
Abstract
Objective
Human bone marrow-derived mesenchymal stromal cells (hBM-MSCs) are well known to modulate T cells. However, the molecular mechanisms that mark hBM-MSCs immunomodulation of T cells are not fully resolved.
Materials and methods
hBM-MSCs harvested from sternum or iliac crest of five healthy donors and characterized in accordance with the International Society of Cellular Therapy (ISCT) guidelines are co-cultured with T cells. Additionally, modulatory effects of MSCs on T-cell viability, proliferation, cytokine profile, co-stimulatory pathway, activation and immunomodulation are also determined.
Results
hBM-MSCs significantly reduced the expression of T-cell activation marker CD38 as well as co-stimulatory markers CD134 and CD154, whilst that of CD27 remained unchanged. BrdU, CFSE and Ki67 proliferation assays showed that hBM-MSCs reduced T-cell proliferation. Moreover, viability of T cells remained unchanged when co-cultured with hBM-MSCs. Finally, T cells when co-cultured with hBM-MSCs showed increased secretion of IL-10 and IL-11.
Conclusion
Collectively, hBM-MSCs are able to modulate the main steps involved in T-cell response toward a tolerogenic state. Thus, establishing immunobiological criteria defining the immunosuppressive effect of hBM-MSCs is of importance to reach efficient immunotherapeutic intervention.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human bone marrow contains a subpopulation of self-renewing, multipotent mesenchymal stromal cells collectively named bone marrow-derived mesenchymal stromal cells (hBM-MSCs) [1]. Due to their multilineage capacities, hBM-MSCs are able to differentiate into different cell lineages including osteoblasts, chondrocytes, adipocytes and many others opening thus great perspective in the field of regenerative medicine [2–9]. Moreover, hBM-MSCs act as a supporting stroma and secrete a wide variety of factors required to maintain hematopoietic niche essential for proper hematopoietic stem cell (HSC) maintenance and differentiation in the bone marrow [10]. Protocols for isolation, culture and expansion of MSCs from bone marrow are well established and were subjected to extensive research [11]. In addition to their regenerative properties, hBM-MSCs showed promising immunomodulatory potential fueling the research toward using these cells in cell therapy as immunomodulatory cells. As such, hBM-MSCs can suppress variety of immune cells including but not limited to lymphocytes [12, 13], dendritic cells [14], natural killer cells [15], B lymphocytes [16] and natural killer T and γδ T cells [17]. Accordingly, multiple clinical attempts documented that injection of MSCs can suppress the immune system and hence be used to treat graft versus host diseases following allogeneic hematopoietic stem cell transplantation [18, 19]. HBM-MSCs are shown to modulate immune cells activation including T cells through various mechanisms. However, no clear consensus has been established to document criteria of hBM-MSCs mediated immunosuppression of T cells. Previously, the ISCT described that to define MSCs, the cells must fulfill several cellular and molecular criteria including, adherence to plastic, expression of several mesenchymal markers whilst lacking hematopoietic markers, and able to differentiate into at least three cell lineages including osteoblasts, chondrocytes and adipocytes when cultured in osteogenic, chondrogenic and adipogenic environment, respectively [1]. Interestingly, the characteristics of the immunosuppressive properties of hBM-MSCs on T cells do not follow known criteria and a clear consensus is yet to be defined. It is worthy to note that the immunosuppressive effect of hBM-MSCs on T cells is achieved through various signaling mechanisms. For instance, hBM-MSCs are shown to suppress the immune system through induction of T-cell apoptosis by activating PD-1/B7-H1 programmed cell death pathway [20]. Moreover, hBM-MSCs mediated immunosuppression is achieved either through the release of soluble regulatory factors by contact dependent mechanisms or by inducing Treg generation [21, 22]. Activation, co-stimulatory pathway, proliferation and cytokine secretion are critical steps in the immunobiology of the lymphocyte response [23]. Considering that T cells play an important role in immune response, effective immunosuppression can be achieved through modulation of these steps. Therefore, we propose that defining the immunomodulatory properties of MSCs should take into account several immunobiological criteria that include both cellular and molecular markers which illustrate the impact of MSCs on T-cell proliferation, activation and survival.
Materials and methods
Isolation and culture of hBM-MSCs
This study was conducted in accordance with the Declaration of Helsinki (1964) and approved by the local ethics committee of the “Institut Jules Bordet” (Belgium). BM was harvested from the sternum or iliac crest of healthy donors after giving informed written consent (n = 5). hBM-MSCs were isolated using the classical adhesion method. Briefly, mononuclear cells (MNCs) were isolated by density gradient centrifugation (LinfoSep, Biomedics, Madrid, Spain), washed in Hanks’ Balanced Salt Solution (HBSS, Lonza Europe, Verviers, Belgium) and seeded at 2 × 104 cells/cm2 in Dulbecco’s Modified Eagle’s Medium with low glucose (DMEM-LG, Lonza) supplemented with 15 % fetal bovine serum (FBS, Sigma-Aldrich, Bornem, Belgium), 2 mM l-glutamine and 50 U/ml penicillin (both from Lonza). Cell cultures were incubated at 37 °C in a 5 % CO2 humidified atmosphere. After 48 h, non-adherent cells were removed by washing, and the medium was changed twice a week. When subconfluence (80–90 %) was achieved, adherent cells were trypsinized (Lonza) and expanded by replating at a lower density (1000 cells/cm2). hBM-MSCs were evaluated at passage 2 (P2).
