
Abstract
Poly(ADP-ribose) polymerase-2 (PARP-2) activity contributes to a cells’ poly(ADP-ribosyl)ating potential and like PARP-1, has been implicated in several DNA repair pathways including base excision repair and DNA single strand break repair. Here the consequences of its stable depletion in HeLa, U20S, and AS3WT2 cells were examined. All three PARP-2 depleted models showed increased sensitivity to the cell killing effects on ionizing radiation as reported in PARP-2 depleted mouse embryonic fibroblasts providing further evidence for a role in DNA strand break repair. The PARP-2 depleted HeLa cells also showed both higher constitutive and DNA damage-induced levels of polymers of ADP-ribose (PAR) associated with unchanged PARP-1 protein levels, but higher PARP activity and a concomitant lower PARG protein levels and activity. These changes were accompanied by a reduced maximal recruitment of PARP-1, XRCC1, PCNA, and PARG to DNA damage sites. This PAR-associated phenotype could be reversed in HeLa cells on re-expression of PARP-2 and was not seen in U20S and AS3WT2 cells. These results highlight the complexity of the relationship between different members of the PARP family on PAR metabolism and suggest that cell model dependent phenotypes associated with the absence of PARP-2 exist within a common background of radiation sensitivity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The observation that polymers of ADP-ribose (PAR) could be synthesised in response to DNA-damaging agents in cells from Poly(ADP-ribose) polymerase 1 (Parp1) knockout (KO) mice [1, 2] led to the isolation of a second poly(ADP)ribose polymerase, PARP-2 [1]. In mouse models, the KO of either Parp1 or Parp2 has no impact on viability; however, embryonic lethality is seen in the double KO mice suggesting that there is some overlap in the cellular activities of these two members of the PARP family as DNA damage sensors and signal transducers in single strand break repair (SSBR), base excision repair (BER), and double strand break repair (DSB) [3, 4] (for reviews see [5, 6]). Both PARP-1 and PARP-2 are activated through their binding to DNA strand interruptions, and catalyze the transfer of ADP-ribose moieties from NAD+ to acceptor proteins, resulting in the attachment of linear or branched PAR. PARP-1 and PARP-2 can form homodimers and heterodimers [4] and are themselves targets for poly(ADP-ribosyl)ation with this auto-modification playing a key role in the control of their activity and localization. Both proteins are recruited to sites of laser-induced DNA damage, albeit with different kinetics: PARP-1 was shown to accumulate quickly and transiently, while, in comparison to PARP-1, PARP-2 showed a delayed and persistent accumulation at repair sites in both MEFs and HeLa cells [7]. Based on these kinetic studies and the affinities of PARP-2 compared to PARP-1 for different DNA structures, it was proposed that PARP-2 had a role in the later steps of the BER process [7]. In addition to members of the PARP family, other proteins have a high affinity for poly(ADP-ribosyl)ated proteins or the polymers themselves which is exploited in certain mechanisms, such as DNA repair, for the control of the localization of proteins involved in these processes (see [8–10] and references there in). One such protein is XRCC1 that interacts physically and functionally with practically all the actors of the BER and SSBR pathways [4, 11, 12] and in particular with those implicated in the Short Patch (SP) BER sub-pathway. Intriguingly, although XRCC1 is recruited very slowly to sites of laser-induced DNA damage in MEFs lacking Parp1 in contrast to its normal recruitment in Parp2 deleted MEFS [7], XRCC1 negatively regulates the activity of both PARP-1 and PARP-2 by limiting their auto-modification and thus forcing them to reside on the DNA damage site [4]. Taken together these results would suggest that each PARP contributes differently to the SSBR and BER mechanisms. Extensive studies on PARP-1 have allowed its role in these two repair mechanisms to be defined (for a review see [13, 14]) in addition to its involvement in other repair mechanisms such as the alternative non-homologous end-joining (NHEJ) [15–17] and nucleotide excision repair [18, 19]. In contrast, although biochemical studies have revealed that PARP-2, like PARP-1, interacts with the SSBR/BER factors XRCC1, DNA polymerase beta and DNA ligase III [1, 4], its role in both SP and Long Patch (LP) BER sub-pathways remains to be fully elucidated and in particular in human cells.
In the present study, the impact of the stable depletion of PARP-2 in three cell models was examined. In all an increased sensitivity to the cell killing effects of ionizing radiation was detected supporting a role of PARP-2 in DNA strand break repair. However, only in the PARP-2 depleted HeLa cells was a phenotype observed of higher PARP activity and lower PARG protein levels associated with higher basal and DNA induced levels of PAR and sub-maximal recruitment of PARP-1 and BER proteins to sites of laser-induced DNA damage. These results highlight the complexity of the relationship between PARP-1, PARP-2, and PARG on the status quo of polymer levels and suggest that certain phenotypes associated with the depletion of PARP-2 are cell line dependent.
Materials and methods
Cell lines, gene silencing, and drugs
shRNA design and cloning in pEBVsiRNA vectors carrying a hygromycin B resistance cassette and establishment of stable knockdown clones for PARP-1 (PARP-1KD), PARP-2 (PARP-2KD), and PARG (PARGKD) and Control clones by selection and amplification of resistant colonies to hygromycin B was carried out as previously described [1, 20, 21]. HeLa cells carrying the pBD650 plasmid that expressed an inefficient shRNA sequence were used as Controls [21]. The RNAi targeted sequences for parp1 (NM_001618), and parg (NM_003631) were nucleotides 2,068–2,086 and nucleotides 2,325–2,343, respectively. For parp2 (NM_005484) two sequences were targeted nucleotides 829–847 (clones1,470/1 and 1470/7) and 1,077–1,095 (clone 1,471/13). All HeLa cells clones were grown in Dulbecco’s modified Eagle’s medium supplemented with 10 % FCS, 100 units/ml penicillin and 100 mg/ml streptomycin in the presence of 125 µg/ml hygromycin B at 37 °C under 5 % CO2. The control and PARP-2 depleted cell populations derived from the osteosarcoma cells U2OS and the SV40 immortalized fibroblast AS3WT2 cells from a healthy donor [22], were obtained by transfecting the pEBV vectors [21] expressing the control shRNA sequence (U2OS CTL and AS3WT2 CTL cells) or an shRNA that targeted parp2 at nucleotides 829–847 (U2OS PARP-2KD and AS3WT2 PARP-2KD cells) unless otherwise indicated. Transfected cells were selected by supplementing the standard growth media with 150 μg/ml hygromycin B. All tissue culture reagents were from Life Technologies (Cergy-Pontoise, France). The neocarzinostatin (NCS) holoprotein, prepared and titrated as described [23], was stored as a sterile 1 mM stock solution in 2 mM sodium formate buffer, pH 4.0, in liquid nitrogen.
