
Abstract
Multiple sclerosis (MS) is an autoimmune disease, characterized by multiple demyelination of axons in both white and gray matter in the Central Nervous System (CNS). There is increasing evidence to support the notion that angiogenesis and chronic inflammation are mutually related. Different immune cells, including monocytes–macrophages, lymphocytes, neutrophils, mast cells (MCs) and dendritic cells are able to secrete an array of angiogenic cytokines, which promote growth, migration, and activation of endothelial cells. MCs play various roles in MS pathogenesis, influencing the innate immune response in peripheral tissues and in CNS. The aim of this review article is to discuss the role of MCs in MS pathogenesis with particular reference to the involvement of these inflammatory cells in the angiogenic processes occurring during MS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is an autoimmune disease, characterized by multiple demyelination of axons in both white and gray matter in the Central Nervous System (CNS). The course of the disease involves an early, predominantly inflammatory demyelinating disease phase of relapsing remitting MS (RRMS), accounting for ~ 85% of cases, which evolves into a progressively degenerative stage associated with axonal loss and scar formation, causing physical and cognitive disability. MS has been considered an adaptive immune response through the activation of CD4+ and CD8+ T cells, specifically identifying myelin fragments that induce tissue damage [1]. Myelin destruction is initiated by myelin-reactive T cells, and is further amplified by the inflammatory response of myeloid cells, including brain-resident microglia and infiltrating inflammatory macrophages [2].
There is increasing evidence to support the notion that angiogenesis and chronic inflammation are mutually related. Different immune cells, including monocytes–macrophages, lymphocytes, neutrophils, mast cells (MCs) and dendritic cells are able to secrete an array of angiogenic cytokines, which promote growth, migration, and activation of endothelial cells.
MCs have been identified in different components of the CNS, including leptomeninges, where MCs play a role in limiting infections of the brain parenchyma [3], thalamus and hypothalamus the dura mater of the spinal cord the infundibulum, pineal organ, area postrema, choroid plexuses, supraoptic crest, the subfornical organ and the ventricles [4, 5].
The aim of this review article is to discuss the role of MCs in MS pathogenesis with particular reference to the involvement of these inflammatory cells in the angiogenic processes occurring during MS.
Mast cells and MS
Most of our knowledge concerning MS immunopathogenesis derives from studies conducted utilizing the experimental autoimmune encephalomyelitis (EAE) experimental model, which is induced in rodents via immunization with CNS-derived antigens [myelin basic protein (MBP); proteolipid protein (PLP); myelin oligodendrocyte glycoprotein (MOG] emulsified with complete Freund’s adjuvant in the presence of pertussis toxin [6,7,8]. These antigens induce an acute demyelinating process mediated by T cells and macrophages which can have a chronic relapsing course similar to MS. Also, similar to MS in EAE, the blood brain barrier (BBB) is altered, allowing infiltration of inflammatory cells in the brain parenchyma and destruction of myelin and oligodendrocytes.
MCs play a critical role in both initiation and progression of MS and EAE, and accumulate in the demyelinating plaques found in the CNS associated with immune cell infiltrates, but also in the CNS parenchyma and the leptomeninges of MS patients [9]. Increased numbers of MCs and their activation products are detected in the brain and spinal cord of MS patients and mice with EAE [10].
In EAE, meningeal MCs promote cellular influx into the CNS by altering BBB integrity through release of histamine, leukotriens, and chemokines, and expression of tumor necrosis factor alpha (TNF-α), which recruits neutrophils and leads to increased permeability and number of cells that invade CNS. Rats and mice with EAE exhibit increased degranulation of their brain MCs [11] and elevated levels of the demyelinating MC protease [12]. EAE autoimmune demyelination of CNS is accompanied by an increased density of both total MCs and degranulated MCs upon recognition of MBP and purinergic P2 receptor [13, 14]. MBP could induce MC activation and superstimulation of activated T cells [15, 16].
Activators/receptors involved in mast cell stimulation in MS
MC degranulation occurs within minutes of activation and results in the rapid release of substances from pre-formed granules (Table 1). Degranulation begins with the activation of receptors with high affinity for IgE (FcεR1) and Fcγ RIIA and their cross-linking with a rearrangement of F-actin and microtubule formation. As a late response, MCs release cytokines and chemokines that are synthesized de novo (Table 1). The mechanism for activation involves IgE receptors in cooperation with the toll-like receptor (TLR). TLRs also recruit CD14 or CD48 for the effect of TLR ligands [17].
