
Abstract
Sepsis refers to host response disorders caused by infection, leading to life-threatening organ dysfunction. RNA-binding motif protein 3 (RBM3) is an important cold-shock protein that is upregulated in response to mild hypothermia or hypoxia. In this study, we aimed to investigate whether RBM3 is involved in sepsis-associated acute lung injury (ALI). Intraperitoneal injection of LPS (10 mg/kg) was performed in wild type (WT) and RBM3 knockout (KO, RBM3−/−) mice to establish an in vivo sepsis model. An NLRP3 inflammasome inhibitor, MCC950 (50 mg/kg), was injected intraperitoneally 30 min before LPS treatment. Serum, lung tissues, and BALF were collected 24 h later for further analysis. In addition, we also collected serum from sepsis patients and healthy volunteers to detect their RBM3 expression. The results showed that the expression of RBM3 in the lung tissues of LPS-induced sepsis mice and the serum of patients with sepsis was significantly increased and positively correlated with disease severity. In addition, RBM3 knockout (KO) mice had a low survival rate, and RBM3 KO mice had more severe lung damage, inflammation, lung cell apoptosis, and oxidative stress than WT mice. LPS treatment significantly increased the levels of nucleotide binding and oligomerization domain-like receptor family 3 (NLRP3) inflammasomes and mononuclear cell nuclear factor-κB (NF-κB) in the lung tissues of RBM3 KO mice. However, these levels were only slightly elevated in WT mice. Interestingly, MCC950 improved LPS-induced acute lung injury in WT and RBM3 KO mice but inhibited the expression of NLRP3, caspase-1, and IL-1β. In conclusion, RBM3 was overexpressed in sepsis patients and LPS-induced mice. RBM3 gene deficiency aggravated sepsis-associated ALI through the NF-κB/NLRP3 pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sepsis is a severe systemic inflammatory response syndrome caused by infection, with a high global mortality rate. Sepsis can lead to multiple organ dysfunction syndrome (MODS) [1]. The lung is often the first organ to undergo dysfunction in sepsis. Sepsis-induced acute lung injury may occur, and some patients develop acute respiratory distress syndrome (ARDS). Excessive inflammation is one of the main causes of lung injury in sepsis [2].
RNA-binding motif protein 3 (RBM3) is an RNA-binding protein synthesized in response to low temperature exposure. The RBM3 gene is located on the X chromosome and contains 157 amino acids [3]. RBM3 promotes mRNA stability through a variety of mechanisms, enhances the translation of a number of proteins, and even regulates microRNA biosynthesis [4]. In addition, RBM3 also regulates the Wnt/β-catenin signaling pathway [5] and the cell cycle; RBM3 promotes cell proliferation while inhibiting cell apoptosis [6]. Extensive evidence has confirmed that RBM3 plays an important role in neuroprotection and tumor development. In previous studies, we found elevated RBM3 expression in ALI mouse models. In the study, we used LPS for the treatment of human pulmonary microvascular endothelial cells (HPMVECs) to simulate an in vitro ALI model and found that RBM3 overexpression improved cell survival but interrupted the tight junctions of microvascular endothelial cells [7]. However, the exact role of RBM3 in sepsis-induced acute lung injury in vivo remains to be observed.
Many studies have shown that inflammasome nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) is critically involved in the inflammatory response [8]. Foreign substances such as lipopolysaccharides can activate NF-κB signaling, causing phosphorylation and translocation of NF-κB p65, resulting in transcriptional upregulation of inflammasome-related components (NLRP3, pro-IL-1β, and pro-IL-18), which in turn activate caspase-1 [9], followed by apoptosis-associated speck-like protein (ASC) and caspase-1 recruitment and their assembly into a cytoplasmic complex, leading to proteolytic IL-1β and IL-18 activation and subsequent pyroptosis induction [10]. Growing evidence has accumulated that NLRP3 is involved in a variety of inflammation-related diseases, such as gout, atherosclerosis, and acute lung injury [11]. NLRP3 was recognized to be a key factor for the occurrence and development of sepsis-induced lung injury, which has a potentially beneficial effect on sepsis-induced ALI after the use of drugs inhibiting NLRP3 activation or gene knockout [12, 13]. Therefore, targeted inhibition of NLRP3 inflammasome activation may be a possible strategy for ALI treatment.
Studies have shown that cold-inducible RNA-binding protein (CIRBP) is similar to RBM3, and its expression is also upregulated by hypoxia and mild hypothermia [14]. During sepsis, CIRP is transferred from the nucleus to the cytoplasm and is released into the circulatory system, increasing the severity and mortality of sepsis [15, 16]. However, no reports on the role of RBM3 in the inflammatory response have been published thus far. Therefore, in this study, we aimed to investigate the effects of RBM3 on NLRP3 inflammasome activation and on sepsis-induced acute lung injury in an in vivo model. In addition, we also conducted a clinical trial in which we studied RBM3 expression changes in the blood of patients with sepsis and their correlation with disease severity.
Materials and methods
Human samples
This clinical observational study was conducted in the Affiliated Hospital of Southwest Medical University (Luzhou, Sichuan, China). We selected 16 adult patients diagnosed with sepsis in the intensive care unit (ICU) and 14 healthy volunteers. The characteristics of the patient population are presented in Table 1. Enzyme-linked immunosorbent assay (ELISA) was employed to detect RBM3 protein expression in the serum of sepsis patients and healthy volunteers. We also assessed the correlation between the RBM3 level and the APACHE II score, which was used to assess sepsis severity. This study was approved by the Clinical Trial Ethics Committee of the Affiliated Hospital of Southwest Medical University (Number: KY2020126), and informed consent was obtained from each patient or immediate family member before the trial. The clinical trial was registered in the Chinese Clinical Trial Registration Center (Number: ChiCTR2100050990).