Western blotting
Total protein extraction was obtained from the different cell lines using the M-PER (Pierce, Perbio Science, Brebières, France) reagent supplemented with a cocktail of protease and phosphatase inhibitors. Protein concentrations were measured by the Bradford method using the Bio-Rad protein assay. Following denaturing extracts (40 µg/lane, 95 °C, 5 min) in Laemmli loading buffer and separation on 7.5 % SDS-PAGE gels, proteins were electro-transferred to nitrocellulose membranes. The membranes were blocked with 5 % non-fat milk for 1 h at room temperature, incubated with the primary antibodies (ALX-210-303-Yuc anti-PARP-2 from ALEXIS Biochemicals, Villeurbanne, France, B01P anti-PARG from Abnova, Heidelberg, Germany, and C2-10 anti-PARP-1 from Trevigen, Gaithersburg, USA) overnight at 4 °C and then with secondary antibodies coupled to horseradish peroxidase (Jackson Immuno Research Laboratories, Soham, Cambridgeshire, UK) for 1 h at room temperature. Membranes were probed with an enhanced chemiluminescence reagent and re-hybridized with an anti-α-tubulin antibody clone DM1A (Sigma-Aldrich, Saint-Quentin Fallavier, France). For PAR level analysis by western blot 1.5 × 106 cells were harvested in cold PBS and re-suspended in 50 μl of lysis buffer (62.5 mM Tris–HCl pH 6.8, 6 M urea, 10 % glycerol, 2 % SDS, 5 % 2-mercaptoethanol). The DNA was shredded by sonication and the samples were denatured by heating (65 °C, 15 min) prior loading onto a 7.5 % SDS-PAGE gel. After migration proteins were transferred onto Hybond Nylon membrane (GE Healthcare Lifescience, Orsay, France). The membrane was blocked in PBS containing 0.05 % Tween-20 and 5 % non-fat milk for 1 h at room temperature and incubated overnight at 4 °C with the anti-PAR polyclonal antibody (BD 551813) BD Biosciences, Le Pont-de-Claix, France) and then with secondary antibodies coupled to horseradish peroxidase (Jackson ImmunoResearch Laboratories, Soham, Cambridgeshire, UK) for 1 h at room temperature. Membranes were probed with an enhanced chemiluminescence reagent ECL-prime (GE Healthcare Lifescience, Orsay, France) and the signal was detected using the G: Box Chemi XL1.4 (Syngene, Frederick, USA).
RNA extraction, reverse transcription, and real-time quantitative PCR analysis
Total RNA was extracted using Rneasy mini kit (Qiagen). 1 μg RNA was reverse transcribed using the High Capacity RNA to cDNA kit (Applied Biosystems). To quantify the relative expression level of gapdh, parp1, parp2, and parg Taqman gene expression assays were used (Applied Biosystems). Briefly, cDNA was diluted in Taqman gene expression master mix with the specific Taqman assay for gapdh (Hs99999905_m1), parp1 (Hs00242302_m1), parp2 (Hs00193931_m1) or parg (Hs00608256_m1), and real-time qPCR was carried out using a StepOnePlus (Applied Biosystems). The cycle conditions were 50 °C for 2 min, 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. The threshold crossing value was obtained for each transcript and normalized to gapdh. The relative quantitation of each mRNA was performed using the comparative Ct method. Expression profiles were measured in three independent RNA samples.
Colony formation survival assays
To assess the clonogenic survival Control (1 × 103) and PARP-2KD (2 × 103) cells were plated in 25 cm2 flasks in triplicate, and allowed to adhere for 4–5 h at 37 °C before treatment. Flasks were then either exposed to ionizing radiation or treated with increasing concentrations of NCS. Aliquots of NCS were thawed just before use and diluted to the required treatment concentration in fresh complete medium adjusted to pH 7.0. The whole treatment was performed in dim light to avoid photo-induced degradation of the drug. Irradiations were carried out at room temperature using an IBL-637 (137Cs) γ-ray irradiator (CIS-Biointernational, Gif-sur-Yvette, France) at a dose rate of 0.7 Gy/min or with a low-energy Philips MCN-323 X-ray generator (200-kVp, 0.3-mm copper and 1-mm aluminium additional filtration, 80-keV effective energy) operating at 21 mA with a dose rate of 1 Gy/min. To assess whether survival differed between the Control and PARP-2KD cells when irradiated in S-phase, cells were synchronized at the G1–S junction using a double thymidine block and γ-irradiated 2 h after release from the block. Cell cycle progression was monitored by dual parameter flow cytometry using a FACSCalibur cytofluorometer (Becton–Dickinson Biosciences) with BrdUrd pulse labeling (10 µM, 15 min) of S-phase cells as described previously [24]. Colonies were allowed to grow for 10–15 days, before fixing with methanol, staining with Coomassie, and counting. Small colonies (less than 50 cells) were disregarded. The colony count relative to mock-treated cells (S) was adjusted for best fit to the classical linear-quadratic equation (Ln S = −αD−βD 2) where D is the radiation dose and α and β adjustable parameters characterizing the response. Calculations were made through non-linear least-squares regression taking all data points into account, using Kaleidagraph software (Synergy Software, Reading, Pennsylvania).
Expression plasmids and transfection
pEGFP-PARP-1 and pEGFP-PARP-2, pEGFP-NLS-PCNA, pXRCC1-YFP, pEGFP-PARG plasmids were a kind gift from Drs. V. Schreiber (UMR7175CNRS, ESBS, Illkirch, France), P. Kannouche (UMR 8200 CNRS, Gustave Roussy, Paris XI University, Villejuif, France), and P. Radicella (IRCM, CEA-DSV, Fontenay-aux-Roses, France), respectively. Plasmids were purified using a Macherey–Nagel NucleoBond® PC 500 EF kit (Düren, Germany). For live-cell experiments cells were plated at 50 % confluence onto 25 mm round glass coverslips and then transfected 24 h later using JETPEI (Polyplus transfection, Ozyme, St. Quentin en Yvelines, France) according to the manufacturer’s recommendations and cultured for a further 36 h before use.
