
Abstract
Background
The number of heatstroke victims hit record numbers in 2022 as global warming continues. In heat-induced injuries, circulatory shock is the most severe and deadly complication. This review aims to examine the mechanisms and potential approaches to heat-induced shock and the life-threatening complications of heatstroke.
Methods
A computer-based online search was performed using the PubMed database and Web of Science database for published articles concerning heatstroke, shock, inflammation, coagulopathy, endothelial cell, cell death, and heat shock proteins.
Results
Dehydration and heat-induced cardiomyopathy were reported as the major causes of heat-induced shock, although other heat-induced injuries are also involved in the pathogenesis of circulatory shock. In addition to dehydration, the blood volume decreases considerably due to the increased vascular permeability as a consequence of endothelial damage. Systemic inflammation is induced by factors that include elevated cytokine and chemokine levels, dysregulated coagulation/fibrinolytic responses, and the release of damage-associated molecular patterns (DAMPs) from necrotic cell death that cause distributive shock. The cytoprotective heat shock proteins can also facilitate circulatory disturbance under excess heat stress.
Conclusions
Multiple mechanisms are involved in the pathogenesis of heat-induced shock. In addition to dehydration, heat stress-induced cardiomyopathy due to the thermal damage of mitochondria, upregulated inflammation via damage-associated molecular patterns released from oncotic cells, unbalanced coagulation/fibrinolysis, and endothelial damage are the major factors that are related to circulatory shock.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Climate change significantly affects human health and nature. In 2022, because of the historic heatwave, more than 800 deaths potentially linked to the punishing heat were reported in Oregon, Washington, and western Canada. Besides, western wildfires have burned nearly 700,000 acres of dried fields and forests. https://www.washingtonpost.com/climate-environment/2021/07/03/climate-change-heat-dome-death/. The warming is not just a North American issue, but also worldwide. A record-breaking heatwave with temperatures as high as 47.4 °C hit Spain, and the WHO announced that more than 1,700 deaths were recorded in Spain and Portugal. Again, thousands of acres of forests were lost by the wildfire. https://www.who.int/europe/news/item/22-07-2022-heatwave-in-europe--local-resilience-saves-lives---global-collaboration-will-save-humanity Also, other parts of the world are experiencing excessively hot seasons.
Dehydration is a major threat during heatstroke which may cause hypovolemia, but it also increases osmolarity, blood viscosity, and microcirculatory injury [1]. However, even when hydration is maintained, heat-related illness can occur due to the host’s inflammatory and coagulation responses to heat stress [2]. A baboon model of heatstroke demonstrated that microvascular damage, thrombus formation, inflammation, and apoptosis are involved in the pathophysiology of heatstroke [3]. Furthermore, pathological examination of dogs with heatstroke revealed multiorgan coagulative necrosis with hemorrhagic tendency [4]. These observations suggest that heat-induced disseminated intravascular coagulation (DIC) and systemic inflammation are also critical mechanisms of multiorgan failure and death.
High temperatures increase cardiovascular mortality via elevated blood pressure, increased blood viscosity, and changes in heart rate, especially in vulnerable individuals such as elderly people and patients with preexisting cardiovascular diseases [1]. In heatstroke models, animals subjected to the ambient temperature of 43.0 °C showed a sudden drop in blood pressure immediately after the core body temperature exceeded 42.0 °C, which mimics hyperdynamic shock [5]. In a rat model, circulatory shock was attenuated, and recovery occurred if the animals were cooled down immediately after reaching 42.0 °C [6]. Other than small animal experiments, the primate model of heatstroke constantly reproduces circulatory shock when the core body temperature reaches 43.0 °C, and a significant correlation between body temperature and the decrease in systolic arterial pressure is reported [3, 7, 8]. As a result, there are likely temperature and time thresholds that increase the risk of mortality. In this review, we will explore the pathogenesis of heat-induced shock and examine the critical factors that determine unfavorable outcomes.
Methods
We conducted a computer-based search on PubMed and Web of Science databases for publications up to August 2022 using the search MeSH (Medical Subject Headings) terms “heat stroke,” “shock,” “inflammation,” “coagulopathy,” “endothelial cell,” “cell death,” and “heat shock protein.” First, 943 articles were screened with “heat stroke,” AND “shock.” Then, we identified the related articles by limiting the terms including “inflammation,” “coagulopathy,” “endothelial cell,” “cell death,” or “heat shock protein.” Finally, we selected 94 highly related articles and included them in this review.
Heat-induced shock
Previous reports note that heat stress increases blood pressure and heart rate until the body temperature reaches a certain level, but the subjects progress to circulatory shock and die if the temperature surpasses that limit [1]. Figure 1 demonstrates the typical changes in blood pressure and heart rate of a rat subjected to heat. When placed in a climate chamber at 43.0 °C and relative humidity of 60%, the core body temperature reached 41.0 °C 1 hour later, blood pressure and heart rate rose ~ 1.6 times higher than baseline when body temperature was 42.0 °C and heart rate increased. Then, blood pressure and heart rate gradually decrease, and when the body temperature exceeds 42.5 °C, circulatory shock follows [9]. Similar observations were repeatedly reported in the other animals as well as in humans. A large cohort of over 3,000 cases of heat-related illness in patients, Glasgow coma scale < 15, body temperature ≥ 38 °C (100.4℉), systolic blood pressure ≤ 100 mmHg, heart rate ≥ 100/min, and age ≥ 65 years are associated with increased incidence of hospital admission [10]. Thus, “four 100s” (GCS < 100%, temperature ≥ 100℉, blood pressure ≤ 100 mmHg, heart rate ≥ 100/min in elderly people) indicates higher disease severity. Heat-induced shock is hypothesized to be caused by multiple factors that include impairment of cardiovascular function [11], excess inflammation [12], unbalanced coagulation/fibrinolysis [13], and endothelial dysfunction [14]. It has been shown that hyperthermia can impair homeostasis by disrupting the integrity of the vessels, interfering with cytokine production and nitric oxide synthesis [1]. The effects of various agents and substances, i.e., free radical scavengers, recombinant protein C, hyperbaric oxygen, glucocorticoids, interleukin (IL)-1 receptor antagonists, L-arginine, and estrogen were examined; however, none of them has shown robust evidence of clinical benefit [15].

