
Abstract
Platelets primarily mediate hemostasis and thrombosis, whereas leukocytes are responsible for immune responses. Since platelets interact with leukocytes at the site of vascular injury, thrombosis and vascular inflammation are closely intertwined and occur consecutively. Recent studies using real-time imaging technology demonstrated that platelet–neutrophil interactions on the activated endothelium are an important determinant of microvascular occlusion during thromboinflammatory disease in which inflammation is coupled to thrombosis. Although the major receptors and counter receptors have been identified, it remains poorly understood how heterotypic platelet–neutrophil interactions are regulated under disease conditions. This review discusses our current understanding of the regulatory mechanisms of platelet–neutrophil interactions in thromboinflammatory disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
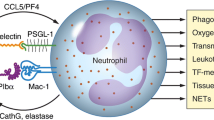
Cardiovascular disease is the number one killer in Western society. Activated platelets, leukocytes, and endothelial cells (ECs) contribute to the pathogenesis of the disease. During vascular inflammation, neutrophils roll over the activated ECs through the interaction between selectins and their ligands, followed by adhesion to the ECs, which is mediated by the interaction between β2 integrins and ICAM-1 [1]. Subsequently, activated αLβ2 and αMβ2 integrins interact with intercellular adhesion molecule-1 (ICAM-1) on the activated endothelium, thereby inducing neutrophil adhesion and crawling, respectively [2, 3]. In the presence of chemotactic stimuli, adherent neutrophils rapidly transmigrate across ECs. It was reported that several EC surface molecules, including platelet-EC adhesion molecule-1 (PECAM-1), CD99, EC-selective adhesion molecule (ESAM), and junctional adhesion molecules (JAMs), control this process [3–7]. Recent intravital microscopic studies have provided compelling evidence that activated neutrophils adherent to inflamed ECs can support homotypic and heterotypic cell–cell interactions and that platelet–neutrophil aggregation on activated ECs is the crucial determinant of microvascular occlusion during vascular inflammation (Fig. 1) [8, 9]. The heterotypic platelet–neutrophil interactions are mainly mediated by binding of neutrophil P-selectin glycoprotein ligand-1 (PSGL-1) and αMβ2 integrin to platelet P-selectin and glycoprotein Ibα (GPIbα), respectively [8–12]. While neutrophils attach to the inflamed venules under low blood shear, platelets adhere to activated ECs and sub-endothelial matrix proteins such as collagen and von Willebrand factor (vWF) under high blood shear and then support neutrophil rolling and adhesion as well as platelet accumulation following arterial injury [13–15]. Although neutrophils and platelets preferentially adhere to the site of vascular injury under low and high shear conditions, respectively, the receptors and counter receptors for heterotypic cell–cell interactions are similar under both conditions [16].

Heterotypic cell–cell interactions during vascular inflammation. a During vascular inflammation, neutrophil rolling over and adhesion to the activated ECs are mediated by selectins-their ligands and β2 integrins-ICAM-1, respectively. These adherent and crawling neutrophils allow for platelet adhesion and accumulation. b During arterial thrombosis, platelets adhere to vWF and collagen through GPIb/IX/V complex and GPVI, respectively, thereby inducing platelet aggregation. The adherent platelets support neutrophil rolling and adhesion via the receptor–counter receptor interaction. c The receptor and counter receptors of heterotypic neutrophil–platelet interactions. Heterotypic interactions are mainly mediated by the interactions of P-selectin with PSGL-1 and αMβ2 integrin with GPIbα. Other molecules also contribute to heterotypic interactions, such as platelet JAM-3 binding to neutrophil αMβ2 integrin. Platelet αIIbβ3 integrin can interact with neutrophil αMβ2 integrin through fibrinogen. d Heterotypic EC–neutrophil–platelet interactions can lead to occlusion in microvessels during thromboinflammation. In addition to EC–neutrophil–platelet interactions, RBCs may be trapped and incorporated into cell–cell aggregates
Tissue and vascular injuries activate ECs, resulting in not only the expression of adhesion molecules including P- and E-selectins, ICAMs, and vascular cell adhesion molecule-1 (VCAM-1) but also the production and release of vWF, reactive oxygen species (ROS), and inflammatory cytokines [3, 17–19]. Neutrophil recruitment to inflamed ECs is critical for vascular inflammation since activated neutrophils are the main source of ROS [19] and numerous enzymes including peptidylarginine deiminase 4 (PAD4), elastase, and cathepsin G [20, 21], thereby linking inflammatory and thrombotic responses and aggravating the disease conditions. Our recent studies showed that in addition to neutrophil adhesion to ECs, platelet–neutrophil interactions play a crucial role in slowing blood flow rates and mediating occlusion of inflamed venules [8]. Activated and adherent platelets express numerous surface receptors (GPIbα, αIIbβ3, CD40, and toll-like receptors) and release diverse granular molecules (P-selectin, ADP, and platelet factor 4) and cytokines (interleukin-1, RANTES, platelet-derived growth factor, transforming growth factor-β, and epidermal growth factor) [22–25], which enhance inflammatory responses. Further, platelets are enriched with numerous chemokines (CXCL4, CXCL8, and CCL2) and express their corresponding receptors [25, 26]. Therefore, platelets and neutrophils cooperate to propagate the pathogenesis of thromboinflammation.
Unlike arterial thrombosis, which is primarily mediated by platelet aggregation and well inhibited by antagonists of αIIbβ3 integrin [27], preclinical and clinical studies have suggested that coagulation cascades, platelets, and leukocytes should be considered for intervention in thromboinflammatory disease [28–34]. Since thrombosis and inflammation occur in numerous vessels and organs under several pathological conditions, including vascular inflammation [8, 35], ischemia/reperfusion injury [36, 37], transfusion-related acute lung injury [38], atherosclerosis [39, 40], and cell transplantation and therapies [41], understanding of the detailed mechanisms mediating thromboinflammation could lead to the identification of an effective therapeutic target. In this review, we will focus on major surface receptors and signaling pathways that regulate heterotypic platelet–neutrophil interactions under thromboinflammatory conditions and also summarize how the platelet–neutrophil interaction participates in the initiation and propagation of the disease.
Surface receptors mediating platelet–neutrophil interactions
P-selectin and PSGL-1
As an important marker of platelet activation, P-selectin is stored in α-granules of resting platelets and exposed on the surface upon agonist stimulation [42]. Binding of platelet P-selectin to neutrophil PSGL-1 is required for the initial contact between both cells. In addition, neutrophil PSGL-1 interacts with EC P- and E-selectins, which mediates tethering and rolling of neutrophils and induces activation of protein kinases such as Syk and phosphoinositide-3-kinase (PI3K), integrin activation, and cytoskeletal remodeling [43]. Furthermore, circulating microparticles mainly derived from monocytes binds to platelet P-selectin through PSGL-1, thereby accumulating tissue factor and generating thrombin [44]. Studies with mice producing high levels of soluble P-selectin demonstrated that binding of P-selectin to leukocyte PSGL-1 enhances the plasma concentration of procoagulant microparticles [45]. These results suggest that the interaction between P-selectin and PSGL-1 is critical for promoting thrombus formation. Biochemical studies revealed that the sulfation of the N-terminal Tyr 46, 48, and 51 residues and O-glycosylation of Thr 57 in PSGL1 are important for the interaction with the C-type lectin domain of selectins [46, 47]. The sialyl Lewis X moiety synthesized by fucosyltransferase VII is also required for the binding of PSGL-1 to selectins [48]. Further, the phosphorylation of the N-terminal Tyr residues is important for PSGL-1 binding to selectins under shear conditions [49].
Intravital microscopic studies with P-selectin- or PSGL-1-deficient mice demonstrated that the P-selectin–PSGL-1 interaction is crucial for leukocyte rolling over platelet thrombi at the site of arteriolar injury [15] and that EC P-selectin also mediates initial rapid rolling of leukocytes through PSGL-1 [10, 50]. Consistently, in vitro studies also suggested that inhibition and deletion of P-selectin and PSGL-1 abolish the initial interaction between neutrophils and platelets and the subsequent activation of signaling molecules in both cells [51–54]. Studies with P-selectin-deficient mice revealed that monocyte-derived microparticles incorporate into the developing thrombus through the P-selectin–PSGL-1 interaction following arteriolar injury [55], which further mediates fibrin generation. Recently, Sreeramkumar and colleagues [35] have reported that inhibition and deletion of PSGL-1 impair platelet attachment at the uropod and that platelet–neutrophil interactions through PSGL-1 and P-selectin are critical for triggering vascular disease such as ischemic stroke. Therefore, there is no doubt that platelet P-selectin and neutrophil PSGL-1 contribute to the initial association between both cells, thereby transducing signaling pathways and activating other surface molecules such as integrins during thrombosis and vascular inflammation.
Glycoprotein Ibα (GPIbα) and αMβ2 integrin
The GPIb/IX/V complex is a key platelet receptor binding to vWF at the site of vascular injury, thereby initiating platelet adhesion and accumulation under high shear conditions [56, 57]. It comprises four transmembrane proteins: GPIbα, GPIbβ, GPIX and GPV (2:2:2:1 ratio). The N-terminal region of GPIbα binds to several ligands including vWF, αMβ2 integrin, thrombin, and P-selectin [12, 58–61]. Neutrophil αMβ2 (macrophage-1 antigen, Mac-1) is a promiscuous integrin interacting with numerous ligands including ICAMs on ECs, plasma proteins (fibrinogen and factor X), complement pathway product (C3bi), extracellular matrix proteins (fibronectin, laminin, collagen, and vitronectin), and platelet GPIbα [62]. αMβ2 integrin regulates a variety of neutrophil functions such as crawling [63, 64], chemotaxis [65], survival [66], apoptosis [67], and neutrophil extracellular trap (NET) formation [68]. The major binding site for most ligands is located in the inserted (I) domain in the αM subunit. Consistently, in vitro studies suggested that the N-terminal region (Phe201-Gly268) of GPIbα binds to the I domain of αMβ2 integrin, thereby inducing stable and firm association between platelets and neutrophils [12]. In vitro and in vivo studies demonstrated that deletion or inhibition of αMβ2 integrin and GPIbα abolishes the interaction of platelets with neutrophils or monocytes under inflammatory conditions [9, 12, 69]. Recently, we demonstrated using real-time intravital microscopy that αM deletion improves blood flow rates during TNF-α-induced cremaster venular inflammation [8]. Thus, GPIbα–αMβ2 association is critical for platelet–neutrophil interactions and microvessel occlusion during vascular inflammation. Although the ligand binding and downstream signaling pathway through the GPIb/IX/V complex and αMβ2 integrin have been studied [58, 62, 70, 71], it remains elusive how the ligand-binding function of both molecules is regulated during thromboinflammatory disease.