Live-cell videomicroscopy
The recruitment and the real-time follow-up of the protein of interest was carried out through a Confocal Leica SP5 system attached to a DMI6000 stand using 63×/1.4 objective as previously described [25]. During experiments glass coverslips containing transfected cells were mounted in a Ludin Chamber and maintained at 37 °C. All recordings were made using the appropriate sampling frequency (512 × 512 images, line average of 4 and zooming set to 7.94). DNA damage was generated using a 405 nm laser line (diode) focused onto a single spot of constant size (176 nm) within the nucleus to generate a point of damage with a constant amount of energy. Fluorophores were excited using a 488 nm (EGFP) or 514 nm (YFP) Argon laser line. Recruitment of the protein of interest was then monitored by fluorescence using the same setting as for the pre-damage sequence. Nomarski contrast images were taken in parallel. Images were captured at 2–5 s interval. Cell motion during the acquisition was compensated with the StackReg plugin (Philippe Thévenaz, Biomedical Imaging Group, Swiss Federal Institute of Technology, Lausanne) and the kinetics of the recruitment of proteins were analyzed under the ImageJ software (Rasband, W.S., ImageJ, U.S. National Institutes of Health, http://imagej.nih.gov/ij/, 1997–2011) using an in-house developed macro. Data extraction is a semi-automated process where the user is first invited to draw two regions of interest (ROI): a nuclear region and the site of damage. The background ROI is automatically set to the inverse of the nuclear ROI. Intensity from the damage site is corrected for both the observational photo-bleaching and background, and normalized so that its intensity at start-up equals 1: corrected intensity = [(focus intensity(t) − background intensity(t) × focus area/background area) × nuclear intensity(t)][focus intensity (t 0) × nuclear intensity (t 0). The time point of induction of the laser-induced damage is extracted from the normalized curve by determining when the maximum change in intensity is achieved between two successive images. The normalized time course from the damage on was fitted to a mono-exponential recovery of the form y = a(1−exp(−k × t)) + b which allows the estimation of the half-time maximum recruitment (t 1/2 = (ln2)/k) as well as the expected maximum recruitment (max = a+b).
Immunofluorescence detection of PAR formation
For the detection of polymer formation after exposure to γ-rays cells were plated onto glass coverslips in 6-well plates and 48 h later were exposed to 8 Gy. Three min after the end of the irradiation cells were fixed (4 % formaldehyde in PBS for 25 min). Cells were then washed 3 times with PBS containing 0.1 % Triton X-100, rinsed with PBS alone and re-washed once with PBSMT (PBS with 2 % milk, 0.5 % Triton X-100). After blocking for 30 min at RT with PBSMT, cells were incubated with a primary antibody against poly (ADP-ribose) (Trevigen, clone 10HA, 1/400 dilution in PBSMT) for 1 h at 37 °C. Following washing with PBSMT, slides were incubated with AlexaFluor 488 goat anti rabbit IgG (Molecular Probes, 1/500 dilution in PBSMT) for 1 h at RT. After washing, once with PBSMT and twice with PBS, coverslips were mounted with Vectashield containing DAPI 1.5 µg/ml (Vector Laboratories). Images of fields were acquired automatically on a Leica confocal SP5 system, using its “tile scan” option, with a 63x objective (line average of 4 and zooming set to 4). 300 cells, at least, were analyzed per experimental condition. The global fluorescence intensity by nucleus was quantified in arbitrary units (AU) using Image J software.
Measurements of PARP and PARG activity
Total PARP activity was measured in triplicate as described by Zaremba et al. [26] using 5 × 103 digitonin-permeabilized cells in a reaction mixture containing 350 μM NAD+ and 10 μg/ml oligonucleotide. After blotting onto a nylon membrane, the PAR was detected following incubation with the primary anti-PAR 10H antibody (ALEXIS Biochemicals, Villeurbanne, France), then with secondary antibodies coupled to horseradish peroxidase (Jackson ImmunoResearch Laboratories) and finally visualization using the ECL Plus detection kit (GE Healthcare Lifescience, Orsay, France). The luminescent signal was detected and quantified using a G: Box Chemi XL1.4 (Syngene, Frederick, USA). Alternatively, PARP activity of adherent cells was also determined by measuring the incorporation of isotope from 32P-NAD+ into trichloroacetic acid (TCA)-precipitable proteins, as previously described [27]. Briefly cells were seeded in a 6 well plate 24 h prior to the activity assay, the culture media was then removed and the adherent cells overlaid with 1 ml of reaction buffer (56 mM Hepes pH 7.5, 28 mM KCl, 28 mM NaCl, 2 mM MgCl2, 0.01 % digitonin, 200 μg/ml of annealed oligonucleotide (CGGAATTCCG) and 0.125 μM NAD supplemented with 1.25 μCi/ml of 32P-NAD. Following a 10 min incubation at 37 °C, the cells were scraped on ice and the proteins were precipitated by adding 200 μl of ice cold 50 % TCA for 4 h. Cells were pelleted, samples were washed three times with 500 μl of ice cold 5 % TCA and solubilized overnight at 37 °C in 500 μl of 2 % SDS/0.1 N NaOH and the radioactivity counted in a liquid scintillation counter (TriCarb 2910TR, Perkin Elmer, USA). The PARP activity in the PARP depleted cells in the different cell lines is expressed as a % of the incorporated 32P-NAD seen in control cells of the corresponding cell line from at least three independent experiments. PARG activity was measured in cell extracts using a commercially available kit (HT Chemiluminescent PARG Assay Kit, Trevigen) that quantitates the loss of biotinylated PAR from histones according to the manufacturers’ instructions.
Statistics
Statistical analyses were performed using StatEL software (AD Science, Paris, France) or the VassarStats website (http://vassarstats.net/).
Results
Higher levels of polymer detected in PARP-2 depleted HeLa cells
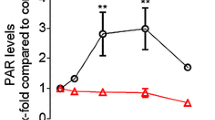
To investigate the impact of PARP-2 depletion in human cells, we first established three stably depleted PARP-2 (PARP-2KD) HeLa clones using an shRNA expression system that targeted two different nucleotide regions. These PARP-2KD cells were cultivated for more than 80 days in culture with levels of the targeted proteins remaining below 1 % of that seen in Control cells, as assessed by the quantification of western blots. The level of PARP-1 protein was unchanged in the three PARP-2KD clones; however, PARG mRNA (Supplementary Fig. 1a) and protein levels (Fig. 1a) were lower in all 3 clones. PARG protein levels were 53 ± 5 % in PARP-2KD clone 1,470/1, 52 ± 7 % in PARP-2KD clone 1,470/7 and 58 ± 9 % in PARP-2KD clone 1,471/13 compared to Control cells based on four independent protein extracts. A reduction in the level of the PARG protein in HeLa PARP-1KD cells (60 ± 8 % compared to Control cells based on 4 independent protein extracts) (Fig. 1a) was also noted in accordance with other studies [28, 29]. Basal PAR levels, as assessed by western blot (Fig. 1b) or immunofluorescence (Fig. 1c) were significantly higher in the PARP-2KD cells compared to Control cells. Induced PAR levels after exposure to IR were also statistically higher in the PARP-2KD cells (clones 1,470/7 and 1,471/13) compared to the HeLa Control cells (p < 10−5, n = 500 cells). This higher PAR level was associated with lower PARG activity in the 3 PARP-2KD clones (p = 0.0002 to p = 0.001 depending on the PARP-2KD clone) (Fig. 1d). Under these same experimental conditions, HeLa PARP-1 KD cells that have reduced PARG protein levels have a lower PARG activity compared to the Control cells (p = 0.0015). We used two different in vitro assays to measure the maximum PARP activity in digitonin-permeabilized cells. With both approaches, we observed that the depletion of PARP-2 resulted in higher levels of PARP activity compared to Control cells (Fig. 1e, f). The difference observed in the PARP activity using a PAR immunodetection-based approach (Fig. 1f) compared to measuring the incorporation of 32P-NAD+ into TCA-precipitable proteins in the HeLa PARP-1KD cells might reflect a difference in assay sensitivity. This low PARP activity in the PARP-1KD cells is less than the 10–15 % contribution to the poly(ribosy)lating activity of PARP-2 reported in PARP-1−/− MEFs [4] and PARP-1−/− 3T3 cells [1] and may reflect the nature of the DNA substrate used to activate the different PARPs in in vitro assays that assess the total poly(ribosy)lating potential of a cell [1, 26].

