
Abstract
Amphiregulin (AREG) is a well-characterized member of the epidermal growth factor (EGF) family and is one of the ligands of the EGF receptor (EGFR). AREG plays a key role in mammalian development and in the control of branching morphogenesis in various organs. Furthermore, AREG participates in a wide range of physiological and pathological processes activating the major intracellular signalling cascades governing cell survival, proliferation and motility. In this article, we review current advances in exocrine glands morphogenesis, focusing on the salivary gland, and discuss the essential aspects of AREG structure, function and regulation, and its differential role within the EGFR family of ligands. Finally, we identify emerging aspects in AREG research applied to mammary gland development and the salivary gland autoimmune disease, Sjögren’s syndrome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The epidermal growth factor receptor (EGFR) is a transmembrane tyrosine kinase receptor that is activated by several ligands acting as a switchboard for the activation of various signalling pathways mainly controlling proliferation, differentiation, and survival. Signalling through the EGF/EGFR pathway is complex and is intricately involved in several pathological conditions in the context of chronic inflammation. Accumulating evidence suggests that deregulation of the dialog between the immune system and EGFR signalling pathways contributes to promote tumorigenesis and metastasis in a variety of cancer types (Hanahan and Weinberg 2011; Mendelsohn and Baselga 2006). A number of reports in the past few years, have described EGFR signalling effects on mammalian development and demonstrated essential key roles for these proteins in the control of tissue morphogenesis (Segatto et al. 2011; Wieduwilt and Moasser 2008; Yarden and Sliwkowski 2001). Moreover, EGFR signalling is critical for almost all aspects of exocrine gland morphogenesis, including branching morphogenesis, epithelial maturation and postnatal ductal elongation (Hauser and Hoffman 2015; Hogan 1999; Iber and Menshykau 2013; Okumura et al. 2012; Sternlicht and Sunnarborg 2008; Wang and Laurie 2004). In recent years, important, interesting studies have revealed that branching morphogenesis of the exocrine glands appears to be controlled by a crosstalk between the ectodermal epithelium and endodermal mesenchymal tissues and some instructive or permissive epithelial signals are provided by members of the EGF/EGFR family (Hennighausen and Robinson 2001; Lu and Werb 2008; Wang and Laurie 2004). Meanwhile, great advances in the understanding of the mechanisms of salivary gland morphogenesis have been achieved; the salivary glands (SGs) provide an excellent model for clarifying the cellular signalling triggered by the EGF system and have been used as a good experimental sample in developmental biology for over 50 years (Andrew and Ewald 2010; Davies 2002; Gresik et al. 2009; Grobstein 1953; Harunaga et al. 2011; Kadoya and Yamashina 2005; Melnick and Jaskoll 2000; Patel et al. 2006; Sequeira et al. 2010; Tucker 2007). Amphiregulin (AREG), a protein required for epithelial differentiation, appears to be an important constituent of the morphogenesis and development program (Berasain and Avila 2010, 2014; Johnson et al. 1993; Lu et al. 2006; Wang and Laurie 2004). In fact, through EGFR-binding, AREG activates major intracellular signalling cascades governing cell survival, proliferation and motility (Busser et al. 2009, 2011). The roles of AREG are not limited to the control of morphogenesis but as recently established, include promotion of the growth of normal epithelial cells, orchestration of immunity, inflammation and tissue repair and the regulation of tumor progression. Based on the current findings, here we review the different cellular and molecular mechanisms that can drive the dynamic nature of development and branching morphogenesis of exocrine glands. We focus in particular on the SGs and also discuss the recent findings linking the activities of the EGFR system with its ligand AREG. The final section is dedicated to a discussion of the recent advances and new emerging evidence supporting the role of AREG in mammary gland development and the SGs autoimmune disease Sjögren’s syndrome (SS).