Characterization of hBM-MSCs
HBM-MSCs were characterized by establishing their flow cytometry immunophenotype and their differentiation potential according to ISCT criteria [1]. The immunophenotype of hBM-MSCs was established by flow cytometry (MACSQuant®, Miltenyi Biotec, Netherlands) using the following monoclonal antibodies: anti-CD45-FITC and anti-HLA-DR-PE (Exalpha Biologicals, Maynard, MA), anti-CD34-PE and anti-CD73-PE (BD Biosciences, San Diego, CA, USA), anti-CD14-PE, anti-CD19-PE, anti-CD105-FITC and anti-CD90-PE (R&D systems, Minneapolis, MN, USA). Adherent cells were harvested with the TrypLE Select (Lonza) solution washed by centrifugation in PBS (Lonza) and finally resuspended in Miltenyi Biotec buffer. Then, harvested cells were incubated for 30 min at room temperature (RT) with conjugated primary antibody. After the labeling period, the cells were again washed, resuspended in PBS and fixed with 4 % paraformaldehyde. After cell staining, the data were acquired and analyzed on a MacsQuant analyzer (Miltenyi Biotec, Netherlands).The trilineage potential of hBM-MSCs was confirmed through inducing differentiation into adipogenic, osteogenic and chondrogenic lineages using the appropriate culture conditions (NH media, Miltenyi Biotec) as previously described [24].
Osteogenic differentiation
5000 cells/well were plated in a 24 well plate with culture medium. After 5 days, medium was completely discarded and replaced by osteogenic medium (StemMACS OsteoDiff Media, Miltenyi Biotec). Cells were fed weekly with complete replacement of osteogenic medium. After 21 days, the mineralization of the extracellular matrix was assessed by Alizarin Red staining. Cells were washed in phosphate-buffered saline (PBS) and fixed in 70 % ethanol at room temperature for 5 min followed by several washes in H2O. Cells were stained in 40 mM Alizarin red (Sigma-Aldrich) pH = 4.2 for 15 min at room temperature, rinsed in H2O, and then air-dried. The red staining was examined by light microscopy.
Adipogenic differentiation
5000 cells/well were plated in a 24 well plate with culture medium. After 5 days, medium was completely discarded and replaced with adipogenic medium (StemMACS AdipoDiff Media, Miltenyi Biotec). Cells were fed weekly with complete replacement of adipogenic medium. At day 7, cells were stained with Oil Red O solution (Sigma) after fixing (8 % formaldehyde). Lipid vacuoles were then observed by light microscopy.
Chondrogenic differentiation
150,000 cells were cultured in the tip of a 15 ml conical tube (Greiner) to enable cell culture in micromass with chondrogenic medium (StemMACS ChondroDiff Media, Miltenyi Biotec). Cells were resuspended carefully and cultured at 37 °C, 5 % CO2, humidified atmosphere with cap slightly screwed. Half of the chondrogenic medium was replaced weekly. At day 21, aggregates were stained with Alcian blue (Sigma) to highlight cartilage proteoglycans. In some cases, cryosectioned pellets were stained with Alcian blue to confirm chondrogenic differentiation.
Isolation of T-lymphocytes and co-culture with hBM-MSCs
Peripheral blood mononuclear cells (PBMCs) were obtained from the peripheral blood (PB) of healthy donors (n = 7) after Ficoll-Hypaque gradient centrifugation. T-lymphocytes were purified (>95 % purity) by positive selection using the MACS system (Miltenyi Biotec) as previously described [25]. T-lymphocytes were activated with 5 µg/ml phytohemagglutinin (PHA, Remel Europe, Kent, UK) and 20 U/ml of interleukin 2 (IL2, Biotest AG, Dreieich, Germany). Co-cultures of activated T-lymphocytes (1 × 105) in the presence of hBM-MSCs (1/4 cell ratio) were performed in flat-bottomed well plates during 5 days.
