
Abstract
Objective
Porphyromonas gingivalis is involved in the pathogenesis of chronic inflammatory periodontal disease. Recent studies have suggested that the NLRP3 inflammasome plays an important role in the development of chronic inflammation. We investigated a possible association between the inflammasome in gingival inflammation and bone loss induced by P. gingivalis infection using NLRP3-deficient mice.
Methods
Wild-type and NLRP3-deficient mice were injected orally with P. gingivalis. We assessed alveolar bone loss, expression of pro-interleukin (IL)-1β, pro-IL-18, receptor activator of nuclear factor kappa-B ligand (RANKL), and osteoprotegerin (OPG) in gingival tissue, as well as IL-1β, IL-18, and IL-6 production and caspase-1 activity in peritoneal macrophages.
Results
Porphyromonas gingivalis challenge significantly increased alveolar bone loss; gingival gene expression of pro-IL-1β, pro-IL-18, and RANKL; production of IL-1β, IL-18, and IL-6; and caspase-1 activity in peritoneal macrophages of wild-type mice, but did not affect NLRP3-deficient mice. Meanwhile, OPG mRNA expression in gingival tissue and peritoneal IL-6 production were significantly higher in NLRP3-knockout mice.
Conclusions
Porphyromonas gingivalis activated innate immune cells via the NLRP3 inflammasome. These results suggest that the NLRP3 inflammasome, followed by a response from the IL-1 family, is critical in periodontal disease induced by wild-type P. gingivalis challenge via sustained inflammation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Periodontal disease, a chronic, destructive, and inflammatory condition that affects many adults, is a major cause of tooth loss and is characterized by chronic infection associated with Gram-negative anaerobic bacteria in the dental biofilm. It leads to the irreversible destruction of tissues supporting the teeth and is clinically detectable by periodontal pockets and alveolar bone loss [1, 2]. Because periodontitis often causes bacteremia, a relationship between periodontitis and systemic diseases caused by periodontal pathogens has been explored [3].
Periodontitis is a multifactorial disease, and the genetic background of patients as well as the presence of pathogenic bacteria and immune mechanisms are important elements. During the course of inflammation in response to dental plaque bacterial species, gingival fibroblasts produce interleukin (IL)-1, IL-6, IL-8, tumor necrosis factor-alpha (TNF-α), and transforming growth factor-beta (TGF-β) [4–6]. Although cytokine secretion at inflammatory sites is initially protective in eliminating infectious bacteria, excessive or sustained proinflammatory cytokine production is related to periodontal destruction, with periodontal attachment loss and alveolar bone resorption triggered by the receptor activator of nuclear factor kappa-B ligand (RANKL)/osteoprotegerin (OPG) signaling pathway. Cytokine concentrations in gingival crevicular fluid (GCF) and saliva are higher in patients with aggressive periodontitis than in healthy subjects, and decrease after periodontal treatment [7, 8]. Macrophages represent an important source of pro-inflammatory cytokines, including IL-1β and TNF-α; under dysregulation, these cytokines contribute to the destruction of host tissue. Therefore, understanding the mechanisms of periodontal pathogen-induced immune cell signaling could provide useful information for the prevention and treatment of periodontitis.
In this respect, the NLRP3 inflammasome may play an important role in IL-1β production in response to several bacterial ligands, including lipopolysaccharides (LPSs), peptidoglycan, and bacterial RNA [9–12]. Peripheral blood mononuclear cell-derived macrophages reportedly express NALP3 mRNA [13] to release proinflammatory IL-1β, which is highly induced by bacterial LPS [14]. In contrast, NALP3-deficient macrophages do not produce active IL-1β in response to bacterial infection [9, 10].
Porphyromonas gingivalis, an anaerobic, Gram-negative bacterium, is a periodontal pathogen that possesses an array of virulence factors involved in the pathogenesis of chronic periodontitis [15]. Recent evidence suggests that it contributes to periodontitis by functioning as a keystone pathogen [7, 8]. Porphyromonas gingivalis virulence factors, such as LPSs, hemagglutinins, gingipains, and fimbriae, are important in the induction of immune responses, including inflammatory cytokine production, such as IL-1, and inflammation-related signaling pathway activation [15]. Moreover, IL-1 mediates periodontal tissue destruction by stimulating alveolar bone resorption. However, the mechanism by which P. gingivalis participates in this inflammatory response via IL-1 is unknown. Therefore, we examined the effects of the NLRP3 inflammasome on inflammatory response and bone metabolism accelerated by P. gingivalis infection using inflammasome-deficient mice.