The changes of vital signs in a rat model of heat stroke. The body temperature increased after being subjected to heat, and reached approximately 41 ℃ at 1 h, and heart rate and blood pressure started to rise thereafter. The hyperdynamic state continued until the body temperature reached 42 ℃. Rats suddenly fell into shock when the body temperatures approached 43 ℃
Other than the above mechanisms, damage to the vascular endothelium increases vascular permeability and exacerbates hypovolemia, further worsened by dehydration. Fluid resuscitation is therefore the primary initial treatment for heatstroke-induced shock. However, with respect to the choice of fluids, the effects of various fluids have been examined. For example, an infusion of 3% hypertonic saline solution attenuated the heatstroke-induced hypotension more effectively than 0.9% saline [16]. Hydroxyethyl starch (HES) was also reported to improve circulatory shock by maintaining colloid osmotic pressure [17]. Furthermore, a combination of 6% HES plus 7.2% saline solution showed better performance in the resuscitation of hypotension compared to 7.2% saline solution or HES alone [18]. It is important to consider that fluid therapy, in combination with cooling, is the only established treatment for heat-induced shock currently available.
Heat-induced cardiomyopathy
In response to hyperthermia, our body increases cardiac output to decrease the body temperature by increasing the blood flow to the body surface [19]. This response increases cardiac workload, oxygen consumption, and the risk of heart failure. Decompensated heart failure due to increased cardiac output and hypertension has been thought to be the major mechanism of cardiogenic shock [20]. Bathini et al. [21, 22] surveyed the incidence of myocardial infarction in heatstroke using the National Inpatient Sample dataset from 2003 to 2014. As a result, over 3000 heatstroke cases were recorded, and among those patients, 12% were complicated by circulatory failure, and 7% were complicated by myocardial infarction.
Besides the excess workload, heat stress-induced cardiomyopathy due to the sustained hyper-adrenergic state is another explanation for circulatory collapse [11]. In animal models of heatstroke, blood pressure and heart rate increase, and this hyperdynamic state continues until the body temperature reaches the threshold point. Increased cardiac troponin I was observed with the increased heart rate and blood pressure in a rat model of heatstroke [23]. In addition, increased body temperature was associated with high B-type natriuretic peptides (BNP) [24].
Finally, direct thermal injury to the myocardium is also suspected. Nakagawa et al. [25] examined mRNA expression of the various cardiac muscle-related proteins in rats subjected to heat and concluded that hyperthermia induces myocardial damage shortly after exposure to heat. They also have shown that the changes were more prominent in the ventricle than in the atrium. The mechanism of myocardial injury is thought to be the imbalance of energy production and expenditure, e.g., supply and demand imbalance. Mitochondrial membrane function plays a key role in maintaining cell homeostasis due to its vital role in adenosine 5′-triphosphate (ATP) synthesis. It is reported that heat stress activates the lysosomal-mitochondrial apoptotic pathway and increases intracellular reactive oxygen species, which results in increased lysosomal membrane permeability with mitochondrial depolarization and cytochrome C release to the cytosol [26].
Αlpha-lipoic (α-Lipoic) acid is a cofactor of mitochondrial enzymes and exerts antioxidant properties. In a rat model of heatstroke, α-lipoic acid was shown to reduce cardiac superoxide anion formation and protein expression of cleaved caspase 3 and resulted in the suppression of myocardial injury [27].
Taken together, heat-induced shock is considered to be primarily a combination of cardiogenic shock due to heat-induced cardiomyopathy and hypovolemic shock. However, factors including excess inflammation, hypercoagulation, microthrombosis, and apoptotic and necrotic cell death can be involved and we will examine their association with heat-induced shock as follows.
Heat-induced inflammation
Complex mechanisms regulate inflammation in heatstroke, and it is generally accepted that excessive activation of inflammation contributes to organ dysfunction and death. Bouchama et al. [28] reported that the circulating IL-6, IL-1β, and interferon-γ increased in the primate model of heatstroke, and the levels were 220 ± 44 pg/ml, 42 ± 14 pg/ml, and 1,180 ± 879 pg/ml, respectively. They also reported that IL-6 concentrations correlated with the severity of illness. Similar to those seen in sepsis, the overactivated or dysregulated inflammatory responses contribute to tissue injury and death in heatstroke. Using the same model, the same group evaluated the counter-action to the upregulated inflammation and showed the early release of anti-inflammatory cytokines and chemokines such as IL-10, IL-1 receptor antagonist, soluble tumor necrosis factor (TNF) receptor I, II, and IL-8, while circulating levels of regulatory cytokine IL-12p40 were significantly decreased [8]. These results suggest that complex cytokine networks regulate the inflammatory reactions to heat; however, excess heat stress disturbs those delicate regulation systems [29]. Of note, the increased cytokine release is suspected to be partially of gut origin [30]. Endotoxin released from the damaged intestine is considered to be responsible for the increased levels of cytokines. Welc et al. [31] reported that skeletal muscles were another source of cytokine release. The skeletal muscles are known to be susceptible to heat and a variety of reactions, including the rapid rises in mRNA of IL-6 and IL-10 and upregulation of toll-like receptor-4 (TLR-4), and heat shock protein (HSP)-72 mRNA were reported.
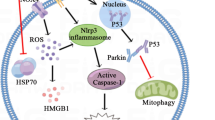
The levels of inflammatory and pyrogenic cytokines are thought to participate in the pathogenesis of heatstroke-induced shock; however, their levels are not high enough to induce shock alone. In this respect, the roles of damage-associated molecular patterns (DAMPs) [32], inflammasomes [33], and gut-derived endotoxin [34] should be considered (Fig. 2). Together with increased cytokines, high-mobility group box 1 (HMGB-1) and inflammasome lead to excess inflammation and organ dysfunction [35]. In hospitalized patients with exertional heatstroke, it was reported that 84% met the diagnostic criteria for systemic inflammatory response syndrome and those patients required longer periods of hospitalization [36].

Events in the vasculature in heatstroke. Heat injures directly and indirectly various cells including monocytes, neutrophils, platelets, and endothelial cells. Activated monocytes express tissue factor and activate coagulation system and release proinflammatory cytokines such as tumor necrosis factor α (TNF α) and interleukin (IL)-1β, and IL-6. Damaged leukocytes fall into oncosis and release various damage-associated molecular patterns (DAMPs) including high-mobility group box 1 (HMGB-1), histone and cell-free DNA. Platelets are activated directly by heat and indirectly by heme released from damaged muscle. Damage of endothelial cell results in increased vascular permeability and elevated production of plasminogen activator inhibitor 1 and von Willebrand factor and induces microthrombosis which disturbs microcirculation
As for the treatment, only a few studies have examined the effects of cytokine antagonism on heat-induced damage. Shen et al. [37] examined the effect of IL-1 receptor antagonist (IL-1ra) in the rabbit model of heatstroke. As a result, IL-1ra dose-dependently attenuated heatstroke-induced hypotension and organ dysfunction, which led to improved survival time. Umemura et al. [38] have reported the anti-inflammatory effects of a bone marrow-derived mononuclear cell in severe heatstroke. Further studies regarding the implication of anti-inflammatory therapies for heatstroke-induced shock are warranted.