Animals and sepsis model
Wild-type C57BL/6 male mice (8–12 weeks old) were purchased from the Southwest Medical University Animal Center (Luzhou, Sichuan, China). RBM3 KO (RBM3−/−) male mice (8–12 weeks old) were purchased from Cyagen Biosciences Inc. (Guangzhou, China). All mice were fed in a laminar flow in a specific pathogen-free atmosphere at Southwest Medical University. All experimental procedures were performed in accordance with the National Institutes of Health guidelines for the use of experimental animals and approved by the Animal Ethics Committee of Southwest Medical University.
All WT and RBM3−/− C57BL/6 mice were randomly divided into six groups: Control WT group, LPS WT group, LPS + MCC950 WT group, Control RBM3−/− group, LPS RBM3−/− group, and LPS + MCC950 RBM3−/− group. In short, the mice in the LPS group were injected intraperitoneally with LPS (10 mg/kg weight) dissolved in normal saline to establish an in vivo model of sepsis, whereas all mice in the control group were intraperitoneally injected with the same amount of normal saline. The survival rate of some mice was calculated within seven days after the model was established, and the other mice were sacrificed 24 h after the model was established. We collected samples of eyeball blood, lung tissue, and bronchoalveolar lavage fluid from the mice for subsequent experiments. In addition, we established an LPS + MCC950 model based on the LPS-induced sepsis model. MCC950 dissolved in saline (50 mg/kg weight) was intraperitoneally injected as a specific NLRP3 inhibitor 30 min before LPS injection.
Bronchoalveolar lavage fluid (BALF) collection
After the mouse was anaesthetized, a needle was inserted into the exposed trachea and fixed with a 4–0 silk ligation. Then, threefold lavage was performed with 1 mL of precooled sterile normal saline. The collected bronchoalveolar lavage fluid (BALF) was centrifuged at 400×g for 10 min at 4 °C.
Histological score of lung injury
Lung tissue specimens were collected 24 h post-LPS treatment, fixed with 4% paraformaldehyde for 24 h, embedded in paraffin, sliced into 4-μm microsections, and hematoxylin–eosin-stained. Lung injury was then assessed blindly under a light microscope (× 200 times), and each histological feature was scored based on the sum of the scores of the degree of injury (such as the number of infiltrating cells, alveolar wall thickening, hemorrhage, and interstitial edema). The range was from 0 (normal) to 4 (maximum).
Lung wet/dry weight ratio
Furthermore, we weighed the dissected fresh lung sample to obtain the wet weight. The lungs were then placed in an incubator at 65 °C to dry. Twenty-four hours later, they were removed, and the dry weight was measured, followed by calculations of the wet/dry lung weight ratio, which indicated the degree of pulmonary edema.
ELISA
The collected human and mouse whole-blood samples were centrifuged at 4 °C for 15 min, and the separated serum was collected. A Human RBM3 ELISA Kit (Andy Gene Biotechnology Co., Ltd, Beijing, China) was used to detect the RBM3 content in human serum. The alanine aminotransferase (ALT) and procalcitonin (PCT) levels in the mouse serum were determined with mouse-specific ELISA reagents (Meimian, Jiangsu, China). In addition, we also measured the IL-1β, IL-6, and TNF-α levels in lung tissues and BALF with an enzyme-linked immunosorbent assay kit, following the manufacturer's instructions (Meimian, Jiangsu, China).
qRT‒PCR analysis
Total RNA was extracted from the lung tissues using an RNA simple total RNA kit (Tiangen, Beijing, China) following the standard protocol. This isolated RNA was reverse-transcribed into cDNA using ReverTra Ace qPCR RT Master Mix (Toyobo, Japan) according to the manufacturer's protocol. Then, the cDNA was amplified using SuperReal PreMix Plus (SYBR Green) (Tiangen, Beijing, China) on a real-time PCR system (Roche). The relative gene expression of RBM3 mRNA was analyzed using the 2−∆∆CT method, with β-actin employed as the internal reference gene. The following primer sequences were used: RBM3, forwards, 5′-AGGGCTGAGTTTTGACACCAA-3′, and reverse, 5′-ACAAACCCAAATCCCCGAGAT-3′; β-actin forwards, 5′-TTTGCAGCTCCTTCGTTGC-3′, and reverse, 5′-TCGTCATCCATGGCGAACT-3′.
Western blotting
RIPA lysis buffer (50 mM Tris–HCl pH 7.5, 1% NP-40, 0.5 mM ethylenediaminetetraacetic acid [EDTA], 0.1% SDS, 150 mM NaCl, 0.5% sodium deoxycholate) containing PMSF and protein phosphatase inhibitors was used to extract proteins from lung tissues, and then a BCA protein assay (Beyotime, China) was used to determine the protein concentration of the supernatant. The protein samples were separated on SDS‒PAGE gels and then transferred to NC membranes (PALL, USA). After blocking with 5% skimmed milk in TBST at room temperature for 2 h, the membrane was incubated overnight at 4 °C with primary antibodies against RBM3 (1: 500), Bax (1: 5000), Bcl-2 (1: 1000) (Proteintech, China), caspase-1 (1: 500), caspase-3 (1: 500), phospho-NF-κB p65 (Ser536, 1: 200), NF-κB p65 (1: 500), IκBα (1: 500) (Santa Cruz Biotechnology, CA, USA), phospho-IκBα (1: 1000), IL-1β (1: 1000) (Affinity, Biosciences, China), NLRP3 (1: 500) and β-actin (1: 5000) (Beyotime, China). Appropriate secondary antibodies in TBST buffer with 5% skim milk were used to incubate the membranes at room temperature for 1 h, and then immunoblotting bands were detected via BeyoECL Star chemiluminescent substrate (Beyotime, China). Finally, protein bands were analyzed with ImageJ software (version 1.31, USA).