Modification of PAR levels, PARG, and PARP activity in HeLa PARP-2KD cells. a Representative levels of expression of the PARP-2, PARG, and PARP-1 proteins in the different HeLa cell cultures as assessed by western blotting. b Polymer levels as assessed by western blotting from untreated cell cultures. c Representative images of PAR immunofluorescence in Control cells or PARP-2KD cells before and after exposure to 8 Gy of γ-rays IR and the quantification of the PAR staining. Data represents the mean ± SD based on the fluorescence intensity in 500 cells/treatment group in two independent experiments. d PARG activity measured in protein extracts from the different cell lines, expressed as a % of the activity seen in the Control cells. Data are median ±95 % confidence interval for four independent experiments in triplicate and shows that in PARP-2KD cells PARG activity is significantly reduced compared to the Control cells. e Maximum PARP activity in permeabilized cells expressed as a % of the activity detected in Control cells showing that the PARP-2KD clones have a significantly higher activity that the Control cells. Data are median ±95 % confidence interval for three independent experiments in triplicate. f PARP activity in the HeLa PARP-2KD clone 1,470/7 assessed by the incorporation of isotope from 32P-NAD+ into trichloroacetic acid-precipitable proteins, expressed as a % of the activity detected in Control cells, was higher than in Control cells. Data represents the mean ± SD of four independent experiments
The depletion of PARP-2 in HeLa cells modulates the recruitment of PARP-1 and XRCC1
To investigate the impact of higher polymer levels in the HeLa PARP-2 depleted cells, we next investigated the recruitment to and the dissociation from sites of laser-induced damage of fluorescent-tagged PARP-1, XRCC1, PCNA, and PARG in real time. The EGFP-PARP-1 protein shows a rapid and transient accumulation at the site of damage immediately after micro-irradiation in Control cells (Fig. 2a) as previously described by several groups [7, 25, 30, 31]. In contrast, EGFP-PARP-1 shows a slower and reduced recruitment in the three HeLa PARP-2 depleted cells (57 s for PARP-2KD clone 1,470/1, 58 s for PARP-2KD clone 1,470/7 and 62 s for PARP-2KD 1,471/13 with a maximum relative intensity of 2.43 ± 0.07, 2.32 ± 0.07, and 2.26 ± 0.08, respectively, vs. a maximum relative intensity of 2.96 ± 0.07 reached at 50 s for Control cells p < 10−3) (Fig. 2a). These results would suggest that in HeLa cells PARP-2 is necessary for a maximal PARP-1 recruitment to DNA damage sites.

Recruitment of PARP-1, XRCC1, PCNA, and PARG in HeLa Control and PARP-2KD cells to UVA laser-induced damage sites. Quantitative analysis of the relative spot intensity with time of a EGFP-PARP-1, b XRCC1-YFP, c EGFP-PCNA, and d EGFP-PARG recruitment in Control cells (closed circles) and in the three PARP-2KD clones (open triangles, circles, and squares). For comparative purposes the recruitment of XRCC1-YFP in HeLa PARP-1KD cells (panel b) and EGFP-PCNA in HeLa PARGKD cells (panel c) are shown. Data represents mean relative spot intensity ± SEM, n = 5–23 individual cells from a minimum of two independent experiments
Under the same experimental conditions, XRCC1-YFP recruitment was initially slower and reached lower maximal levels in all three HeLa PARP-2KD clones compared to Control cells (maximal relative spot intensity reached was 4.40 ± 0.16 in PARP2KD clone 1,470/1 (n = 13), 4.52 ± 0.15 in PARP2KD clone 1,470/7 (n = 9) and 4.34 ± 0.18 in PARP2KD 1,471/13 (n = 13) vs. 6.86 ± 0.16 (n = 18) for Control cells). This reduction was not as marked as seen for XRCC1-YFP recruitment in PARP-1KD cells (maximal relative spot intensity reached was 1.83 ± 0.06) (Fig. 2b) but would suggest that the presence of PARP-2 is necessary to obtain the recruitment profile of XRCC1 seen in Control cells. The reasons behind this reduction remain unclear: it could be related to the lower PARP-1 recruitment that impacts directly on XRCC1 recruitment or that the higher nuclear PAR levels in these PARP-2KD cells could impede XRCC1 recruitment to DNA damage sites. A reduced XRCC1 recruitment was also seen in Cdk5-depleted cells that have elevated basal polymer levels [32]. Another possibility is that PARP-2 itself is involved in the recruitment of XRCC1 as PARP-2 has been shown to interact with XRCC1 [4] and thus in its absence, XRCC1 recruitment would be reduced.
In addition to a reduction in the recruitment of these key BER/SSBR proteins in the absence of PARP-2 in this HeLa cell model, PARP-2 depletion significantly reduced (p < 0.0001) the recruitment of GFP-PCNA with recruitment not reaching the levels seen in Control cells (relative spot intensity reached at 300 s was 2.03 ± 0.11 in PARP2KD clone 1,470/1 (n = 13), 1.95 ± 0.05 in PARP2KD clone 1,470/7 (n = 16), and 2.06 ± 0.08 in PARP2KD 1,471/13 (n = 5) vs. 3.53 ± 0.24 (n = 16) for Control cells) (Fig. 2c).