Viability and apoptosis
Lymphocyte viability was assessed using the 7-AAD BD Via-Probe™ viability staining solution (BD Biosciences) combined with CD45 labeling. After co-culture, T cells were first stained with an anti-CD45-VioBlue (Miltenyi Biotec) monoclonal antibody to exclude hBM-MSCs and then stained with 7-AAD solution. Absolute volumetric cell counting was performed by flow cytometry (Miltenyi Biotec) and viability expressed as the % of negative 7-AAD cells. Lymphocyte apoptosis was quantified by double staining of the cells with Annexin V-FITC (Biosource International, Camarillo, CA, USA) and propidium iodide (PI), according to the manufacturer’s instructions and analyzed by flow cytometer (Miltenyi Biotec).
T-cell immunophenotype
The expression profile of the CD38 activation marker as well as the co-stimulatory pathway of T cells was investigated by flow cytometry using the following antibodies: anti-CD38-PE (Miltenyi Biotec), anti CD27-PerCP, anti-CD134-FITC and anti-CD154-Pe/cy5 (Biolegend). T cells were incubated for 30 min at room temperature (RT) with conjugated primary antibody. After the labeling period, the cells were again washed, resuspended in PBS and fixed with 4 % paraformaldehyde. After cell staining, the data were acquired and analyzed on a MacsQuant analyzer (Miltenyi Biotec).
T-cell proliferation assays
Lymphocyte proliferation was evaluated by comparing three methods: BrdU incorporation, CFSE labeling and Ki-67 staining according to the respective manufacturer’s instructions. For BrdU incorporation (Roche Applied Science, Germany), the assay is based on the detection of the thymidine analog, 5-bromo-2-deoxyuridine (BrdU) incorporated into the genomic DNA of proliferating cells. This assay implies that hBM-MSCs have to be γ-irradiated (15 Gy) using a 57Co source to avoid BrdU incorporation. 50 µM of BrdU dye was added to the co-cultures at day four. T-cell proliferation was evaluated by measuring BrdU incorporation in a colorimetric assay according to the manufacturer’s instructions. Lymphocyte proliferation index is defined as the ratio between the optical density (OD) of proliferative stimulated T cells and the OD of unstimulated T cells after eliminating backgrounds of the other cell type controls. For CFSE labeling (Invitrogen, USA), the CFDA-SE (Carboxyfluorescein diacetate succinimidyl ester) is used as a cell-tracing reagent. The CFDA-SE dye passively diffuses into cells where the acetate groups are removed by intracellular esterases to yield highly fluorescent CFSE. A cell dye concentration of 5 µM CFDA-SE is used to label purified T-lymphocytes to avoid cell cytotoxicity and to allow long-term culture (5 days). The labeling (10 min) was performed out of the culture before co-incubation with hBM-MSCs. After 5 days, T-cell suspensions were harvested and CFSE fluorescence was visualized by flow cytometer (Miltenyi Biotec). CFSE profile of labeled cells is composed by several distinctive peaks representing the number of divisions or generations that proliferated lymphocytes have undergone. For Ki-67 staining, after the 5 days of culture in the presence or not of hBM-MSCs, T cells were recovered, underwent steps of fixation and permeabilization and finally stained with anti-human PE-Ki-67 antibodies or corresponding isotype control (BD Pharmingen, USA). The Ki67 monoclonal antibody recognizes a nuclear antigen which is only expressed during all active phases of cell division, but is absent in resting cells. Using flow cytometry (Miltenyi Biotec), we evaluated lymphocyte proliferation by determining Ki-67 positive expression [26, 27].
IL-10 and IL-11 ELISA quantification
IL-10 and IL-11 levels were measured in cell culture supernatants using the ELISA technique, according to the manufacturer’s instructions (R&D Systems Europe Ltd, United Kingdom). The amounts of IL-10 and IL-11 found in co-culture supernatants are expressed as fold increase compared with controls.
Statistical analysis
The data were collected from five independent experiments. The results were expressed as the mean ± SEM. Statistical comparisons were performed using the t test for paired samples. A p value less than 0.05 was considered statistically significant and noted as *p < 0.0003 and **p < 0.00001.
Results
Characterization of hBM-MSCs
The characteristics of hBM-MSCs were determined according to the ISCT guidelines. As shown in Fig. 1a, isolated mesenchymal stromal cells were able to adhere to plastic culture dish, and successfully differentiated into osteoblasts, adipocytes and chondrocytes when cultured in appropriate induction medium (Fig. 1a–d). In term of phenotype, these cells expressed mesenchymal markers including CD73, CD90 and CD105, but were negative for CD45, CD14, CD19, CD34 and HLA-DR (Fig. 1c). As such, this pool of cells was used for all subsequent experiments (Fig. 1b, c).