αIIbβ3 integrin
αIIbβ3 integrin is the most abundant platelet receptor (80,000–100,000 copies per platelet) and necessary for platelet aggregation via interaction with fibrinogen. Studies with blocking antibodies and platelets from a patient with Glanzmann’s thrombasthenia revealed that the interaction of αMβ2 integrin with fibrinogen bound to αIIbβ3 integrin mediates neutrophil adhesion to adherent platelets under flow conditions [72]. However, we and others showed that inhibition of αIIbβ3 integrin does not reduce neutrophil–platelet interactions under static and stirring conditions [8, 52, 73]. Thus, despite its necessity during platelet thrombus formation, it is controversial whether platelet αIIbβ3 integrin is required for platelet–neutrophil interactions under inflammatory conditions.
Junctional adhesion molecule-3 (JAM-3)
JAM-3 (also known as JAM-C) is expressed on human platelets and ECs in the vasculature [74–76]. It was reported that inhibition of platelet JAM-3 with blocking antibodies impairs its binding to leukocyte αMβ2 integrin [76]. Interestingly, a combination of anti-GPIbα and anti-JAM-3 antibodies showed additive inhibitory effects on platelet–neutrophil interactions under static conditions, suggesting that both JAM-3 and GPIbα are important for the interaction with neutrophil αMβ2 integrin. Intravascular JAM-3 also regulates leukocyte recruitment and transmigration during inflammation, arthritis, and atherosclerosis [75, 77–80].
CD40 ligand (CD40L) and CD40
CD40L (also known as CD154), a transmembrane protein of the TNF-α family, plays a critical role during inflammation and thrombosis upon the interaction with CD40, a member of the TNF receptor family [81, 82]. CD40L and CD40 are expressed in many cell types including leukocytes and ECs [83]. Because of the enrichment in platelets, most circulating CD40L is derived from activated platelets through shedding by an unidentified enzyme(s) [84, 85]. Since soluble CD40L is biologically active and retains the ability to bind CD40, it has been thought to be a biomarker for vascular diseases [86, 87]. It was reported that circulating soluble CD40L mediates neutrophil–platelet interactions in acute coronary syndrome [88]. Consistently, studies using blocking antibodies and CD40L-deficient mice revealed that the interaction of soluble CD40L with platelet and neutrophil CD40 enhances the expression of P-selectin and αMβ2 integrin [89–91] and mediates platelet–neutrophil aggregation. Further, the interaction between platelet CD40L and neutrophil CD40 enhances ROS generation and thus aggravates oxidative stress [92]. Previous studies demonstrated that platelet CD40L also binds to EC CD40, thereby up-regulating the expression of E-selectin, VCAM-1, and ICAM-1 and inducing the secretion of chemokines such as interleukin-8 [81]. Thus, the CD40L–CD40 interactions increase the surface expression of P-selectin and αMβ2 integrin via granular secretion, thereby regulating platelet–neutrophil association during thromboinflammatory disease.
Toll-like receptors (TLRs)
Toll-like receptors are a family of innate immune system receptors that mediate the host response to infection [93]. Among many isoforms, previous studies using a TLR4 receptor antagonist and blocking antibodies suggested that platelet TLR4 is required for LPS-induced platelet–neutrophil interactions and that this interaction enhances neutrophil activation and NET formation, thereby trapping bacteria in the vasculature [94].
Consistently, other studies showed that Shiga toxin and LPS induce tissue factor expression and platelet–leukocyte aggregation in hemolytic uremic syndrome [95]. LPS–TLR4 binding causes the release of sCD40L from platelets [96, 97], which is inhibited by blocking anti-TLR4 antibodies [97]. However, earlier studies by Montrucchio and colleagues [98] suggested that LPS directly binds to leukocytes but not platelets and that LPS-induced platelet–leukocyte interactions are mediated by leukocyte activation.
It was also reported that platelet TLR2 is important for platelet–neutrophil aggregation in response to gram-negative bacteria and periodontitis [99, 100]. Recent studies suggested that injection of a TLR7 agonist into mice results in platelet–neutrophil aggregation through activation of platelet TLR7 [101]. In neutrophils, most TLRs except TLR3 are expressed [102]. Similar to platelet TLRs, neutrophil TLR isoforms recognize numerous microbial molecules including LPS (for TLR4) or peptidoglycans (for TLR2), thereby enhancing the production of ROS, cytokines, and chemokines [103]. Although each platelet and neutrophil TLR isoform may play a distinct role during inflammatory diseases, the detailed mechanisms of TLR signaling in inducing platelet–neutrophil interactions remains to be determined.
αLβ2 integrin
αLβ2 integrin is a key receptor for neutrophil adhesion to and emigration across activated ECs during inflammation [63, 104]. In vitro studies using blocking antibodies showed that binding of neutrophil αLβ2 integrin to platelet ICAM-2 regulates neutrophil-platelet attachment under flow conditions [105]. Nevertheless, the importance of αLβ2-ICAM-2 binding for platelet–neutrophil interactions has not yet been determined in vivo.
CD24
CD24 is a mucin-type glycosylphosphatidylinositol-linked protein expressed on the surface of neutrophils and tumor cells [106, 107]. While one study showed that neutrophil CD24 is not essential for the initial binding to P-selectin [108], other studies implicated that neutrophil and tumor cell CD24 bind to platelet or EC P-selectin in a manner dependent on divalent cations and that CD24-P-selectin binding may cause platelet–neutrophil interactions under disease conditions such as inflammation and cancer metastasis [106].
Molecules regulating the function of surface receptors required for platelet–neutrophil interactions
Following vascular injury, platelets and neutrophils are activated by soluble agonists and adhesive proteins via their surface receptors (Figs. 2, 3) [102, 109]. Ligand–receptor interactions stimulate diverse signaling pathways such as activation of G-proteins, phospholipase C, protein and lipid kinases, Ca2+ mobilization, and cytoskeletal rearrangement. Importantly, those signaling molecules regulate the function of surface receptors, thereby affecting platelet–neutrophil interactions. Thus, understanding how intracellular signaling modulates platelet–neutrophil interactions will help identify novel therapeutic targets to prevent and treat vascular occlusion in thromboinflammatory disease.

Receptor-mediated signaling pathways in neutrophils. Under inflammatory conditions, neutrophils can be activated by initial rolling on activated endothelium and by soluble ligands such as TNF-α. TNFR signaling stimulates NF-κB-mediated gene transcription, regulating the expression of numerous pro-inflammatory proteins. GPCR stimulation leads to inhibition of adenylyl cyclase (AC) through Gαi and activation of PI3K and PLCβ through Gβγi. Rac1/2 activity and activated Akt and PKC phosphorylate p47phox, inducing ROS generation through the NOX2 complex. PLCβ hydrolyzes PIP2 to form IP3 and DAG which in turn mediates calcium release from the ER and DAG-sensitive PKC activation, respectively. Depletion of ER calcium induces STIM1 clustering at the ER-PM junction, interacting with ORAI1, and allowing calcium influx. FcγR signaling leads to ITAM- and Syk-mediated PLCγ2 activation that induces similar downstream effects to those seen from GPCRs. TLR activation leads to NF-κB activation, as seen in TNFR activation. The signaling pathways of GPCRs, FcγR, and TLR enhance MAPK activity and cytosolic calcium levels. During cell activation, exocytosis-mediated granular secretion also induces the membrane translocation of αMβ2 integrin. Neutrophil activation further release histones and form neutrophil extracellular traps (NETs). In addition to DNA and histones, NETs form a scaffold for proteases and trap circulating bacteria and platelets

Receptor-mediated signaling pathways in platelets. Numerous agonists stimulate platelets through their receptors. Gqα is able to stimulate both Lyn and PLCβ. Lyn then phosphorylates PI3K, leading to PIP3 generation and AKT phosphorylation. Activated AKT stimulates NOS-PKG-MAPK signaling. PLCβ activation allows for generation of DAG and IP3 from PIP2. IP3 then binds to the IP3R on the dense tubular system, inducing calcium release into the cytosol. Both DAG and calcium can activate PKC, leading to αIIbβ3 integrin activation. Increases in cytosolic calcium also lead to integrin activation through activation of CalDAG-GEF1. Other GPCRs on the platelet membrane are the P2Y receptors for ADP, the 5-HT receptor for serotonin, and the thromboxane receptor for TXA2. Stimulation of these receptors also induces platelet aggregation through their corresponding G-proteins. Platelet ITAMs, such as FcγR and GPVI, signal through Syk-SLP-76-ADAP and Lyn/Fyn, respectively. Both of these receptors lead to activation of PLCγ2, thus generating IP3 and DAG and inducing platelet aggregation and granule secretion as described above
Src family kinases (SFKs)
It is known that there are 7–8 members of SFKs in human platelets and neutrophils [110, 111]. Inhibition of SFKs with PP2, a broad-spectrum SFK inhibitor, partially impaired thrombin-mediated P-selectin exposure and αIIbβ3 integrin activation [112]. Further, studies with SFK inhibitors and isoform-specific KO mice suggested that SFKs such as Lyn and Fyn affect vWF-GPIbα binding and thus regulate platelet activation including Ca2+ mobilization and cytoskeletal reorganization [113–115]. SFKs are also activated by the initial platelet–neutrophil attachment, and activated SFKs stabilize the cell–cell interaction [116, 117]. Studies with mice lacking double (Hck and Fgr) or triple (Hck, Fgr, and Lyn) SFKs suggested that the SFK-Pyk2 signaling axis modulates αMβ2 integrin function and thus is important for neutrophil accumulation to adherent platelets in vitro and in vivo [118]. More recent studies demonstrated that mice deficient in Hck, Fgr, and Lyn show remarkable defects in neutrophil recruitment as shown in β2 integrin-null mice, suggesting the critical role of the three SFKs in neutrophil adhesive function [119]. Inhibition of SFKs with PP1 impaired αMβ2 integrin clustering and cytoskeletal reorganization in neutrophils [116]. Further, P-selectin-mediated crosslinking of PSGL-1 results in αMβ2 integrin activation in a SFKs-dependent manner [117]. Interestingly, it was reported that the plasma levels of soluble P-selectin increases in patients with arterial occlusive disease [120], which would enhance platelet–neutrophil aggregation during vascular disease. Although SFKs play important roles in regulating the function of platelet and neutrophil surface receptors, most studies have been carried out using non-specific inhibitors. Therefore, studies using isoform-specific and multiple KO mice are required to determine the distinct and redundant role of each isoform in platelet–neutrophil interactions.