Determination of MDA content and SOD activity
Following the kit instructions (Nanjing Jiancheng Biotechnology Research Institute, Nanjing, China), the content of malondialdehyde (MDA) and the activity of superoxide dismutase (SOD) in lung tissue homogenate supernatants were determined.
TUNEL staining
A terminal deoxynucleotidyl transferase-mediated dUDP-biotin nick-end labeling (TUNEL) method with a commercial kit manufactured by Roche (Roche, Indianapolis, IN, USA) was used to detect lung cell apoptosis. We calculated the total number of cells and the number of positive cells in each microscopic field (×400). Apoptosis index = 100 × TUNEL-positive cells/total cells%.
Immunofluorescence
The sections used for immunofluorescence staining were obtained from paraffin-embedded tissues. Lung tissue was immersed, embedded, sectioned, deparaffinized, and rehydrated. After antigen retrieval and goat serum blockade at room temperature for 15 min, the sections were incubated with anti-NLRP3 antibody (1:100; Beyotime, China) at 4 °C overnight. Next, FITC-labeled goat anti-mouse IgG secondary antibody (1:100; Beyotime; green fluorescence) was added, and the samples were incubated at room temperature for 90 min. The nucleus was stained with DAPI (4′,6-diamidino-2-phenylindole) for cellular localization. Finally, we observed NLRP3 localization under a fluorescence microscope (×200 magnification).
Statistical analysis
All data were expressed as the mean ± standard deviation (SD) and analyzed by t test or one-way ANOVA, followed by Tukey's multiple comparison test. Survival curves were measured by the Mantel–Cox test. Correlation analysis was performed using Spearman's correlation test. A value of P < 0.05 was considered to indicate statistically significant differences. All statistical analyses were performed using GraphPad Prism 8.0 and SPSS 17.0 software.
Results
RBM3 expression was increased in the lung tissues of septic mice and in the blood of septic patients
Currently, the role of RBM3 in sepsis is unclear. We established a sepsis model by intraperitoneal injection of LPS and conducted comparisons with the control group to detect the changes in the expression of RBM3 in the lung tissue. The results showed that RBM3 protein content and mRNA expression in the lung tissue of the LPS group was significantly higher than that of the control group (Fig. 1A and B).

The expression of RBM3 in the lung tissues of the LPS-induced sepsis model of mice and the blood of patients with sepsis. After 24 h of LPS treatment of mice, western blotting (A) and RT‒qPCR (B) were used to detect the expression of RBM3 in lung tissue, n = 5–6 per group. ELISA (C) was used to detect the serum RBM3 content of patients with sepsis, and the correlation between the serum RBM3 content of patients with sepsis and Acute Physiologic and Chronic Health Evaluation II scores was evaluated (D). *P < 0.05 vs the control group
Elevated plasma concentrations of CIRP, which is also a cold-shock protein, were detected in patients with sepsis and were significantly associated with a poor prognosis [15]. Similarly, in our clinical observational study, we found that the expression of RBM3 in the serum of sepsis patients was higher than that in the serum of nonsepsis patients (Fig. 1C). In addition, the analysis of the relationship between the APACHE II score and the expression of RBM3 in patients with sepsis established that the expression of RBM3 was positively correlated with sepsis severity (Fig. 1D).
RBM3 knockout increased sepsis severity
An earlier study revealed that CIRP increased the severity and mortality of sepsis [17]. However, in our experiments, RBM3 acted differently from CIRP in sepsis. To determine the effect of RBM3 on sepsis severity, we injected LPS into the abdominal cavity of WT and RBM3 KO mice and calculated the 7-day survival rate of the mice after the induction of ALI. Next, we observed 12- and 24-h body temperature changes and measured the serum levels of procalcitonin (PCT) and alanine transaminase (ALT). These two markers are used to evaluate the degree of systemic inflammation and damage in sepsis [18, 19]. In clinical practice, PCT is a biomarker of bacterial infection, and ALT is a marker of liver damage. We found that all WT and KO mice in the control group survived for 7 days. The 7-day survival rate of the WT mice was significantly higher than that of the KO mice after the completion of LPS-induced ALI (Fig. 2A). We also established that the body temperature of the septic RBM3 KO mice was lower than that of the septic WT mice at 12 and 24 h postoperatively (Fig. 2B). In addition, 24 h post-LPS treatment, the serum PCT and ALT levels of the septic WT mice were higher than those of the control mice, and the serum PCT and ALT levels of the septic RBM3 KO mice were higher than those of the WT mice. The serum PCT and ALT levels in the two control groups of RBM3 KO and WT mice were similar (Fig. 2C and D). These results indicate that RBM3 deficiency can aggravate the systemic inflammatory response and damage caused by sepsis.