Materials and methods
Bacterial strain
A wild-type (WT) P. gingivalis strain (ATCC 33,277) was cultured on anaerobic blood agar plates (Becton–Dickinson, Sunnyvale, CA) in a model 1024 anaerobic system (Forma Scientific, Marietta, OH) with 10 % H2, 80 % N2, and 10 % CO2 for 3–5 days. Cultures were inoculated into brain heart infusion broth (Difco Laboratories, Detroit, MI) supplemented with 5 μg/mL of hemin and 0.4 μg/mL of menadione and grown for 2 days until reaching an optical density of 0.8 at 660 nm, corresponding to 109 colony forming units (CFU)/mL. Cultured cells were centrifuged at 8000 × g for 15 minutes at 4 °C and diluted with 5 % carboxymethyl cellulose (CMC) for oral infection.
Mice and oral infection
Eight-week-old male C57BL/6 mice (WT), obtained from Japan SLC, Inc. (Hamamatsu, Japan), and eight-week-old male C57BL/6 background NLRP3 knockout (KO) mice [16] were maintained in an experimental facility under pathogen-free conditions and fed a regular diet and water ad libitum (Fig. 1). At week ten, mice received sulfamethoxazole and trimethoprim (final concentrations, 1 mg/mL and 200 μg/mL, respectively) in water bottles ad libitum for 7 days. After 2 days, the WT and NLRP3 KO mice were divided randomly into two groups (n = 6 per group). Each group was orally challenged with 0.1 mL of 5 % CMC or live P. gingivalis (109 CFU/mouse) five times per week for 3 weeks. At 2 days and 4 weeks after the final infection, the mice were euthanized by intraperitoneal injection of Somnopentyl (Kyoritsu Seiyaku, Tokyo, Japan) and bone and tissue samples were collected. All mice were housed in temperature- and humidity-controlled clean racks with a 12-hour light/dark cycle. All mice were maintained in a barriered animal facility under pathogen-free conditions, monitored daily until sacrifice, and appeared to be healthy throughout the course of the experiment. The Institutional Animal Care and Use Committee of Nihon University approved all animal protocols.

Schematic of the experimental procedure. Eleven-week-old male C57/BL6 and NLRP3 KO mice were randomly divided into two groups: C57/BL6 (group 1) and NLRP3 KO (group 2) mice were challenged with 100 µL of CMC. C57/BL6 (group 3) and NLRP3 KO (group 4) mice were challenged with 100 µL (109 CFU) of WT P. gingivalis. Animals were challenged orally with CMC or P. gingivalis five times per week for 3 weeks and sacrificed 4 weeks after the final challenge
Quantification of alveolar bone loss
Horizontal bone loss around the maxillary molars was assessed using a morphometric method, as described previously [17]. Briefly, after removing gingival tissue, skulls were immersed overnight in 3 % hydrogen peroxide, pulsed for 1 minute in bleach, and stained with 1 % methylene blue. The distance from the cementoenamel junction (CEJ) to the alveolar bone crest (ABC) was measured at 28 buccal sites per mouse [18]. Measurements were taken under a dissecting microscope (50 × magnification) fitted with a video image marker measurement system (VHX-100; KEYENCE, Osaka, Japan), and standardized to give measurements in micrometers. Bone measurements were performed in triplicate by two evaluators using a random and blinded protocol.
Analysis of gene expression by quantitative real-time reverse transcriptase PCR
Total RNA was isolated from gingival tissue (n = 6 per group) using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) and reverse-transcribed with oligo(dT) primers using SuperScript reverse transcriptase (Invitrogen, Carlsbad, CA, USA) to generate complementary DNA. Quantitative real-time reverse transcriptase (RT)-PCR analyses were performed using a Thermal Cycler Dice real-time PCR system (Takara Bio Inc., Otsu, Japan) in accordance with the manufacturer’s protocol. The PCR consisted of an initial denaturation step at 95 °C for 10 minutes, followed by 40 cycles at 95 °C for 15 s, 55 °C for 25 s, and 60 °C for 35 s. The primer sequences were as follows: Pro-IL1β forward (5′-GCGGAGCAGACACACTTACA-3′) and reverse (5′-TTGGCAAACTCCACCACATA-3′); Pro-IL-18 forward (5′-TTCCCATTCCTGTCCTTCAC-3′) and reverse (5′-CCTCTGGCTCTGCATCATTC-3′); RANKL forward (5′-AGTGCCTGTGTCCTCAATCGTC-3′) and reverse (5′-AGGGCCAGCATAGGTGCAAG-3′); OPG forward (5′-TCATGAAGTGTGACGTTGACATCCGT-3′) and reverse (5′-CCTAGAAGCATTTGCGGTGCACGATG-3′); and GAPDH forward (5′-TCATGAAGTGTGACGTTGACATCCGT-3′) and reverse (5′-CCTAGAAGCATTTGCGGTGCACGATG-3′). PCR was performed in duplicate for each gene. Target mRNA levels were normalized to that of GAPDH.