Phospholipase (PLC)
Phospholipase hydrolyzes membrane PIP2 into IP3 and diacylglycerol, thereby increasing the cytosolic Ca2+ level and activating PKC, respectively [121]. Among PLC isoforms that are activated by an agonist, PLCβ2/3 and PLCγ2 are the major isoforms and are activated by Gqα/Giβγ- and immunoreceptor tyrosine-based activation motif (ITAM)-regulated signaling pathways, respectively, which have been described extensively in other reviews [122, 123]. In platelets, binding of thrombin and TXA2 to their receptors activates PLCβ2 through Gqα, whereas the collagen–GPVI interaction stimulates PLCγ2 through scaffold proteins and numerous tyrosine kinases [122]. Due to the effect on cytosolic Ca2+ levels, PLC is critical for regulating the early events of platelet activation. Previous studies suggested that PLCβ-regulated AKT activity may mediate P-selectin exposure on thrombin-stimulated platelets [124] and that PLCγ2 activation through GPVI signaling is critical for the increase in cytosolic Ca2+ and αIIbβ3 integrin activation [125, 126]. Furthermore, studies using PLCγ2 KO mice showed the important role of PLCγ2 in increasing cytosolic Ca2+ through the vWF-GPIb/IX/V interaction [114]. In neutrophils, binding of an agonist, such as fMLF, to the receptor mediates PLCβ2/3 activation through Giβγ [123]. Previous studies demonstrated that deletion of PLCβ2 and/or PLCβ3 abrogates IP3 production, increase in cytosolic Ca2+, PKC activation, αMβ2 up-regulation, and ROS generation following fMLF stimulation [127, 128]. Although the importance of PLC for platelet and neutrophil activation has been clearly reported, it remains unclear how each PLC isoform regulates platelet–neutrophil interactions during thromboinflammatory disease.
Phosphatidylinositol 3-kinase (PI3K)-AKT
Phosphatidylinositol 3-kinase phosphorylates PI on the third carbon and is composed of three different classes, class I-III, based on the structure, lipid substrate specificity, and regulation [129, 130]. PI-3,4,5-trisphosphate (PIP3) is one of the PI3K products which is required for the membrane association and activation of AKT and PLCγ2 [131, 132]. Phosphatase and tensin homolog (PTEN) and SH2 domain-containing inositol 5-phosphatase 1 (SHIP1) regulate the levels of PI3K products [133–135]. Previous studies with PI3K inhibitors revealed that PI3K does not affect P-selectin exocytosis and the kinetics of actin assembly during platelet activation [136–138]. However, PI3K p85α-null platelets showed a defect in P-selectin exposure induced by GPVI- but not Gq- and Gi-mediated signaling [139]. Other studies revealed that PI3K plays an important role in vWF-GPIbα binding, intracellular Ca2+ mobilization, and platelet activation under shear but not static conditions [140], suggesting the important role of PI3K under in vivo pathological conditions. Studies with a cell membrane-permeable, dominant-negative form of the class IA PI3K p85α suggested that PI3K regulates αMβ2-mediated neutrophil adhesion and NADPH oxidase 2 (NOX2) activity [141]. Consistently, inhibition of PI3K with LY294002 significantly impairs neutrophil ROS generation and fMLF-induced neutrophil–platelet interactions [92]. It was reported that PSGL-1-mediated neutrophil rolling over E-selectin activates the spleen tyrosine kinase (Syk), and Syk-mediated integrin function in part requires the PI3Kγ-AKT signaling [142]. Further studies are required to determine the role of each PI3K isoform in regulating P-selectin exposure and αMβ2 integrin function.
AKT is a well-known downstream molecule of PI3K. Each AKT isoform, AKT1-3, plays an overlapping and distinct role during platelet activation [143–145]. Platelet AKT1 regulates thrombin-induced P-selectin exposure and intracellular Ca2+ release, thereby affecting αIIbβ3 integrin activation [8, 143]. Similarly, platelet AKT2 and 3 are important for P-selectin exposure and αIIbβ3 integrin activation induced by low concentrations of thrombin or U46619 [8, 144, 145]. It is of interest to note that platelet AKT isoforms, but not PI3K, regulate P-selectin exposure following thrombin stimulation [8, 136, 138, 143, 144], suggesting that platelet α-granule secretion is differentially regulated by PI3K and AKT. Unlike platelets, neutrophils express only AKT1 and 2, and previous studies showed that AKT2 modulates NOX2 activity and ROS generation during neutrophil activation [146]. PI3K-AKT signaling also mediates P-selectin exposure induced by stimulation of platelet TLR2, thereby affecting platelet–neutrophil interactions [99]. Importantly, our recent studies using Akt isoform-specific KO mice and their bone marrow chimera demonstrated that neutrophil AKT2 plays a critical role in intracellular Ca2+ release and the membrane translocation and activation of αMβ2 integrin, thereby controlling neutrophil–platelet interactions during vascular inflammation [8]. These results indicate that platelet and neutrophil AKT are critical for regulating platelet–neutrophil interactions during vascular disease.
Protein kinase C (PKC)
The PKC family is composed of three subfamilies based on the requirement for second messengers (Ca2+, diacylglycerol, and phospholipids) [147]. A broad-spectrum PKC inhibitor, Ro-31-8220 partially inhibited P-selectin exposure and αIIbβ3 integrin activation in AYPGKF-stimulated P2Y12-deficient platelets [112]. Studies using isoform-specific PKC inhibitors suggested that some of the novel and atypical PKC isoforms regulate P-selectin exposure on thrombin-activated platelets and platelet–neutrophil interactions [138]. Atypical PKCζ colocalizes with αMβ2 integrin in neutrophils and mediates soluble CD40L-induced activation and clustering of the integrin and neutrophil–platelet interactions [90]. Interestingly, PKCδ deletion differentially regulates P-selectin exposure; decreased through PAR4 signaling but increased via GPVI signaling [148]. Moreover, inhibition of PKCδ with a dominant-negative TAT peptide blocks ERK recruitment to p47phox and delays the initiation of TNF-α-induced O ·−2 generation through NOX2 in neutrophils [149]. Since PKC isoforms play a distinct role in regulating platelet and neutrophil functions, future studies using isoform-specific and multiple KO mice are required to determine how each isoform regulates neutrophil–platelet interactions.
Mitogen-activated protein kinases (MAPKs)
Activated MAPKs are crucial for regulating thromboxane A2 production, granule secretion, and αIIbβ3 integrin activation [109]. It was reported that p38 MAPK is not important for Ca2+ mobilization, P-selectin exposure and αIIbβ3 integrin activation in response to thrombin [150]. In contrast, recent studies showed that inhibition of extracellular signal-regulated kinases (ERK) and p38 MAPK significantly impairs P-selectin exposure and αIIbβ3 integrin activation in histone-stimulated platelets [151]. Treatment of neutrophils with platelet-activating factor (PAF) up-regulates αMβ2 integrin expression and stimulates β2 integrin-dependent adhesion through ERK, but not PI3K [152].
Phosphodiesterase 4 (PDE4)
Recent studies using isoform-specific inhibitors suggested that PDE4, but not PDE3 or PDE5, is important for P-selectin-mediated αMβ2 integrin activation, thereby inducing the formation of platelet–neutrophil aggregates in vitro and in vivo [153].
Nuclear factor-κB (NF-κB) signaling
Activation of NF-κB is mediated by the signal-induced phosphorylation and degradation of IκB and regulates transcription of many genes involved in inflammation, immunity, cell proliferation, and survival [154]. It was reported that IκBα is phosphorylated and degraded in thrombin-activated platelets and that IκB kinase inhibitors impair P-selectin exposure, αIIbβ3 integrin activation, and ERK phosphorylation in activated platelets [151, 155, 156]. Recent studies suggested that treatment of platelets with TLR2 and 4 agonists triggers P-selectin exposure through NF-κB signaling [157]. Moreover, the interaction of platelets with hepatic ECs induces activation of NF-κB signaling and promotes adhesion of neutrophils and lymphocytes to P-selectin on both platelets and ECs [158]. Previous studies implicated that inhibition and knockdown of the NF-kB subunits suppress the surface expression of αMβ2 integrin in PMA-stimulated neutrophil-like HL60 cells [159]. Thus, gene regulation through NF-κB signaling plays a crucial role in modulating platelet–neutrophil interactions under inflammatory conditions.
Small GTPases
Small GTPases are important signaling mediators involved in numerous cellular functions [160]. Among several family members, Rho family GTPases including Rac1, Cdc42, and RhoA are the best studied and have been shown to control cytoskeletal rearrangement [161]. Since GTPases are activated and inactivated by binding of GTP and GDP, respectively, they are regulated by GTPase activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs) [162]. It is known that Cdc42 and Rac control the formation of finger-like filopodial protrusions and lamellipodia, respectively, whereas RhoA mediates actin stress fiber formation. In platelets, studies using mice lacking Rac1, Cdc42, or both demonstrated the importance of each GTPase for thrombopoiesis, P-selectin exposure, and αIIbβ3 activation following agonist stimulation [163–165]. Further, Cdc42-null platelets showed defects in platelet GPIb signaling [166, 167]. Deletion of another small GTPase, Rap1b impairs P-selectin exposure on activated platelets [168]. In neutrophils, engagement of PSGL-1 activates Ras activity [169], which may regulate β2 integrin activation. Rap1 is activated by cytosolic Ca2+ and diacylglycerol through PLC activation [170] and controls αMβ2 integrin activation induced by LPS and TNF-α [171]. Studies using Cdc42-null mice suggested that αMβ2 clustering is regulated by Cdc42 during neutrophil migration [172]. Rac1/2, components of the NADPH oxidase 2 complex, regulate ROS generation [173]. Further studies are required to determine the role of each small GTPase and its GAP/GEF in regulating platelet–neutrophil interactions during vascular disease.
Reactive oxygen species (ROS)
Reactive oxygen species are signaling molecules which play important roles during vascular disease and homeostasis [174]. ROS are produced under hypoxic inflammatory conditions [175]. Both ROS and hypoxia can transcriptionally and non-transcriptionally regulate hypoxia-induced factor-1α/2α (HIF-1α/2α) and NF-κB in intravascular cells [176–178], thereby resulting in the expression of vasoactive substances and pro-inflammatory molecules. Previous studies demonstrated that hypoxia significantly induces the gene expression of β2 integrins in leukocytes in a manner dependent on HIF-1 [179]. Biochemical studies with HIF-1-deficient myeloid cells revealed that hypoxia-induced HIF-1α regulates neutrophil survival through NF-κB activity [180]. Conversely, NF-κB also regulates the gene transcription of HIF-1α [181], suggesting the intimate link between the two signaling pathways during hypoxia–ischemia and inflammation.