Morphogenesis of Exocrine Glands and Molecular Regulation of Signalling Pathways Involved in Development
Every exocrine gland is derived from a primitive ingrowth, or budding, from an epithelial surface (Hogan 1999). This ingrowth may possess, at the beginning, a tubular structure, but in other instances, as growth proceeds, the solid column of cells may divide and will subsequently become tubulated. The timing and positioning of the epithelial ingrowth or bud are very precise and depend on changes in the molecular interactions through ductal elongation and beyond. In general, the formation of any branched organ takes place through the following steps: (1) the initial specification and formation of an organ anlage, (2) its invagination, (3) the initiation and outgrowth of its rudimentary branches, (4) the formation of a continuous lumen, and (5) tissue-specific differentiation of the entire network and its terminal structures (Sternlicht et al. 2005; Sternlicht 2006). Even if a conserved mechanism regulating exocrine glands morphogenesis has been individuated, organ-specific variations were recognized (Davies 2002). As regards the lacrimal glands, several authors demonstrated that they begin development at the 22- to 25-mm human embryologic stage as solid epithelial buds that arise from the ectoderm of the superolateral conjunctival fornix (de la Cuadra-Blanco et al. 2003; Hoffman et al. 2002; Keibel and Elze 1908; Tripathi and Tripathi 1990). These buds constitute the glandular primordium located in the superior exterior area of epithelial condensation, surrounded by the mesenchyme (de la Cuadra-Blanco et al. 2003; Hoffman et al. 2002; Keibel and Elze 1908; Tripathi and Tripathi 1990). Tripathi and Tripathi (1990) thought that the lacrimal gland arose from the neural crest; but various authors studying other mammals have since confirmed the ectodermal origin of the lacrimal gland (Lovicu et al. 1999). At this stage, the primordium receives the artery and the lacrimal nerve, and lumina appear within the glandular epithelial buds. During the tenth week of embryo development, the lacrimal gland splits into two portions, the orbital and palpebral glands and continues arborisation with an increase in glandular vascularization; in fact, the formation of epithelial buds at the level of the fornix continues to be observed during the 15th–16th week of human embryo development (de la Cuadra-Blanco et al. 2003).
Similar stages of organogenesis were observed during the development of the human mammary gland. At early stages of morphogenesis, placodes appear and each placode undergoes expansion and invagination into the underlying mesenchyme producing a bud (Cowin and Wysolmerski 2010). The next phase of development involves the organ-typical ductal branching during which epithelial cells emerge from the mammary bud and grows down from the mesenchyme and into the mammary fat pad, a collection of preadipocytes that originates from a mesenchymal condensation (Hens and Wysolmerski 2005; Sakakura 1987). At this stage, the primary bud begins to branch in a characteristic dichotomous fashion to form the ductal tree of the nascent mammary gland (Biggs and Mikkola 2014). The epithelial duct system grows slowly until puberty; at this age a rapid expansion occurs; the juvenile mammary gland, upon stimulus by ovarian hormones during puberty, exhibits ductal elongation and further branching into the mammary fat pad (Cowin and Wysolmerski 2010). During pregnancy, the process of maturation of the gland becomes complete with the formation of mature acinar structures, or alveoli (Oakes et al. 2006) to produce milk during lactation.
Common molecular pathways are beginning to emerge in the organogenesis of several ectodermal organs that might reflect their common evolutionary history. For the mammary gland a wide variety of hormonal and peptide factors has been identified, for their inductive role in the modelling of the gland, such as the EGF and fibroblast growth factors (FGF) families and their receptors, members of the transforming growth factor (TGF) family, insulin-like growth factors (IGFs), and EGF-like family members (Hynes and Watson 2010). Several molecules and mechanisms are being investigated to clarify exocrine gland organogenesis, and genetic studies support the notion that parathyroid-like hormones are essential for the primitive epithelial ingrowth responsible for mammary glands formation. Parathyroid-like hormones are secreted by the early mammary bud epithelium and after release, regulate the cell fate and specify the mammary mesenchyme promoting numerous mesenchymal responses (Hiremath and Wysolmerski 2013). Further studies have evidenced an important role for the cadherins known to be expressed in the mammary gland and regulate the morphologic development and function of lobulo-alveolar structures in the mammary gland (Daniel et al. 1995; Knudsen et al. 2005). Cadherins also play a crucial role in pancreatic ingrowth, that is dependent on mesenchymal aggregation; the epithelial pancreatic cells receive essential signals from the overlying mesenchyme, that regulate pancreatic growth and branching morphogenesis by triggering the proliferation of precursors and differentiation of the cells (Andl et al. 2006).