Morphology and flow cytometry immunophenotyping of hBM-MSCs. a Mesenchymal stromal cells at passage 1 adhering to plastic culture dish. b Representative Flow cytometry characterization of human BM-MSC at P1. Cells were stained with specific MAb (red line) against CD14, CD19, CD34, CD45, HLA-DR, CD73, CD90 and CD105. Gray lines indicate isotype-matched mouse IgG Ab control staining. c Immunophenotyping was done for all donors (n = 5) Results are expressed in percentages of positive cells (mean ± SEM). d hBM-MSCs differentiation into osteoblasts (a), adipocytes (b) and chondroblasts (c). a Calcium mineralization was assessed by Alizarin Red staining; b Adipocytes were colored using Oil Red O staining; c Microtome sample was stained with Alcian Blue
hBM-MSCs inhibited both T-cell activation and co-stimulatory pathway
Activation of T cell is a major hallmark of immune system stimulation and represents a prerequisite step toward inducing T-cell proliferation and clonal expansion. T-cell activation can be achieved through the expression of specific surface receptors or direct cell–cell co-stimulatory mechanism [28]. Since these mechanisms included the expression of activation markers such as CD38, CD134, CD27 and CD154, we therefore assessed the expression level of these markers in T cells co-incubated with hBM-MSCs. In Fig. 2, the expression of CD38, an activation marker, is significantly reduced by around 52 % in T cells co-incubated with hBM-MSCs when compared to the T cells alone. Similarly, in Fig. 2, the expression levels of CD134 and CD154 were also reduced by 48.7 and 63.8 %, respectively, in the T cells co-cultured with hBM-MSCs. No impact of hBM-MSCs on the CD27 expression was noted (Fig. 2).

hBM-MSCs differently modulate activation and co-stimulatory markers on activated T cells. PHA/IL-2-activated T lymphocytes from peripheral blood were co-cultured (direct contact) in the presence of hBM-MSCs (1/4 cell ratio) for 5 days. hBM-MSC decreased the expression of activation markers CD38 on activated T cells in the co-cultures. In b, hBM-MSCs decreased the expression of co-stimulatory markers CD134 and CD154 on activated T cells while the levels of CD27 expression remained unchanged in the co-cultures. Results were obtained by flow cytometry and expressed as the percentage of positive cells from eight independent experiments. Values were statistically significant (**p < 0.00001) compared with control cultures (PHA/IL-2-activated T cells without hBM-MSCs)
hBM-MSCs inhibited T-cell proliferation but had no effect on T-cell survival
To assess the effect of hBM-MSCs on T-cell proliferation, we used 3 different methods including CFSE, BrdU and Ki67 expression assays. Using these experiments, we successfully demonstrated that hBM-MSCs significantly inhibited T-cell proliferation and this inhibition reached levels as high as 68 %. Figure 3A shows that the levels of BrdU incorporation in T cells co-incubated with hBM-MSCs were reduced by 68 % when compared to T cells alone. Similarly, CFSE assay confirmed the results obtained with BrdU experiment and we observed a decrease of CFSE peak generation number by 52.5 % of activated T cells co-incubated with hBM-MSCs compared to T cells alone (Fig. 3b). Moreover, the levels of Ki67 positive T cells were reduced by 48 % when T cells are co-cultured with hBM-MSCs (Fig. 3c). Interestingly, hBM-MSCs did not affect the survival of T cells and no significant difference was obtained in the survival rate between T cells co-incubated or not with hBM-MSCs. (Figure 4).

hBM-MSCs inhibit allogeneic T-cell proliferation. a T cells/hBM-MSCs co-cultures at a ratio (1:4) were used to perform BrdU incorporation assay in activated T cells co-cultured or not with hBM-MSCs during 5 days of culture. A significant inhibition of T-cell proliferation is obtained in co-cultures. b CFSE-labeled T cells were activated by PHA/IL-2 and then co-cultured with hBM-MSCs at a ratio of 1:4 for 5 days in the presence or absence of direct contact. After this period, lymphocyte proliferation was evaluated by measuring CFSE fluorescence, and the data represent the mean percentage of CFSE positive T cells c Activated T-cell cultures in presence or absence of hBM-MSCs are stained with anti-human PE-Ki-67 antibodies or corresponding isotype control. Lymphocyte proliferation is evaluated with flow cytometry which detects positive Ki67 cells. As shown in the c Ki67 incorporation is reduced up to 50 % in PHA/IL-2-activated T lymphocytes co-cultured with hBM-MSCs as compared to PHA/IL-2-activated T lymphocytes alone. Data are expressed as the mean ± SEM percentage of KI67 positive cells from eight independent experiments (**p < 0.00001)

The viability of PHA/IL-2-activated T lymphocytes is not affected in co-cultures with hBM-MSCs. After co-culture, T cells are stained with an anti-CD45-VioBlue monoclonal antibody and then stained with 7-AAD solution. The analysis was performed by flow cytometry and the viability expressed as the % of negative 7-AAD cells among CD45+ population. Lymphocyte apoptosis was quantified by double cell staining with Annexin V-FITC and propidium iodide (PI), and analyzed by flow cytometer. As shown in the Fig. 4, during the co-culture hBM-MSC preserved the viability of PHA/IL-2-activated T lymphocytes