The major source of ROS is membrane NADPH oxidases (NOXs). Combination studies with a NOX1 inhibitor and NOX2 KO mice implicated that platelet-derived ROS do not regulate P-selectin exposure on collagen-related peptide-activated platelets [182]. Interestingly, it was reported that incubation of platelets with H2O2 (>50–100 µM for 1 h) induces shedding of GPIbα by activating TNF-α-converting enzyme (TACE) [183]. Although this in vitro study provides evidence that oxidative stress may attenuate the thrombotic function of platelets and inhibit platelet–neutrophil interactions, it is unclear how much ROS, such as H2O2, would be produced at the site of vascular injury. Platelets from patients with chronic granulomatous disease (X-CGD) that is genetically deficient in NOX2 (gp91phox), showed defects in CD40L expression induced by various agonists [184]. Compared with platelets [182, 185], neutrophils produce larger amounts of extracellular ROS via NOX2 during cell activation [186]. Studies using neutrophils of X-CGD patients and NOX2 KO mice demonstrated that neutrophil NOX2-generated ROS are crucial for killing microbial pathogens [187, 188] and function as signaling molecules that regulate the activity of kinases and phosphatases [189]. Despite the importance of NOXs-generated ROS for thromboinflammation including ischemic stroke [190, 191], it remains poorly understood how ROS mechanistically contribute to the pathogenesis.
Pathological role of platelet–neutrophil interactions under thromboinflammatory conditions
Thromboinflammation is pathological conditions under which thrombotic and immune responses occur together. Previous studies suggested that platelets, leukocytes, and coagulation factors should all be considered to prevent and treat thromboinflammatory diseases [29, 30, 32–34, 192]. Because of the involvement of leukocytes, extracellular ROS produced from activated neutrophils would be a critical factor distinguishing thromboinflammation from arterial thrombosis that results from platelet aggregation [27]. We will briefly summarize how platelet–neutrophil interactions influence thromboinflammatory conditions, focusing on vascular occlusion during ischemic stroke and sickle cell disease (SCD).
Ischemic stroke
Ischemic stroke occurs when an artery to the brain becomes too narrow or is occluded. If fresh blood is not provided to the brain cells for more than a few minutes, ischemic responses are initiated, resulting in necrosis of the cells. The damaged brain tissue rapidly releases a large amount of ROS and proinflammatory mediators including IL-1β and MCP-1 [193]. The cytokines and chemokines trigger the expression of adhesion molecules on cerebral ECs and thus promote adhesion and transendothelial migration of leukocytes [194]. During ischemic stroke, the transmigrated leukocytes also generate an excessive amount of ROS, thereby augmenting the activation and damage of resident cells and amplifying the inflammatory condition [193, 195]. Following ischemic events, changes in microRNA levels in circulating leukocytes may contribute to increased leukocyte activation and thrombus formation [196]. Kleinschnitz and colleagues [29] reported that inhibition of αIIbβ3 integrin has no inhibitory effect on the size of ischemic stroke, but causes intracerebral hemorrhage in the mouse model of transient middle cerebral artery occlusion. Moreover, clinical studies showed that antagonists of αIIbβ3 integrin, abciximab and tirofiban, result in fatal intracerebral hemorrhage in patients with ischemic stroke [30, 197]. Instead, ischemic stroke is initiated by platelet GPIb–IX–V complex and GPVI, causing the interaction between platelets and vessel walls and allowing for further neutrophil recruitment. Binding of platelet GPIbα to vWF on activated/damaged ECs mediates platelet tethering on the vessel wall, and subsequent interaction between GPVI and collagen induces robust platelet activation and platelet thrombus formation [198]. Further, P-selectin expressed on activated platelets and ECs is able to bind to PSGL-1 on neutrophils, leading to neutrophil–platelet–ECs interactions and occluding the microvasculature [199]. We and others found that integrilin potentiates neutrophil-platelet aggregation under in vitro shear conditions [8, 52, 73]. Instead, inhibition of GPIbα with Fab fragments of a blocking anti-GPIbα antibody was a favorable approach to treat ischemic events in a mouse model [29], implicating that the interaction of GPIbα with vWF and/or αMβ2 integrin is crucial for cerebral ischemia/reperfusion injury [8, 200]. Thus, these results indicate that ischemic stroke is not simply mediated by platelet aggregation but by other intravascular cells including neutrophils. Nevertheless, it still remains unclear whether the direct interaction between platelets and neutrophils is critical for the pathogenesis of ischemic stroke. Previous studies showed that platelets also indirectly induce neutrophil recruitment into the ischemic region and thus promote inflammatory conditions. Using IL-1α/β KO mice and isoform-specific antibodies, the authors found that platelets activate brain ECs through secreted IL-1α and enhance ICAM-1 and VCAM-1 expression and CXCL1 release, thereby inducing neutrophil transendothelial migration during ischemic stroke [22]. Further, other studies using NOX inhibitors and KO mice suggested that NOX2-derived ROS are likely to be critical for mediating ischemic stroke [201, 202], and that hematopoietic cell NOX2 contributes more to the pathogenesis of stroke than brain and EC NOX2 [203]. In addition, the importance of NOX4 during oxidative stress induced by ischemic stroke has been reported [204]. Nonetheless, the regulatory mechanisms of oxidative stress in ischemic stroke remain to be determined.
Sickle cell disease (SCD)
Sickle cell disease, an inherited hematological disorder, results from the Glu 6 Val mutation in the β-globin chain [205]. When sickle hemoglobin (HbS) is deoxygenated, it aggregates into large polymers. The polymerized HbS causes a distortion of the shape of red blood cells (RBCs) and a remarkable decrease in its deformability. The sickle-shaped red cells become rigid and stick to the vessel wall. Inflammation is induced by the exposure of negatively charged phospholipids, such as phosphatidylserine, on the abnormal erythrocytes and the presence of chronic hemolysis [206]. Studies using SCD patients and mouse models demonstrated that recurrent vaso-occlusive events are the hallmark of the disease and induced by intravascular cell–cell aggregates [206]. Such vaso-occlusion is the main cause of pain crises and acute chest syndrome, which increase the morbidity and mortality in SCD patients [207]. Since hydroxyurea, the only drug approved by the Food and Drug Administration, minimally alleviates vaso-occlusion, novel therapies are required for SCD patients. Hypoxia/reoxygenation in SCD mice increases platelet–neutrophil interactions in a manner dependent on P-selectin [208]. Studies from Paul Frenette’s group have demonstrated that targeting selectins, PDE9, and NETs have benefits on vaso-occlusive events in a mouse model of SCD [209–211]. Indeed, rivipansel (GMI-1070, a pan selectin inhibitor) significantly decreased levels of biomarkers of endothelial and leukocyte activation and reduced vaso-occlusive events in clinical trials [212, 213]. Nevertheless, selectin inhibitors would block neutrophil rolling over and adhesion to the inflamed endothelium and thus may impair innate immune responses against bacterial pathogens. Since NETs induced by PAD4-citrullinated histones have been recognized as a critical component for venous and arterial thrombosis through the activation of the coagulation cascade and platelet adhesion [214–216], inhibition of NET formation may be beneficial for the intervention of vaso-occlusive events in SCD. Other studies showed that inhibition of histone deacetylase and induction of heme oxygenase-1 reduce vaso-occlusive events in sickle mice [217–219]. Recently, we have demonstrated that AKT2 is a critical regulator for the heterotypic cell–cell interaction and thus could be a potential target for vaso-occlusive events in SCD [8]. Using real-time intravital microscopy in SCD mice, infusion of a selective AKT2 inhibitor resulted in a significant reduction in cell–cell aggregation and improvement of blood flow rates. Importantly, we found that in addition to neutrophil–EC interactions, neutrophil–platelet interactions slow down blood flow rates and induce microvascular occlusion [8]. Further studies are necessary to determine which therapies would efficiently prevent vaso-occlusive events potentially in combination with hydroxyurea.
Concluding remarks
The pathological role of platelet–neutrophil association during vascular disease has long been speculated to be important. Recent in vivo intravital microscopic studies have shown that the interaction between activated neutrophils and platelets is tightly regulated and induces microvascular occlusion during thromboinflammatory disease. Because of the multiple signaling pathways regulating the receptor-counter receptor interaction, the detailed mechanism of the heterotypic cell–cell interaction still remains elusive. Recent results raise the following questions. How would intracellular signaling molecules sequentially and differentially regulate the function of platelet and neutrophil receptors during vascular disease? In addition to the intracellular signaling pathways, are any extracellular molecules required for the ligand-binding function of surface receptors under oxidative stress? Does ROS-mediated oxidative stress affect platelet–neutrophil interactions during thromboinflammation? How are the processes linking thrombosis and inflammation initiated? To answer these questions, specific inhibitors and KO mice are required to investigate the role of signaling molecules including protein kinases during thromboinflammation. Importantly, due to the difficulties of mimicking oxidative stress conditions in vitro, real-time intravital microscopy will be a powerful technique to visualize platelet–neutrophil–EC interactions in microvessels of live animals. A better understanding of the mechanisms mediating the heterotypic platelet–neutrophil interactions could lead to the identification of novel therapeutic targets for the prevention and treatment of thromboinflammatory diseases.