Heat-induced coagulopathy
Similar to the study on cytokines, the primate model provides valuable information about coagulopathy in heatstroke. Using the baboon model of heatstroke as mentioned above, Bouchama et al. [6] reported systemic inflammation and activated coagulation, represented by increased plasma IL-6, prolonged prothrombin time (PT) and activated partial thromboplastin time (aPTT), elevated D-dimer levels, and decreased platelet count, along with multiple organ dysfunction indicated by the increase of creatinine, creatine kinase, lactate dehydrogenase, and transaminases. This picture is highly compatible with disseminated intravascular coagulation (DIC). With respect to the physiological anticoagulants, significant reductions in the levels of antithrombin, proteins C, and protein S were reported [39]. Furthermore, hyperthermia is known to cause conformational changes and intracellular retention of antithrombin [40].
Fibrinolysis is suggested to be upregulated by the reduction of plasminogen activator inhibitor 1 (PAI-1) and increased tissue plasminogen activator (tPA) that increase fibrin/fibrinogen degradation products (FDP) despite no changes in fibrinogen or plasminogen-alpha 2-antiplasmin complex levels [39]. Thus, fibrinolytic status varies depending on the phases, and a biphasic change from an enhanced phase to a suppressed fibrinolytic phase that is seen in sepsis-associated DIC can occur in heatstroke-induced coagulopathy [41]. A mouse model showed that more severe coagulation disorders were associated with poorer outcomes, and coagulation biomarkers could serve as biomarkers for heatstroke [42]. In a human cross-sectional survey from Japan, heatstroke was frequently complicated by disseminated intravascular coagulation (11.6% of heatstroke patients were meeting the criteria for DIC) [43]. Hyperthermia-induced DIC is classified as thrombotic-type DIC in its late phase [44]; however, bleeding complications can also occur [13]. Hyperthermia can directly affect coagulation and fibrinolysis, activate inflammation, and induce cellular damage, which leads to further worsening of heatstroke-induced coagulopathy. Huisse et al. [45] examined the link between coagulopathy and inflammation in 18 critical heatstroke patients and reported high circulating levels of IL-6, IL-8, C5a, IL-1 receptor antagonist, HSP60, and HSP70, findings consistent with activated coagulation. As seen in sepsis, heat-related insult serves as a link between coagulation and inflammation [46]. The microthrombosis formed in DIC can cause a circulatory disturbance, but its direct association with circulatory shock needs further investigation.
For the treatment of heatstroke-induced coagulopathy, the effects of activated protein C were examined in a heatstroke baboon model. Although significant decreases in IL-6, soluble thrombomodulin, and microvesicles were reported, coagulopathy and outcomes were not improved [47].
Heat-induced leukocyte apoptosis and cell death-related factors
Leukocytes are susceptible to heat, and the blood smear examination demonstrated the morphological changes of leukocytes in heatstroke [9] (Fig. 3). Ward et al. [48] demonstrated the botryoid nucleus of neutrophils (radially arranged nuclear segments connected by spoke-like chromatin filaments) and the hyper-lobulated nucleus of lymphocytes in a patient with heatstroke. Similarly, leukopenia due to both mature neutropenia and lymphopenia with increased apoptotic bodies in the background were also reported in dogs with heatstroke [49]. These nuclear changes in the leukocytes are thought to result from nuclear degeneration induced by heat. Lymphocytes are more easily damaged by heat, and they fall into oncosis (formerly necrosis) [50]. Oncosis, but not apoptosis, is known to elicit inflammation by releasing alarmins, such as IL-1, histones, HMGB-1, and HSPs [51, 52]. These mediators stimulate macrophages and dendritic cells to secrete cytokines and upregulate immune responses. Interestingly, Basu et al. [53] reported the potential role of cytoprotective HSPs in facilitating inflammation by activating the nuclear factor (NF)-κB translocation. However, further research is needed to reveal the regulating effect of HSPs on NF-κB pathway.

Morphological changes of the leukocytes subjected to heat stress. The blood smear examination shows a variety of morphological changes in blood cells. When the body temperature reached 41.5 ℃, a neutrophil with a botryoid (hypersegmented) nucleus appeared. Platelets were assembled, but not aggregated (left panel). When the body temperature approaches 43 ℃, ruptured lymphocytes and aggregated platelets were observed (right panel)
Milleron et al. [54] reported that heat stress activates an apical protease that stimulates mitochondrial outer membrane permeabilization and effector caspase-3 processing. They also suggested that heat stress can initiate apoptosis with caspase-like activity by apical protease, independent of an ordinary caspase-dependent pathway. We demonstrated that leukocytes initially showed apoptotic change after being subjected to heat, and turned their cell death style to oncosis when the temperature exceeds 43 °C [9] (Fig. 4). Hirose et al. [55] reported an increase in NETosis, suicidal cell death with neutrophil extracellular traps (NETs) formation, and elevated citrullinated histone H3 levels in patients with heatstroke. Although the detailed mechanism remains to be elucidated, since histones are extremely deleterious and shock inducible, the increase of cell death-related mediators such as histones, HMGB-1, and cell-free DNA due to oncosis and NETosis may have an impact on heat-induced shock.