hBM-MSCs modulated T-cell cytokine secretion
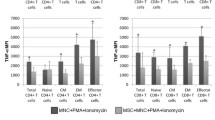
It is well known that MSCs mediated T-cell immunomodulation can be achieved through various mechanisms including the secretion of IL-10, a potent anti-inflammatory cytokine. Therefore, the levels of IL-10 produced by both MSCs and T cells when either co-incubated or cultured alone were assessed by ELISA. As shown in Fig. 5, MSCs did not produce detectable amount of IL-10 whilst activated T-lymphocytes constitutively produced 57.33 ± 6.34 pg/ml of IL-10. The production of IL-10 by activated T-lymphocytes was significantly increased in co-culture with hBM-MSCs reaching a level up to 179.3 ± 34 pg/ml which is threefold higher when compared to levels in cultures of T cells alone. Levels of IL-11 were also assessed to identify if increased production of IL-10 correlated with an increased release of IL-11, a cytokine involved in the proliferation of hematopoietic progenitor cells but also able to modulate cytokine production from activated CD4+ T cells (downregulation of IFN-γ and increased IL-4 production) [29]. Importantly, activated T-lymphocytes did not produce a significant amount of IL-11 (461.1 ± 61.9 pg/ml), whilst MSCs alone significantly a produced higher amount which was further increased up to 1094 ± 151 pg/ml in co-cultures reaching levels more than twofold higher (Fig. 5).

IL-10 and IL-11 are significantly released during the co-cultures of hBM-MSCs/T cells. Cell culture supernatants obtained from 5-day hBM-MSCs/T-lymphocyte co-cultures were used to assess IL-10 and IL-11 secretion by ELISA. The amounts of IL-10 and IL-11 found in co-cultures are expressed as pg/ml. The data are presented as the mean ± SEM of cytokine level from eight independent experiments (*p < 0.0003)
Discussion
Organ transplant rejection [30], autoimmune diseases and inflammatory illnesses such as but not limited to rheumatoid arthritis [31] and inflammatory bowel diseases [32] are characterized by abnormal activation of the immune system that targets healthy tissues causing chronic tissue damage and permanent disability. As such, effective immunosuppression is a cornerstone therapeutic element in treating these disorders. T cells play an important role in humoral immunity, and T-cell inhibition is a primordial target in autoimmune diseases and inflammatory conditions. Several immunosuppressive pharmacologic interventions, such as Muromonab, Basiliximab and Daclizumab, have been developed and target T cells either directly or by blocking the activity of circulating cytokines e.g., involved in T-cell proliferation and/or activation using antithymocyte globulins [33]. Although important, immunosuppressive treatments produce plethora of serious side effects including infection, anaphylaxis and increased risk of cancer [34]. MSCs isolated from various origins including bone marrow, umbilical cord, and adipose tissue have shown promising immunosuppressive properties [35]. While MSCs from various origins showed significant immunomodulatory properties on T cells, the extent to which MSCs suppress T cells is still elusive. The mechanism of hBM-MSC-induced T-cell suppression correlates with the specific properties of isolated MSCs. The results from clinical trials have not been encouraging or reproducible and might be related to the “art” of isolating and propagating MSCs. Therefore, developing a calibration standard, a database and a set of functional tests would be a better quality metric for MSCs [36]. Accordingly, the immunomodulatory properties of MSCs have to be defined according to several immune-biological criteria in a way to ensure their efficiency. Indeed, to induce efficient immunosuppression, hBM-MSCs must first be non-immunogenic as described by the criteria defined by ISCT [1]. Immunotherapy relies on the non-immunogenicity of the cellular product used. Additionally, biologic behavior in culture should be compatible with the criteria set by the ISCT [1]. The ISCT consolidated the characteristics that mesenchymal stromal should acquire to be considered “mesenchymal stromal cells” [1]. In this study, we first reiterated the properties of isolated hBM-MSCs (morphology, phenotype and differentiation potential) and found to be compatible with the minimal criteria listed by the ISCT. Moreover, the isolated MSCs are considered non-immunogenic since they lack HLA-DR expression and thus will not be capable of activating the immune system. Considering that MSCs modulation of T cells can be influenced by the quality of hBM-MSCs, the characteristic of isolated cells according to ISCT was achieved first and for consistency, these pools of ISCT-compatible hBM-MSCs are used for subsequent experimentation. Interestingly, published data focused on identifying the signaling mechanisms of hBM-MSCs that mediate immunosuppression of T cells rather than the “quality” of T-cell suppression. Whether the activation of these signaling pathways will ensure efficient immunosuppression is still elusive [13, 37–39]. Accordingly, Cuerquis et al. showed that hBM-MSC transiently increased IFN-γ and interleukin (IL)-2 by CD3/CD28-activated PBMCs before suppressing T-cell proliferation [40].
