivanov II, Itoh K, Littman DR, Fagarasan S (2008) Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity 29(2):261–271
Masahata K, Umemoto E, Kayama H, Kotani M, Nakamura S, Kurakawa T, Kikuta J, Gotoh K, Motooka D, Sato S et al (2014) Generation of colonic IgA-secreting cells in the caecal patch. Nat Commun 5:3704
Suzuki K, Fagarasan S (2009) Diverse regulatory pathways for IgA synthesis in the gut. Mucosal Immunol 2(6):468–471
Lin M, Du L, Brandtzaeg P, Pan-Hammarstrom Q (2014) IgA subclass switch recombination in human mucosal and systemic immune compartments. Mucosal Immunol 7(3):511–520
Macpherson AJ, Gatto D, Sainsbury E, Harriman GR, Hengartner H, Zinkernagel RM (2000) A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 288(5474):2222–2226
Kroese FG, Butcher EC, Stall AM, Herzenberg LA (1989) A major peritoneal reservoir of precursors for intestinal IgA plasma cells. Immunol Invest 18(1–4):47–58
Kroese FG, Butcher EC, Stall AM, Lalor PA, Adams S, Herzenberg LA (1989) Many of the IgA producing plasma cells in murine gut are derived from self-replenishing precursors in the peritoneal cavity. Int Immunol 1(1):75–84
Macpherson AJ, Slack E (2007) The functional interactions of commensal bacteria with intestinal secretory IgA. Curr Opin Gastroenterol 23(6):673–678
Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, Von Andrian UH (2003) Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature 424(6944):88–93
Mora JR, von Andrian UH (2008) Differentiation and homing of IgA-secreting cells. Mucosal Immunol 1(2):96–109
Hapfelmeier S, Lawson MA, Slack E, Kirundi JK, Stoel M, Heikenwalder M, Cahenzli J, Velykoredko Y, Balmer ML, Endt K et al (2010) Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 328(5986):1705–1709
Mesin L, Di Niro R, Thompson KM, Lundin KE, Sollid LM (2011) Long-lived plasma cells from human small intestine biopsies secrete immunoglobulins for many weeks in vitro. J Immunol 187(6):2867–2874
Wei M, Shinkura R, Doi Y, Maruya M, Fagarasan S, Honjo T (2011) Mice carrying a knock-in mutation of Aicda resulting in a defect in somatic hypermutation have impaired gut homeostasis and compromised mucosal defense. Nat Immunol 12(3):264–270
Tangye SG, Ma CS, Brink R, Deenick EK (2013) The good, the bad and the ugly—TFH cells in human health and disease. Nat Rev Immunol 13(6):412–426
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV et al (2009) Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139(3):485–498
Sczesnak A, Segata N, Qin X, Gevers D, Petrosino JF, Huttenhower C, Littman DR, Ivanov II (2011) The genome of th17 cell-inducing segmented filamentous bacteria reveals extensive auxotrophy and adaptations to the intestinal environment. Cell Host Microbe 10(3):260–272
Hirota K, Turner JE, Villa M, Duarte JH, Demengeot J, Steinmetz OM, Stockinger B (2013) Plasticity of Th17 cells in Peyer’s patches is responsible for the induction of T cell-dependent IgA responses. Nat Immunol 14(4):372–379
Cao AT, Yao S, Gong B, Elson CO, Cong Y (2012) Th17 cells upregulate polymeric Ig receptor and intestinal IgA and contribute to intestinal homeostasis. J Immunol 189(9):4666–4673
Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T, Hori S, Fagarasan S (2009) Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science 323(5920):1488–1492
Cong Y, Feng T, Fujihashi K, Schoeb TR, Elson CO (2009) A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc Natl Acad Sci USA 106(46):19256–19261
Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, Srivastava M, Divekar DP, Beaton L, Hogan JJ et al (2011) Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med 17(8):975–982
Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, Wang YH, Lim H, Reynolds JM, Zhou XH et al (2011) Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med 17(8):983–988
Good-Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM, Shlomchik MJ (2010) PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat Immunol 11(6):535–542
Dullaers M, Li D, Xue Y, Ni L, Gayet I, Morita R, Ueno H, Palucka KA, Banchereau J, Oh S (2009) A T cell-dependent mechanism for the induction of human mucosal homing immunoglobulin A-secreting plasmablasts. Immunity 30(1):120–129
Seo GY, Youn J, Kim PH (2009) IL-21 ensures TGF-beta 1-induced IgA isotype expression in mouse Peyer’s patches. J Leukoc Biol 85(5):744–750
Tezuka H, Abe Y, Asano J, Sato T, Liu J, Iwata M, Ohteki T (2011) Prominent role for plasmacytoid dendritic cells in mucosal T cell-independent IgA induction. Immunity 34(2):247–257
Hu S, Yang K, Yang J, Li M, Xiong N (2011) Critical roles of chemokine receptor CCR10 in regulating memory IgA responses in intestines. Proc Natl Acad Sci USA 108(45):E1035–E1044
Moghaddami M, Cummins A, Mayrhofer G (1998) Lymphocyte-filled villi: comparison with other lymphoid aggregations in the mucosa of the human small intestine. Gastroenterology 115(6):1414–1425
Lugering A, Ross M, Sieker M, Heidemann J, Williams IR, Domschke W, Kucharzik T (2010) CCR6 identifies lymphoid tissue inducer cells within cryptopatches. Clin Exp Immunol 160(3):440–449
Nochi T, Denton PW, Wahl A, Garcia JV (2013) Cryptopatches are essential for the development of human GALT. Cell Rep 3(6):1874–1884
Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H, Koyasu S (2010) Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463(7280):540–544
Kruglov AA, Grivennikov SI, Kuprash DV, Winsauer C, Prepens S, Seleznik GM, Eberl G, Littman DR, Heikenwalder M, Tumanov AV et al (2013) Nonredundant function of soluble LTalpha3 produced by innate lymphoid cells in intestinal homeostasis. Science 342(6163):1243–1246
Kunkel EJ, Kim CH, Lazarus NH, Vierra MA, Soler D, Bowman EP, Butcher EC (2003) CCR10 expression is a common feature of circulating and mucosal epithelial tissue IgA Ab-secreting cells. J Clin Investig 111(7):1001–1010
Bowman EP, Kuklin NA, Youngman KR, Lazarus NH, Kunkel EJ, Pan J, Greenberg HB, Butcher EC (2002) The intestinal chemokine thymus-expressed chemokine (CCL25) attracts IgA antibody-secreting cells. J Exp Med 195(2):269–275
Pabst O, Ohl L, Wendland M, Wurbel MA, Kremmer E, Malissen B, Forster R (2004) Chemokine receptor CCR9 contributes to the localization of plasma cells to the small intestine. J Exp Med 199(3):411–416
Gohda M, Kunisawa J, Miura F, Kagiyama Y, Kurashima Y, Higuchi M, Ishikawa I, Ogahara I, Kiyono H (2008) Sphingosine 1-phosphate regulates the egress of IgA plasmablasts from Peyer’s patches for intestinal IgA responses. J Immunol 180(8):5335–5343
Hieshima K, Kawasaki Y, Hanamoto H, Nakayama T, Nagakubo D, Kanamaru A, Yoshie O (2004) CC chemokine ligands 25 and 28 play essential roles in intestinal extravasation of IgA antibody-secreting cells. J Immunol 173(6):3668–3675
Lazarus NH, Kunkel EJ, Johnston B, Wilson E, Youngman KR, Butcher EC (2003) A common mucosal chemokine (mucosae-associated epithelial chemokine/CCL28) selectively attracts IgA plasmablasts. J Immunol 170(7):3799–3805