Time-lapse changes of leukocytes subjected to heat stress. Leukocytes were subjected to 43 ℃ in vitro, and sequential changes were observed under the microscope with DAPI (4’,6-diamidino-2-phenylindole). Leukocytes (white arrows) were vacuolized or formed apoptotic bodies at 30 min. At 60 min, most leukocytes were dead and nuclei were colored blue by DAPI stain. At 90 min, ballooned necrotic leukocyte was observed and DNA stained blue with DAPI expanded outside the dead leukocytes except for the apoptotic leukocyte (white arrowhead)
Histones also act as the strong initiators of coagulation and inflammation that are released into circulation mainly by NETosis as well as by oncosis with the rupture of nuclear and plasma membranes [56]. Li et al. [57] reported that the elevation of histone H3 was associated with increased disease severity and longer duration of symptoms in heatstroke. Bruchim et al. [33] also reported higher histone H3 levels among non-survived dogs and dogs with hemostatic impairment. Other than histones, HMGB-1 and IL-1β also appeared to be involved [35]. Tong et al. [58] showed HMGB-1 levels in plasma and liver cytoplasm were elevated in the heatstroke model of rats, along with increased transaminase levels. In their study, histologic examination noted that HMGB-1 monoclonal antibody pretreatment reduced the pathologic impairments. The same authors also reported elevated levels of plasma HMGB-1 at an early stage of heatstroke [59]. Dehbi et al. [60] also demonstrated the critical roles of HMGB-1 as an early mediator of inflammation that evokes IL-6 and IL-1βelevation, tissue injury, and lethality in a heatstroke model of rats. HMGB-1 is also reported to induce organ dysfunctions, and mice subjected to the core body temperature of 41 °C exhibited signs of hemorrhage and apoptosis in multiple organs. This model showed significant upregulation of HMGB-1 and its receptor RAGE (receptor for advanced glycation end products) [61].
Heat-induced endotheliopathy and vasculopathy
Previous studies have shown that vascular endothelium is a major target of heat stress. Increased vascular permeability due to endothelial injury leads to decreased blood volume and increased edema and leukocyte transmigration. In addition to dehydration, blood volume loss accelerates the collapse of the systemic circulation (Fig. 5). Bouchama et al. [62] reported increased serum endothelial cell damage biomarkers, i.e., intercellular adhesion molecular-1 (ICAM-1), endothelin, and von Willebrand factor-antigen in patients with heatstroke. In addition, the increase of circulating endothelial glycocalyx component syndecan-1 is also reported [38]. Meanwhile, Tong et al. [63] have shown the potential involvement of the mesenteric lymphoid system in endothelial injury in a rat model subjected to heat. Yamaguchi et al. [64] used a mouse model of heatstroke and cultured endothelial cells subjected to heat and demonstrated the damaged blood–brain barrier integrity that leads to increased microvascular permeability in the brain. Although multiple factors are involved in heat-induced brain damage, brain edema caused by endothelial damage is assumed to be an important factor.

Time-lapse changes of endothelial cell subjected to heat stress. Cultured rat aortic endothelial cells were subjected to 43 ℃ in vitro, and sequential changes were observed under the microscope. The morphological change was minimal until the temperature reached 40 ℃. At 41 ℃, endothelial cells shrank, and the spaces between the cells expanded. No remarkable changes of nuclei were observed until temperature reached 43 ℃; however, the spaces expanded as the temperature rose
Roberts et al. [65] have described that pathological examination revealed that vascular endothelium apoptosis is the mechanism of endothelial damage in the baboon model of heatstroke. Others have also revealed that heat stress induces endothelial cell apoptosis [66, 67]. Gu et al. [67] demonstrated that apoptosis of human umbilical vein endothelial cells apoptosis increased when treated at 43 °C for 2 h in vitro, followed by replacement with fresh media and incubation for 6 h. Chen et al. [68] also reported the highest levels of inflammatory mediators, such as serum levels of TNF‑α, IL‑1β, and IL‑6, and numbers of apoptotic endothelial cells were present at 6 h. It is assumed that endothelial cell death style changes in a time- and temperature-dependent manner. Pei et al. [69] reported that in addition to apoptosis, pyroptosis, a programmed form of inflammation-related cell death, was also seen in heat stroke. Furthermore, Huang et al. [70] have shown that necroptosis, a type of regulated necrosis, have also been recognized in cultured pulmonary vascular endothelial cells. However, programmed cell death is energy-dependent cell death. When the temperature surpasses the threshold point and disturbed oxidative phosphorylation decreases in mitochondrial function, the energy-dependent programmed cell death cannot be completed and turns to unprogrammed oncosis [71].
Heat-induced platelet activation
In addition to the leukocytes, platelets are also considered to participate in the pathogenesis of heat-induced coagulopathy [72]. At first, platelet counts were known to decrease in patients with heatstroke frequently, and platelet counts were negatively correlated with the degree of organ dysfunction and mortality [73]. According to the studies that used platelet-rich plasma obtained from healthy subjects, platelets increased aggregability with increased temperature until 43 °C and lost their function at higher temperatures [74, 75]. On the contrary, a similar study that used platelet-rich plasma from heatstroke patients did not show a consistent result [76]. Wang et al. [77] also explored the direct effects of heat on platelet ranges between 40 and 42 °C, and reported that heat stress elicited caspase-3-dependent apoptosis in platelets by affecting mitochondrial function.
Both the direct and indirect effects of heat on platelets should be considered. As described before, damaged endothelial cells release von Willebrand factor, lose their anticoagulant properties, upregulate the expression of adhesion molecules, and turn antithrombotic properties toward the opposite side [63]. In this progression, the increased count of CD41 (GP IIb–IIIa)-positive platelet microvesicles after exposure to heat stress was reported [78]. Thus, the interaction between platelets and vascular endothelium should be upregulated.
Additional studies reported that heme-activated platelets induce macrophage extracellular traps (METs) and exacerbate rhabdomyolysis-induced acute kidney injury [79]. In this setting, myoglobin and heme released by heat-induced rhabdomyolysis activate platelets by binding to C-type lectin-like receptor-2 (CLEC-2) and glycoprotein VI (GPVI), heme receptors, and activated platelets to stimulate METs release and involved in the exacerbation of acute renal failure [80]. However, inflammation by activated platelets is also expected, and other mechanisms of activated platelet involvement in circulatory shock should be examined.
Heat-induced tissue injury
Heat-induced tissue damage has direct and indirect contributions to the pathophysiology of circulatory shock in patients with heatstroke multiorgan dysfunction, including the central nervous system, renal, hepatic, and coagulation systems [81]. Multiple mechanisms are involved in the pathogenesis of organ failure, including; excess inflammatory/anti-inflammatory cytokines, increased oxygen free radicals, ischemia/reperfusion injury, decreased tissue perfusion, increased HSPs, and necrotic cell death [82, 83]. Regarding cell death in heatstroke, induction of apoptosis with widespread apoptosis in lymphoid systems such as the spleen, lymph nodes, thymus, and the lamina propria in the small bowel are reported [3]. However, since apoptosis is not cytotoxic, it may not directly responsible for heat-induced shock. Necroptosis, a type of programmed necrosis, was recently suggested to participate in heat stress-induced organ damage. Huang et al. [70] showed that heat stress upregulates receptor-interacting protein 1 (RIP1) to induce endothelial necroptosis. Li et al. [84] reported that scavenging reactive oxygen species production can attenuate RIP1 kinase-dependent necroptosis and suppress heatstroke-induced intestinal damage, with NF-κB activation exhibiting a central role in this pathway [85]. HSPs can promote cellular survival via stabilizing and suppressing this NF-κB signaling pathway [86].