It has been previously shown that the recruitment of PARG to DNA damage sites depends not only on PARP-1 and PAR synthesis but also on the interaction of PARG with PCNA [31, 33]. As predicted from the reduced PCNA recruitment observed the maximal levels of EGFP-PARG recruitment at the damage site plateaued in the HeLa PARP-2KD cells was about 50 % of the levels seen in the Control cells post-irradiation (maximum relative spot intensity 5.38 ± 0.18 in Control (n = 16), 2.55 ± 0.02 in PARP2KD clone 1,470/1 (n = 14) and 2.80 ± 0.02 in PARP2KD 1,471/13 (n = 10)) (Fig. 2d). A similar kinetic profile was also seen for the recruitment of EGFP-PCNA in a PARGKD HeLa cell line showing that PARG also is required for PCNA recruitment (Fig. 2e).
The PARP-2 protein expression was restored by cultivating the PARP-2KD cells in the absence of hygromycin B for 20 days with a concomitant increase in the expression of the PARP-2 and PARG proteins (Supplementary Fig. 2a). In addition, the increase in PARP activity (Supplementary Fig. 2b), the higher levels of basal polymer levels (Supplementary Fig. 2c) and the reduced GFP-PARP-1 recruitment to sites of laser-induced DNA damage (Supplementary Fig. 2d) were all restored when the cells were grown in the absence of the selective agent hygromycin. Based on the phenotype observed in HeLa cells, it would suggest that PARP-2 plays a role in the regulation of the PARG protein expression and the regulation of PAR metabolism.
PARP-2 depleted cells are sensitive to DNA single and double strand breaks
We next examined the effect of the depletion of PARP-2 on clonogenic survival after exposure of cells to agents that produce DNA strand breaks (Fig. 3a). In asynchronous cultures, the three HeLa PARP-2KD clones showed a significant increase in sensitivity (p < 10−4) to the cell killing effects of both 137Cs γ -rays (D37CTL/D37PARP-2KD = 1.37 ± 0.21) and 200 kV X-rays (D37CTL/D37PARP-2KD = 1.49 ± 0.20) with 200 kV X-rays being more efficient than 137Cs γ -rays as expected from their known relative biological efficiency, namely, 1.30 vs. 1.07, respectively, relative to 60Co radiation [34, 35]. Both Control and PARP-2KD cells synchronized in S-phase were more radioresistant than asynchronous cells but the ratio of their D37 values (D37CTL/D37PARP-2KD = 1.60 ± 0.35 for 137Cs γ-rays) was not significantly different from that for asynchronous cultures (Supplementary Fig. 1b). These results distinguish cells depleted in PARP-2 from those depleted in PARP-1, as the S-phase radioresistance is negligible in PARP-1 depleted cells as shown by the ratio of the D37 values for S-phase vs. asynchronous cultures of 1.04 ± 0.27 for PARP-1KD cells (Supplementary Fig. 1c) compared to 1.39 ± 0.21 for Control cells and 1.19 ± 0.17 for PARP-2KD cells. PARP-2KD clones compared to control cells were also significantly more sensitive to the cell killing effects of NCS, a protein antibiotic that reportedly induces DSBs only in DNA [36, 37], ([NCS]37CTL/[NCS]37PARP-2KD = 1.39 ± 0.22) (Supplementary Fig. 1b). Sensitivity to NCS has been previously reported for cells depleted in DNA-PKcs [32], ATM [38] and Artemis [39] but not in HeLa PARP-1KD cells or PARP-1KO MEFs [32, 36]. These results raise the intriguing possibility that the decreased PARG expression and increased PAR level in PARP-2 depleted cells impacts not only on SSB and BER pathways but also on the processing of DNA DSBs in mammalian cells.

Impact of the depletion of PARP-2 on radiation susceptibility. Clonogenic assays of Control (open symbols) and HeLa PARP-2KD cells (closed symbols) exposed to increasing doses of a γ-rays or X-rays were performed with asynchronous cell cultures. Data represents mean ± SD from at least two independent experiments in triplicate. The solid lines represent the curves drawn for best fit of data to the linear-quadratic equation (see “Materials and methods”). The dotted lines mark the upper and lower limits of the confidence interval drawn from the SDs calculated for the α and β parameters of the linear-quadratic model. b Clonogenic cell survival was assessed after exposure to 3 Gy γ-rays in HeLa; U2OS and AS3WT2 cells in which PARP-2 was stably depleted. Statistically lower survival was seen compared to the parental line on the depletion of PARP-2. Data represents the mean ± SD of four independent experiments
The phenotype of PARP-2 depleted U20S and AS3WT2 cells
To assess whether a similar phenotype was seen in other cell models when PARP-2 was depleted, we used the same shRNA expression system to stably deplete PARP-2 in U2OS and AS3WT2 cells. The U2OS and AS3WT2 cells were chosen because they express similar levels of the PARP2 mRNA as HeLa Control cells (data not shown). The radiation sensitivity was assessed after exposure to 3 Gy using a clonogenic survival assay. The depletion of PARP-2 in all three models (Fig. 4a) resulted in as significant reduction in cell survival compared to PARP-2 expressing HeLa Control cells (26 % compared to 16 %, p = 0.002), U2OS (20 % compared to 14 %, p < 0.0001) and AS3WT2 (89 % compared to 38 %, p < 0.0001) (Fig. 3a). However, no impact on PARG protein levels (Fig. 4a, b), PARP activity (Fig. 4c) or the recruitment of EGFP-PARP-1 and XRCC1-YFP to sites of laser-induced DNA damage sites (Fig. 4d) were noted in either model in direct contrast to the PAR dependent phenotype seen in the PARP-2 depleted HeLa cell line. These results would suggest that the direct interaction of PARP-1 and XRCC1 with PARP-2 [4] could not account for the reduced recruitment observed of both proteins seen in the HeLa PARP-2KD cells. The PARP activity measured in the two model cell lines PARP-2 depleted is not different than the PARP activity measured in the respective control cells expressing the PARP-2 protein (p < 0.5 and p < 0.4, respectively) (Fig. 4d). These observations suggest that unlike the situation in murine cells where PARP-2 depletion results in a reduced PARP activity measured by a similar method [1, 40] (Fig. 4d), PARP-2 depletion in human cells does not reduce PARP activity.