As extensively studied and demonstrated, the mechanism of tissue induction and specification in the development of exocrine glands requires the activation of transcription factors that act as important competence factors. Pax6, for example, restricts FGF10 responsive epithelia in the lacrimal gland (Makarenkova et al. 2000). In fact, as demonstrated by Makarenkova et al. (2000), the expression of Pax6, a transcription factor that has both paired and homeodomain DNA binding motifs (Mansouri et al. 1994), was required in conjunctival epithelium for the induction of FGF10 expression in adjacent mesenchyme. In the last years, several transcription factors and secreted signalling molecules playing an essential role in ductal elongation have been identified (Ahlgren et al. 1997; Davenport et al. 2003; Gu et al. 2003; Phippard et al. 1996; van Genderen et al. 1994). Particular interest was aroused by the identification of discoidin domain receptor 1 tyrosine kinase (DDR1) and the ErbB receptors as fundamental factors for ductal branching and elongation (Fata et al. 2004). The discoidin domain receptors, DDR1 and DDR2, are two closely related receptor tyrosine kinases that contain a discoidin homology domain in their extracellular regions (Fata et al. 2004). DDR1 was shown to induce signal transducers and activators of transcription (STAT)5 phosphorylation and transcription, triggering the signalling cascade that leads to lactation (Faraci-Orf et al. 2006) and seems to have an important role in differentiation, cell motility, collagen synthesis and signalling. Absence of the Ddr1 gene determines severe defects in mammary gland development and the complete absence of lactation; this is different from what has been observed in mice lacking α2β1 integrin, that show branching abnormalities but normal lactation (Chen et al. 2002). It thus appears that ductal branching involves a number of different basement membrane-dependent adhesion mechanisms, as demonstrated for DDR1, integrin α2, α3, and α6 subunits, and cell surface β1,4-galactosyltransferase (Fata et al. 2004). In particular, DDR1 and α2β1 integrin regulate distinct aspects of the branching process.
Recently, growing interest has been aroused in the EGFR ligand, AREG, in exocrine glands morphogenesis, and many researchers have tried to clarify its role. The effect of estrogen on pubertal expression of AREG is now amply documented, shown to be an important paracrine mediator of ductal elongation during puberty-related mammary gland development. In fact, ductal outgrowth is impaired in AREG-deficient mice but not in mice lacking EGF, TGF-α, heparin-binding EGF-like growth factor, or betacellulin (Sternlicht et al. 2005). The role played by AREG during ductal elongation of several exocrine glands is under the control of growth factors, cytokines, and hormones, as well as depending on interactions with extracellular matrix (ECM) proteins (Simian et al. 2001; Sympson et al. 1994; Witty et al. 1995), because cell signalling through the ECM can have an impact on cell fate decisions, cell proliferation and survival, ECM degradation can also release important molecular factors such as AREG, Wingless/Int-1 (Wnt), TGF-β and FGF, which have been shown to regulate branching (Sternlicht 2006). Furthermore, endogenous inhibitors, such as tissue inhibitors of matrix metalloproteinases (MMPs), were demonstrated to mediate the counterbalance of the MMPs activity during organogenesis (Wang et al. 2000). The importance of AREG in the morphogenesis of exocrine glands will be discussed in paragraph entitled “AREG control of mammary gland morphogenesis”.
From the above-reported notions it can be seen that the process of branching morphogenesis is, a complex phenomenon that requires an intricately regulated network of different signalling factors, depending on the organ, and many coordinated cell functions, including cell adhesion, polarization, proliferation, migration as well as cell–cell and cell–ECM interactions (Pozzi and Zent 2011). Essential diffusible factors are more numerous, perhaps reflecting the enhanced complexity of branching. These include AREG, bone morphogenetic proteins (BMPs), a group of signalling molecules that belongs to the TGF-β superfamily of proteins, initially discovered for their ability to induce bone formation and now recognized to play crucial roles in embryogenesis and development, as well as colony stimulating factor 1, FGF10, TGF-β1-3, and Wnt4 (Wang and Laurie 2004). A cohort of signalling proteins that regulate branching morphogenesis have been well characterized, such as nuclear factor (NF)-κB (Brantley et al. 2001), FGF10 and Wnt, a family of secreted, cysteine-rich glycoproteins that function as short-range signalling factors (Hogan 1999). Wnt proteins, in particular, are associated with the cell surface and extracellular matrix (Parkin et al. 1993) and are now recognized as physiological signals that stimulate ductal morphogenesis (Robinson et al. 2000).