Peritoneal infection and cytokine and caspase-1 assays
After the 3-week oral challenge, the mice were infected intraperitoneally with live P. gingivalis (5 × 107 CFU/mouse) and peritoneal exudate cells were harvested after 2 hours. The peritoneal exudate cells were washed with RPMI 1640 medium, resuspended at a density of 1 × 106 cells/mL in RPMI 1640 containing penicillin and 5 % fetal bovine serum (Gibco supplied by Invitrogen Co., Carlsbad, CA, USA), and stimulated with live P. gingivalis at a multiplicity of infection of 10:1 for 16 hours. IL-1β (Thermo Scientific Co., Rockford, IL, USA), IL-18 (MBL, Nagoya, Japan), and IL-6 (R&D Systems Inc., Minneapolis, MN, USA) levels were measured in the culture supernatant. Caspase-1 activity in the cytoplasmic fraction was measured using a fluorometric caspase-1 assay kit (Abcam Inc., Cambridge, MA, USA). Levels of released 7-amino-4-trifluoromethylcoumarin (AFC) were measured with a micro-plate reader (ARVO MX 1420 ARVO series Multilabel Counter, Perkin-Elmer, Yokohama, Kanagawa, Japan) with excitation at 400 nm and emission at 505 nm.
Statistical analysis
Data are presented as the mean ± standard deviation (SD). Differences in total atherosclerotic plaque accumulation were assessed with a one-way analysis of variance (ANOVA) followed by the Tukey–Kramer multiple comparison test. P-values <0.05 were considered to indicate statistical significance.
Results
The inflammasome is involved in P. gingivalis-induced alveolar bone loss
Since the effect of inflammation on bone loss is mediated by pro-inflammatory cytokines (6), we examined the participation of the inflammasome in P. gingivalis-induced alveolar bone loss. At 18 weeks, alveolar bone loss was significantly higher in P. gingivalis-challenged C57BL/6 mice compared with the CMC-treated control mice (Fig. 2a, b; p < 0.01). In contrast, the increased levels of bone loss induced by P. gingivalis were ameliorated significantly in NLRP3-KO mice (Fig. 2a, b; p < 0.01). Furthermore, the knockdown of NLRP3 without the infection did not influence bone resorption (Fig. 2a, b).

Porphyromonas gingivalis-induced alveolar bone loss and the effect of the NALP3 inflammasome on bone loss. (A and B) C57/BL6 and NLRP3-KO mice were treated with CMC or 109 CFU of P. gingivalis as described in the Materials and Methods. The distance from the CEJ to the ABC at fourteen predetermined sites in the unfleshed maxilla were measured and summed for each mouse. Results are expressed as the means ± SD (n = 6). ## p < 0.01
Porphyromonas gingivalis-challenge downregulates the expression of pro-inflammatory- and bone resorption-related factors in NLRP3-KO mice gingival tissue
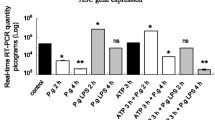
Since the NLRP3–caspase-1 inflammasome promotes maturation of the inflammatory cytokines IL-1β and IL-18, and IL-1β-promotes RANKL followed by periodontal destruction, we investigated whether the NLRP3 inflammasome is also involved in P. gingivalis-induced IL-1β, IL-18, and RANKL expression. At 14 weeks, pro-IL-1β, pro-IL-18, and RANKL mRNA levels were significantly higher in the gingival tissue of P. gingivalis-challenged C57/BL6 mice compared with that of CMC-treated control mice (Fig. 3; p < 0.05). In contrast, the increase in pro-IL-1β, pro-IL-18, and RANKL mRNA levels was decreased significantly in NLRP3 KO mice challenged with P. gingivalis (Fig. 3; p < 0.05). Conversely, OPG expression, an inhibitor of osteoclast development via RANKL, was unaffected in P. gingivalis-challenged C57/BL6 mice compared with CMC-treated control mice, but a remarkable increase in OPG expression was observed in NLRP3 KO mice challenged with P. gingivalis (Fig. 3; p < 0.05).