The MC mediators involved in EAE/MS immunopathogenesis include histamine, TNF-α, interleukin-1 beta and -6 (IL-1β and IL-6), matrix metalloproteinesases (MMPs), tryptase, and protease-activated receptors (PARs).
MC-derived histamine causes the disruption of BBB tightness and passage of lymphocytes and their myelin-directed antibodies, granulocytes that together with microglia/macrophages, and MC-secreted proteases, participate in demyelination and neuronal damage implicated in the pathogenesis of MS [18].
TNFα expression has been correlated with CNS inflammation, degeneration and disease activity in MS [19,20,21]. TNF-α recruits neutrophils and leads to increased permeability and number of cells that invade CNS. The entry of neutrophils into the meninges and the CNS parenchyma was abrogated in the absence of MC-derived TNF-α [16, 22].
Neutrophils and MCs promote B cell proliferation contributing to the organization of B cell follicle-like structures that are found in the meninges of MS patients [23]. MCs influence Th1 differentiation in EAE [15, 24, 25] and meningeal MCs modulate T lymphocyte effector function through their expression of cytokines and other co-stimulating molecules [3].
The increased MC production of IL-1β and IL-6 promotes the shift of regulatory T cells (Tregs) into active T helper type 17 (Th17) lymphocytes producing IL-17 and IL-22 [26,27,28,29]. The small intestine microenvironment is involved in the immunopathogenesis of EAE and MS by promoting Th17 expansion [30]. Expression of CXCL12 MC chemoattractant at lumen surface of endothelial cells facilitates trafficking and accumulation of CXCR4-expressing MCs [31]. Granulocyte–macrophage-colony-stimulating factor (GM-CSF) is involved in neuroinflammation in EAE. Meningeal MCs contribute to myelin-specific T cell accumulation and GM-CSF expression, as T cells neither accumulate in meninges nor produce GM-CSF in the absence of MCs [32].
Elevated levels of tryptase are present in the cerebrospinal fluid of MS patients [33], and can activate peripheral mononuclear cells to secrete TNFα, IL-1β and IL-6 [34] as well as stimulate PARs, that can lead to microvascular leakage and widespread inflammation [35]. Nitric oxide (NO), oxygen radicals, TNFα and proteases have been implicated in neuronal apoptotic cell death [36, 37]. Microarray analysis of MS lesions shows that transcripts encoding tryptase, chymase, histamine, osteopontin, and FcεRI are significantly increased in chronic disease [38,39,40,41,42,43].
Studies in mast cell deficient animal models
MCs depend on c-kit signaling for their growth and long-term survival. In 1978, a MC-deficient mouse (WBX C57 BL/6) F1-W/Wv (WBB6 KitW/Wv) was first described [44], in which ~ 80–90% reduction activity of c-kit signaling was induced. Other MC-deficient mice have since been described, including KitW−sh/W−sh mice, which have a mutation upstream from the Kit promoter that interferes with c-kit expression [45]. Lack of MCs in Kit mutant mice can be selectively repaired by the transfer of genetically compatible wild-type MCs. Cpa3Cre/+ mice contain a transgene encoding Cre-recombinase under the control of the carboxypeptidase 3 (Cpa3) promoter [46]. Cpa3Cre/+ mice are deficient in both mucosal and connective-tissue MCs and has a partial reduction of splenic basophils [46].
These experimental animal models have been used in the study of EAE. In detail, female (in this study were used only female) KitW/Wv develop significantly less severe EAE, in terms of significantly reduced disease incidence, delayed disease onset, and decreased mean clinical scores, than wild-type counterparts [22]; KitW−sh/W−sh mice develop more severe EAE, in terms of the presence of more inflammatory foci in the CNS and increased T cell response against myelin, than wild-type counterparts [47]; no differences in EAE disease severity were observed in side-by-side experiments with wild-type, Cpa3Cre/+ and KitW/Wv mice [46]. These latter authors concluded that “the correlations between the numbers and the distribution of MCs and EAE pathology do not reflect an active role of MCs in this disease” [46].