Mei HE, Yoshida T, Sime W, Hiepe F, Thiele K, Manz RA, Radbruch A, Dorner T (2009) Blood-borne human plasma cells in steady state are derived from mucosal immune responses. Blood 113(11):2461–2469
Pan J, Kunkel EJ, Gosslar U, Lazarus N, Langdon P, Broadwell K, Vierra MA, Genovese MC, Butcher EC, Soler D (2000) A novel chemokine ligand for CCR10 and CCR3 expressed by epithelial cells in mucosal tissues. J Immunol 165(6):2943–2949
Wang W, Soto H, Oldham ER, Buchanan ME, Homey B, Catron D, Jenkins N, Copeland NG, Gilbert DJ, Nguyen N et al (2000) Identification of a novel chemokine (CCL28), which binds CCR10 (GPR2). J Biol Chem 275(29):22313–22323
Feng N, Jaimes MC, Lazarus NH, Monak D, Zhang C, Butcher EC, Greenberg HB (2006) Redundant role of chemokines CCL25/TECK and CCL28/MEC in IgA+ plasmablast recruitment to the intestinal lamina propria after rotavirus infection. J Immunol 176(10):5749–5759
Morteau O, Gerard C, Lu B, Ghiran S, Rits M, Fujiwara Y, Law Y, Distelhorst K, Nielsen EM, Hill ED et al (2008) An indispensable role for the chemokine receptor CCR10 in IgA antibody-secreting cell accumulation. J Immunol 181(9):6309–6315
Jin Y, Xia M, Sun A, Saylor CM, Xiong N (2010) CCR10 Is important for the development of skin-specific {gamma}{delta}T cells by regulating their migration and location. J Immunol 185(10):5723–5731
Wagner N, Lohler J, Kunkel EJ, Ley K, Leung E, Krissansen G, Rajewsky K, Muller W (1996) Critical role for beta7 integrins in formation of the gut-associated lymphoid tissue. Nature 382(6589):366–370
Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, Otipoby KL, Yokota A, Takeuchi H, Ricciardi-Castagnoli P et al (2006) Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 314(5802):1157–1160
Hammerschmidt SI, Friedrichsen M, Boelter J, Lyszkiewicz M, Kremmer E, Pabst O, Forster R (2011) Retinoic acid induces homing of protective T and B cells to the gut after subcutaneous immunization in mice. J Clin Invest 121(8):3051–3061
Hammerschmidt SI, Ahrendt M, Bode U, Wahl B, Kremmer E, Forster R, Pabst O (2008) Stromal mesenteric lymph node cells are essential for the generation of gut-homing T cells in vivo. J Exp Med 205(11):2483–2490
Molenaar R, Greuter M, van der Marel AP, Roozendaal R, Martin SF, Edele F, Huehn J, Forster R, O’Toole T, Jansen W et al (2009) Lymph node stromal cells support dendritic cell-induced gut-homing of T cells. J Immunol 183(10):6395–6402
Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY (2004) Retinoic acid imprints gut-homing specificity on T cells. Immunity 21(4):527–538
Shirakawa AK, Nagakubo D, Hieshima K, Nakayama T, Jin Z, Yoshie O (2008) 1,25-dihydroxyvitamin D3 induces CCR10 expression in terminally differentiating human B cells. J Immunol 180(5):2786–2795
Sigmundsdottir H, Pan J, Debes GF, Alt C, Habtezion A, Soler D, Butcher EC (2007) DCs metabolize sunlight-induced vitamin D3 to ‘program’ T cell attraction to the epidermal chemokine CCL27. Nat Immunol 8(3):285–293
Yanagibashi T, Hosono A, Oyama A, Tsuda M, Suzuki A, Hachimura S, Takahashi Y, Momose Y, Itoh K, Hirayama K et al (2013) IgA production in the large intestine is modulated by a different mechanism than in the small intestine: bacteroides acidifaciens promotes IgA production in the large intestine by inducing germinal center formation and increasing the number of IgA+ B cells. Immunobiology 218(4):645–651