Expression of pro-inflammatory- and bone resorption-related molecules in mouse gingival tissue following P. gingivalis challenge. Relative mRNA levels normalized to GAPDH were determined with real-time RT-PCR. Data are expressed as fold-increases in mRNA levels compared to sham-inoculated negative controls. Results are expressed as the means ± SD (n = 6). ## p < 0.01, # p < 0.05
Porphyromonas gingivalis challenge downregulates inflammasome-related factor secretion in peritoneal macrophages of NLRP3-KO mice
Compared with CMC-treated C57/BL6 mice, peritoneal macrophages from P. gingivalis-challenged C57/BL6 mice secreted more IL-1β (Fig. 4; P. gingivalis, 446.7 ± 33.3 pg/mL, CMC, 18.3 ± 3.3 pg/mL; p < 0.01), IL-18 (Fig. 4; P. gingivalis, 10.5 ± 0.9 pg/mL, CMC, 0.4 ± 1.6 pg/mL; p < 0.05), and IL-6 (Fig. 4; P. gingivalis, 1302 ± 58.5 pg/mL, CMC, 618.3 ± 36.3 pg/mL; p < 0.01) and had higher caspase-1 levels (Fig. 4; P. gingivalis, 1750.3 ± 98.5 AFC, CMC, 250.7 ± 10.1 AFC; p < 0.01). In contrast, P. gingivalis-challenged NLRP3-KO mice exhibited markedly lower IL-1β and IL-18 production and caspase-1 activity compared with P. gingivalis-challenged C57/BL6 mice (p < 0.01; Fig. 4).

IL-1β, IL-18, and IL-6 production and caspase-1 activity in mouse macrophages following P. gingivalis challenge. Macrophages from P. gingivalis-challenged mice were exposed to live P. gingivalis. IL-1β, IL-18, and IL-6 levels in culture supernatant were analyzed with ELISAs. Caspase-1 activity in the cytoplasmic fraction was measured using a fluorometric caspase-1 assay kit. The data are expressed as the mean ± SD (n = 6). ## p < 0.01, # p < 0.05
IL-6 secretion was markedly higher in NLRP3 KO mice challenged with P. gingivalis (Fig. 4; p < 0.01).
Discussion
We found that oral challenge with P. gingivalis accelerated alveolar bone resorption in C57/BL6 mice, which did not affect bone loss in NLRP3 KO mice. Furthermore, although P. gingivalis-challenged mice had significantly higher gingival pro-IL-1β, pro-IL-18, and RANKL mRNA; IL-1β and IL-18 protein; and caspase-1 AFC levels in peritoneal macrophages compared with the CMC-treated controls, these increases were suppressed significantly in the P. gingivalis-challenged NLRP3 KO mice. These results suggest that the NLRP3 inflammasome is involved in bone metabolism and resorption induced by P. gingivalis infection.
In complex tissues such as periodontal tissue, innate signals can originate from several sources and promote periodontitis in association with pattern-recognition receptors (PRRs). These signals include various extracellular signaling pathways and facilitate infectious agent clearance and inflammatory response induction [19]. PRRs are important in the recognition of pathogen- and danger-associated molecular patterns. Subsets of PRRs belonging to the NOD-like receptor (NLR) family detect molecular patterns in the cytosol and activate the formation of a multi-protein signaling platform, termed the inflammasome. The NLRP3 inflammasome, which is the best-characterized member of the inflammasome family, is a multi-protein complex that induces IL-1β and IL-18 inflammatory cytokine maturation by activating caspase-1 [20]. Periodontal diseases have been associated with increased bone resorption, and various studies support the relationship between inflammatory cytokines and RANKL-stimulated osteoclast activity [21]. IL-1β and IL-18 are strong stimulators of in vivo and in vitro bone resorption via RANKL upregulation that stimulate osteoclastogenesis [22, 23]. Meanwhile, OPG inhibits osteoclast differentiation by binding to RANKL [24]. NLRP3 proteins are upregulated in gingival tissues from periodontitis patients [12].