RBM3 gene knockout reduced the survival rate and aggravated systemic inflammatory damage in septic mice. (A) Seven-day survival curves of RBM3 KO and WT mice treated with LPS. n = 20 per group, *P < 0.05 vs the control WT group, #P < 0.05 vs the LPS WT group. (B) The rectal temperature of mice was measured at 12 h and 24 h after LPS treatment. n = 6 per group. #P < 0.05 vs the LPS WT group. (C) and (D). The levels of PCT and ALT in serum were detected by ELISA. n = 5–6 per group. *P < 0.05 vs the control WT group, #P < 0.05 vs the LPS WT group
RBM3 knockout aggravated sepsis-induced ALI
Twenty-four hours after the intraperitoneal injection of LPS, mouse lung tissue was subjected to HE staining for the detection of histomorphological changes. The results showed that acute lung injury occurred in the lung tissue of mice, which was more serious in the RBM3 KO mice in the LPS group, with more obvious inflammatory cell infiltration, interstitial edema, and alveolar and interstitial hemorrhage (Fig. 3A and B). The assessment of pulmonary edema changes revealed that LPS significantly increased the lung wet weight/dry weight ratio, but no significant difference in the lung wet weight/dry weight ratio between the WT and RBM3 KO mice was observed (Fig. 3C). The total protein concentration in BALF is an indicator of capillary permeability. We established that the protein concentration in BALF of the WT mice in the LPS group was significantly higher than that in the control group, whereas the loss of RBM3 led to a further increase in the protein concentration in BALF in the RBM3 KO mice (Fig. 3D). Meanwhile, an enzyme-linked immunosorbent assay was used to determine the expression levels of the inflammatory factors TNF-α, IL-6, and IL-1β in BALF and lung tissues in each of the groups. The levels of the inflammatory factors TNF-α, IL-6, and IL-1β in the lung tissues of the mice in the LPS group were significantly higher than those in the control group. The levels of the inflammatory factors in the lung tissue of the RBM3 KO mice were even more increased compared to those of the WT mice. These results indicate that RBM3 is related to acute lung injury, and the absence of RBM3 can aggravate acute lung injury caused by sepsis in mice (Fig. 3E–J).

RBM3 deficiency leads to more severe acute lung injury (ALI). A and B Evaluation of lung injury in WT and KO mice by HE staining, representative images of lung tissue sections and corresponding lung injury score histograms. n = 6–7 per group. *P < 0.05 vs the control WT group, #P < 0.05 vs the LPS WT group. Scale bars, 100 µm. C Lung wet/dry weight ratio. n = 5 per group. *P < 0.05 vs the control WT group. No significant difference was found between lungs from the LPS WT group and LPS KO group. D The protein concentration in BALF was determined by the BCA method. n = 8 per group. *P < 0.05 vs the control WT group, #P < 0.05 vs the LPS WT group. E–G The expression of the cytokines IL-1β, TNF-α and IL-6 in BALF was detected by ELISA. n = 4–5 per group. *P < 0.05 vs the control WT group, #P < 0.05 vs the LPS WT group. H–J The expression of the cytokines IL-1β, TNF-α and IL-6 in lung tissues was detected by ELISA. n = 5–10 per group. *P < 0.05 vs the control WT group, #P < 0.05 vs the LPS WT group
RBM3 alleviated apoptosis and oxidative stress in ALI
Apoptosis is an important pathological process in acute lung injury [20], and previous studies have shown that RBM3 alleviates apoptosis [7, 21]. Therefore, to determine whether RBM3 is involved in apoptosis during acute lung injury, we used Western blotting to detect the levels of apoptosis-related proteins in the lung tissues of the mice in each group. The expression levels of Bax and caspase-3 in the lung tissues of the RBM3 KO mice in the LPS group were higher than those in the WT mice, but the expression of Bcl-2 was lower (Fig. 4A). In addition, TUNEL staining showed that the apoptotic cell number in the RBM3 KO mice was higher than that in the WT mice 24 h post-LPS treatment (Fig. 4B and C).

RBM3 deficiency aggravated apoptosis and oxidative stress in lung tissue. A Representative western blot bands of Bax, caspase-3, Bcl-2 and β-actin in lung tissues of WT mice and RBM3 KO mice after LPS or saline treatment and the corresponding histogram of changes in Bax, caspase-3 and Bcl-2. n = 5 per group. *P < 0.05 vs the control WT group, #P < 0.05 vs the LPS WT group. B and C TUNEL staining detected lung cell apoptosis in WT and KO mice. Representative staining of TUNEL and DAPI in the lung. The ratio of TUNEL-positive/DAPI-positive cells represents the corresponding TUNEL staining histogram. n = 5 per group. *P < 0.05 vs the control WT group, #P < 0.05 vs the LPS WT group. Scale bars, 20 µm. D and F Comparison of the levels of MDA and SOD activity in lung tissues. n = 5 per group. *P < 0.05 vs the control WT group, #P < 0.05 vs the LPS WT group
In addition, the activation of oxidative stress-responsive signaling pathways also occurs during sepsis, inducing organ and tissue damage. MDA is the main product of the lipid peroxidation reaction of oxygen free radicals, and SOD is the main oxygen free radical scavenger of the body. The level of the two can indirectly reflect the degree of lipid peroxidation. Our study found that LPS treatment increased the level of MDA and reduced the activity of SOD compared with the control group; LPS-treated RBM3 KO mice had higher MDA levels but lower SOD activity than WT mice (Fig. 4D and E).
RBM3 knockout increased NLRP3 expression and activated the NF-κB pathway in LPS-treated mice
NF-κB is known to be a key transcription factor in inflammatory responses [22] that can induce NLRP3 and IL-1β transcription, playing an important role in inflammatory responses. Therefore, we first investigated whether RBM3 could alter postmodelling NF-κB phosphorylation. In this study, the expression of P-IκBα and P-NF-κB in the lung tissues of mice was significantly increased by LPS treatment. The expression of p-IκBα and p-NF-κB in the lung tissues of RBM3 KO mice was further increased compared with that of WT mice (Fig. 5A). In addition, we detected the expression of NLRP3 pathway-related proteins in lung tissues. LPS treatment significantly increased NLRP3, caspase-1, and IL-1β levels. NLRP3, caspase-1, and IL-1β expression levels in the lung tissues of RBM3 KO mice were higher than those of the WT mice (Fig. 5B). Similarly, immunostaining showed more NLRP3-positive cells in the lung tissues of the RBM3 KO mice (Fig. 5C). These results suggest that RBM3 deficiency may further promote LPS-induced activation of the NF-κB/NLRP3 signaling pathway.