References
Zarbock A, Ley K (2009) Neutrophil adhesion and activation under flow. Microcirculation 16(1):31–42
Henderson RB, Lim LH, Tessier PA, Gavins FN, Mathies M, Perretti M, Hogg N (2001) The use of lymphocyte function-associated antigen (LFA)-1-deficient mice to determine the role of LFA-1, Mac-1, and alpha4 integrin in the inflammatory response of neutrophils. J Exp Med 194(2):219–226
Phillipson M, Kubes P (2011) The neutrophil in vascular inflammation. Nat Med 17(11):1381–1390
Muller WA, Weigl SA, Deng X, Phillips DM (1993) PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med 178(2):449–460
Khandoga A, Huettinger S, Khandoga AG, Li H, Butz S, Jauch KW, Vestweber D, Krombach F (2009) Leukocyte transmigration in inflamed liver: a role for endothelial cell-selective adhesion molecule. J Hepatol 50(4):755–765
Lou O, Alcaide P, Luscinskas FW, Muller WA (2007) CD99 is a key mediator of the transendothelial migration of neutrophils. J Immunol 178(2):1136–1143
Bixel MG, Li H, Petri B, Khandoga AG, Khandoga A, Zarbock A, Wolburg-Buchholz K, Wolburg H, Sorokin L, Zeuschner D et al (2010) CD99 and CD99L2 act at the same site as, but independently of, PECAM-1 during leukocyte diapedesis. Blood 116(7):1172–1184
Li J, Kim K, Hahm E, Molokie R, Hay N, Gordeuk VR, Du X, Cho J (2014) Neutrophil AKT2 regulates heterotypic cell–cell interactions during vascular inflammation. J Clin Invest 124(4):1483–1496
Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS (2009) Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med 15(4):384–391
Yang J, Hirata T, Croce K, Merrill-Skoloff G, Tchernychev B, Williams E, Flaumenhaft R, Furie BC, Furie B (1999) Targeted gene disruption demonstrates that P-selectin glycoprotein ligand 1 (PSGL-1) is required for P-selectin-mediated but not E-selectin-mediated neutrophil rolling and migration. J Exp Med 190(12):1769–1782
Wang Y, Sakuma M, Chen Z, Ustinov V, Shi C, Croce K, Zago AC, Lopez J, Andre P, Plow E et al (2005) Leukocyte engagement of platelet glycoprotein Ibalpha via the integrin Mac-1 is critical for the biological response to vascular injury. Circulation 112(19):2993–3000
Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, Ballantyne CM, Zhang L, Furman MI, Berndt MC et al (2000) Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med 192(2):193–204
Wagner DD, Burger PC (2003) Platelets in inflammation and thrombosis. Arterioscler Thromb Vasc Biol 23(12):2131–2137
Yang J, Furie BC, Furie B (1999) The biology of P-selectin glycoprotein ligand-1: its role as a selectin counterreceptor in leukocyte–endothelial and leukocyte–platelet interaction. Thromb Haemost 81(1):1–7
Gross PL, Furie BC, Merrill-Skoloff G, Chou J, Furie B (2005) Leukocyte-versus microparticle-mediated tissue factor transfer during arteriolar thrombus development. J Leukoc Biol 78(6):1318–1326
Zarbock A, Polanowska-Grabowska RK, Ley K (2007) Platelet–neutrophil–interactions: linking hemostasis and inflammation. Blood Rev 21(2):99–111
Wagner DD, Frenette PS (2008) The vessel wall and its interactions. Blood 111(11):5271–5281
Sprague AH, Khalil RA (2009) Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol 78(6):539–552
Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, Shah AM (2006) NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal 8(5–6):691–728
Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y (2010) PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 207(9):1853–1862
Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov K, Khandagale AB et al (2010) Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med 16(8):887–896
Thornton P, McColl BW, Greenhalgh A, Denes A, Allan SM, Rothwell NJ (2010) Platelet interleukin-1alpha drives cerebrovascular inflammation. Blood 115(17):3632–3639
Gear AR, Camerini D (2003) Platelet chemokines and chemokine receptors: linking hemostasis, inflammation, and host defense. Microcirculation 10(3–4):335–350
Steinhubl SR, Moliterno DJ (2005) The role of the platelet in the pathogenesis of atherothrombosis. Am J Cardiovasc Drugs 5(6):399–408
Semple JW, Italiano JE Jr, Freedman J (2011) Platelets and the immune continuum. Nat Rev Immunol 11(4):264–274
Gleissner CA, von Hundelshausen P, Ley K (2008) Platelet chemokines in vascular disease. Arterioscler Thromb Vasc Biol 28(11):1920–1927
Bledzka K, Smyth SS, Plow EF (2013) Integrin alphaIIbbeta3: from discovery to efficacious therapeutic target. Circ Res 112(8):1189–1200
Kenne E, Renne T (2014) Factor XII: a drug target for safe interference with thrombosis and inflammation. Drug Discovery Today 19(9):1459–1464
Kleinschnitz C, Pozgajova M, Pham M, Bendszus M, Nieswandt B, Stoll G (2007) Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation 115(17):2323–2330
Adams HP Jr, Effron MB, Torner J, Davalos A, Frayne J, Teal P, Leclerc J, Oemar B, Padgett L, Barnathan ES et al (2008) Emergency administration of abciximab for treatment of patients with acute ischemic stroke: results of an international phase III trial: Abciximab in Emergency Treatment of Stroke Trial (AbESTT-II). Stroke 39(1):87–99
Li J, Kim K, Hahm E, Molokie R, Hay N, Gordeuk VR, Du X, Cho J (2014) Neutrophil Akt2 regulates heterotypic cell–cell interactions during vascular inflammation. J Clin Investig 124:1483–1496
Thiagarajan RR, Winn RK, Harlan JM (1997) The role of leukocyte and endothelial adhesion molecules in ischemia-reperfusion injury. Thromb Haemost 78(1):310–314
Coller BS (2005) Leukocytosis and ischemic vascular disease morbidity and mortality: is it time to intervene? Arterioscler Thromb Vasc Biol 25(4):658–670
Yonekawa K, Harlan JM (2005) Targeting leukocyte integrins in human diseases. J Leukoc Biol 77(2):129–140
Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, Nacher M, Pitaval C, Radovanovic I, Fukui Y et al (2014) Neutrophils scan for activated platelets to initiate inflammation. Science 346(6214):1234–1238
Nieswandt B, Kleinschnitz C, Stoll G (2011) Ischaemic stroke: a thrombo-inflammatory disease? J Physiol 589(Pt 17):4115–4123
Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA, Pina E, Geller DA (2008) Factors in the pathophysiology of the liver ischemia-reperfusion injury. J Surg Res 147(1):153–159
Looney MR, Gilliss BM, Matthay MA (2010) Pathophysiology of transfusion-related acute lung injury. Curr Opin Hematol 17(5):418–423
Lievens D, von Hundelshausen P (2011) Platelets in atherosclerosis. Thromb Haemost 106(5):827–838
Woollard KJ, Geissmann F (2010) Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol 7(2):77–86
Nilsson B, Teramura Y, Ekdahl KN (2014) The role and regulation of complement activation as part of the thromboinflammation elicited in cell therapies. Mol Immunol 61(2):185–190
Furie B, Furie BC, Flaumenhaft R (2001) A journey with platelet P-selectin: the molecular basis of granule secretion, signalling and cell adhesion. Thromb Haemost 86(1):214–221
Carlow DA, Gossens K, Naus S, Veerman KM, Seo W, Ziltener HJ (2009) PSGL-1 function in immunity and steady state homeostasis. Immunol Rev 230(1):75–96
Furie B, Furie BC (2004) Role of platelet P-selectin and microparticle PSGL-1 in thrombus formation. Trends Mol Med 10(4):171–178
Andre P, Hartwell D, Hrachovinova I, Saffaripour S, Wagner DD (2000) Pro-coagulant state resulting from high levels of soluble P-selectin in blood. Proc Natl Acad Sci USA 97(25):13835–13840
Liu W, Ramachandran V, Kang J, Kishimoto TK, Cummings RD, McEver RP (1998) Identification of N-terminal residues on P-selectin glycoprotein ligand-1 required for binding to P-selectin. J Biol Chem 273(12):7078–7087
McEver RP, Cummings RD (1997) Perspectives series: cell adhesion in vascular biology. Role of PSGL-1 binding to selectins in leukocyte recruitment. J Clin Investig 100(3):485–491
Martinez M, Joffraud M, Giraud S, Baisse B, Bernimoulin MP, Schapira M, Spertini O (2005) Regulation of PSGL-1 interactions with L-selectin, P-selectin, and E-selectin: role of human fucosyltransferase-IV and -VII. J Biol Chem 280(7):5378–5390
Xiao B, Tong C, Jia X, Guo R, Lu S, Zhang Y, McEver RP, Zhu C, Long M (2012) Tyrosine replacement of PSGL-1 reduces association kinetics with P- and L-selectin on the cell membrane. Biophys J 103(4):777–785
Jung U, Ley K (1999) Mice lacking two or all three selectins demonstrate overlapping and distinct functions for each selectin. J Immunol 162(11):6755–6762
Evangelista V, Manarini S, Sideri R, Rotondo S, Martelli N, Piccoli A, Totani L, Piccardoni P, Vestweber D, de Gaetano G et al (1999) Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: role of PSGL-1 as a signaling molecule. Blood 93(3):876–885
Caron A, Theoret JF, Mousa SA, Merhi Y (2002) Anti-platelet effects of GPIIb/IIIa and P-selectin antagonism, platelet activation, and binding to neutrophils. J Cardiovasc Pharmacol 40(2):296–306
Konstantopoulos K, Neelamegham S, Burns AR, Hentzen E, Kansas GS, Snapp KR, Berg EL, Hellums JD, Smith CW, McIntire LV et al (1998) Venous levels of shear support neutrophil-platelet adhesion and neutrophil aggregation in blood via P-selectin and beta2-integrin. Circulation 98(9):873–882
Lam FW, Burns AR, Smith CW, Rumbaut RE (2011) Platelets enhance neutrophil transendothelial migration via P-selectin glycoprotein ligand-1. Am J Physiol Heart Circ Physiol 300(2):H468–H475
Falati S, Liu Q, Gross P, Merrill-Skoloff G, Chou J, Vandendries E, Celi A, Croce K, Furie BC, Furie B (2003) Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J Exp Med 197(11):1585–1598
Lopez JA, Dong JF (1997) Structure and function of the glycoprotein Ib–IX–V complex. Curr Opin Hematol 4(5):323–329
Li R, Emsley J (2013) The organizing principle of the platelet glycoprotein Ib–IX–V complex. J Thromb Haemost 11(4):605–614
Andrews RK, Berndt MC (2013) Bernard–Soulier syndrome: an update. Semin Thromb Hemost 39(6):656–662
Huizinga EG, Tsuji S, Romijn RA, Schiphorst ME, de Groot PG, Sixma JJ, Gros P (2002) Structures of glycoprotein Ibalpha and its complex with von Willebrand factor A1 domain. Science 297(5584):1176–1179
Ruggeri ZM, Zarpellon A, Roberts JR, McClintock RA, Jing H, Mendolicchio GL (2010) Unravelling the mechanism and significance of thrombin binding to platelet glycoprotein Ib. Thromb Haemost 104(5):894–902