Recently, inflammasome-independent sources of IL-1β have been suggested to contribute to the pathogenesis of inflammatory disease [25, 26]. Therefore, we evaluated the involvement of the NLRP3 inflammasome in P. gingivalis-induced bone loss using NLRP3-KO mice. Alveolar bone loss induced by infection was suppressed significantly in NLRP3-KO mice, suggesting that the production of inflammatory cytokines mediated by the NLRP3 inflammasome is important for P. gingivalis-induced bone loss. Conversely, gingival OPG expression was significantly higher in P. gingivalis-challenged NLRP3 KO mice, although it was not influenced by P. gingivalis infection in the WT mice. This confirmed observations that OPG levels in the GCF were lower in periodontitis patients than gingivitis patients [27]. Recent research has also indicated that OPG gene therapy prevents periodontitis-induced bone loss [28]. Therefore, knockdown of NLRP3 may be involved in preventing bone resorption by suppressing the production of inflammasome-related inflammatory cytokines and by activating OPG. Knockdown of NLRP3 did not influence IL-6 production in P. gingivalis-induced macrophages. A previous report indicated that IL-6 is an inflammatory marker, but does not directly participate with inflammasomes [29]. Since IL-6 production was higher in the NLRP3-KO mice than the WT mice, NLRP3 may control IL-6 production due to P. gingivalis exposure, although more evidence is required to clarify this possibility. We also tried to detect IL-1β, IL-6, IL-18 and Caspase-1 in gingival tissue at protein levels, but all values were lower than detection limit. It is thought that there is considerably less number of gingival macrophages than macrophages existing in peritoneal exudate cell, otherwise, there was a time-lag between gene expression and protein secretion. We would like to inspect them more in future.
In summary, this study provides the first evidence that P. gingivalis promotes periodontal disease through the inflammatory response mediated by NLRP3 inflammasome activation. Furthermore, our data suggest that NLRP3 inflammasome activation is involved in bone metabolism after P. gingivalis challenge.
References
Loesche WJ, Grossman NS. Periodontal disease as a specific, albeit chronic, infection: diagnosis and treatment. Clin Microbiol Rev. 2001;14(4):727–52.
Burt B, Research S. Therapy Committee of the American Academy of P. Position paper: epidemiology of periodontal diseases. J Periodontol. 2005;76(8):1406–19.
Olsen I. Update on bacteraemia related to dental procedures. Transfus Apher Sci. 2008;39(2):173–8.
Imatani T, Kato T, Okuda K. Production of inflammatory cytokines by human gingival fibroblasts stimulated by cell-surface preparations of Porphyromonas gingivalis. Oral Microbiol Immunol. 2001;16(2):65–72.
Morandini AC, Sipert CR, Ramos-Junior ES, Brozoski DT, Santos CF. Periodontal ligament and gingival fibroblasts participate in the production of TGF-beta, interleukin (IL)-8 and IL-10. Braz Oral Res. 2011;25(2):157–62.
Yucel-Lindberg T, Brunius G. Epidermal growth factor synergistically enhances interleukin-8 production in human gingival fibroblasts stimulated with interleukin-1beta. Arch Oral Biol. 2006;51(10):892–8.
Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 2011;10(5):497–506.
Darveau RP, Hajishengallis G, Curtis MA. Porphyromonas gingivalis as a potential community activist for disease. J Dent Res. 2012;91(9):816–20.
Becker CE, O’Neill LA. Inflammasomes in inflammatory disorders: the role of TLRs and their interactions with NLRs. Semin Immunopathol. 2007;29(3):239–48.
Kummer JA, Broekhuizen R, Everett H, Agostini L, Kuijk L, Martinon F, van Bruggen R, Tschopp J. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem. 2007;55(5):443–52.
Pontillo A, Girardelli M, Agostinis C, Masat E, Bulla R, Crovella S. Bacterial LPS differently modulates inflammasome gene expression and IL-1beta secretion in trophoblast cells, decidual stromal cells, and decidual endothelial cells. Reprod Sci. 2013;20(5):563–6.
Park E, Na HS, Song YR, Shin SY, Kim YM, Chung J. Activation of NLRP3 and AIM2 inflammasomes by Porphyromonas gingivalis infection. Infect Immun. 2014;82(1):112–23.