For managing tissue injury, Bouchama et al. [87] reported that glucocorticoids attenuated the activation of complement and suppressed decreases in arterial blood pressure. However, neither tissue damage nor the outcome was improved in the primate heatstroke model.
Heat shock proteins (HSPs)
HSPs were first recognized as proteins produced in response to hyperthermal stress. Most genes are suppressed when subjected to heat, but the genes encoding HSPs are activated, and HSPs are also produced in response to other stresses such as ischemia and chemical exposure. HSPs function as molecular chaperones that control conformational changes by folding and unfolding proteins to maintain cellular homeostasis, thereby acting for host protection in general [88]. Huisse et al. [45] reported increased levels of inflammation and stress mediators such as IL-6, IL-8, and HSP60 and HSP70 in critically ill patients. The ability of HSPs to avoid apoptosis and promote cell survival is thought to be an inhibition of caspase activation-dependent, but other mechanisms, such as mitochondria-dependent and death-receptor-mediated mechanisms, are also proposed [86]. HSP27, 32, 70, 72, and 90 are the representatives of inducible cytoprotective HSPs [89], and the effects of HSP32, also known as heme oxygenase-1, were revealed in a heatstroke model [90]. The protective effects of HSP27 and HSP90 are the regulations of the stability and activity of various components of the NF-κB activation pathway and regulate TNF-mediated cellular survival. However, if the thermal stress is too intense, signals that initiate apoptosis are activated by HSPs. Baba et al. [91] demonstrated the association between HSP72 overexpression and apoptosis, cardiovascular decompensation, and poor outcome. However, their experimental model was hypovolemic sheep with or without endotoxin.
Nevertheless, since HSPs are primarily cytoprotective, they are promising candidates for therapeutics. Hsu et al. [92] showed that pre-induction of cardiac HSP72 improved hypotension in heatstroke rats by increasing cardiac mechanical efficiency and arterial elastance.
Despite the progress in research on pathophysiology, only rapid cooling and fluid resuscitation are the current practice. Intensive research on cardiac and vascular protection, regulation of inflammation, and management of coagulation disorder is warranted.
Conclusions
Complex mechanisms exist as underlying conditions of heatstroke-induced shock. Although dehydration and cardiomyopathy due to mitochondrion-linked pathologies are considered to be the direct inducers, activated inflammation, coagulation, oncotic cell death, damaged vascular endothelium, and cellular/tissue damage are involved in the pathogenesis of circulatory shock. In this sense, heatstroke-induced shock is a mixture of cardiogenic, hypovolemic, and distributive shock. Since heatstroke-induced shock occurs suddenly and is often deadly, it is important to predict it by measuring troponin, cytokines, DAMPs, and HSPs. Except for rapid cooling and fluid resuscitation, there is no established treatment; however, anti-inflammatory and anticoagulant therapy may have potential benefits. However, further investigation is warranted for this and other therapeutic modalities with the increasing specter of global warming.
References
Gostimirovic M, Novakovic R, Rajkovic J, Djokic V, Terzic D, Putnik S, Gojkovic-Bukarica L. The influence of climate change on human cardiovascular function. Arch Environ Occup Health. 2020;75(7):406–14.
Bouchama A, Abuyassin B, Lehe C, Laitano O, Jay O, O’Connor FG, Leon LR. Classic and exertional heatstroke. Nat Rev Dis Primers. 2022;8(1):8.
Roberts GT, Ghebeh H, Chishti MA, Al-Mohanna F, El-Sayed R, Al-Mohanna F, Bouchama A. Microvascular injury, thrombosis, inflammation, and apoptosis in the pathogenesis of heatstroke: a study in baboon model. Arterioscler Thromb Vasc Biol. 2008;28(6):1130–6.
Bruchim Y, Loeb E, Saragusty J, Aroch I. Pathological findings in dogs with fatal heatstroke. J Comp Pathol. 2009;140(2–3):97–104.
Wang JL, Ke DS, Lin MT. Heat shock pretreatment may protect against heatstroke-induced circulatory shock and cerebral ischemia by reducing oxidative stress and energy depletion. Shock. 2005;23(2):161–7.
Chou YT, Lai ST, Lee CC, Lin MT. Hypothermia attenuates circulatory shock and cerebral ischemia in experimental heatstroke. Shock. 2003;19(4):388–93.
Bouchama A, Roberts G, Al Mohanna F, El-Sayed R, Lach B, Chollet-Martin S, Ollivier V, Al Baradei R, Loualich A, Nakeeb S, Eldali A, de Prost D. Inflammatory, hemostatic, and clinical changes in a baboon experimental model for heatstroke. J Appl Physiol. 2005;98(2):697–705.
Bouchama A, Ollivier V, Roberts G, Al Mohanna F, de Prost D, Eldali A, Saussereau E, El-Sayed R, Chollet-Martin S. Experimental heatstroke in baboon: analysis of the systemic inflammatory response. Shock. 2005;24(4):332–5.
Iba T, Sawada T, Kondo Y, Kondo K, Levy JH. Morphological changes of blood cells in a rat model of heatstroke: a pilot study. J Clin Med. 2022;11:4821.
Hayashida K, Kondo Y, Hifumi T, Shimazaki J, Oda Y, Shiraishi S, Fukuda T, Sasaki J, Shimizu K. A novel early risk assessment tool for detecting clinical outcomes in patients with heat-related illness (J-ERATO score): development and validation in independent cohorts in Japan. PLoS ONE. 2018;13(5):e0197032.
Chen WT, Lin CH, Hsieh MH, Huang CY, Yeh JS. Stress-induced cardiomyopathy caused by heat stroke. Ann Emerg Med. 2012;60(1):63–6.
Leon LR, Helwig BG. Role of endotoxin and cytokines in the systemic inflammatory response to heat injury. Front Biosci. 2010;2(3):916–38.