Romo GM, Dong JF, Schade AJ, Gardiner EE, Kansas GS, Li CQ, McIntire LV, Berndt MC, Lopez JA (1999) The glycoprotein Ib–IX–V complex is a platelet counterreceptor for P-selectin. J Exp Med 190(6):803–814
Mayadas TN, Cullere X (2005) Neutrophil beta2 integrins: moderators of life or death decisions. Trends Immunol 26(7):388–395
Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P (2006) Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med 203(12):2569–2575
Sumagin R, Prizant H, Lomakina E, Waugh RE, Sarelius IH (2010) LFA-1 and Mac-1 define characteristically different intralumenal crawling and emigration patterns for monocytes and neutrophils in situ. J Immunol 185(11):7057–7066
McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P (2010) Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330(6002):362–366
Whitlock BB, Gardai S, Fadok V, Bratton D, Henson PM (2000) Differential roles for alpha(M)beta(2) integrin clustering or activation in the control of apoptosis via regulation of akt and ERK survival mechanisms. J Cell Biol 151(6):1305–1320
Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN (1996) A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity 5(6):653–666
Behnen M, Leschczyk C, Moller S, Batel T, Klinger M, Solbach W, Laskay T (2014) Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via FcgammaRIIIB and Mac-1. J Immunol 193(4):1954–1965
Corken A, Russell S, Dent J, Post SR, Ware J (2014) Platelet glycoprotein Ib-IX as a regulator of systemic inflammation. Arterioscler Thromb Vasc Biol 34(5):996–1001
Du X (2007) Signaling and regulation of the platelet glycoprotein Ib–IX–V complex. Curr Opin Hematol 14(3):262–269
Li Z (1999) The alphaMbeta2 integrin and its role in neutrophil function. Cell Res 9(3):171–178
Weber C, Springer TA (1997) Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J Clin Investig 100(8):2085–2093
Scholz T, Zhao L, Temmler U, Bath P, Heptinstall S, Losche W (2002) The GPIIb/IIIa antagonist eptifibatide markedly potentiates platelet–leukocyte interaction and tissue factor expression following platelet activation in whole blood in vitro. Platelets 13(7):401–406
Bazzoni G (2003) The JAM family of junctional adhesion molecules. Curr Opin Cell Biol 15(5):525–530
Chavakis T, Keiper T, Matz-Westphal R, Hersemeyer K, Sachs UJ, Nawroth PP, Preissner KT, Santoso S (2004) The junctional adhesion molecule-C promotes neutrophil transendothelial migration in vitro and in vivo. J Biol Chem 279(53):55602–55608
Santoso S, Sachs UJ, Kroll H, Linder M, Ruf A, Preissner KT, Chavakis T (2002) The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J Exp Med 196(5):679–691
Aurrand-Lions M, Lamagna C, Dangerfield JP, Wang S, Herrera P, Nourshargh S, Imhof BA (2005) Junctional adhesion molecule-C regulates the early influx of leukocytes into tissues during inflammation. J Immunol 174(10):6406–6415
Weber C, Fraemohs L, Dejana E (2007) The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol 7(6):467–477
Palmer G, Busso N, Aurrand-Lions M, Talabot-Ayer D, Chobaz-Peclat V, Zimmerli C, Hammel P, Imhof BA, Gabay C (2007) Expression and function of junctional adhesion molecule-C in human and experimental arthritis. Arthritis Res Ther 9(4):R65
Shagdarsuren E, Djalali-Talab Y, Aurrand-Lions M, Bidzhekov K, Liehn EA, Imhof BA, Weber C, Zernecke A (2009) Importance of junctional adhesion molecule-C for neointimal hyperplasia and monocyte recruitment in atherosclerosis-prone mice-brief report. Arterioscler Thromb Vasc Biol 29(8):1161–1163
Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek RA (1998) CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 391(6667):591–594
Andre P, Prasad KS, Denis CV, He M, Papalia JM, Hynes RO, Phillips DR, Wagner DD (2002) CD40L stabilizes arterial thrombi by a beta3 integrin–dependent mechanism. Nat Med 8(3):247–252
Schonbeck U, Libby P (2001) The CD40/CD154 receptor/ligand dyad. Cell Mol Life Sci 58(1):4–43
Lievens D, Eijgelaar WJ, Biessen EA, Daemen MJ, Lutgens E (2009) The multi-functionality of CD40L and its receptor CD40 in atherosclerosis. Thromb Haemost 102(2):206–214
Rahman M, Zhang S, Chew M, Syk I, Jeppsson B, Thorlacius H (2013) Platelet shedding of CD40L is regulated by matrix metalloproteinase-9 in abdominal sepsis. J Thromb Haemost 11(7):1385–1398
Pamukcu B, Lip GY, Snezhitskiy V, Shantsila E (2011) The CD40-CD40L system in cardiovascular disease. Ann Med 43(5):331–340
Antoniades C, Bakogiannis C, Tousoulis D, Antonopoulos AS, Stefanadis C (2009) The CD40/CD40 ligand system: linking inflammation with atherothrombosis. J Am Coll Cardiol 54(8):669–677
Setianto BY, Hartopo AB, Gharini PP, Anggrahini DW, Irawan B (2010) Circulating soluble CD40 ligand mediates the interaction between neutrophils and platelets in acute coronary syndrome. Heart Vessels 25(4):282–287
Zhao Z, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huber HE, Duggan ME, Lindsley CW (2008) Development of potent, allosteric dual Akt1 and Akt2 inhibitors with improved physical properties and cell activity. Bioorg Med Chem Lett 18(1):49–53
Jin R, Yu S, Song Z, Zhu X, Wang C, Yan J, Wu F, Nanda A, Granger DN, Li G (2013) Soluble CD40 ligand stimulates CD40-dependent activation of the beta2 integrin Mac-1 and protein kinase C zeda (PKCzeta) in neutrophils: implications for neutrophil–platelet interactions and neutrophil oxidative burst. PLoS ONE 8(6):e64631
Rahman M, Zhang S, Chew M, Ersson A, Jeppsson B, Thorlacius H (2009) Platelet-derived CD40L (CD154) mediates neutrophil upregulation of Mac-1 and recruitment in septic lung injury. Ann Surg 250(5):783–790
Vanichakarn P, Blair P, Wu C, Freedman JE, Chakrabarti S (2008) Neutrophil CD40 enhances platelet-mediated inflammation. Thromb Res 122(3):346–358
Armant MA, Fenton MJ (2002) Toll-like receptors: a family of pattern-recognition receptors in mammals. Genome Biol. 3(8):REVIEWS:3011
Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD et al (2007) Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 13(4):463–469
Stahl AL, Sartz L, Nelsson A, Bekassy ZD, Karpman D (2009) Shiga toxin and lipopolysaccharide induce platelet–leukocyte aggregates and tissue factor release, a thrombotic mechanism in hemolytic uremic syndrome. PLoS ONE 4(9):e6990
Lievens D, Zernecke A, Seijkens T, Soehnlein O, Beckers L, Munnix IC, Wijnands E, Goossens P, van Kruchten R, Thevissen L et al (2010) Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood 116(20):4317–4327
Cognasse F, Lafarge S, Chavarin P, Acquart S, Garraud O (2007) Lipopolysaccharide induces sCD40L release through human platelets TLR4, but not TLR2 and TLR9. Intensive Care Med 33(2):382–384
Montrucchio G, Bosco O, Del Sorbo L, Fascio Pecetto P, Lupia E, Goffi A, Omede P, Emanuelli G, Camussi G (2003) Mechanisms of the priming effect of low doses of lipopoly-saccharides on leukocyte-dependent platelet aggregation in whole blood. Thromb Haemost 90(5):872–881
Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, Hayashi C, Genco CA, Iafrati M, Freedman JE (2009) Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res 104(3):346–354
Assinger A, Laky M, Schabbauer G, Hirschl AM, Buchberger E, Binder BR, Volf I (2011) Efficient phagocytosis of periodontopathogens by neutrophils requires plasma factors, platelets and TLR2. J Thromb Haemost 9(4):799–809
Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, Kurt-Jones EA, Ravid K, Freedman JE (2014) Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood 124(5):791–802
Futosi K, Fodor S, Mocsai A (2013) Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol 17(3):638–650
Hayashi F, Means TK, Luster AD (2003) Toll-like receptors stimulate human neutrophil function. Blood 102(7):2660–2669
Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, Dai XY, Bromley SK, Dustin ML, Entman ML et al (1999) Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol 163(9):5029–5038
Kuijper PH, Gallardo Tores HI, Lammers JW, Sixma JJ, Koenderman L, Zwaginga JJ (1998) Platelet associated fibrinogen and ICAM-2 induce firm adhesion of neutrophils under flow conditions. Thromb Haemost 80(3):443–448
Aigner S, Sthoeger ZM, Fogel M, Weber E, Zarn J, Ruppert M, Zeller Y, Vestweber D, Stahel R, Sammar M et al (1997) CD24, a mucin-type glycoprotein, is a ligand for P-selectin on human tumor cells. Blood 89(9):3385–3395
Aigner S, Ramos CL, Hafezi-Moghadam A, Lawrence MB, Friederichs J, Altevogt P, Ley K (1998) CD24 mediates rolling of breast carcinoma cells on P-selectin. FASEB J 12(12):1241–1251
de Bruijne-Admiraal LG, Modderman PW, Von dem Borne AE, Sonnenberg A (1992) P-selectin mediates Ca(2+)-dependent adhesion of activated platelets to many different types of leukocytes: detection by flow cytometry. Blood 80(1):134–142
Li Z, Delaney MK, O’Brien KA, Du X (2010) Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol 30(12):2341–2349
Senis YA, Mazharian A, Mori J (2014) Src family kinases: at the forefront of platelet activation. Blood 124(13):2013–2024
Okutani D, Lodyga M, Han B, Liu M (2006) Src protein tyrosine kinase family and acute inflammatory responses. Am J Physiol Lung Cell Mol Physiol 291(2):L129–L141
Xiang B, Zhang G, Stefanini L, Bergmeier W, Gartner TK, Whiteheart SW, Li Z (2012) The Src family kinases and protein kinase C synergize to mediate Gq-dependent platelet activation. J Biol Chem 287(49):41277–41287
Falati S, Edmead CE, Poole AW (1999) Glycoprotein Ib-V-IX, a receptor for von Willebrand factor, couples physically and functionally to the Fc receptor gamma-chain, Fyn, and Lyn to activate human platelets. Blood 94(5):1648–1656
Mangin P, Yuan Y, Goncalves I, Eckly A, Freund M, Cazenave JP, Gachet C, Jackson SP, Lanza F (2003) Signaling role for phospholipase C gamma 2 in platelet glycoprotein Ib alpha calcium flux and cytoskeletal reorganization. Involvement of a pathway distinct from FcR gamma chain and Fc gamma RIIA. J Biol Chem 278(35):32880–32891
Yin H, Liu J, Li Z, Berndt MC, Lowell CA, Du X (2008) Src family tyrosine kinase Lyn mediates VWF/GPIb-IX-induced platelet activation via the cGMP signaling pathway. Blood 112(4):1139–1146