Zheng F, Xing S, Gong Z, Xing Q. NLRP3 inflammasomes show high expression in aorta of patients with atherosclerosis. Heart Lung Circ. 2013;22(9):746–50.
Yamaguchi Y, Kurita-Ochiai T, Kobayashi R, Suzuki T, Ando T. Activation of the NLRP3 inflammasome in Porphyromonas gingivalis-accelerated atherosclerosis. Pathog Dis. 2015;73(4). doi:10.1093/femspd/ftv011.
Bostanci N, Belibasakis GN. Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol Lett. 2012;333(1):1–9.
Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440(7081):233–6.
Klausen B, Evans RT, Sfintescu C. Two complementary methods of assessing periodontal bone level in rats. Scand J Dent Res. 1989;97(6):494–9.
Kobayashi R, Kono T, Bolerjack BA, Fukuyama Y, Gilbert RS, Fujihashi K, Ruby J, Kataoka K, Wada M, Yamamoto M, et al. Induction of IL-10-producing CD4+ T-cells in chronic periodontitis. J Dent Res. 2011;90(5):653–8.
Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22(2):240–73.
Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411.
Cochran DL. Inflammation and bone loss in periodontal disease. J Periodontol. 2008;79(8 Suppl):1569–76.
Ruscitti P, Cipriani P, Carubbi F, Liakouli V, Zazzeroni F, Di Benedetto P, Berardicurti O, Alesse E, Giacomelli R. The role of IL-1beta in the bone loss during rheumatic diseases. Mediators Inflamm. 2015;2015:782382.
Dai SM, Nishioka K, Yudoh K. Interleukin (IL) 18 stimulates osteoclast formation through synovial T cells in rheumatoid arthritis: comparison with IL1 beta and tumour necrosis factor alpha. Ann Rheum Dis. 2004;63(11):1379–86.
Polzer K, Joosten L, Gasser J, Distler JH, Ruiz G, Baum W, Redlich K, Bobacz K, Smolen JS, van den Berg W, et al. Interleukin-1 is essential for systemic inflammatory bone loss. Ann Rheum Dis. 2010;69(1):284–90.
Netea MG, van de Veerdonk FL, van der Meer JW, Dinarello CA, Joosten LA. Inflammasome-independent regulation of IL-1-family cytokines. Annu Rev Immunol. 2015;33:49–77.
Lukens JR, Gross JM, Calabrese C, Iwakura Y, Lamkanfi M, Vogel P, Kanneganti TD. Critical role for inflammasome-independent IL-1beta production in osteomyelitis. Proc Natl Acad Sci USA. 2014;111(3):1066–71.
Babur C, Ozcan G, Cebi DU, Pervane B, Ozdemir B, Yucel A, Biri AA, Babur C. Gingival crevicular fluid levels of osteoprotegerin (OPG) in premenopausal and postmenopausal women with or without chronic periodontitis. J Dent. 2012;40(5):364–71.
Tang H, Mattheos N, Yao Y, Jia Y, Ma L, Gong P. In vivo osteoprotegerin gene therapy preventing bone loss induced by periodontitis. J Periodontal Res. 2015;50(4):434–43.
McGeough MD, Pena CA, Mueller JL, Pociask DA, Broderick L, Hoffman HM, Brydges SD. Cutting edge: IL-6 is a marker of inflammation with no direct role in inflammasome-mediated mouse models. J Immunol. 2012;189(6):2707–11.
Acknowledgments
The authors are grateful to Drs. Tomomi Hashizume-Takizawa and Mio Hagiwara for their technical assistance. This study was supported by the Grants-in-Aid for Scientific Research (26463145) from the Japan Society for the Promotion of Science, “Strategic Research Base Development” Program (Japan [MEXT], 2010-2014 [S1001024]) for Private Universities of the Ministry of Education, Culture, Sports, Science and Technology, Japan. (3107), as well as the Nihon University Multidisciplinary Research Grant for 2014.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors report no conflicts of interest.
Additional information
Responsible Editor: John Di Battista.
Rights and permissions
About this article

Cite this article
Yamaguchi, Y., Kurita-Ochiai, T., Kobayashi, R. et al. Regulation of the NLRP3 inflammasome in Porphyromonas gingivalis-accelerated periodontal disease. Inflamm. Res. 66, 59–65 (2017). https://doi.org/10.1007/s00011-016-0992-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-016-0992-4