Impact of the depletion of PARP-2 in U2OS and AS3WT2 cells The depletion of PARP-2 in U2OS and AS3WT2 cells had no impact on the relative PARG protein level compared to the parental line transfected with a control shRNA a was quantified b from three independent extracts, or on c PARP activity as measured by the incorporation of isotope from 32P-NAD+ into trichloroacetic acid-precipitable proteins, or d the recruitment of EGFP-PARP-1 or XRCC1-YFP to sites of laser-induced DNA damage sites in AS3WT2 PARP-2KD cells or EGFP-PARP-1 in U2OS PARP-2KD cells. The PARP-2 depleted cells used were generated using the shRNA that targeted PARP2 at nucleotides 829–847 (AS3WT2 PARP-2KD 1470) or at nucleotides 1,077–1,095 (AS3WT2 PARP-2KD 1471)
Discussion
Poly(ADP-ribosyl)ation regulates the activity of many proteins involved in different cellular processes including DNA repair and cell death. While the involvement of PARP-1 in both the BER and SSBR pathways as well as DNA DSB repair has been examined, the role of PARP-2 in these processes is less well characterized. In the present study, we have found that the depletion of PARP-2 using a shRNA construct results in radiosensitivity in the three models examined but that PARP-2 depleted Hela cells have additional phenotypes that reflect a change in the status quo of the polymer levels that is not seen in the other two models examined. The PAR-related phenotype seen in HeLa cells is robust: it was seen in 3 independent clones cultured for over 20 passages and the concomitant changes associated with increased PAR levels can be reversed on re-expression of PARP-2.
In the HeLa PARP-2KD cells the increased PAR levels are associated with a small but reproducible increase in PARP activity and a decrease in PARG activity associated with a decrease in PARG protein and mRNA levels. An increase in PARP activity in the absence of PARP-2 has previously been reported in thymocytes and stimulated B lymphocytes from Parp2 −/− mice with a 20 % increase in PAR synthesis compared to Control cells [17, 41, 42]. In contrast while no changes in PARP activity were seen in the U2OS and AS3WT2 cells when PARP-2 was depleted reduced PAR activity was found in PARP2−/− immortalized 3T3 fibroblasts (this study and [1]) and in PARP2−/− MEF cells [1, 4] and PARP-2 depleted MOVAS cells [40]. These results suggest that additional mechanisms exist that regulate PARP activity that involve PARP-2 and appear to be cell type specific. The observation that the in vitro PAR synthesis rate catalyzed in the presence of both PARP-1 and PARP-2 was lower than in the case of PARP-1 alone [4, 43], could suggest the PARP-1/PARP-2 homodimer can downregulate PARP-1 activity or that following activation through binding to DNA structures the poly(ADP-ribosyl)ation of PARP-1 by PARP-2 impacts on its enzymatic activity [4, 43].
The HeLa PARP-2KD cells also show a robust reduction in PARG expression. It has also been reported that the constitutive suppression of the Parg gene in cells using a shRNA approach caused the accumulation of PAR levels [1, 28] and that Parp1 gene expression and PARP-1 protein expression is downregulated by the knockdown of the Parg gene [44, 45]. The balance between these proteins has been shown to influence transcriptional regulation of many genes with PARP-1 and PARG enzymatic activities required for some but not all target genes [29]. Our results would suggest that the impact of PARP-2 on the expression of PARG at the transcriptional level may be cell type specific.
Taken together the results from the three cell models highlight the complex interplay between the PARP-1, PARP-2, and PARG proteins both in terms of their expression and enzymatic activity. Why this PAR-related phenotype is only seen in the HeLa cells and not in the AS3WT2 or U20S cells is not clear. One possibility is that it is the balance between the expression of the 3 proteins or their mRNAs that is essential for the observed response and that the depletion of PARP-2 perturbs this balance triggering other changes. In the AS3WT2 or U20S cells, although the levels of the PARG transcript and protein are lower than the reduced level seen after PARP-2 depletion in HeLa cells this lower level is not sufficient to cause an overall increase in polymer levels suggesting that additional factors control this status quo. Transcriptome analysis of the different PARP-2 depleted clones could potentially help to unravel these observations. PARP-1 activity can also be modulated by post-translational modifications. For instance, we have previously shown in HeLa cells that the depletion of the Cdk5 kinase is associated with increased PARP-1 activity resulting in increased polymer levels [32]. Whether differences in such modifications could be underlying these variations could be addressed using an analysis of the PARP-1 protein in the different models by mass spectrometry.
One consequence of PARP-2 depletion that was shared between the three lines was an increased sensitivity to the cell killing effects of IR. While this has not yet been further investigated in the AS3WT2 or U20S cells, the observation that HeLa PARP-2KD cells show S-phase radioresistance distinguishes them from HeLa PARP-1KD cells [25, 36]. This would suggest that PARP-2 might be implicated in not only the processing of DNA SSBs but also of DSBs generated throughout the cell cycle supported by the observation that the HeLa PARP-2KD cells were sensitive to NCS. Such a role for PARP-2 is supported from animal models. In Parp2 −/− MEF model, a significant delay in DNA strand break sealing after exposure to MNU, as measured by the Comet assay, was found and Parp2 KO mice and Parp2 −/− mouse 3T3 fibroblasts were sensitive to the cell killing effects of IR [3, 46]. While PARP-1 clearly has a role in replication fork restart by homologous recombination and NHEJ (see Robert et al. [13]), the experimental evidence for a role of PARP-2 in DSB repair is sparse [17, 42]. PARP-2 was reported to suppress translocations between c-myc and IgH, which are induced during class switching [17] and impaired T cell receptor rearrangements in mice [42]. In addition, it cannot be ruled out that the increased sensitivity is not, at least in part, a consequence of the deregulated PAR levels found in the absence of PARP-2 that impacts on the choice of DNA repair pathways.
In conclusion, these results highlight the cell type and species-specific phenotypes associated with the depletion of PARP-2 and identify a common feature namely radiation sensitivity. Whether the differences reflect the presence or absence of other enzymatic activities that might modulate overall PARP activity remains to be established but clearly care must be taken on the extrapolation of cellular phenotypes between different cell models.