SGs Development
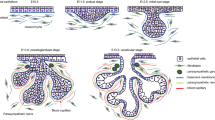
Morphogenesis of complex organs such as the SGs requires the cooperation and coordination of large network signalling pathways to control cell proliferation, quiescence, apoptosis, and histodifferentiation (Davidson et al. 2002; Gardner et al. 2003; Melnick and Jaskoll 2000; Melnick et al. 2001a, b, c, d). SGs develop as highly branched structures and offer an excellent model system used to delineate branching morphogenesis. The development is regulated by multiple stage-specific growth factors, cytokines, and transcription factors which are expressed at specific times, triggering the organogenesis process (Jaskoll and Melnick 1999; Jaskoll et al. 2002; Kashimata and Gresik 1997; Kashimata et al. 2000a, b; Melnick et al. 2001a, b, c, d). SGs organogenesis involves various cell types and their stem/progenitor cells, including epithelial, mesenchymal, neuronal, lymphatic, and endothelial cells. Many complex interactions among these cell types and their extracellular matrix microenvironment arise during the development of the salivary epithelium. During oral cavity development, a transient formation appears, that initially defines the boundaries of the ectoderm and endoderm and that separates the oral cavity from the cavity of the primordial pharynx (Patel and Hoffman 2014). The submandibular gland (SMG) placode is visible as a localized thickening of the oral epithelium adjacent to the tongue around at embryonic day (ED) 11.5 of development, known as the prebud stage (Tucker 2007). By ED12, the salivary proof has enlarged and invaginated into the underlying mesenchyme which begins to condense, resulting in the formation of a primary bud linked to the oral surface by a duct that will become the major secretory duct. By ED13, known as the pseudoglandular stage, the final part of the bud has grown in size and undergone a process of cluster formations resulting in approximately 3–5 epithelial buds. Lumen formation of the primary duct occurs by ED13.5, and the majority of the ducts develop a lumen at the canalicular stage, as from about ED15.5. Around ED17.5, the branches and terminal buds are delved to form the ductal and acinar system and at this point, the terminal bud stage is completed and exhibits distinct lumina and presumptive ducts (Melnick and Jaskoll 2000) (Fig. 1).

Schematic representation and hematoxylin and eosin (H&E) staining of stages of human salivary gland morphogenesis. a Pre bud: salivary gland development begins at gestational age 10/11 (GA 10/11) as an epithelial thickening. b Initial bud: compact cluster of epithelial cells derived from the oral surface epithelium that invade the surrounding mesenchyme. c Pseudoglandular stage: in this stage a network of epithelial stalks with a ductal system forms and begins the lumen formation. d Canalicular stage: formation of complex architecture of ductal branches producing a multi-lobed gland. e Terminal bud: differentiation of terminal buds into acinar lobules. Salivary gland sections stained with H&E (Fischer et al. 2008); GA, gestational age; W, week
Cellular Signalling Mechanisms Involved in Salivary Gland Development
Formation of the SGs involves a coordinated development mechanism triggered by multiple reciprocal interactions among the epithelium and its surrounding mesenchyme; recent studies have also revealed what signals spatio-temporally drive the migrating neural crest cells to control placode initiation in mice SGs, and identified multiple molecules, including components of the extracellular matrix, cell adhesion receptors, proteases, and growth factors, that mediate these instructive interactions. It is clear from experiments using heterotypic tissue recombination that specific SMG mesenchyme-secreted factors are required to induce the epithelium to form a salivary gland (Patel et al. 2006).