Piccardoni P, Sideri R, Manarini S, Piccoli A, Martelli N, de Gaetano G, Cerletti C, Evangelista V (2001) Platelet/polymorphonuclear leukocyte adhesion: a new role for SRC kinases in Mac-1 adhesive function triggered by P-selectin. Blood 98(1):108–116
Xu T, Zhang L, Geng ZH, Wang HB, Wang JT, Chen M, Geng JG (2007) P-selectin cross-links PSGL-1 and enhances neutrophil adhesion to fibrinogen and ICAM-1 in a Src kinase-dependent, but GPCR-independent mechanism. Cell Adhes Migr 1(3):115–123
Evangelista V, Pamuklar Z, Piccoli A, Manarini S, Dell’elba G, Pecce R, Martelli N, Federico L, Rojas M, Berton G et al (2007) Src family kinases mediate neutrophil adhesion to adherent platelets. Blood 109(6):2461–2469
Kovacs M, Nemeth T, Jakus Z, Sitaru C, Simon E, Futosi K, Botz B, Helyes Z, Lowell CA, Mocsai A (2014) The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. J Exp Med 211(10):1993–2011
Woollard KJ, Kling D, Kulkarni S, Dart AM, Jackson S, Chin-Dusting J (2006) Raised plasma soluble P-selectin in peripheral arterial occlusive disease enhances leukocyte adhesion. Circ Res 98(1):149–156
Cockcroft S (2006) The latest phospholipase C, PLCeta, is implicated in neuronal function. Trends Biochem Sci 31(1):4–7
Stalker TJ, Newman DK, Ma P, Wannemacher KM, Brass LF (2012) Platelet signaling. Handb Exp Pharmacol 210:59–85
Futosi K, Fodor S, Mocsai A (2013) Reprint of Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol 17(4):1185–1197
Resendiz JC, Kroll MH, Lassila R (2007) Protease-activated receptor-induced Akt activation–regulation and possible function. J Thromb Haemost 5(12):2484–2493
Pasquet JM, Bobe R, Gross B, Gratacap MP, Tomlinson MG, Payrastre B, Watson SP (1999) A collagen-related peptide regulates phospholipase Cgamma2 via phosphatidylinositol 3-kinase in human platelets. Biochem J 342(Pt 1):171–177
Asselin J, Gibbins JM, Achison M, Lee YH, Morton LF, Farndale RW, Barnes MJ, Watson SP (1997) A collagen-like peptide stimulates tyrosine phosphorylation of syk and phospholipase C gamma2 in platelets independent of the integrin alpha2beta1. Blood 89(4):1235–1242
Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D (2000) Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science 287(5455):1046–1049
Jiang H, Kuang Y, Wu Y, Xie W, Simon MI, Wu D (1997) Roles of phospholipase C beta2 in chemoattractant-elicited responses. Proc Natl Acad Sci USA 94(15):7971–7975
Laurent PA, Severin S, Gratacap MP, Payrastre B (2014) Class I PI 3-kinases signaling in platelet activation and thrombosis: PDK1/Akt/GSK3 axis and impact of PTEN and SHIP1. Adv Biol Regul 54:162–174
Hawkins PT, Stephens LR, Suire S, Wilson M (2010) PI3K signaling in neutrophils. Curr Topics Microbiol Immunol 346:183–202
Bhaskar PT, Hay N (2007) The two TORCs and Akt. Dev Cell 12(4):487–502
Watson SP, Auger JM, McCarty OJ, Pearce AC (2005) GPVI and integrin alphaIIb beta3 signaling in platelets. J Thromb Haemost 3(8):1752–1762
Weng Z, Li D, Zhang L, Chen J, Ruan C, Chen G, Gartner TK, Liu J (2010) PTEN regulates collagen-induced platelet activation. Blood 116(14):2579–2581
Lioubin MN, Algate PA, Tsai S, Carlberg K, Aebersold A, Rohrschneider LR (1996) p150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes Dev 10(9):1084–1095
Mondal S, Subramanian KK, Sakai J, Bajrami B, Luo HR (2012) Phosphoinositide lipid phosphatase SHIP1 and PTEN coordinate to regulate cell migration and adhesion. Mol Biol Cell 23(7):1219–1230
Chen M, Geng JG (2001) Inhibition of protein tyrosine phosphatases suppresses P-selectin exocytosis in activated human platelets. Biochem Biophys Res Commun 286(4):831–838
Kovacsovics TJ, Bachelot C, Toker A, Vlahos CJ, Duckworth B, Cantley LC, Hartwig JH (1995) Phosphoinositide 3-kinase inhibition spares actin assembly in activating platelets but reverses platelet aggregation. J Biol Chem 270(19):11358–11366
Libersan D, Merhi Y (2003) Platelet P-selectin expression: requirement for protein kinase C, but not protein tyrosine kinase or phosphoinositide 3-kinase. Thromb Haemost 89(6):1016–1023
Watanabe N, Nakajima H, Suzuki H, Oda A, Matsubara Y, Moroi M, Terauchi Y, Kadowaki T, Suzuki H, Koyasu S et al (2003) Functional phenotype of phosphoinositide 3-kinase p85alpha-null platelets characterized by an impaired response to GP VI stimulation. Blood 102(2):541–548
Yap CL, Anderson KE, Hughan SC, Dopheide SM, Salem HH, Jackson SP (2002) Essential role for phosphoinositide 3-kinase in shear-dependent signaling between platelet glycoprotein Ib/V/IX and integrin alpha(IIb)beta(3). Blood 99(1):151–158
Gao XP, Zhu X, Fu J, Liu Q, Frey RS, Malik AB (2007) Blockade of class IA phosphoinositide 3-kinase in neutrophils prevents NADPH oxidase activation- and adhesion-dependent inflammation. J Biol Chem 282(9):6116–6125
Mueller H, Stadtmann A, Van Aken H, Hirsch E, Wang D, Ley K, Zarbock A (2010) Tyrosine kinase Btk regulates E-selectin-mediated integrin activation and neutrophil recruitment by controlling phospholipase C (PLC) gamma2 and PI3Kgamma pathways. Blood 115(15):3118–3127
Chen J, De S, Damron DS, Chen WS, Hay N, Byzova TV (2004) Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood 104(6):1703–1710
Woulfe D, Jiang H, Morgans A, Monks R, Birnbaum M, Brass LF (2004) Defects in secretion, aggregation, and thrombus formation in platelets from mice lacking Akt2. J Clin Investig 113(3):441–450
O’Brien KA, Stojanovic-Terpo A, Hay N, Du X (2011) An important role for Akt3 in platelet activation and thrombosis. Blood 118(15):4215–4223
Chen J, Tang H, Hay N, Xu J, Ye RD (2010) Akt isoforms differentially regulate neutrophil functions. Blood 115(21):4237–4246
Nishizuka Y (1995) Protein kinase C and lipid signaling for sustained cellular responses. FASEB J 9(7):484–496
Chari R, Getz T, Nagy B Jr, Bhavaraju K, Mao Y, Bynagari YS, Murugappan S, Nakayama K, Kunapuli SP (2009) Protein kinase C[delta] differentially regulates platelet functional responses. Arterioscler Thromb Vasc Biol 29(5):699–705
Kilpatrick LE, Sun S, Li H, Vary TC, Korchak HM (2010) Regulation of TNF-induced oxygen radical production in human neutrophils: role of delta-PKC. J Leukoc Biol 87(1):153–164
Begonja AJ, Geiger J, Rukoyatkina N, Rauchfuss S, Gambaryan S, Walter U (2007) Thrombin stimulation of p38 MAP kinase in human platelets is mediated by ADP and thromboxane A2 and inhibited by cGMP/cGMP-dependent protein kinase. Blood 109(2):616–618
Carestia A, Rivadeneyra L, Romaniuk MA, Fondevila C, Negrotto S, Schattner M (2013) Functional responses and molecular mechanisms involved in histone-mediated platelet activation. Thromb Haemost 110(5):1035–1045
Khreiss T, Jozsef L, Chan JS, Filep JG (2004) Activation of extracellular signal-regulated kinase couples platelet-activating factor-induced adhesion and delayed apoptosis of human neutrophils. Cell Signal 16(7):801–810
Totani L, Piccoli A, Dell’Elba G, Concetta A, Di Santo A, Martelli N, Federico L, Pamuklar Z, Smyth SS, Evangelista V (2014) Phosphodiesterase type 4 blockade prevents platelet-mediated neutrophil recruitment at the site of vascular injury. Arterioscler Thromb Vasc Biol 34(8):1689–1696
Oeckinghaus A, Ghosh S (2009) The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 1(4):a000034
Malaver E, Romaniuk MA, D’Atri LP, Pozner RG, Negrotto S, Benzadon R, Schattner M (2009) NF-kappaB inhibitors impair platelet activation responses. J Thromb Haemost 7(8):1333–1343
Lee HS, Kim SD, Lee WM, Endale M, Kamruzzaman SM, Oh WJ, Cho JY, Kim SK, Cho HJ, Park HJ et al (2010) A noble function of BAY 11-7082: inhibition of platelet aggregation mediated by an elevated cAMP-induced VASP, and decreased ERK2/JNK1 phosphorylations. Eur J Pharmacol 627(1–3):85–91
Rivadeneyra L, Carestia A, Etulain J, Pozner RG, Fondevila C, Negrotto S, Schattner M (2014) Regulation of platelet responses triggered by Toll-like receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappaB. Thromb Res 133(2):235–243
Lalor PF, Herbert J, Bicknell R, Adams DH (2013) Hepatic sinusoidal endothelium avidly binds platelets in an integrin-dependent manner, leading to platelet and endothelial activation and leukocyte recruitment. Am J Physiol Gastrointest Liver Physiol 304(5):G469–G478
Lee DH, Tam SS, Wang E, Taylor GR, Plante RK, Lau CY (1996) The NF-kappa B inhibitor, tepoxalin, suppresses surface expression of the cell adhesion molecules CD62E, CD11b/CD18 and CD106. Immunol Lett 53(2–3):109–113
Takai Y, Sasaki T, Matozaki T (2001) Small GTP-binding proteins. Physiol Rev 81(1):153–208
Aslan JE, McCarty OJ (2013) Rho GTPases in platelet function. J Thromb Haemost 11(1):35–46
Cherfils J, Zeghouf M (2013) Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev 93(1):269–309
Akbar H, Kim J, Funk K, Cancelas JA, Shang X, Chen L, Johnson JF, Williams DA, Zheng Y (2007) Genetic and pharmacologic evidence that Rac1 GTPase is involved in regulation of platelet secretion and aggregation. J Thromb Haemost 5(8):1747–1755
Akbar H, Shang X, Perveen R, Berryman M, Funk K, Johnson JF, Tandon NN, Zheng Y (2011) Gene targeting implicates Cdc42 GTPase in GPVI and non-GPVI mediated platelet filopodia formation, secretion and aggregation. PLoS ONE 6(7):e22117
Pleines I, Dutting S, Cherpokova D, Eckly A, Meyer I, Morowski M, Krohne G, Schulze H, Gachet C, Debili N et al (2013) Defective tubulin organization and proplatelet formation in murine megakaryocytes lacking Rac1 and Cdc42. Blood 122(18):3178–3187
Pleines I, Eckly A, Elvers M, Hagedorn I, Eliautou S, Bender M, Wu X, Lanza F, Gachet C, Brakebusch C et al (2010) Multiple alterations of platelet functions dominated by increased secretion in mice lacking Cdc42 in platelets. Blood 115(16):3364–3373
Bialkowska K, Zaffran Y, Meyer SC, Fox JE (2003) 14-3-3 zeta mediates integrin-induced activation of Cdc42 and Rac. Platelet glycoprotein Ib-IX regulates integrin-induced signaling by sequestering 14-3-3 zeta. J Biol Chem 278(35):33342–33350
Zhang G, Xiang B, Ye S, Chrzanowska-Wodnicka M, Morris AJ, Gartner TK, Whiteheart SW, White GC 2nd, Smyth SS, Li Z (2011) Distinct roles for Rap1b protein in platelet secretion and integrin alphaIIbbeta3 outside-in signaling. J Biol Chem 286(45):39466–39477