While hBM-MSCs mediated T-cell modulation is a well-established fact, the identity of an effective T-cell suppression is still lacking. To date, all reports provided evidence on four separate major concepts of T cells modulation including T-cell activation, co-stimulation, proliferation and cytokine production. hBM-MSCs mediated T-cell modulation can be regrouped into two major categories. The first category consists in inhibition of T-cell proliferation through a general reduction in T-cell number, whilst in the second category, hBM-MSCs target specific T-cell population by modulating specific T-cell activation markers including CD38 and CD154 [41]. This dual mechanism achieved by hBM-MSCs will further lead, in addition to a decrease in total T-cell number, to the reduction in T-cell subpopulations that can play a major role in immune recognition. Accordingly, to be considered efficient, hBM-MSCs must inhibit markers that crosslink both T-cell proliferation and activation such as CD38 and CD154 [41]. CD38 is of particular interest since the subset of circulated T cells expressing CD38 belongs to the early activated pool of T cells. Moreover, CD38+ T cells are shown to produce IL-2 and IFN-γ [42]. The CD27/CD70 pathway is majorly involved in T-cell activation, T-cell development, and T-cell dependent antibody production by B cells. These pathways that co-stimulate and activate T cells are well known to be involved in organ rejection and reduce graft survival [43]. Interestingly, our data showed that hBM-MSCs caused significant reduction in CD38 and CD154 expression in T cells but the levels of CD27 remained unchanged. IL-10 and IL-11 are two important cytokines known for their immunomodulation properties. IL-10 acts to reduce and control inflammation and does so by inducing anti-inflammatory responses through activating IL-10 receptor. Once activated, IL-10 receptor mitigates inflammatory signaling in many innate and adaptive immune cells [44]. Moreover, in addition to IL-10, IL-11 is another cytokine with a pleiotropic effect on various immune cells and can abrogate pro-inflammatory factors release including nitric oxide, TNF-α and IL-1β by activated macrophages, and inhibits production of IL-2 and IFN-γ. Therefore, some of the immunomodulatory properties of hBM-MSCs can be mediated through the concomitant production of IL-10 and IL-11. Interestingly, the levels of both cytokines were significantly increased in hBM-MSC and T-cell co-cultures [29], leading to the assumption that hBM-MSCs partly immunomodulate T cells through increased release of IL-10 and IL-11. We previously reported that IL-10 secretion, mainly from activated T cells, was stimulated during co-culture with MSCs [24, 45]. In contrast, IL-11 is essentially produced by MSCs and no IL-11 secretion was observed for T cells even after their activation. Taken together all these data, we can conclude that, although exist, no clear immunosuppressive profile has been determined for hBM-MSCs over T cells so far. Accordingly, T cells can still escape the immunosuppressive properties of hBM-MSCs even though some of the above criteria existed. It is noteworthy to mention that in our cell model, hBM-MSCs did not affect T-cell viability. This observation is compatible with previous reports documenting that hBM-MSCs can modulate T cells whilst producing anti-apoptotic effect through targeting T-cell death receptor pathway of apoptosis [46]. Additionally, we were able to reproduce the immunosuppressive properties obtained with hBM-MSCs. These findings are of considerable importance since the immunosuppressive properties of hBM-MSCs are not necessarily mediated through induction of T-cell cell death. Alternatively, keeping and maintaining T cells t at a low number and a quiescent statelong enough can induce efficient immunosuppression. The novelty of this study is that it provides new criteria for effective hBM-MSCs mediated immunosuppression of T cells largely ignored in the literature. We were the first to show that acceptable criteria for immunosuppression must at least involve well-designed protocols that include in addition to characterized MSCs, the immunosuppression experiments listed in the study. To be immunologically efficient, MSCs have to be first of all, non-immunogenic by lacking HLA-DR expression, and further by altering main steps involved in T-cell response: from reducing T cell activation by down-regulating CD38 expression, altering T cell co-stimulatory pathway through modulation of key co-stimulatory molecules (CD134 and CD154), promoting an anti-inflammatory cytokine profile by increasing IL-10 and IL-11, and finally inhibiting T-cell proliferation. We believe that all the above immunomodulatory effects obtained with hBM-MSCs when co-incubated with T cells represent the minimum acceptable profile to ensure effective T-cell immunosuppression at least in vitro. Noteworthy to mention that the immunosuppressive profile of hBM-MSCs isolated from various sources is not homogeneous. Interestingly, we were the first to report and compare the immunosuppressive properties of MSCs isolated from differences sources on T cells [47]. Moreover, immuno-tolerogenic environment created by hBM-MSCs continues to be a complex biological process requiring in part the generation of specific regulatory immune cells through the induction of IL-10 [48]. Considering a clear criterion for effective hBM-MSCs mediated immunosuppression of T cell is still lacking at least in vitro, thus comparing the effectiveness of immunosuppression among MSCs from different sources will be an elusive task. Accordingly, the molecular basis upon which a MSC pool is selected from a particular source for immunosuppressive clinical endpoint will continue to represent a major challenge. Hence, setting immune-biological criteria for MSC mediated immunosuppression of T cell is a key element in moving a step forward toward achieving MSCs mediated optimal immunosuppression on T cells.