Overall, even if the results of these studies are not univocal, they support the involvement of MCs in the EAE mouse model of MS. Nevertheless, more studies are needed to exclude that other variables may be involved; for example, it may be that there is a difference between induction of disease effects and other factors involved in disease progression. Moreover, the discrepant results obtained by different groups may be related to different protocols of immunization used to elicit EAE.
Mast cells and angiogenesis
Several lines of evidence indicate that MCs synthesize and release potent angiogenic cytokines, including vascular endothelial growth factor (VEGF), fibroblast growth factor-2 (FGF-2), the serine proteases tryptase and chymase, IL-8, transforming growth factor beta (TGF-β), TNF-α and nerve growth factor (NGF) [48]. MC-secreted molecules facilitate neo-vascularization not only by a direct angiogenic effect, but also by stimulating other inflammatory cells of the microenvironment to release angiogenic mediators and cytokines as well as extracellular matrix-degrading proteases. MCs store in their secretory granules pre-formed active serine proteases, including tryptase and chymase [49]. Tryptase stimulates the proliferation of endothelial cells, promotes vascular tube formation in vitro, is angiogenic in vivo in the chick embryo chorioallantoic membrane (CAM) assay [50] degrades connective-tissue matrix, and activates MMPs and plasminogen activator (PA), which induce the release of VEGF or FGF-2 from their extracellular matrix-bound state [51].
MCs are recruited early in tissue microenvironment and play a critical role in both angiogenesis and tissue remodeling. In turn, MCs recruit eosinophils and neutrophils and activate T and B cell immune responses [52], and MC-derived MMPs degrade the interstitial stroma and, hence, release extracellular matrix-bound angiogenic factors.
Mast cells and angiogenesis in MS
Several evidences indicate a role for angiogenesis in MS [53, 54]. Indeed, increased blood vessel density and endothelial cell proliferation have been demonstrated in MS white matter (WM) [55], elevated VEGF expression has been detected in reactive astrocytes of both active and inactive chronic demyelinated lesions [56, 57] in normal-appearing WM from postmortem MS brains [58, 59] and also in sera of MS patients during clinical relapses [60, 61]. In the late MS phase, VEGF acting as a neuroprotective agent, is decreased in cerebrospinal fluid (CSF) of MS patients and also in peripheral blood mononuclear cells from secondary progressive MS patients [62, 63]. Finally, the first in vivo demonstration of a pro-angiogenic activity of CSF from MS patients has been reported using the chick embryo CAM assay (Fig. 1) [64].

(Reproduced from [64])
12-day-old chick embryo chorioallantoic membranes (CAMs) incubated on day 8 for4 days with gelatin sponges loaded with different CSF samples and50 ng VEGF used as positive control. Note that samples from relapsing–remitting (RR), secondary progressive (SP) and primary progressive (PP) patients induce a strong angiogenic response, inform of new-forming blood vessels radially converging toward the gelatin sponges, comparable to the angiogenic response induced by VEGF, while samples from neurological control (NC) and clinical isolated syndrome (CIS) induce a lower angiogenic response.
Several studies demonstrated that osteopontin (OPN) is highly expressed within MS lesions [65, 66] and significantly high levels of OPN are detected in MS blood and CSF samples [67,68,69]. OPN correlated with disease activity and progression [67], and it was found that high CSF levels of OPN also correlated with disease severity in primary progressive MS [70]. Baseline plasma OPN levels and vessel density were significantly correlated in active RRMS [71]. OPN is produced by a variety of immune cells including macrophages, activated T cells, dendritic cells, and MCs, can induce VEGF [72] or VEGF can induce OPN [73] and both molecules can function in synergy. Natalizumab treatment induces a significant reduction of the pro-angiogenic activity in sera from highly active MS patients tested in the CAM assay, indicating that NTZ could exert its anti-inflammatory properties also by inhibiting the angiogenetic mechanisms in RRMS patients [71].
Therefore, two major roles of the angiogenic response in MS may be hypothesized: early in the disease course, the increase of angiogenesis may allow greater peripheral immune/inflammatory response leading to a worse disease activity; later in the disease course, the increased angiogenesis may represent a rescue attempt to address the hypoperfusion due to the reduction in vascular permeation into the CNS.
Therapeutic strategies
Available anti-MC drugs are restricted to Imatinib and other c-kit-related antibodies, MC stabilizers (sodium cromoglycate or ketotifen), or MC mediator antagonists (anti-histamines, anti-leukotrienes).