Hidari KI, Weyrich AS, Zimmerman GA, McEver RP (1997) Engagement of P-selectin glycoprotein ligand-1 enhances tyrosine phosphorylation and activates mitogen-activated protein kinases in human neutrophils. J Biol Chem 272(45):28750–28756
M’Rabet L, Coffer P, Zwartkruis F, Franke B, Segal AW, Koenderman L, Bos JL (1998) Activation of the small GTPase rap1 in human neutrophils. Blood 92(6):2133–2140
Caron E, Self AJ, Hall A (2000) The GTPase Rap1 controls functional activation of macrophage integrin alphaMbeta2 by LPS and other inflammatory mediators. Curr Biol 10(16):974–978
Kumar S, Xu J, Perkins C, Guo F, Snapper S, Finkelman FD, Zheng Y, Filippi MD (2012) Cdc42 regulates neutrophil migration via crosstalk between WASp, CD11b, and microtubules. Blood 120(17):3563–3574
Anderson KE, Chessa TA, Davidson K, Henderson RB, Walker S, Tolmachova T, Grys K, Rausch O, Seabra MC, Tybulewicz VL et al (2010) PtdIns3P and Rac direct the assembly of the NADPH oxidase on a novel, pre-phagosomal compartment during FcR-mediated phagocytosis in primary mouse neutrophils. Blood 116(23):4978–4989
Lassegue B, Griendling KK (2010) NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 30(4):653–661
Peyssonaux C, Johnson RS (2004) An unexpected role for hypoxic response: oxygenation and inflammation. Cell Cycle 3(2):168–171
Dewhirst MW, Cao Y, Moeller B (2008) Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 8(6):425–437
Thompson AA, Elks PM, Marriott HM, Eamsamarng S, Higgins KR, Lewis A, Williams L, Parmar S, Shaw G, McGrath EE et al (2014) Hypoxia-inducible factor 2alpha regulates key neutrophil functions in humans, mice, and zebrafish. Blood 123(3):366–376
Haddad JJ (2002) Antioxidant and prooxidant mechanisms in the regulation of redox(y)-sensitive transcription factors. Cell Signal 14(11):879–897
Kong T, Eltzschig HK, Karhausen J, Colgan SP, Shelley CS (2004) Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc Natl Acad Sci USA 101(28):10440–10445
Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM, Cowburn AS, Johnson N et al (2005) Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med 201(1):105–115
van Uden P, Kenneth NS, Rocha S (2008) Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem J 412(3):477–484
Walsh TG, Berndt MC, Carrim N, Cowman J, Kenny D, Metharom P (2014) The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol 2:178–186
Brill A, Chauhan AK, Canault M, Walsh MT, Bergmeier W, Wagner DD (2009) Oxidative stress activates ADAM17/TACE and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc Res 84(1):137–144
Pignatelli P, Sanguigni V, Lenti L, Ferro D, Finocchi A, Rossi P, Violi F (2004) gp91phox-dependent expression of platelet CD40 ligand. Circulation 110(10):1326–1329
Begonja AJ, Gambaryan S, Geiger J, Aktas B, Pozgajova M, Nieswandt B, Walter U (2005) Platelet NAD(P)H-oxidase-generated ROS production regulates alphaIIbbeta3-integrin activation independent of the NO/cGMP pathway. Blood 106(8):2757–2760
Dinauer MC, Orkin SH, Brown R, Jesaitis AJ, Parkos CA (1987) The glycoprotein encoded by the X-linked chronic granulomatous disease locus is a component of the neutrophil cytochrome b complex. Nature 327(6124):717–720
Quie PG, White JG, Holmes B, Good RA (1967) In vitro bactericidal capacity of human polymorphonuclear leukocytes: diminished activity in chronic granulomatous disease of childhood. J Clin Investig 46(4):668–679
Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC (1995) Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet 9(2):202–209
Fialkow L, Wang Y, Downey GP (2007) Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic Biol Med 42(2):153–164
Carbone F, Camillo Teixeira P, Braunersreuther V, Mach F, Vuilleumier N, Montecucco F (2014) Pathophysiology and treatments of oxidative injury in ischemic stroke: focus on the phagocytic NADPH oxidase 2. Antioxid Redox Signal
Radermacher KA, Wingler K, Langhauser F, Altenhofer S, Kleikers P, Hermans JJ, Hrabe de Angelis M, Kleinschnitz C, Schmidt HH (2013) Neuroprotection after stroke by targeting NOX4 as a source of oxidative stress. Antioxid Redox Signal 18(12):1418–1427
Hagedorn I, Schmidbauer S, Pleines I, Kleinschnitz C, Kronthaler U, Stoll G, Dickneite G, Nieswandt B (2010) Factor XIIa inhibitor recombinant human albumin Infestin-4 abolishes occlusive arterial thrombus formation without affecting bleeding. Circulation 121(13):1510–1517
Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT (2009) Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. FEBS J 276(1):13–26
Yilmaz G, Granger DN (2008) Cell adhesion molecules and ischemic stroke. Neurol Res 30(8):783–793
Jin R, Yang G, Li G (2010) Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 87(5):779–789
Jickling GC, Ander BP, Zhan X, Noblett D, Stamova B, Liu D (2014) microRNA expression in peripheral blood cells following acute ischemic stroke and their predicted gene targets. PLoS ONE 9(6):e99283
Kellert L, Hametner C, Rohde S, Bendszus M, Hacke W, Ringleb P, Stampfl S (2013) Endovascular stroke therapy: tirofiban is associated with risk of fatal intracerebral hemorrhage and poor outcome. Stroke 44(5):1453–1455
Stoll G, Kleinschnitz C, Nieswandt B (2008) Molecular mechanisms of thrombus formation in ischemic stroke: novel insights and targets for treatment. Blood 112(9):3555–3562
Carvalho-Tavares J, Hickey MJ, Hutchison J, Michaud J, Sutcliffe IT, Kubes P (2000) A role for platelets and endothelial selectins in tumor necrosis factor-alpha-induced leukocyte recruitment in the brain microvasculature. Circ Res 87(12):1141–1148
Soriano SG, Coxon A, Wang YF, Frosch MP, Lipton SA, Hickey PR, Mayadas TN (1999) Mice deficient in Mac-1 (CD11b/CD18) are less susceptible to cerebral ischemia/reperfusion injury. Stroke 30(1):134–139
Walder CE, Green SP, Darbonne WC, Mathias J, Rae J, Dinauer MC, Curnutte JT, Thomas GR (1997) Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke 28(11):2252–2258
De Silva TM, Brait VH, Drummond GR, Sobey CG, Miller AA (2011) Nox2 oxidase activity accounts for the oxidative stress and vasomotor dysfunction in mouse cerebral arteries following ischemic stroke. PLoS ONE 6(12):e28393
Tang XN, Zheng Z, Giffard RG, Yenari MA (2011) Significance of marrow-derived nicotinamide adenine dinucleotide phosphate oxidase in experimental ischemic stroke. Ann Neurol 70(4):606–615
Kleinschnitz C, Grund H, Wingler K, Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft P et al (2010) Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol 8(9)
Bunn HF (1997) Pathogenesis and treatment of sickle cell disease. N Engl J Med 337(11):762–769
Frenette PS, Atweh GF (2007) Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Investig 117(4):850–858
Ballas SK, Gupta K, Adams-Graves P (2012) Sickle cell pain: a critical reappraisal. Blood 120(18):3647–3656
Polanowska-Grabowska R, Wallace K, Field JJ, Chen L, Marshall MA, Figler R, Gear AR, Linden J (2010) P-selectin-mediated platelet–neutrophil aggregate formation activates neutrophils in mouse and human sickle cell disease. Arterioscler Thromb Vasc Biol 30(12):2392–2399
Chang J, Patton JT, Sarkar A, Ernst B, Magnani JL, Frenette PS (2010) GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood 116(10):1779–1786
Almeida CB, Scheiermann C, Jang JE, Prophete C, Costa FF, Conran N, Frenette PS (2012) Hydroxyurea and a cGMP-amplifying agent have immediate benefits on acute vaso-occlusive events in sickle cell disease mice. Blood 120(14):2879–2888
Chen G, Zhang D, Fuchs TA, Manwani D, Wagner DD, Frenette PS (2014) Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 123(24):3818–3827
Wun T, Styles L, DeCastro L, Telen MJ, Kuypers F, Cheung A, Kramer W, Flanner H, Rhee S, Magnani JL et al (2014) Phase 1 study of the E-selectin inhibitor GMI 1070 in patients with sickle cell anemia. PLoS ONE 9(7):e101301
Okpala I. Investigational selectin-targeted therapy of sickle cell disease. Expert Opin Investig Drugs. 2014:1–10
Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, Hu J, Wang Y, Wagner DD (2013) Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci USA 110(21):8674–8679
Savchenko AS, Martinod K, Seidman MA, Wong SL, Borissoff JI, Piazza G, Libby P, Goldhaber SZ, Mitchell RN, Wagner DD (2014) Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J Thromb Haemost 12(6):860–870
Knight JS, Luo W, O’Dell AA, Yalavarthi S, Zhao W, Subramanian V, Guo C, Grenn RC, Thompson PR, Eitzman DT et al (2014) Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res 114(6):947–956
Hebbel RP, Vercellotti GM, Pace BS, Solovey AN, Kollander R, Abanonu CF, Nguyen J, Vineyard JV, Belcher JD, Abdulla F et al (2010) The HDAC inhibitors trichostatin A and suberoylanilide hydroxamic acid exhibit multiple modalities of benefit for the vascular pathobiology of sickle transgenic mice. Blood 115(12):2483–2490
Belcher JD, Young M, Chen C, Nguyen J, Burhop K, Tran P, Vercellotti GM (2013) MP4CO, a pegylated hemoglobin saturated with carbon monoxide, is a modulator of HO-1, inflammation, and vaso-occlusion in transgenic sickle mice. Blood 122(15):2757–2764
Vercellotti GM, Khan FB, Nguyen J, Chen C, Bruzzone CM, Bechtel H, Brown G, Nath KA, Steer CJ, Hebbel RP et al (2014) H-ferritin ferroxidase induces cytoprotective pathways and inhibits microvascular stasis in transgenic sickle mice. Front Pharmacol. 5:79
Acknowledgments
We apologize to our colleagues whose works are not cited in this review due to space restrictions. We thank Soochong Kim for his helpful comments on the manuscript. This work was supported by grants from National Institutes of Health. J.L. and K.K. are recipients of the American Heart Association postdoctoral fellowship award.
Conflict of interest
All authors declare no competing financial interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. Li, K. Kim, and A. Barazia contributed equally to this review.
Rights and permissions
About this article

Cite this article
Li, J., Kim, K., Barazia, A. et al. Platelet–neutrophil interactions under thromboinflammatory conditions. Cell. Mol. Life Sci. 72, 2627–2643 (2015). https://doi.org/10.1007/s00018-015-1845-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-1845-y