References
Ame JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Hoger T, Menissier-de Murcia J, de Murcia G (1999) PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem 274(25):17860–17868
Shieh WM, Ame JC, Wilson MV, Wang ZQ, Koh DW, Jacobson MK, Jacobson EL (1998) Poly(ADP-ribose) polymerase null mouse cells synthesize ADP-ribose polymers. J Biol Chem 273(46):30069–30072
Menissier de Murcia J, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Ame JC, Dierich A, LeMeur M, Sabatier L, Chambon P, de Murcia G (2003) Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J 22(9):2255–2263
Schreiber V, Ame JC, Dolle P, Schultz I, Rinaldi B, Fraulob V, Menissier-de Murcia J, de Murcia G (2002) Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem 277(25):23028–23036
Beck C, Robert I, Reina-San-Martin B, Schreiber V, Dantzer F (2014) Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp Cell Res. doi:10.1016/j.yexcr.2014.07.003
Yelamos J, Schreiber V, Dantzer F (2008) Toward specific functions of poly(ADP-ribose) polymerase-2. Trends in mol med 14(4):169–178
Mortusewicz O, Ame JC, Schreiber V, Leonhardt H (2007) Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res 35(22):7665–7675
Althaus FR, Kleczkowska HE, Malanga M, Muntener CR, Pleschke JM, Ebner M, Auer B (1999) Poly ADP-ribosylation: a DNA break signal mechanism. Mol Cell Biochem 193(1–2):5–11
Malanga M, Althaus FR (2004) Poly(ADP-ribose) reactivates stalled DNA topoisomerase I and Induces DNA strand break resealing. J Biol Chem 279(7):5244–5248
Schreiber V, Dantzer F, Ame JC, de Murcia G (2006) Poly(ADP-ribose): novel functions for an old molecule. Nat Rev 7(7):517–528
Caldecott KW (2003) XRCC1 and DNA strand break repair. DNA Repair 2(9):955–969
Keil C, Grobe T, Oei SL (2006) MNNG-induced cell death is controlled by interactions between PARP-1, poly(ADP-ribose) glycohydrolase, and XRCC1. J Biol Chem 281(45):34394–34405
Robert I, Karicheva O, Reina San Martin B, Schreiber V, Dantzer F (2013) Functional aspects of PARylation in induced and programmed DNA repair processes: preserving genome integrity and modulating physiological events. Mol Aspects Med 34(6):1138–1152. doi:10.1016/j.mam.2013.02.001
Woodhouse BC, Dianov GL (2008) Poly ADP-ribose polymerase-1: an international molecule of mystery. DNA Repair 7(7):1077–1086
Audebert M, Salles B, Weinfeld M, Calsou P (2006) Involvement of polynucleotide kinase in a poly(ADP-ribose) polymerase-1-dependent DNA double-strand breaks rejoining pathway. J Mol Biol 356(2):257–265
Mansour WY, Rhein T, Dahm-Daphi J (2010) The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res 38(18):6065–6077
Robert I, Dantzer F, Reina-San-Martin B (2009) Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. J Exp Med 206(5):1047–1056. doi:10.1084/jem.20082468
Ghodgaonkar MM, Zacal N, Kassam S, Rainbow AJ, Shah GM (2008) Depletion of poly(ADP-ribose) polymerase-1 reduces host cell reactivation of a UV-damaged adenovirus-encoded reporter gene in human dermal fibroblasts. DNA Repair 7(4):617–632. doi:10.1016/j.dnarep.2008.01.001
Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, Hensbergen P, Deelder A, de Groot A, Matsumoto S, Sugasawa K, Thoma N, Vermeulen W, Vrieling H, Mullenders L (2012) PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J cell Biol 199(2):235–249. doi:10.1083/jcb.201112132
Biard DS (2007) Untangling the relationships between DNA repair pathways by silencing more than 20 DNA repair genes in human stable clones. Nucleic Acids Res 35(11):3535–3550
Biard DS, Despras E, Sarasin A, Angulo JF (2005) Development of new EBV-based vectors for stable expression of small interfering RNA to mimick human syndromes: application to NER gene silencing. Mol Cancer Res 3(9):519–529
Le Chalony C, Hoffschir F, Gauthier LR, Gross J, Biard DS, Boussin FD, Pennaneach V (2012) Partial complementation of a DNA ligase I deficiency by DNA ligase III and its impact on cell survival and telomere stability in mammalian cells. Cell Mol Life Sci 69(17):2933–2949. doi:10.1007/s00018-012-0975-8
Favaudon V (1983) Gamma-radiolysis study of the reductive activation of neocarzinostatin by the carboxyl radical. Biochimie 65(11–12):593–607
Fernet M, Megnin-Chanet F, Hall J, Favaudon V (2010) Control of the G2/M checkpoints after exposure to low doses of ionising radiation: implications for hyper-radiosensitivity. DNA Repair 9(1):48–57. doi:10.1016/j.dnarep.2009.10.006
Godon C, Cordelieres FP, Biard D, Giocanti N, Megnin-Chanet F, Hall J, Favaudon V (2008) PARP inhibition versus PARP-1 silencing: different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic Acids Res 36(13):4454–4464
Zaremba T, Thomas HD, Cole M, Coulthard SA, Plummer ER, Curtin NJ (2011) Poly(ADP-ribose) polymerase-1 (PARP-1) pharmacogenetics, activity and expression analysis in cancer patients and healthy volunteers. Biochem J 436(3):671–679. doi:10.1042/BJ20101723
Virag L, Scott GS, Cuzzocrea S, Marmer D, Salzman AL, Szabo C (1998) Peroxynitrite-induced thymocyte apoptosis: the role of caspases and poly (ADP-ribose) synthetase (PARS) activation. Immunology 94(3):345–355
Erdelyi K, Bai P, Kovacs I, Szabo E, Mocsar G, Kakuk A, Szabo C, Gergely P, Virag L (2009) Dual role of poly(ADP-ribose) glycohydrolase in the regulation of cell death in oxidatively stressed A549 cells. FASEB J 23(10):3553–3563. doi:10.1096/fj.09-133264
Frizzell KM, Gamble MJ, Berrocal JG, Zhang T, Krishnakumar R, Cen Y, Sauve AA, Kraus WL (2009) Global analysis of transcriptional regulation by poly(ADP-ribose) polymerase-1 and poly(ADP-ribose) glycohydrolase in MCF-7 human breast cancer cells. J Biol Chem 284(49):33926–33938. doi:10.1074/jbc.M109.023879
Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, Wilson SH, Yasui A (2004) In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc Natl Acad Sci USA 101(38):13738–13743
Mortusewicz O, Fouquerel E, Ame JC, Leonhardt H, Schreiber V (2011) PARG is recruited to DNA damage sites through poly(ADP-ribose)- and PCNA-dependent mechanisms. Nucleic Acids Res 39(12):5045–5056
Bolin C, Boudra MT, Fernet M, Vaslin L, Pennaneach V, Zaremba T, Biard D, Cordelieres FP, Favaudon V, Megnin-Chanet F, Hall J (2011) The impact of cyclin-dependent kinase 5 depletion on poly(ADP-ribose) polymerase activity and responses to radiation. Cell Mol Life Sci 69:951–962
Slade D, Dunstan MS, Barkauskaite E, Weston R, Lafite P, Dixon N, Ahel M, Leys D, Ahel I (2011) The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature 477(7366):616–620. doi:10.1038/nature10404