The severity of EAE can be reduced by preventing activation of MCs by administration of the MC stabilizer picroximil, the H1 receptor antagonist hydroxyzine, or intracisternal administration of C48/80, a systemic MC degranulating agent, before immunization [74, 75]. Histamine H3 receptor blockade with GSK239512 enhances lesion remyelination in MS patients [76], and hydroxysine, a first generation anti-histamine molecule controlled the initiation and progression of an EAE experimental model [75]. The administration of cladribine, an apoptotic drug that reduces the numbers of MCs, T and B cells, exerts positive effects in both clinical and neuroimaging settings in RRMS [77]. Patients showed significant lower relapse rate, higher relapse free rates, and a significant reduction of brain lesions determined by MRI. Pinke et al. [78], investigated the effect of ketotifen fumarate (Ket), a stabilizer of MC activity and a second-generation anti-histamine substance used in the treatment of allergic disorders, on EAE development and demonstrated that Ket significantly reduced disease prevalence and severity, and restored BBB permeability levels. Ket through MC stabilization may inhibit the release of chemotactic mediators from neutrophils [79].
Long-term treatment of patients with chronic myeloid leukemia with Imatinib (Gleevec), a receptor tyrosine kinase inhibitor, resulted in a reduction of bone marrow MC number to 5% of pre-treatment values [80]. Imatinib blocks MC proliferation in vitro and reduces the severity of EAE through reduction of CNS inflammation, demyelination, T cell recruitment, and enhancement of BBB integrity [81, 82].
MCs generated Sphingosine-1-phosphate (S1P) [83], which plays an important role in MS [84]. The S1P1 modulator FTY720 is currently used as MS-immunosuppressant therapeutic agent potentially able to decrease MC trafficking into CNS as it reduces the number of MCs in the intestinal mucosa and prevents allergic diarrhea in mice [85].
Overall, these evidences indicate that inhibition of the release of MC mediators may be suggested as a therapeutic strategy in the treatment of MS patients. However, while most of these effects responsible of BBB disruption and recruitment of inflammatory cells in the CNS, have been attributed to soluble factors released by MC granules, their contribution to other stages of EAE/MS pathogenesis, should be further clarified. The definition of this matter is fundamental to define how MCs contribute to each of the pathological steps of EAE/MS development to define the most responsive disease stages.
Concluding remarks
Innate immune cells, including neutrophils and macrophages, are involved in initiation and progression of EAE/MS. More recently, an important role in neuroinflammation and neurodegeneration occurring in the course of the disease has been attributed to MCs [54, 86].
MCs play various roles in MS pathogenesis as it has been extensively investigated using the EAE model, influencing the innate immune response in peripheral tissues and in CNS. These cells are activated in early disease and express mediators that affect BBB integrity and recruit T cells for pathogenic activation [8]. In addition to their ability to express many mediators including TNF-α, IL-6, and IL-1β, that promote the pathogenic immune response in MS and EAE, MCs can also directly provoke demyelination in vitro [87]. Increased vascular permeability has been found in the CNS where MC degranulation compromises the BBB and allows further entry of inflammatory substances into the brain [88].
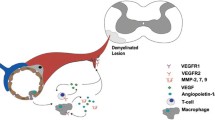
MCs could, therefore, participate in the pathogenesis of MS different ways: they could be stimulated to release cytokines/chemokines inducing inflammatory cells recruitment and activation; disrupt BBB and permit entry of active T cells in brain parenchyma; stimulate angiogenesis (Fig. 2).

Interactions between mast cells and other cells of brain microenvironment involved in brain angiogenesis and alterations of blood–brain barrier permeability
Although angiogenesis is likely not the first event in the pathogenesis of MS, its role in the different phases of disease progression makes it an important target in therapeutic options. The advantages of inhibiting angiogenesis in the early phase of MS are related to reduction of the vascular supply of nutrients and inflammatory cells to the demyelinating lesions, inhibiting the production of endothelial-derived pro-inflammatory molecules. Blocking VEGF signaling and angiogenesis reduced clinical and pathological signs of disease in the early phase in an animal model of MS [89].
Here, we have suggested a potential role of MCs in the regulation of angiogenic processes occurring during MS progression. MCs are considered a potentially useful target for pharmacological strategies in the treatment of MS, through the selective inhibition of angiogenesis and preventing MC-mediated immune suppression.