References
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–7.
Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418:41–9.
Makino S, Fukuda K, Miyoshi S, Konishi F, Kodama H, Pan J, et al. Cardiomyocytes can be generated from marrow stromal cells in vitro. J Clin Invest. 1999;103:697–705.
Noth U, Osyczka AM, Tuli R, Hickok NJ, Danielson KG, Tuan RS. Multilineage mesenchymal differentiation potential of human trabecular bone-derived cells. J Orthop Res. 2002;20:1060–9.
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–7.
Song L, Tuan RS. Transdifferentiation potential of human mesenchymal stem cells derived from bone marrow. Faseb J. 2004;18:980–2.
Tuan RS, Boland G, Tuli R. Adult mesenchymal stem cells and cell-based tissue engineering. Arthritis Res Ther. 2003;5:32–45.
Wang G, Bunnell BA, Painter RG, Quiniones BC, Tom S, Lanson NA Jr, et al. Adult stem cells from bone marrow stroma differentiate into airway epithelial cells: potential therapy for cystic fibrosis. Proc Natl Acad Sci USA. 2005;102:186–91.
Woodbury D, Schwarz EJ, Prockop DJ, Black IB. Adult rat and human bone marrow stromal cells differentiate into neurons. J Neurosci Res. 2000;61:364–70.
Tavassoli M, Friedenstein A. Hemopoietic stromal microenvironment. Am J Hematol. 1983;15:195–203.
Caterson EJ, Nesti LJ, Danielson KG, Tuan RS. Human marrow-derived mesenchymal progenitor cells: isolation, culture expansion, and analysis of differentiation. Mol Biotechnol. 2002;20:245–56.
Glennie S, Soeiro I, Dyson PJ, Lam EW, Dazzi F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood. 2005;105:2821–7.
Krampera M, Glennie S, Dyson J, Scott D, Laylor R, Simpson E, et al. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood. 2003;101:3722–9.
Jiang XX, Zhang Y, Liu B, Zhang SX, Wu Y, Yu XD, et al. Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood. 2005;105:4120–6.
Spaggiari GM, Capobianco A, Becchetti S, Mingari MC, Moretta L. Mesenchymal stem cell-natural killer cell interactions: evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood. 2006;107:1484–90.
Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F, et al. Human mesenchymal stem cells modulate B-cell functions. Blood. 2006;107:367–72.
Prigione I, Benvenuto F, Bocca P, Battistini L, Uccelli A, Pistoia V. Reciprocal interactions between human mesenchymal stem cells and gammadelta T cells or invariant natural killer T cells. Stem Cells. 2009;27:693–702.
Gao L, Liu F, Tan L, Liu T, Chen Z, Shi C. The immunosuppressive properties of non-cultured dermal-derived mesenchymal stromal cells and the control of graft-versus-host disease. Biomaterials. 2014;35:3582–8.
Hematti P. Role of mesenchymal stromal cells in solid organ transplantation. Transplant Rev (Orlando). 2008;22:262–73.
Yan Z, Zhuansun Y, Liu G, Chen R, Li J, Ran P. Mesenchymal stem cells suppress T cells by inducing apoptosis and through PD-1/B7-H1 interactions. Immunol Lett. 2014;162:248–55.
Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13:392–402.
Le Blanc K, Davies LC. Mesenchymal stromal cells and the innate immune response. Immunol Lett. 2015;168:140–6.
Clarkson MR, Sayegh MH. T-cell costimulatory pathways in allograft rejection and tolerance. Transplantation. 2005;80:555–63.
Najar M, Raicevic G, Fayyad-Kazan H, De Bruyn C, Bron D, Toungouz M, et al. Bone marrow mesenchymal stromal cells induce proliferative, cytokinic and molecular changes during the T cell response: the importance of the IL-10/CD210 Axis. Stem Cell Rev. 2015;11:442–52.
Najar M, Rouas R, Raicevic G, Boufker HI, Lewalle P, Meuleman N, et al. Mesenchymal stromal cells promote or suppress the proliferation of T lymphocytes from cord blood and peripheral blood: the importance of low cell ratio and role of interleukin-6. Cytotherapy. 2009;11:570–83.
Bryant RJ, Banks PM, O’Malley DP. Ki67 staining pattern as a diagnostic tool in the evaluation of lymphoproliferative disorders. Histopathology. 2006;48:505–15.