Fu KK, Phillips TL, Heilbron DC, Ross G, Kane LJ (1979) Relative biological effectiveness of low- and high-LET radiotherapy beams for jejunal crypt cell survival at low doses per fraction. Radiology 132(1):205–209. doi:10.1148/132.1.205
Nikjoo H, Lindborg L (2010) RBE of low energy electrons and photons. Phys Med Biol 55(10):R65–R109. doi:10.1088/0031-9155/55/10/R01
Noel G, Giocanti N, Fernet M, Megnin-Chanet F, Favaudon V (2003) Poly(ADP-ribose) polymerase (PARP-1) is not involved in DNA double-strand break recovery. BMC cell biol 4:7. doi:10.1186/1471-2121-4-7
Ohtsuki K, Ishida N (1975) Neocarzinostatin-induced breakdown of deoxyribonucleic acid in HeLa-S3 cells. J antibiot 28(2):143–148
Ziv Y, Bar-Shira A, Pecker I, Russell P, Jorgensen TJ, Tsarfati I, Shiloh Y (1997) Recombinant ATM protein complements the cellular A-T phenotype. Oncogene 15(2):159–167. doi:10.1038/sj.onc.1201319
Mohapatra S, Kawahara M, Khan IS, Yannone SM, Povirk LF (2011) Restoration of G1 chemo/radioresistance and double-strand-break repair proficiency by wild-type but not endonuclease-deficient Artemis. Nucleic Acids Res 39(15):6500–6510. doi:10.1093/nar/gkr257
Szanto M, Rutkai I, Hegedus C, Czikora A, Rozsahegyi M, Kiss B, Virag L, Gergely P, Toth A, Bai P (2011) Poly(ADP-ribose) polymerase-2 depletion reduces doxorubicin-induced damage through SIRT1 induction. Cardiovasc Res 92(3):430–438. doi:10.1093/cvr/cvr246
Ambrose HE, Willimott S, Beswick RW, Dantzer F, de Murcia JM, Yelamos J, Wagner SD (2009) Poly(ADP-ribose) polymerase-1 (Parp-1)-deficient mice demonstrate abnormal antibody responses. Immunology 127(2):178–186. doi:10.1111/j.1365-2567.2008.02921.x
Yelamos J, Monreal Y, Saenz L, Aguado E, Schreiber V, Mota R, Fuente T, Minguela A, Parrilla P, de Murcia G, Almarza E, Aparicio P, Menissier-de Murcia J (2006) PARP-2 deficiency affects the survival of CD4+CD8+ double-positive thymocytes. EMBO J 25(18):4350–4360. doi:10.1038/sj.emboj.7601301
Kutuzov MM, Khodyreva SN, Ame JC, Ilina ES, Sukhanova MV, Schreiber V, Lavrik OI (2013) Interaction of PARP-2 with DNA structures mimicking DNA repair intermediates and consequences on activity of base excision repair proteins. Biochimie 95(6):1208–1215. doi:10.1016/j.biochi.2013.01.007
Shirai H, Poetsch AR, Gunji A, Maeda D, Fujimori H, Fujihara H, Yoshida T, Ogino H, Masutani M (2013) PARG dysfunction enhances DNA double strand break formation in S-phase after alkylation DNA damage and augments different cell death pathways. Cell Death Dis 4:e656. doi:10.1038/cddis.2013.133
Uchiumi F, Watanabe T, Ohta R, Abe H, Tanuma S (2013) PARP1 gene expression is downregulated by knockdown of PARG gene. Oncol Rep 29(5):1683–1688. doi:10.3892/or.2013.2321
Huber A, Bai P, de Murcia JM, de Murcia G (2004) PARP-1, PARP-2 and ATM in the DNA damage response: functional synergy in mouse development. DNA Repair 3(8–9):1103–1108
Acknowledgments
Research in Inserm U612 is supported by funding from Institut Curie and Inserm. MTB had a PhD fellowship from the Presidency of the University of Paris-Sud XI, SC and AF have PhD fellowships from the French Ministry of Research. CB, TZ and MTB were supported by Institute Curie’s International Post-doctoral fellow program and TZ by a fellowship from the Fondation Pierre-Gilles de Gennes. The assistance of members of the Institut Curie Small Animal Radiation Platform is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
18_2014_1765_MOESM1_ESM.tif
Supplementary Fig. 1 Impact of PARP-2 depletion in HeLa cells on mRNA expression profiles and cell survival. The depletion of PARP-2 was associated with (a) a reduction in parg mRNA in the three HeLa PARP-2KD clones. Data represents mean ± SD from at least three independent experiments. (b) Lower clonogenic survival was observed in HeLa PARP-2KD cells compared to Control cells (Control cells—open symbols, PARP-2KD cells—closed symbols) after exposure to increasing doses of γ-rays in S-phase (b-left panel) or neocarzinostatin in asynchronous cultures (b-right panel). Data represents mean ± SD from at least two independent experiments in triplicate.(c) Clonogenic survival of HeLa PARP-1KD cells compared to Control cells (Control cells—open symbols, PARP-1KD cells—filled symbols) after exposure to increasing doses of γ-rays in S-phase (c-left panel) and in asynchronous cultures (c-right panel). Data represents mean ± SD from one experiment in triplicate. In panels b and c, the solid lines represent the curves drawn for best fit of data to the linear-quadratic equation (see Materials and Methods). The dotted lines mark the upper and lower limits of the confidence interval drawn from the SDs calculated for the α and β parameters of the linear-quadratic model (TIFF 46907 kb)
18_2014_1765_MOESM2_ESM.tif
Supplementary Fig. 2 Restoring PARP-2 protein expression leads to restoration of PARG protein expression. The expression of PARP-1, PARG, and PARP-2 protein for HeLa PARP-2KD depleted cells (clones 1,470/1, 1,470/7, and 1,471/13) and the Control cells grown in the presence on hygromycin B (a-upper panel) were compared the protein expression to the same clones grown for 20 days in the absence of hygromycin B (a-lower panel). Growth in the absence of hygromycin B resulted in a concomitant increase in the expression of the PARP-2 and PARG proteins (a), a lowering of PARP activity to levels seen in Control cells as assessed by the incorporation of isotope from 32P-NAD+ into trichloroacetic acid-precipitable proteins (b) and levels of polymers that were similar to that seen in Control cells as assessed by western blotting from untreated cell cultures (c). (d) Quantitative analysis of the relative spot intensity with time for the recruitment of EGFP-PARP-1 to sites of laser-induced damage in Control cells (closed circles), the PARP-2KD clone 1,470/7 grown in the presence of hygromycin (open triangles) or the absence of hygromycin (closed triangles). Data represents mean relative spot intensity ± SEM, n = 40–57 individual cells (TIFF 46,903 kb)
Rights and permissions
About this article

Cite this article
Boudra, MT., Bolin, C., Chiker, S. et al. PARP-2 depletion results in lower radiation cell survival but cell line-specific differences in poly(ADP-ribose) levels . Cell. Mol. Life Sci. 72, 1585–1597 (2015). https://doi.org/10.1007/s00018-014-1765-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-014-1765-2