References
Babbe H, Roers A, Waisman A, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404.
Sospedra M, Martin R. Immunology of multiple sclerosis. Semin Neurol. 2016;36:115–27.
Russi AE, Brown MA. The meninges: new therapeutic targets for multiple sclerosis. Transl Res. 2015;165:255–69.
Dropp JJ. Mast cells in the human brain. Cells Tissues Organs. 1979;105:505–13.
Johnson D, Krenger W. Interactions of mast cells with the nervous system? Recent advances. Neurochem Res. 1992;17:939–51.
Ansari KA, Yokoyama MM, Rand A. Circulating IgE, allergy and multiple sclerosis. Serum levels of IgE, other immunoglobulins and complement (C's) in patients with multiple sclerosis in exacerbation and other neurologic diseases. Acta Neurol Scand. 1976;53:39–50.
Terry RL, Ifergan I, Miller SD. Experimental autoimmune encephalomyelitis in mice. Methods Mol Biol. 2016;1304:145–60.
Brown MA, Weinberg RB. Mast cells and innate lymphoid cells: underappreciated players in CNS autoimmune demyelinating disease. Front Immunol. 2018;9:514.
Kruger PG. Mast cells and multiple sclerosis: a quantitative analysis. Neuropathol Appl Neurobiol. 2001;27:275–80.
Sayed BA, Christy A, Quirion MR, Brown MA. The master switch: the role of mast cells in autoimmunity and tolerance. Annu Rev Immunol. 2008;26:705–39.
Kim DY, Jeoung D, Ro JY. Signaling pathways in the activation of mast cells cocultured with astrocytes and colocalization of both cells in experimental allergic encephalomyelitis. J Immunol. 2010;185:273–83.
Rouleau A, Dimitriadou V, Trung Tuong MD, et al. Mast cell specific proteases in rat brain: changes in rats with experimental allergic encephalomyelitis. J Neural Transm. 1997;104:399–417.
Brenner T, Soffer D, Shalit M, Levi-Schaffer F. Mast cells in experimental allergic encephalomyelitis: characterization, distribution in the CNS and in vitro activation by myelin basic protein and neuropeptides. J Neurol Sci. 1994;122:210–3.
Johnson D, Seeldrayers PA, Weiner HL. The role of mast cells in demyelination. 1. Myelin proteins are degraded by mast cell proteases and myelin basic protein and P2 can stimulate mast cell degranulation. Brain Res. 1988;444:195–8.
Kempuraj D, Tagen M, Iliopoulou BP, et al. Luteolin inhibits myelin basic protein-induced human mast cell activation and mast cell-dependent stimulation of Jurkat T cells. Br J Pharmacol. 2008;155:1076–84.
Sayed BA, Christy AL, Walker ME, Brown MA. Meningeal mast cells affect early T cell central nervous system infiltration and blood-brain barrier integrity through TNF: a role for neutrophil recruitment? J Immunol. 2010;184:6891–900.
Marshall JS. Mast-cell responses to pathogens. Nat Rev Immunol. 2004;4:787–99.
Zappulla JP, Arock M, Mars LT, Liblau RS. Mast cells: new targets for multiple sclerosis therapy? J Neuroimmunol. 2002;131:5–20.
Cocchiara R, Bongiovanni A, Albeggiani G, Azzolina A, Geraci D. Evidence that brain mast cells can modulate neuroinflammatory responses by tumour necrosis factor-α production. NeuroReport. 1998;9:95–8.
Probert L, Selmaj K. TNF and related molecules: trends in neuroscience and clinical applications. J Neuroimmunol. 1997;72:113 7 (1This is a Meeting report from the 6th International TNF Congress: TNF and Related Molecules, Scientific Trends and Clinical Applications. Abstracts are published in European Cytokine Network 7 (1996) 143–346.1).
Rieckmann P, Albrecht M, Kitze B, et al. Cytokine mRNA levels in mononuclear blood cells from patients with multiple sclerosis. Neurology. 1994;44:1523.
Secor VH, Secor WE, Gutekunst CA, Brown MA. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J Exp Med. 2000;191:813–22.
Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14:164–74.
Gregory GD, Raju SS, Winandy S, Brown MA. Mast cell IL-4 expression is regulated by Ikaros and influences encephalitogenic Th1 responses in EAE. J Clin Invest. 2006;116:1327–36.