Progressive advances have demonstrated that the EGF system is a physiologic regulator of development of mouse SMG and of the synthesis of specific integrins as Alpha6 integrin. Synthesis of alpha6 integrin, in cultured SMG, was increased by EGF added and drastically reduced by tyrphostin. Since integrins mediate a number of interactions between epithelial cells and the ECM, it has demonstrated that EGF system may regulate expression of integrins (Kashimata and Gresik 1997). In EGFR mutant mice, the SGs have a reduced number of terminal buds indicating that the EGFR system is necessary for normal SMG development (Jaskoll and Melnick 1999). Molecular and genetic studies have demonstrated that branching morphogenesis appears to be controlled by a conserved set of molecules, including the FGF family. Indeed, the FGF/FGFR system has a crucial role in the development processes of branching morphogenesis of the SGs (Hoffman et al. 2002), as demonstrated by the evidence that FGF/FGFR transgenic mice show an altered MSG phenotype, for example (De Moerlooze et al. 2000; Jaskoll et al. 2004; Ohuchi et al. 2000; Ornitz and Itoh 2001). Furthermore, FGFR cleavage seems to be boosted by the activity of MMPs allowing localized spread of the epithelium at sites where proliferation occurs (Simian et al. 2001). These interesting data suggest that FGFR signalling involves a regulatory network that induces bud formation and duct elongation during branching morphogenesis (Steinberg et al. 2005). In human patients, mutations in the FGF/FGFR pathway are associated with aplasia of the SGs indicating that the normal development of the glands depends on a balanced of signalling triggered by this system (Shams et al. 2007). Advanced research highlighted the essential role of BMPs (2, 4, 7) into regulating initial stages of embryonic SMG morphogenesis and during embryonic SMG branching morphogenesis. In particular, BMP7 knockout mice exhibit an abnormal phenotype, and the mesenchyme of the salivary gland is disorganized with reduced branching and lumen formation (Jaskoll et al. 2002). In the last years, Melnick et al. (2001c), have demonstrated that the tumor necrosis factor (TNF)/TNF-R1 signal transduction pathway also plays an important role in balancing increased proliferation and decreased apoptosis during SMG duct and acini formation. In particular, a loss or reduction in lumen formation is evident in the ectodysplasin (EDA) mutant mouse. EDA and its receptor EDAR are members of the TNF superfamily, that show a critical involvement during the development of all ectodermal derivatives (teeth, hair and sweat glands) (Monreal et al. 1998; Srivastava et al. 1997). Mutations of EDA in humans lead to hypohydrotic ectodermal dysplasia, a disorder characterized by defects in hair, teeth, sweat glands and SGs (Kere et al. 1996). EDA is expressed in the mesenchyme of the SMG while its receptor, EDAR, is localized in the gland epithelium (Mikkola 2008). EDA and EDAR mutant mice have hypoplastic and dysplastic glands that lack lumens and acini (Melnick et al. 2009), but when EDA is supplemented in SMG organotypic cultures branching is increased, while a soluble form of EDAR added in embryonic SMG cultures abrogates EDA/EDAR signalling, resulting in a significant decrease in branching morphogenesis. The absence of SMG ducts and acini in SMG suggests that EDA/EDAR signalling plays multiple roles in salivary gland development, affecting lumina formation and glandular histodifferentiation (Melnick et al. 2009). Interestingly, another essential signal that is triggered in the final stages of SMG development is the EGF/TGF-α/EGFR pathway that regulates the rate of branching and histodifferentiation and progression from the canalicular stage to the terminal bud stage. The increased expression of TGF-α and EGFR supports the importance of this signalling pathway during the formation of distinct lumina in the terminal bud stage (Melnick and Jaskoll 2000). From all the data above it is clear that, while many details regarding cell physiology of adult acinar and ductal SGs cells have been identified, a universally accepted model of saliva secretion at the molecular level still remains to be fully characterised. There is much to be gained from the application of innovative systems biology-based approaches to the study of SGs development and disease. Recently the research community has been facing the challenge of developing sophisticated computational methods that have been successful in deciphering a large amount of data, and mathematical models are being employed to identify the molecular network involved in the complicated process of SGs morphogenesis (Larsen et al. 2010) (Fig. 2).