RBM3 deficiency increases activation of the NLRP3 and NF-κB pathways. A Representative western blot bands of NLRP3, caspase-1, IL-1β and β-actin in lung tissues of WT mice and RBM3 KO mice after LPS treatment and the corresponding histogram of changes in NLRP3, caspase-1 and IL-1β. n = 5 per group. *P < 0.05 vs the control WT group, #P < 0.05 vs the LPS WT group. B Representative western blot bands of NF-κB p-p65, NF-κB p65, p-IκB-α, IκB-α and β-actin in lung tissues of WT mice and RBM3 KO mice after LPS treatment and corresponding histogram of changes in NF-κB p-p65, NF-κB p65, p-IκB-α and IκB-α. n = 5 per group. *P < 0.05 vs the control WT group, #P < 0.05 vs the LPS WT group. C Detection of the expression of NLRP3 in lung tissue by immunofluorescence. Representative staining of DAPI and NLRP3 in lung tissue. Scale bars, 50 µm
RBM3 deficiency aggravated LPS-induced ALI via the NLRP3 inflammasome pathway
To verify whether the effect of RBM3 on LPS-induced ALI depends on the activation of the NLRP3 inflammasome, mice were intraperitoneally injected with the NLRP3 inhibitor MCC950 30 min before LPS administration. Similar to previous results, LPS treatment increased NLRP3 activation and the maturation of caspase-1 and IL-1β. Meanwhile, it also aggravated lung tissue damage and elevated the expression of lung inflammatory factors in the WT and RBM3 KO mice. RBM3 deficiency resulted in higher expression of NLRP3, caspase-1, and IL-1β and lung tissue damage. However, in LPS-treated WT and RBM3 KO mice, MCC950 effectively reduced NLRP3, caspase-1, and IL-1β expression in mouse lung tissue (Fig. 6A). In addition, MCC950 lowered the LPS-induced acute lung injury scores (Fig. 6B and C) and the expression of the inflammatory factors TNF-α, IL-6, and IL-1β in the lung tissues and BALF (Fig. 6D–I).

RBM3 affects LPS-induced sepsis-associated ALI through the NLRP3 pathway. WT mice and RBM3 KO mice were injected with MCC950 (50 mg/kg) into the abdominal cavity before LPS treatment, and lung tissue and BALF were collected 24 h later. A Representative western blot bands of NLRP3, caspase-1, IL-1β and β-actin in lung tissues of WT mice and RBM3 KO mice and the corresponding histogram of changes in NLRP3, caspase-1 and IL-1β. n = 5 per group. *P < 0.05 vs the LPS WT group, #P < 0.05 vs the LPS KO group. &P < 0.05 vs the LPS + MCC950 WT group. B and C Representative images of HE-stained sections of lung tissue and corresponding lung injury score histograms. n = 5 per group. *P < 0.05 vs the LPS WT group, #P < 0.05 vs the LPS KO group. &P < 0.05 vs the LPS + MCC950 WT group. Scale bars, 50 µm. D–F. The expression of the cytokines IL-1β, TNF-α and IL-6 in BALF was detected by ELISA. n = 5 per group. *P < 0.05 vs the LPS WT group, #P < 0.05 vs the LPS KO group. &P < 0.05 vs the LPS + MCC950 WT group. G–I The expression of the cytokines IL-1β, TNF-α and IL-6 in lung tissues was detected by ELISA. n = 5 per group. *P < 0.05 vs the LPS WT group, #P < 0.05 vs the LPS KO group. &P < 0.05 vs the LPS + MCC950 WT group
Discussion
RBM3 and CIRP are cold-shock proteins located in the nucleus [6], and they also shuttle between the nucleus and cytoplasm and even to the outside of the cell to play their physiological roles. Despite the large number of their similarities, particularly regarding their sequence homology, expression, and inducibility, their biological functions are different. For example, although the expression of the two proteins is upregulated in cancer tissues, RBM3 has been consistently used as a biomarker for better cancer prognosis, whereas CIRP indicates the opposite tendency [6]. Similarly, extracellular CIRP can act as a proinflammatory mediator to increase disease severity and mortality in sepsis and septic shock [15]. In our study, we found that the absence of RBM3 could aggravate the severity of sepsis, suggesting that RBM3 may play a protective role against sepsis, in contrast to the activity of CIRP.
Our findings provide novel insights into the effect of RBM3 on sepsis-induced acute lung injury. In our previous studies, the overexpression of RBM3 was found to inhibit the apoptosis of LPS-treated human pulmonary microvascular endothelial cells, but at the same time, it reduced the ZO-1 level by stabilizing MMP9 mRNA and destroying cell tight junctions [7]. Therefore, the specific effect of RBM3 on ALI has remained unclear. In this study, we treated WT and RBM3 KO mice with LPS to establish an in vivo sepsis model. Our results further confirmed that RBM3 deficiency can increase sepsis severity, lung inflammation, apoptosis, and oxidative damage. Moreover, we provide additional evidence for the protective effect of RBM3 against sepsis. Interestingly, RBM3 deficiency can increase the formation of NLRP3 inflammatory bodies and activate the NF-κB pathway to significantly aggravate LPS-induced sepsis-associated acute lung injury.