Gregory GD, Robbie-Ryan M, Secor VH, Sabatino JJ, Brown MA. Mast cells are required for optimal autoreactive T cell responses in a murine model of multiple sclerosis. Eur J Immunol. 2005;35:3478–86.
Codarri L, Gyülvészi G, Tosevski V, et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–7.
Dudeck A, Suender CA, Kostka SL, von Stebut E, Maurer M. Mast cells promote Th1 and Th17 responses by modulating dendritic cell maturation and function. Eur J Immunol. 2011;41:1883–93.
El-Behi M, Ciric B, Dai H, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–75.
Ganeshan K, Bryce PJ. Regulatory T cells enhance mast cell production of IL-6 via surface-bound TGF-β. J Immunol. 2012;188:594–603.
Cosorich I, Dalla-Costa G, Sorini C, et al. High frequency of intestinal TH17 cells correlates with microbiota alterations and disease activity in multiple sclerosis. Sci Adv. 2017;3:e1700492.
McCandless EE, Piccio L, Woerner BM, et al. Pathological expression of CXCL12 at the blood-brain barrier correlates with severity of multiple sclerosis. Am J pathol. 2008;172:799–808.
Russi AE, Walker-Caulfield ME, Guo Y, Lucchinetti CF, Brown MA. Meningeal mast cell-T cell crosstalk regulates T cell encephalitogenicity. J Autoimmun. 2016;73:100–10.
Rozniecki JJ, Hauser SL, Stein M, Lincoln R, Theoharides TC. Elevated mast cell tryptase in cerebrospinal fluid of multiple sclerosis patients. Ann Neurol. 1995;37:63–6.
Malamud V, Vaaknin A, Abramsky O, et al. Tryptase activates peripheral blood mononuclear cells causing the synthesis and release of TNF-α, IL-6 and IL-1β: possible relevance to multiple sclerosis. J Neuroimmunol. 2003;138:115–22.
Bunnett N. Protease-activated receptors: how proteases signal to cells to cause inflammation and pain. Semin Thromb Hemost. 2006;32:039–48.
Bjartmar C, Wujek JR, Trapp BD. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci. 2003;206:165–71.
Steinman MDL. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell. 1996;85:299–302.
Bomprezzi R, Ringnér M, Kim S, et al. Gene expression profile in multiple sclerosis patients and healthy controls: identifying pathways relevant to disease. Hum Mol Genet. 2003;12:2191–9.
Couturier N, Zappulla JP, Lauwers-Cances V, et al. Mast cell transcripts are increased within and outside multiple sclerosis lesions. J Neuroimmunol. 2008;195:176–85.
Kallweit U, Aritake K, Bassetti CL, et al. Elevated CSF histamine levels in multiple sclerosis patients. Fluids Barriers CNS. 2013;10:19.
Steinman L. A molecular trio in relapse and remission in multiple sclerosis. Nat Rev Immunol. 2009;9:440–7.
Tuomisto L, Kilpeläinen H, Riekkinen P. Histamine and histamine-N-methyltransferase in the CSF of patients with multiple sclerosis. Agents Actions. 1983;13:255–7.
Rafiee Zadeh A, Falahatian M, Alsahebfosoul F. Serum levels of histamine and diamine oxidase in multiple sclerosis. Am J Clin Exp Immunol. 2018;7:100–5.
Kitamura Y, Go S, Hatanaka K. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood. 1978;52:447–52.
Lyon MF, Glenister PH. A new allele sash (Wsh) at the W-locus and a spontaneous recessive lethal in mice. Genet Res. 1982;39:315–22.
Feyerabend TB, Weiser A, Tietz A, et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity. 2011;35:832–44.
Piconese S, Costanza M, Musio S, et al. Exacerbated experimental autoimmune encephalomyelitis in mast-cell-deficient Kit W-sh/W-sh mice. Lab Invest. 2011;91:627–41.
Ribatti D, Crivellato E. Mast Cells and Tumours. Netherlands: Springer; 2011.
Metcalfe DD, Baram D, Mekori YA. Mast cells. Physiol Rev. 1997;77:1033–79.
Ribatti D, Ranieri G, Nico B, Benagiano V, Crivellato E. Tryptase and chymase are angiogenic in vivo in the chorioallantoic membrane assay. Int J Dev Biol. 2011;55:99–102.