Schematic overview of molecules required for exocrine gland organogenesis. Multiple proteins are necessary during embryogenesis for successful branching morphogenesis. These proteins include extracellular matrix proteins and proteoglycans such as Laminins, Collagens and Fibronectin; many growth factors and ligands (Shh sonic hedgehog, Eda ectodysplasin-A, AREG amphiregulin, EGF epithelial growth factors, FGF fibroblast growth factor, TGF-β transforming growth factor β, HGF hepatocyte growth factor, BMP7 bone morphogenetic protein 7). All these ligands require the activation of a variety of transmembrane receptors, adhesion molecules (EGFR EGF receptor, PDGFR PDGF receptor, EdaR ectodysplasin-A receptor) and transcription factors that lead to the expression of specific genes involved in exocrine glands development
Clinical Conditions Affecting Salivary Gland Function
In the adult, the SGs function is complex, involving the activity of multiple epithelial cell types to produce saliva, that work in response to stimuli originating outside the endocrine system. Saliva production begins in the basic secretory units of SGs, the submandibular acinar cells, leading to the secretion of incomplete saliva; serous and mucous acinar cells also contribute to this production. In a second phase, this primary fluid is modified passing through the striated and excretory ducts, and then, the final saliva reaches the oral cavity, where it mixes with secretions from minor SGs, which are found throughout the oral cavity, to generate whole saliva. Saliva secretion is under the control of the autonomic nervous system, which controls the volume and the composition of saliva secreted (Proctor and Carpenter 2007). Adult SGs can be affected by infection, inflammation, autoimmune disease, and tumorigenesis. Sialolithiasis and sialadenitis are problems that can occur in the salivary glands. Sialolithiasis is a condition where calcium-rich stones form inside the salivary glands. The cause is unknown but dehydration, a decreased food intake or medications that decrease saliva production seem to be related to the formation of stones; stones often sit inside the gland without causing any symptoms but, occasionally, block the gland’s secretion. Then, the gland appears typically painful and swollen, and the saliva secretion is partially or completely inhibited (Williams 1999). This could be followed by an infection called sialadenitis. Sialadenitis (or sialoadenitis) is an infection involving a salivary gland that can be acute chronic or recurrent. Sialolithiasis could be a predisposing factor but Staphylococcus or Streptococcus bacteria can cause this infection. Sialadenitis is a common condition in older adults and infants (Wilson et al. 2014).
Salivary gland neoplasms consist of a heterogeneous group of lesions with complex clinic-pathologic characteristics that account for approximately 3–10% of head and neck region tumors. According to the World Health Organization, the annual global incidence, considering all SGs neoplasms, ranges 0.4–13.5 cases per 100,000 inhabitants (Fonseca et al. 2012). In the last years, few improvements have been made in the understanding of the molecular mechanisms that act in the development of the lesions; the chemotherapeutic protocols have not been shown to improve survival and surgery remains the main therapeutic approach for patients affected by SGs neoplasms that include a wide variety of both cancerous and non-cancerous tumors. Non-cancerous tumors that can affect the parotid glands include pleomorphic adenomas and Warthin’s tumors. Pleomorphic adenoma is one of the most common benign tumors affecting SGs. It occours mainly in the parotid, but can also grow in the SMG and the minor SGs, although rarely (Rao et al. 2012). Warthin’s tumor is the second most common benign salivary gland tumor, located almost exclusively in the parotid gland and accounting for approximately 15% of all parotid epithelial tumors (Chulam et al. 2013). Only 20% of salivary gland tumors are malignant. On the basis of clinical practice, the smaller the salivary gland, the more likely it is to be a malignant tumor. In the parotid glands, 20–25% of the tumors are malignant. The incidence of malignant tumors increases to 40% for the SMGs, and more than 90% for the sublingual gland (Speight and Barrett 2002).