RBM3 is upregulated in response to cold stimulation, hypoxia [6, 23], and other stress factors, but the role of RBM3 in inflammation-related diseases has rarely been reported. Our previous study found that LPS stimulation increased the expression of RBM3 and its transfer from the nucleus to the cytoplasm. An elevated RBM3 level was found in the lung tissue of septic mice and in the blood of septic patients, which is consistent with our previous study. Rosenthal et al. [24] detected RBM3 expression in human serum samples for the first time and found that it was elevated in the blood samples of patients undergoing cardiopulmonary bypass. Similarly, in this study, RBM3 expression increased in the blood of sepsis patients. A positive correlation between the level of RBM3 expression and the severity of sepsis was observed, which may suggest that RBM3 is translocated from the nucleus to the cytoplasm and released into the extracellular space under the stress state of systemic inflammation, participating in the pathological process of sepsis. Therefore, RBM3 may be used as a new biomarker to reflect sepsis severity, but subsequent clinical studies with more samples should be performed to confirm this hypothesis. Clinically, patients with sepsis often have high fever because of virulent pathogens or inflammatory responses. In some cases, sepsis caused by Gram-negative bacilli is characterized by hypothermia, low leukocyte count, and hypotension, followed by a severe septic shock reaction [25]. LPS is a component of the cell membrane of Gram-negative bacilli. Although hypothermia in mice can also lead to RBM3 upregulation after the establishment of a mouse sepsis model, most of the cases we investigated did not show obvious hypothermia, indicating that the increase in the RBM3 level depends not only on hypothermia but also on the cellular stress response caused by systemic inflammation, which may also promote the expression of RBM3.
The pathogenesis of ALI caused by sepsis involves a multifactorial network of processes and elements, including the expression of inflammatory cytokines, apoptosis, oxidative damage, and mitochondrial apoptosis [26]. This is the first study of sepsis-associated acute lung injury in mice lacking RBM3. After establishing a sepsis model in mice treated with LPS, we found that RBM3 deficiency increased sepsis severity in RBM3 KO mice more significantly than in WT mice, which was evaluated by their mortality and serum PCT and ALT levels. PCT is a biomarker of bacterial infection, ALT is a marker of liver damage, and they reflect the systemic inflammatory response and important organ damage in sepsis [18, 19]. In addition, we also found that RBM3 affected the typical lung histological features of sepsis-associated ALI, namely, septal thickening, hyaline membrane, protein exudate, microthrombosis, and neutrophil infiltration [27]. The loss of RBM3 exacerbated lung tissue injury and increased the levels of the proinflammatory factors IL-1β, IL-6, and TNF-α in the lung tissue and BALF. This discovery opens up a new field for RBM3 application in acute inflammation-related diseases.
Pulmonary endothelial cell apoptosis is the main cause of increased vascular permeability in sepsis-associated ALI, leading to edema, thrombosis, and neutrophil migration [20, 28]. RBM3 has been confirmed to have a good anti-apoptotic effect in neuronal, cancer, and muscle cells [21, 29, 30]. RBM3 inhibits PERK phosphorylation by coordinating with nf90 and protects cells from endoplasmic reticulum stress [31]. Notably, RBM3 can also exert its cytoprotective function through the p38 and JNK pathways [32]. Interestingly, we obtained similar results in the model of sepsis-associated ALI. RBM3 KO mice had more TUNEL-positive cells and higher expression of the proapoptotic proteins Bax and caspase-3 but lower expression of the anti-apoptotic protein bcl-2. These findings suggest that RBM3 contributes to the protection of ALI mice from apoptosis. In addition, oxidative stress is an important mechanism of sepsis-associated ALI. The imbalance between high-level oxygen free radicals and insufficient antioxidants is the main cause of cell damage caused by the accumulation of a large number of proinflammatory cytokines [33]. Therefore, we also detected the levels of MDA and SOD to assess the effect of RBM3 on oxidative stress in ALI. The results showed that the RBM3 KO mice had more severe oxidative stress damage than the wild-type mice.
Inflammation is one of the main pathophysiological processes in sepsis. NLRP3 can be activated by various stimuli and molecular and cellular events, including LPS, and subsequently promotes the maturation and secretion of proinflammatory cytokines and initiates pyroptosis [34, 35]. It is well known that NLRP3 inflammasome-mediated IL-1β production requires activation of the NF-κB pathway [36]. Many microbial components or cytokines can activate NF-κB and subsequently upregulate NLRP3 and IL-1β [35]. In sepsis induced by Gram-negative bacilli, LPS promotes the activation of NF-κB, which leads to the production of inflammatory mediators [37]. P65 is a subunit of an NF-kB heterodimer that is translated into the nucleus, where it induces the transcription of NLRP3 and IL-1β [38]. Previous studies have found that upregulating the activity of NF-κB can increase the expression of RBM3, thereby exerting a cytoprotective effect [7, 39]. These results suggest that NF-κB signaling is a regulator of RBM3 expression. Our study revealed that RBM3 gene deficiency significantly upregulated the expression of phosphorylated NF-κB induced by LPS, indicating that there may be a feedback regulatory pathway between RBM3 and NF-κB. In addition, the NLRP3, caspase1, and IL-1β levels in the lung tissues in the RBM3 KO mice were also more significantly increased by LPS treatment than those in the WT mice. Subsequently, we further treated mice with MCC950, which is a specific inhibitor of NLRP3 [40]. The results showed that the NLRP3 inflammasome pathway-related protein in mice was inhibited and the damage to the lung tissue was reduced. Meanwhile, the aggravation of lung tissue injury caused by RBM3 deletion was also reversed by MCC950. The aforementioned results suggest that RBM3 may affect LPS-induced ALI through the NF-κB and NLRP3 pathways. RBM3 knockout further promotes LPS-induced transcription factor NF-κB activation, increases the transcription of various proinflammatory factors, and upregulates NLRP3, promoting IL-1β production. However, this pathway does not seem to affect pro-caspase1 expression [41]. Therefore, RBM3 may also regulate the activation of NLRP3 inflammation by regulating transcription and translation processes and even the transfer from the nucleus to the cytoplasm, which may be related to the posttranslational modification of RNA-binding proteins. However, the specific molecular mechanisms involved need to be further studied in our future work.