Blair RJ, Meng H, Marchese MJ, et al. Human mast cells stimulate vascular tube formation. Tryptase is a novel, potent angiogenic factor. J Clin Invest. 1997;99:2691–700.
Kinet J-P. The essential role of mast cells in orchestrating inflammation. Immunol Rev. 2007;217:5–7.
Claudio L, Raine CS, Brosnan CF. Evidence of persistent blood-brain barrier abnormalities in chronic-progressive multiple sclerosis. Acta Neuropathol. 1995;90:228–38.
Girolamo F, Coppola C, Ribatti D, Trojano M. Angiogenesis in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol Commun. 2014;2:84.
Kirk J, Plumb J, Mirakhur M, McQuaid S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood-brain barrier leakage and active demyelination. J Pathol. 2003;201:319–27.
Proescholdt MA, Jacobson S, Tresser N, Oldfield EH, Merrill MJ. Vascular endothelial growth factor is expressed in multiple sclerosis plaques and can induce inflammatory lesions in experimental allergic encephalomyelitis rats. J Neuropathol Exp Neurol. 2002;61:914–25.
van Horssen J, Bö L, Vos CMP, Virtanen I, de Vries HE. Basement membrane proteins in multiple sclerosis-associated inflammatory cuffs: potential role in influx and transport of leukocytes. J Neuropathol Exp Neurol. 2005;64:722–9.
Alvarez JI, Cayrol R, Prat A. Disruption of central nervous system barriers in multiple sclerosis. Biochim Biophys Acta. 2011;1812:252–64.
Graumann U, Reynolds R, Steck AJ, Schaeren-Wiemers N. Molecular changes in normal appearing white matter in multiple sclerosis are characteristic of neuroprotective mechanisms against hypoxic insult. Brain Pathol. 2006;13:554–73.
Clauss M, Gerlach M, Gerlach H, et al. Vascular permeability factor: a tumor-derived polypeptide that induces endothelial cell and monocyte procoagulant activity, and promotes monocyte migration. J Exp Med. 1990;172:1535–45.
Theoharides TC. Corticotropin-releasing hormone and the blood-brain-barrier. Front Biosci. 2007;12:1615.
Iacobaeus E, Amoudruz P, Strom M, et al. The expression of VEGF-A is down regulated in peripheral blood mononuclear cells of patients with secondary progressive multiple sclerosis. PLoS ONE. 2011;6:e19138.
Tham E, Gielen AW, Khademi M, Martin C, Piehl F. Decreased expression of VEGF-A in rat experimental autoimmune encephalomyelitis and in cerebrospinal fluid mononuclear cells from patients with multiple sclerosis. Scand J Immunol. 2006;64:609–22.
Ribatti D, Iaffaldano P, Marinaccio C, Trojano M. First evidence of in vivo pro-angiogenic activity of cerebrospinal fluid samples from multiple sclerosis patients. Clin Exp Med. 2016;16:103–7.
Chabas D. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science. 2001;294:1731–5.
Sinclair C, Mirakhur M, Kirk J, Farrell M, McQuaid S. Up-regulation of osteopontin and alphaBeta-crystallin in the normal-appearing white matter of multiple sclerosis: an immunohistochemical study utilizing tissue microarrays. Neuropathol Appl Neurobiol. 2005;31:292–303.
Comabella M, Pericot I, Goertsches R, et al. Plasma osteopontin levels in multiple sclerosis. J Neuroimmunol. 2005;158:231–9.
Iaffaldano P, Ruggieri M, Viterbo RG, Mastrapasqua M, Trojano M. The improvement of cognitive functions is associated with a decrease of plasma Osteopontin levels in Natalizumab treated relapsing multiple sclerosis. Brain Behav Immun. 2014;35:176–81.
Vogt MHJ, Lopatinskaya L, Smits M, Polman CH, Nagelkerken L. Elevated osteopontin levels in active relapsing-remitting multiple sclerosis. Ann Neurol. 2003;53:819–22.
Börnsen L, Khademi M, Olsson T, Sørensen PS, Sellebjerg F. Osteopontin concentrations are increased in cerebrospinal fluid during attacks of multiple sclerosis. Mult Scler. 2010;17:32–42.