Mustafa KY, Omer O, Khogali M, Jamjoom A, Gumaa KA, El-Nasr NA, Gader MA. Blood coagulation and fibrinolysis in heat stroke. Br J Haematol. 1985;61(3):517–23.
DuBose DA, Hinkle JR, Morehouse DH, Ogle PL. Model for environmental heat damage of the blood vessel barrier. Wilderness Environ Med. 1998;9(3):130–6.
Chang CK, Chang CP, Chiu WT, Lin MT. Prevention and repair of circulatory shock and cerebral ischemia/injury by various agents in experimental heatstroke. Curr Med Chem. 2006;13(26):3145–54.
Kuo JR, Lin CL, Chio CC, Wang JJ, Lin MT. Effects of hypertonic (3%) saline in rats with circulatory shock and cerebral ischemia after heatstroke. Intensive Care Med. 2003;29(9):1567–73.
Liu CC, Ke D, Chen ZC, Lin MT. Hydroxyethyl starch produces attenuation of circulatory shock and cerebral ischemia during heatstroke. Shock. 2004;22(3):288–94.
Liu CC, Cheng BC, Lin MT, Lin HJ. Small volume resuscitation in a rat model of heatstroke. Am J Med Sci. 2009;337(2):79–87.
Sankoff J. Heat illnesses: a hot topic in the setting of global climate change. Aust Fam Physician. 2015;44(1–2):22–6.
Marchand M, Gin K. The cardiovascular system in heat stroke. CJC Open. 2021;4(2):158–63.
Bathini T, Thongprayoon C, Petnak T, Chewcharat A, Cheungpasitporn W, Boonpheng B, Chokesuwattanaskul R, Prasitlumkum N, Vallabhajosyula S, Kaewput W. Circulatory failure among hospitalizations for heatstroke in the united states. Medicines. 2020;7(6):32.
Bathini T, Thongprayoon C, Chewcharat A, Petnak T, Cheungpasitporn W, Boonpheng B, Prasitlumkum N, Chokesuwattanaskul R, Vallabhajosyula S, Kaewput W. Acute myocardial infarction among hospitalizations for heat stroke in the united states. J Clin Med. 2020;9(5):1357.
Quinn CM, Duran RM, Audet GN, Charkoudian N, Leon LR. Cardiovascular and thermoregulatory biomarkers of heat stroke severity in a conscious rat model. J Appl Physiol. 2014;117(9):971–8.
Kang R, Nagoshi T, Kimura H, Tanaka TD, Yoshii A, Inoue Y, Morimoto S, Ogawa K, Minai K, Ogawa T, Kawai M, Yoshimura M. Possible association between body temperature and B-Type natriuretic peptide in patients with cardiovascular diseases. J Card Fail. 2021;27(1):75–82.
Nakagawa Y, Inoue H, Shinone K, Ikemura M, Nata M. Molecular biological analysis of cardiac effect of high temperature in rats. Leg Med. 2012;14(2):63–8.
Yi G, Li L, Luo M, He X, Zou Z, Gu Z, Su L. Heat stress induces intestinal injury through lysosome- and mitochondria-dependent pathway in vivo and in vitro. Oncotarget. 2017;8(25):40741–55.
Shen HH, Tseng YS, Kuo NC, Kung CW, Amin S, Lam KK, Lee YM. Alpha-lipoic acid protects cardiomyocytes against heat stroke-induced apoptosis and inflammatory responses associated with the induction of hsp70 and activation of autophagy. Mediators Inflamm. 2019;2019:8187529.
Bouchama A, Al-Sedairy S, Siddiqui S, Shail E, Rezeig M. Elevated pyrogenic cytokines in heatstroke. Chest. 1993;104(5):1498–502.
Leon LR, Bouchama A. Heat stroke. Compr Physiol. 2015;5(2):611–47.
Lian P, Braber S, Garssen J, Wichers HJ, Folkerts G, Fink-Gremmels J, Varasteh S. Beyond heat stress: intestinal integrity disruption and mechanism-based intervention strategies. Nutrients. 2020;12(3):734.
Welc SS, Clanton TL, Dineen SM, Leon LR. Heat stroke activates a stress-induced cytokine response in skeletal muscle. J Appl Physiol. 2013;115(8):1126–37.
Bruchim Y, Ginsburg I, Segev G, Mreisat A, Avital Y, Aroch I, Horowitz M. Serum histones as biomarkers of the severity of heatstroke in dogs. Cell Stress Chaperones. 2017;22(6):903–10.
Zhang ZT, Gu XL, Zhao X, He X, Shi HW, Zhang K, Zhang YM, Su YN, Zhu JB, Li ZW, Li GB. NLRP3 ablation enhances tolerance in heat stroke pathology by inhibiting IL-1β-mediated neuroinflammation. J Neuroinflammation. 2021;18(1):128.
Xia ZN, Zong Y, Zhang ZT, Chen JK, Ma XJ, Liu YG, Zhao LJ, Lu GC. Dexmedetomidine protects against multi-organ dysfunction induced by heatstroke via sustaining the intestinal integrity. Shock. 2017;48(2):260–9.
Geng Y, Ma Q, Liu YN, Peng N, Yuan FF, Li XG, Li M, Wu YS, Li BL, Song WB, Zhu W, Xu WW, Fan J, Su L. Heatstroke induces liver injury via IL-1β and HMGB1-induced pyroptosis. J Hepatol. 2015;63(3):622–33.
Zeller L, Novack V, Barski L, Jotkowitz A, Almog Y. Exertional heatstroke: clinical characteristics, diagnostic and therapeutic considerations. Eur J Intern Med. 2011;22(3):296–9.
Shen KH, Chang CK, Lin MT, Chang CP. Interleukin-1 receptor antagonist restores homeostatic function and limits multiorgan damage in heatstroke. Eur J Appl Physiol. 2008;103(5):561–8.
Umemura Y, Ogura H, Matsuura H, Ebihara T, Shimizu K, Shimazu T. Bone marrow-derived mononuclear cell therapy can attenuate systemic inflammation in rat heatstroke. Scand J Trauma Resusc Emerg Med. 2018;26(1):97.
Al-Mashhadani SA, Gader AG, Al Harthi SS, Kangav D, Shaheen FA, Bogus F. The coagulopathy of heat stroke: alterations in coagulation and fibrinolysis in heat stroke patients during the pilgrimage (Haj) to Makkah. Blood Coagul Fibrinolysis. 1994;5(5):731–6.