Conclusion
In summary, as shown in Fig. 7, the findings of this study confirmed for the first time that RBM3 deficiency can aggravate the inflammatory response, apoptosis, and oxidative stress in LPS-induced sepsis-associated ALI, which may be achieved through the activation of the NF-κB and NLRP3 signaling pathways. Interestingly, the expression level of RBM3 was higher in patients with sepsis and was positively correlated with sepsis severity. This is an important finding, indicating that RBM3 may be used as a novel biomarker for judging the severity of sepsis and can serve as a potential target for the treatment of sepsis-associated ALI.

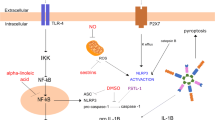
The effect of RBM3 gene knockout on LPS-induced sepsis-associated ALI. RBM3 knockout can activate the NF-κB/NLRP3 signaling pathway in LPS-induced acute lung injury in mice, thereby aggravating the inflammatory response in ALI
References
Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801–10. https://doi.org/10.1001/jama.2016.0287.
Ding X, Tong Y, Jin S, et al. Mechanical ventilation enhances extrapulmonary sepsis-induced lung injury: role of WISP1-αvβ5 integrin pathway in TLR4-mediated inflammation and injury. Crit Care. 2018;22(1):302. https://doi.org/10.1186/s13054-018-2237-0.
Danno S, Nishiyama H, Higashitsuji H, et al. Increased transcript level of RBM3, a member of the glycine-rich RNA-binding protein family, in human cells in response to cold stress. Biochem Biophys Res Commun. 1997;236(3):804–7. https://doi.org/10.1006/bbrc.1997.7059.
Sureban SM, Ramalingam S, Natarajan G, et al. Translation regulatory factor RBM3 is a proto-oncogene that prevents mitotic catastrophe. Oncogene. 2008;27(33):4544–56. https://doi.org/10.1038/onc.2008.97.
Venugopal A, Subramaniam D, Balmaceda J, et al. RNA binding protein RBM3 increases β-catenin signaling to increase stem cell characteristics in colorectal cancer cells. Mol Carcinog. 2016;55(11):1503–16. https://doi.org/10.1002/mc.22404.
Chip S, Zelmer A, Ogunshola OO, et al. The RNA-binding protein RBM3 is involved in hypothermia induced neuroprotection. Neurobiol Dis. 2011;43(2):388–96. https://doi.org/10.1016/j.nbd.2011.04.010.
Feng J, Pan W, Yang X, et al. RBM3 increases cell survival but disrupts tight junction of microvascular endothelial cells in acute lung injury. J Surg Res. 2021;261:226–35. https://doi.org/10.1016/j.jss.2020.12.041.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–32. https://doi.org/10.1016/j.cell.2010.01.040.
An Y, Zhang H, Wang C, et al. Activation of ROS/MAPKs/NF-κB/NLRP3 and inhibition of efferocytosis in osteoclast-mediated diabetic osteoporosis. FASEB J. 2019;33:12515–27.
Chauhan D, Vande Walle L, Lamkanfi M. Therapeutic modulation of inflammasome pathways. Immunol Rev. 2020;297(1):123–38. https://doi.org/10.1111/imr.12908.
Mcgettrick AF, O’Neill LA. NLRP3 and IL-1β in macrophages as critical regulators of metabolic diseases. Diabetes Obes Metab. 2013;15(Suppl 3):19–25. https://doi.org/10.1111/dom.12169.
Chen G, Hou Y, Li X, Pan R, Zhao D. Sepsis-induced acute lung injury in young rats is relieved by calycosin through inactivating the HMGB1/MyD88/NF-κB pathway and NLRP3 inflammasome. Int Immunopharmacol. 2021;96:107623. https://doi.org/10.1016/j.intimp.2021.107623.
Ying Y, Mao Y, Yao M. NLRP3 inflammasome activation by microRNA-495 promoter methylation may contribute to the progression of acute lung injury. Mol Ther Nucleic Acids. 2019;18:801–14. https://doi.org/10.1016/j.omtn.2019.08.028.
Wellmann S, Bührer C, Moderegger E, et al. Oxygen-regulated expression of the RNA-binding proteins RBM3 and CIRP by a HIF-1-independent mechanism. J Cell Sci. 2004;117(Pt 9):1785–94. https://doi.org/10.1242/jcs.01026.
Qiang X, Yang WL, Wu R, et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med. 2013;19(11):1489–95. https://doi.org/10.1038/nm.3368.
Zhou Y, Dong H, Zhong Y, Huang J, Lv J, Li J. The cold-inducible rna-binding protein (CIRP) level in peripheral blood predicts sepsis outcome. PLoS ONE. 2015;10(9):e0137721. https://doi.org/10.1371/journal.pone.0137721.
Khan MM, Yang WL, Brenner M, Bolognese AC, Wang P. Cold-inducible RNA-binding protein (CIRP) causes sepsis-associated acute lung injury via induction of endoplasmic reticulum stress. Sci Rep. 2017;7:41363. https://doi.org/10.1038/srep41363.
Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/sis international sepsis definitions conference. Crit Care Med. 2003;31(4):1250–6. https://doi.org/10.1097/01.CCM.0000050454.01978.3B.
Hirano Y, Aziz M, Yang WL, et al. Neutralization of osteopontin attenuates neutrophil migration in sepsis-induced acute lung injury. Crit Care. 2015;19(1):53. https://doi.org/10.1186/s13054-015-0782-3.
Gill SE, Rohan M, Mehta S. Role of pulmonary microvascular endothelial cell apoptosis in murine sepsis-induced lung injury in vivo. Respir Res. 2015;16(1):109. https://doi.org/10.1186/s12931-015-0266-7.