Soares A, Govender L, Hughes J, Mavakla W, de Kock M, Barnard C, et al. Novel application of Ki67 to quantify antigen-specific in vitro lymphoproliferation. J Immunol Methods. 2010;362:43–50.
Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9:271–85.
Bozza M, Bliss JL, Dorner AJ, Trepicchio WL. Interleukin-11 modulates Th1/Th2 cytokine production from activated CD4+ T cells. J Interferon Cytokine Res. 2001;21:21–30.
Chong AS, Perkins DL. Transplantation: molecular phenotyping of T-cell-mediated rejection. Nat Rev Nephrol. 2014;10:678–80.
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–19.
Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–78.
Bakr MA, Nagib AM, Donia AF. Induction immunosuppressive therapy in kidney transplantation. Exp Clin Transplant. 2014;12(Suppl 1):60–9.
O’Dell JR. Therapeutic strategies for rheumatoid arthritis. N Engl J Med. 2004;350:2591–602.
Siegel G, Schafer R, Dazzi F. The immunosuppressive properties of mesenchymal stem cells. Transplantation. 2009;87:S45–9.
Tanavde V, Vaz C, Rao MS, Vemuri MC, Pochampally RR. Research using Mesenchymal Stem/Stromal Cells: quality metric towards developing a reference material. Cytotherapy. 2015;17:1169–77.
Chabannes D, Hill M, Merieau E, Rossignol J, Brion R, Soulillou JP, et al. A role for heme oxygenase-1 in the immunosuppressive effect of adult rat and human mesenchymal stem cells. 2007.
Li M, Sun X, Kuang X, Liao Y, Li H, Luo D. Mesenchymal stem cells suppress CD8+ T cell-mediated activation by suppressing natural killer group 2, member D protein receptor expression and secretion of prostaglandin E2, indoleamine 2, 3-dioxygenase and transforming growth factor-β. Clin Exp Immunol. 2014;178:516–24.
Ling W, Zhang J, Yuan Z, Ren G, Zhang L, Chen X, et al. Mesenchymal stem cells use IDO to regulate immunity in tumor microenvironment. Cancer Res. 2014;74:1576–87.
Cuerquis J, Romieu-Mourez R, François M, Routy JP, Young YK, Zhao J, et al. Human mesenchymal stromal cells transiently increase cytokine production by activated T cells before suppressing T-cell proliferation: effect of interferon-γ and tumor necrosis factor-α stimulation. Cytotherapy. 2014;16:191–202.
Quarona V, Zaccarello G, Chillemi A, Brunetti E, Singh VK, Ferrero E, et al. CD38 and CD157: a long journey from activation markers to multifunctional molecules. Cytom B Clin Cytom. 2013;84:207–17.
Sandoval-Montes C, Santos-Argumedo L. CD38 is expressed selectively during the activation of a subset of mature T cells with reduced proliferation but improved potential to produce cytokines. J Leukoc Biol. 2005;77:513–21.
Rothstein DM, Sayegh MH. T-cell costimulatory pathways in allograft rejection and tolerance. Immunol Rev. 2003;196:85–108.
Ni G, Wang T, Walton S, Zhu B, Chen S, Wu X, et al. Manipulating IL-10 signalling blockade for better immunotherapy. Cell Immunol. 2015;293:126–9.
Busser H, Najar M, Raicevic G, Pieters K, Velez Pombo R, Philippart P, et al. Isolation and characterization of human mesenchymal stromal cell subpopulations: comparison of bone marrow and adipose tissue. Stem Cells Dev. 2015;24:2142–57.
Benvenuto F, Ferrari S, Gerdoni E, Gualandi F, Frassoni F, Pistoia V, et al. Human mesenchymal stem cells promote survival of T cells in a quiescent state. Stem Cells. 2007;25:1753–60.
Najar M, Raicevic G, Fayyad-Kazan H, De Bruyn C, Bron D, Toungouz M, et al. Impact of different mesenchymal stromal cell types on T-cell activation, proliferation and migration. Int Immunopharmacol. 2013;15:693–702.
Najar M, Raicevic G, Fayyad-Kazan H, Bron B, Toungouz M, Lagneaux L. Mesenchymal stromal cells and immunomodulation: a gathering of regulatory immune cells. Cytotherapy. 2016;18:160–71.
Acknowledgments
M. Najar is a Télévie research fellow of “Le Fonds National de la Recherche Scientifique”.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Author disclosure statement
No competing financial interests exist.
Additional information
Responsible Editor: John Di Battista.
H. Fayyad-Kazan, W. H. Faour contributed equally to this work.
Rights and permissions
About this article

Cite this article
Fayyad-Kazan, H., Faour, W.H., Badran, B. et al. The immunomodulatory properties of human bone marrow-derived mesenchymal stromal cells are defined according to multiple immunobiological criteria. Inflamm. Res. 65, 501–510 (2016). https://doi.org/10.1007/s00011-016-0933-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-016-0933-2