Hernández-Espinosa D, Mota R, Miñano A, Ordóñez A, Yélamos J, Vicente V, Corral J. In vivo effects of hyperthermia on the functional and conformational characteristics of antithrombin. J Thromb Haemost. 2007;5(5):963–70.
Matsumoto H, Takeba J, Umakoshi K, Nakabayashi Y, Moriyama N, Annen S, Ohshita M, Kikuchi S, Sato N, Aibiki M. Successful treatment for disseminated intravascular coagulation (DIC) corresponding to phenotype changes in a heat stroke patient. J Intensive Care. 2019;7:2.
Proctor EA, Dineen SM, Van Nostrand SC, Kuhn MK, Barrett CD, Brubaker DK, Yaffe MB, Lauffenburger DA, Leon LR. Coagulopathy signature precedes and predicts severity of end-organ heat stroke pathology in a mouse model. J Thromb Haemost. 2020;18(8):1900–10.
Shimazaki J, Hifumi T, Shimizu K, et al. Clinical characteristics, prognostic factors, and outcomes of heat-related illness (Heatstroke Study 2017–2018). Acute Med Surg. 2020;7:e516.
Iba T, Levi M, Thachil J, Levy JH. Disseminated intravascular coagulation: the past, present, and future considerations. Semin Thromb Hemost. 2022;48(8):978–87.
Huisse MG, Pease S, Hurtado-Nedelec M, et al. Leukocyte activation: the link between inflammation and coagulation during heatstroke A study of patients during the heat wave in Paris. Crit Care Med. 2008;36:2288–95.
Iba T, Connors JM, Levi M, Levy JH. Heatstroke-induced coagulopathy: Biomarkers, mechanistic insights, and patient management. EClinicalMedicine. 2022;44:101276.
Bouchama A, Kunzelmann C, Dehbi M, Kwaasi A, Eldali A, Zobairi F, Freyssinet JM, de Prost D. Recombinant activated protein C attenuates endothelial injury and inhibits procoagulant microparticles release in baboon heatstroke. Arterioscler Thromb Vasc Biol. 2008;28(7):1318–25.
Ward PC, McKenna RW, Kroft SH. White blood cell changes in hyperthermia. Br J Haematol. 2007;138(2):130.
Mastrorilli C, Welles EG, Hux B, Christopherson PW. Botryoid nuclei in the peripheral blood of a dog with heatstroke. Vet Clin Pathol. 2013;42(2):145–9.
Majno G, Joris I. Apoptosis, oncosis, and necrosis An overview of cell death. Am J Pathol. 1995;146(1):3–15.
Yan YE, Zhao YQ, Wang H, Fan M. Pathophysiological factors underlying heatstroke. Med Hypotheses. 2006;67:609–17.
Epstein Y, Yanovich R. Heatstroke. Reply. N Engl J Med. 2019;381(12):1187.
Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol. 2000;12(11):1539–46.
Milleron RS, Bratton SB. Heat shock induces apoptosis independently of any known initiator caspase-activating complex. J Biol Chem. 2006;281(25):16991–7000.
Hirose T, Hamaguchi S, Matsumoto N, et al. Presence of neutrophil extracellular traps and citrullinated histone H3 in the bloodstream of critically ill patients. PLoS ONE. 2014;9(11):e111755.
Allam R, Kumar SV, Darisipudi MN, Anders HJ. Extracellular histones in tissue injury and inflammation. J Mol Med. 2014;92(5):465–72.
Li Y, Liu Z, Shi X, Tong H, Su L. Prognostic value of plasma exosomal levels of histone H3 protein in patients with heat stroke. Exp Ther Med. 2021;22(3):922.
Tong H, Tang Y, Chen Y, Yuan F, Liu Z, Peng N, Tang L, Su L. HMGB1 activity inhibition alleviating liver injury in heatstroke. J Trauma Acute Care Surg. 2013;74(3):801–7.
Tong HS, Tang YQ, Chen Y, Qiu JM, Wen Q, Su L. Early elevated HMGB1 level predicting the outcome in exertional heatstroke. J Trauma. 2011;71(4):808–14.
Dehbi M, Uzzaman T, Baturcam E, Eldali A, Ventura W, Bouchama A. Toll-like receptor 4 and high-mobility group box 1 are critical mediators of tissue injury and survival in a mouse model for heatstroke. PLoS ONE. 2012;7(9):e44100.
Xue L, Guo W, Li L, Ou S, Zhu T, Cai L, Ding W, Wu W. Metabolomic profiling identifies a novel mechanism for heat stroke-related acute kidney injury. Mol Med Rep. 2021;23(4):241.
Bouchama A, Hammami MM, Haq A, Jackson J, Al-Sedairy S. Evidence for endothelial cell activation/injury in heatstroke. Crit Care Med. 1996;24(7):1173–8.
Tong H, Wan P, Zhang X, Duan P, Tang Y, Chen Y, Tang L, Su L. Vascular endothelial cell injury partly induced by mesenteric lymph in heat stroke. Inflammation. 2014;37(1):27–34.
Yamaguchi T, Shimizu K, Kokubu Y, Nishijima M, Takeda S, Ogura H, Kawabata K. Effect of heat stress on blood-brain barrier integrity in iPS cell-derived microvascular endothelial cell models. PLoS ONE. 2019;14(9):e0222113.
Roberts GT, Chishti MA, Al-Mohanna FH, El-Sayed RM, Bouchama A. Vascular endothelium is severely perturbed and undergoes apoptosis in experimental heatstroke in primates. Blood. 2005;106(11):3972.
Brinton MR, Tagge CA, Stewart RJ, Cheung AK, Shiu YT, Christensen DA. Thermal sensitivity of endothelial cells on synthetic vascular graft material. Int J Hyperthermia. 2012;28(2):163–74.
Gu ZT, Wang H, Li L, Liu YS, Deng XB, Huo SF, Yuan FF, Liu ZF, Tong HS, Su L. Heat stress induces apoptosis through transcription-independent p53-mediated mitochondrial pathways in human umbilical vein endothelial cell. Sci Rep. 2014;4:4469.
Chen F, Li H, Zhu G, Chen X, Tang Z. Sodium tanshinone IIA sulfonate improves inflammation, aortic endothelial cell apoptosis, disseminated intravascular coagulation and multiple organ damage in a rat heat stroke model. Mol Med Rep. 2017;16(1):87–94.
Pei Y, Geng Y, Su L. Pyroptosis of HUVECs can be induced by heat stroke. Biochem Biophys Res Commun. 2018;506(3):626–31.