Iaffaldano P, Ribatti D, Trojano M. Natalizumab reduces serum pro-angiogenic activity in MS patients. Neurol Sci. 2018;39:725–31.
Chakraborty G, Jain S, Kundu GC. Osteopontin promotes vascular endothelial growth factor dependent breast tumor growth and angiogenesis via autocrine and paracrine mechanisms. Cancer Res. 2008;68:152–61.
Li X-D, Chen J, Ruan C-C, Zhu D-L, Gao P-J. Vascular endothelial growth factor-induced osteopontin expression mediates vascular inflammation and neointima formation via Flt-1 in adventitial fibroblasts. Arterioscler Thromb Vasc Biol. 2012;32:2250–8.
Brown MA, Hatfield JK. Mast Cells are important modifiers of autoimmune disease: with so much evidence, why is there still controversy? Front Immunol. 2012;3:147.
Dimitriadou V, Pang X, Theoharides TC. Hydroxyzine inhibits experimental allergic encephalomyelitis (EAE) and associated brain mast cell activation. Int J Immunopharmacol. 2000;22:673–84.
Schwartzbach CJ, Grove RA, Brown R, Tompson D, Then Bergh F, Arnold DL. Lesion remyelinating activity of GSK239512 versus placebo in patients with relapsing-remitting multiple sclerosis: a randomised, single-blind, phase II study. J Neurol. 2017;264:304–15.
Giovannoni G, Comi G, Cook S, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362:416–26.
Pinke KH, Zorzella-Pezavento SFG, de Campos Fraga-Silva TF, et al. Calming down mast cells with ketotifen: a potential strategy for multiple sclerosis therapy? Neurotherapeutics. 2020;17:218–34.
Christy AL, Walker ME, Hessner MJ, Brown MA. Mast cell activation and neutrophil recruitment promotes early and robust inflammation in the meninges in EAE. J Autoimmun. 2013;42:50–61.
Cerny-Reiterer S, Rabenhorst A, Stefanzl G, et al. Long-term treatment with imatinib results in profound mast cell deficiency in Ph+ chronic myeloid leukemia. Oncotarget. 2015;6:3071–84.
Azizi G, Mirshafiey A. Imatinib mesylate: an innovation in treatment of autoimmune diseases. Recent Pat Inflamm Allergy Drug Discov. 2013;7:259–67.
Crespo O, Kang SC, Daneman R, et al. Tyrosine kinase inhibitors ameliorate autoimmune encephalomyelitis in a mouse model of multiple sclerosis. J Clin Immunol. 2011;31:1010–20.
Kulinski JM, Munoz-Cano R, Olivera A. Sphingosine-1-phosphate and other lipid mediators generated by mast cells as critical players in allergy and mast cell function. Eur J Pharmacol. 2016;778:56–67.
Halmer R, Walter S, Faßbender K. Sphingolipids: important players in multiple sclerosis. Cell Physiol Biochem. 2014;34:111–8.
Kurashima Y, Kunisawa J, Higuchi M, et al. Sphingosine 1-phosphate-mediated trafficking of pathogenic Th2 and mast cells for the control of food allergy. J Immunol. 2007;179:1577–85.
Girolamo F, Coppola C, Ribatti D. Immunoregulatory effect of mast cells influenced by microbes in neurodegenerative diseases. Brain Behav Immun. 2017;65:68–89.
Theoharides TC, Dimitriadou V, Letourneau R, Rozniecki JJ, Vliagoftis H, Boucher W. Synergistic action of estradiol and myelin basic protein on mast cell secretion and brain myelin changes resembling early stages of demyelination. Neuroscience. 1993;57:861–71.
Zhuang X, Silverman AJ, Silver R. Brain mast cell degranulation regulates blood-brain barrier. J Neurobiol. 1996;31:393–403.
MacMillan CJ, Furlong SJ, Doucette CD, Chen PL, Hoskin DW, Easton AS. Bevacizumab diminishes experimental autoimmune encephalomyelitis by inhibiting spinal cord angiogenesis and reducing peripheral T-cell responses. J Neuropathol Exp Neurol. 2012;71:983–99.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ribatti, D., Tamma, R. & Annese, T. Mast cells and angiogenesis in multiple sclerosis. Inflamm. Res. 69, 1103–1110 (2020). https://doi.org/10.1007/s00011-020-01394-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-020-01394-2