Zhu X, Yan J, Bregere C, et al. RBM3 promotes neurogenesis in a niche-dependent manner via IMP2-IGF2 signaling pathway after hypoxic-ischemic brain injury. Nat Commun. 2019;10(1):3983. https://doi.org/10.1038/s41467-019-11870-x.
Sun SC. The noncanonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17(9):545–58. https://doi.org/10.1038/nri.2017.52.
Zhu X, Bührer C, Wellmann S. Cold-inducible proteins CIRP and RBM3, a unique couple with activities far beyond the cold. Cell Mol Life Sci. 2016;73(20):3839–59. https://doi.org/10.1007/s00018-016-2253-7.
Rosenthal LM, Tong G, Wowro S, et al. A prospective clinical trial measuring the effects of cardiopulmonary bypass under mild hypothermia on the inflammatory response and regulation of cold-shock protein RNA-binding motif 3. Ther Hypothermia Temp Manag. 2020;10(1):60–70. https://doi.org/10.1089/ther.2018.0038.
Kushimoto S, Abe T, Ogura H, et al. Impact of body temperature abnormalities on the implementation of sepsis bundles and outcomes in patients with severe sepsis: a retrospective sub-analysis of the focused outcome research on emergency care for acute respiratory distress syndrome, sepsis and trauma study. Crit Care Med. 2019;47(5):691–9. https://doi.org/10.1097/CCM.0000000000003688.
Qian M, Lou Y, Wang Y, et al. PICK1 deficiency exacerbates sepsis-associated acute lung injury and impairs glutathione synthesis via reduction of xCT. Free Radic Biol Med. 2018;118:23–34. https://doi.org/10.1016/j.freeradbiomed.2018.02.028.
Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295(3):L379–99. https://doi.org/10.1152/ajplung.00010.2008.
Aschkenasy G, Bromberg Z, Raj N, Deutschman CS, Weiss YG. Enhanced Hsp70 expression protects against acute lung injury by modulating apoptotic pathways. PLoS ONE. 2011;6(11):e26956. https://doi.org/10.1371/journal.pone.0026956.
Al-Astal HI, Massad M, AlMatar M, Ekal H. Cellular functions of RNA-binding motif protein 3 (RBM3): clues in hypothermia, cancer biology and apoptosis. Protein Pept Lett. 2016;23(9):828–35. https://doi.org/10.2174/0929866523666160628090340.
Ferry AL, Vanderklish PW, DuPont-Versteegden EE. Enhanced survival of skeletal muscle myoblasts in response to overexpression of cold shock protein RBM3. Am J Physiol Cell Physiol. 2011;301(2):C392–402. https://doi.org/10.1152/ajpcell.00098.2011.
Zhu X, Zelmer A, Kapfhammer JP, Wellmann S. Cold-inducible RBM3 inhibits PERK phosphorylation through cooperation with NF90 to protect cells from endoplasmic reticulum stress. FASEB J. 2016;30(2):624–34. https://doi.org/10.1096/fj.15-274639.
Zhuang RJ, Ma J, Shi X, et al. Cold-inducible protein RBM3 protects UV irradiation-induced apoptosis in neuroblastoma cells by affecting p38 and JNK pathways and Bcl2 family proteins. J Mol Neurosci. 2017;63(2):142–51. https://doi.org/10.1007/s12031-017-0964-3.
Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348(2):138–50. https://doi.org/10.1056/NEJMra021333.
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–5. https://doi.org/10.1038/nature09663.
Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci. 2019;20(13):3328. https://doi.org/10.3390/ijms20133328.
Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. https://doi.org/10.1038/nri3452.
Opal SM. Endotoxins and other sepsis triggers. Contrib Nephrol. 2010;167:14–24. https://doi.org/10.1159/000315915.
Lin CC, Hsieh HL, Shih RH, Chi PL, Cheng SE, Yang CM. Upregulation of COX-2/PGE2 by endothelin-1 via MAPK-dependent NF-κB pathway in mouse brain microvascular endothelial cells. Cell Commun Signal. 2013;11(1):8. https://doi.org/10.1186/1478-811X-11-8.
Ushio A, Eto K. RBM3 expression is upregulated by NF-κB p65 activity, protecting cells from apoptosis, during mild hypothermia. J Cell Biochem. 2018;119(7):5734–49. https://doi.org/10.1002/jcb.26757.
Xu KY, Wu CY, Tong S, Xiong P, Wang SH. The selective Nlrp3 inflammasome inhibitor Mcc950 attenuates lung ischaemia-reperfusion injury. Biochem Biophys Res Commun. 2018;503(4):3031–7. https://doi.org/10.1016/j.bbrc.2018.08.089.
Liu MH, Lin AH, Lee HF, Ko HK, Lee TS, Kou YR. Paeonol attenuates cigarette smoke-induced lung inflammation by inhibiting ROS-sensitive inflammatory signaling. Mediators Inflamm. 2014;2014:651890. https://doi.org/10.1155/2014/651890.
Funding
The work was supported by the Sichuan Science and Technology Program (2022YFS0632) and the joint foundation of the Luzhou government and Southwest Medical University (2021LZXNYD-J28).
Author information
Authors and Affiliations
Contributions
Contribution: Feiyu Long and Liren Hu performed the experiments and preparing the first draft of the manuscript.Feiyu Long and Yingxu Chen assisted to establish the sepsis models and samples collection. Xiaoxia Duan analysized the data. Keliang Xie, Jianguo Feng and Maohua Wang supervised, prepared, and finalized the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
We declare that there are no conflicts of interest.
Additional information
Responsible Editor: Artur Bauhofer.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Long, F., Hu, L., Chen, Y. et al. RBM3 is associated with acute lung injury in septic mice and patients via the NF-κB/NLRP3 pathway. Inflamm. Res. 72, 731–744 (2023). https://doi.org/10.1007/s00011-023-01705-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-023-01705-3