Huang W, Xie W, Gong J, Wang W, Cai S, Huang Q, Chen Z, Liu Y. Heat stress induces RIP1/RIP3-dependent necroptosis through the MAPK, NF-κB, and c-Jun signaling pathways in pulmonary vascular endothelial cells. Biochem Biophys Res Commun. 2020;528(1):206–12.
Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516.
Iba T, Helms J, Levi M, Levy JH. The role of platelets in heat-related illness and heat-induced coagulopathy. Thromb Res. 2022. https://doi.org/10.1016/j.thromres.2022.08.009.
Zhong L, Wu M, Ji J, Wang C, Liu Z. Association Between Platelet Levels on Admission and 90-day Mortality in Patients With Exertional Heatstroke, a 10 Years Cohort Study. Front Med. 2021;11(8):716058.
Rao GH, Smith CM, Escolar G, White JG. Influence of heat on platelet biochemistry, structure, and function. J Lab Clin Med. 1993;122(4):455–64.
Gader AM, Al-Mashhadani SA, Al-Harthy SS. Direct activation of platelets by heat is the possible trigger of the coagulopathy of heat stroke. Br J Haematol. 1990;74(1):86–92.
Al-Mashhadani SA, Gader AM, Al HS. The role of platelets in the coagulopathy of heatstroke- a study of platelet aggregation in heatstroke patients during the Makkah pilgrimage (Haj) to Makkah. Platelets. 1997;8(1):37–42.
Wang Z, Shi Q, Li S, Du J, Liu J, Dai K. Hyperthermia induces platelet apoptosis and glycoprotein Ibalpha ectodomain shedding. Platelets. 2010;21(3):229–37.
Wilhelm EN, González-Alonso J, Chiesa ST, Trangmar SJ, Kalsi KK, Rakobowchuk M. Whole-body heat stress and exercise stimulate the appearance of platelet microvesicles in plasma with limited influence of vascular shear stress. Physiol Rep. 2017;5(21):e13496.
Okubo K, Kurosawa M, Kamiya M, et al. Macrophage extracellular trap formation promoted by platelet activation is a key mediator of rhabdomyolysis-induced acute kidney injury. Nat Med. 2018;24(2):232–8.
Oishi S, Tsukiji N, Otake S, Oishi N, Sasaki T, Shirai T, Yoshikawa Y, Takano K, Shinmori H, Inukai T, Kondo T, Suzuki-Inoue K. Heme activates platelets and exacerbates rhabdomyolysis-induced acute kidney injury via CLEC-2 and GPVI/FcRγ. Blood Adv. 2021;5(7):2017–26.
Mozzini C, Xotta G, Garbin U, Fratta Pasini AM, Cominacini L. Non-Exertional Heatstroke: a case report and review of the literature. Am J Case Rep. 2017;18:1058–65.
Papathanassoglou ED, Moynihan JA, Ackerman MH. Does programmed cell death (apoptosis) play a role in the development of multiple organ dysfunction in critically ill patients? a review and a theoretical framework. Crit Care Med. 2000;28(2):537–49.
Yang CY, Lin MT. Oxidative stress in rats with heatstroke-induced cerebral ischemia. Stroke. 2002;33(3):790–4.
Li L, Tan H, Zou Z, Gong J, Zhou J, Peng N, Su L, Maegele M, Cai D, Gu Z. Preventing necroptosis by scavenging ROS production alleviates heat stress-induced intestinal injury. Int J Hyperthermia. 2020;37(1):517–30.
Kumar A, Takada Y, Boriek AM, Aggarwal BB. Nuclear factor-kappaB: its role in health and disease. J Mol Med. 2004;82(7):434–48.
Beere HM. “The stress of dying”: the role of heat shock proteins in the regulation of apoptosis. J Cell Sci. 2004;117(13):2641–51.
Bouchama A, Kwaasi A, Dehbi M, Al Mohanna F, Eldali A, El-Sayed R, Tbakhi A, Alzahrani AS, Roberts AG. Glucocorticoids do not protect against the lethal effects of experimental heatstroke in baboons. Shock. 2007;27(5):578–83.
Pirkkala L, Nykänen P, Sistonen L. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J. 2001;15(7):1118–31.
Dehbi M, Baturcam E, Eldali A, Ahmed M, Kwaasi A, Chishti MA, Bouchama A. Hsp-72, a candidate prognostic indicator of heatstroke. Cell Stress Chaperones. 2010;15(5):593–603.
Wen YT, Liu TT, Lin YF, Chen CC, Kung WM, Huang CC, Lin TJ, Wang YH, Wei L. Heatstroke Effect on Brain Heme Oxygenase-1 in Rats. Int J Med Sci. 2015;12(9):737–41.
Baba HA, Wohlschlaeger J, Stubbe HD, Grabellus F, Aken HV, Schmitz KJ, Otterbach F, Schmid KW, August C, Levkau B, Hinder F. Heat shock protein 72 and apoptosis indicate cardiac decompensation during early multiple organ failure in sheep. Intensive Care Med. 2004;30(7):1405–13.
Hsu SF, Chao CM, Chang CP, Lin MT, Cheng BC. Heat shock protein 72 may improve hypotension by increasing cardiac mechanical efficiency and arterial elastance in heatstroke rats. Int J Cardiol. 2016;219:63–9.
Funding
This work was supported in part by a Grant-in-Aid for Special Research in Subsidies for ordinary expenses of private schools from The Promotion and Mutual Aid Corporation for Private Schools of Japan.
Author information
Authors and Affiliations
Contributions
Iba T. and Levy J.H. wrote the main manuscript text and Iba T. prepared figures 1-5. Helms J. and Levi M. revised the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Iba T. has received a research grant from Japan Blood Products Organization and JIMRO. Helms J. has received honoraria from Diagnostica Stago, Pfizer PFE France and Sanofi Aventis France, MSD, Shionogi and Inotrem. Levi M. has received grants and participated in the advisory boards of NovoNordisk, Eli Lilly, Asahi Kasei Pharmaceuticals America, and Johnson & Johnson. Levy JH. serves on the Steering Committees for Instrumentation Laboratories, Merck, and Octapharma.
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Iba, T., Helms, J., Levi, M. et al. Inflammation, coagulation, and cellular injury in heat-induced shock. Inflamm. Res. 72, 463–473 (2023). https://doi.org/10.1007/s00011-022-01687-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-022-01687-8