Lecuyer E, Rakotobe S, Lengline-Garnier H, Lebreton C, Picard M, Juste C, Fritzen R, Eberl G, McCoy KD, Macpherson AJ et al (2014) Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity 40(4):608–620
Agnello D, Denimal D, Lavaux A, Blondeau-Germe L, Lu B, Gerard NP, Gerard C, Pothier P (2013) Intrarectal immunization and IgA antibody-secreting cell homing to the small intestine. J Immunol 190(9):4836–4847
Cerutti A (2011) Immunology. IgA changes the rules of memory. Science 328(5986):1646–1647
Yuvaraj S, Dijkstra G, Burgerhof JG, Dammers PM, Stoel M, Visser A, Kroese FG, Bos NA (2009) Evidence for local expansion of IgA plasma cell precursors in human ileum. J Immunol 183(8):4871–4878
Lindner C, Wahl B, Fohse L, Suerbaum S, Macpherson AJ, Prinz I, Pabst O (2012) Age, microbiota, and T cells shape diverse individual IgA repertoires in the intestine. J Exp Med 209(2):365–377
Lycke N, Holmgren J (1987) Long-term cholera antitoxin memory in the gut can be triggered to antibody formation associated with protection within hours of an oral challenge immunization. Scand J Immunol 25(4):407–412
Lycke N, Holmgren J (1989) Adoptive transfer of gut mucosal antitoxin memory by isolated B cells 1 year after oral immunization with cholera toxin. Infect Immun 57(4):1137–1141
Vajdy M, Lycke N (1993) Stimulation of antigen-specific T- and B-cell memory in local as well as systemic lymphoid tissues following oral immunization with cholera toxin adjuvant. Immunology 80(2):197–203
Williams MB, Rose JR, Rott LS, Franco MA, Greenberg HB, Butcher EC (1998) The memory B cell subset responsible for the secretory IgA response and protective humoral immunity to rotavirus expresses the intestinal homing receptor, alpha4beta7. J Immunol 161(8):4227–4235
Tengvall S, Lundgren A, Quiding-Jarbrink M, Svennerholm AM (2010) BAFF, stimulatory DNA and IL-15 stimulates IgA(+) memory B cells and provides a novel approach for analysis of memory responses to mucosal vaccines. Vaccine 28(33):5445–5450
Bemark M, Boysen P, Lycke NY (2012) Induction of gut IgA production through T cell-dependent and T cell-independent pathways. Ann N Y Acad Sci 1247:97–116
Huard B, McKee T, Bosshard C, Durual S, Matthes T, Myit S, Donze O, Frossard C, Chizzolini C, Favre C et al (2008) APRIL secreted by neutrophils binds to heparan sulfate proteoglycans to create plasma cell niches in human mucosa. J Clin Invest 118(8):2887–2895
Cassese G, Arce S, Hauser AE, Lehnert K, Moewes B, Mostarac M, Muehlinghaus G, Szyska M, Radbruch A, Manz RA (2003) Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J Immunol 171(4):1684–1690
Chu VT, Beller A, Nguyen TT, Steinhauser G, Berek C (2011) The long-term survival of plasma cells. Scand J Immunol 73(6):508–511
Pinto D, Montani E, Bolli M, Garavaglia G, Sallusto F, Lanzavecchia A, Jarrossay D (2013) A functional BCR in human IgA and IgM plasma cells. Blood 121(20):4110–4114
Acknowledgments
The work was partly supported by a grant from National Institute of Health (NIH) and institutional funds of the Pennsylvania State University (to Na Xiong). We thank Micha Davila for editorial help.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xiong, N., Hu, S. Regulation of intestinal IgA responses. Cell. Mol. Life Sci. 72, 2645–2655 (2015). https://doi.org/10.1007/s00018-015-1892-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-1892-4