Over the years, there has been some progress in clarifying specific causes of salivary gland cancer but the etiological agents of such cancers remain unclear. Radiation exposure is considered the best known risk factor, as evident in the increased risk in atomic bomb survivors and in patients subjected to therapeutic radiation for other head and neck cancers. This was confirmed by the observation of an increased occurrence in children with leukemias treated with multiagent chemotherapy and prophylactic cranial irradiation (Prasannan et al. 1999). Obviously, genomic abnormalities in the salivary gland cancers were also documented (Headington et al. 1977). In addition, a history of previous cancers, related to Epstein-Barr virus infection, is implicated in the pathogenesis of salivary lymphoepithelial-like carcinoma (Iezzoni et al. 1995), the risk of salivary gland cancer was increased fourfold in Hodgkin’s lymphoma patients (Dong and Hemminki 2001) and HIV infection was also demonstrated to increase the risk of salivary gland cancers (Jeffers and Webster-Cyriaque 2011). Alcohol and smoking abuse, strongly related to other head and neck cancers, do not seem to be associated with an increased risk for developing salivary gland neoplasms with the exception of an association of smoking with Warthin’s tumor (Kotwall 1992). One interesting finding, but that still requires confirmation, is that occupational exposure in rubber manufacturing and woodworking, and also employment at hairdressers or beauty shops showed an elevated risk of salivary gland cancer (Maślińska et al. 2015; Swanson and Burns 1997).
Overall, other than malignant cancers, the most serious problems for patients with SGs disease result from loss of SGs function, which is an important medical problem associated with SS, collateral damage from head and neck radiation therapy, adverse effects of medication use, and age-related atrophy. Hyposalivation causes a “dry mouth” affecting additional physiological processes, such as mastication and swallowing, and may be associated to secondary oral disease states including sialadenitis, dental caries, acid erosion, periodontal disease, oral candidiasis, mucosal ulcerations and fissures, and dysphagia (Napenas et al. 2009). As a result of these secondary conditions, the quality of life of such patients is very seriously affected. Although the molecular basis for SGs hypofunction and the mechanism of the selective destruction of salivary acinar cells remain poorly understood, new findings in the last years promise to aid the development of satisfactory therapeutic options for these patients.
Focus on Sjögren’s Syndrome
Sjogren’s syndrome is a complex systemic autoimmune disease that primarily affects the SGs and the lacrimal glands, with a multifactorial aetiology, influenced by genetics, environmental and hormonal factors, infections (Delaleu et al. 2005; Lee et al. 2009; Vitali 2003). During the slow course of development, the disease is characterized by CD4+ T lymphocytic infiltration of the exocrine glands, primarily salivary and lacrimal glands causing dysfunction and structural damage (Moutsopoulos 1994). Autoimmunity develops, and 90% of patients produce autoantibodies targeting nuclear autoantigens (Mathews et al. 2008). Using clinical, laboratory and molecular parameters, most researchers agree that patients with this syndrome have a high incidence of non-Hodgkin’s, mucosa-associated lymphoid tissue B lymphoma (Kovacs et al. 2010). When the disease occurs in the absence of another underlying rheumatic disorder it is denominated primary SS (pSS), whereas secondary SS is associated with another underlying rheumatic disease, such as systemic lupus erythematosus, rheumatoid arthritis, or scleroderma. The stimulation of immune mechanisms is thought to play a central role in the pathogenesis of this chronic disorder, as illustrated by several indices of a hyperactive immunologic state, including serum detection of various autoantibodies, in particular those directed against Ro/SS-A and La/SS-B antigens (Tzioufas and Voulgarelis 2007). There is no single disease-specific diagnostic criterion for SS, and indeed, 11 classification/criteria were published between 1965 and 2002 (Shiboski et al. 2012). Sjogren’s syndrome typically presents as dry eyes (xerophthalmia or keratoconjunctivitis sicca) and dry mouth (xerostomia) (Tzioufas and Voulgarelis 2007). In addition to these symptoms, the following items should be present: positive ocular signs by Schirmer’s I test and/or Rose Bengal score; focal lymphocytic sialoadenitis evaluated by histopathology with a focus score ≥1; salivary gland involvement defined by a positive result for at least one among salivary scintigraphy, parotid sialography or unstimulated salivary flow; and the presence in serum of anti-Ro/SSA, anti-La/SSB autoantibodies or both (Fig. 3). The immune-mediated damage appears in the form of apoptosis of glandular epithelial cells (Tzioufas and Voulgarelis 2007) and seems to be mediated by several pro-inflammatory T helper 1-type cytokines (Vitali 2003). The epithelial cells of salivary glands (SGEC) from patients with SS also display alterations in cell adhesion and shape (Gonzalez et al. 2011). SS patients manifest an increased epithelial expression of several inflammatory proteins in the lymphoepithelial lesion (Manoussakis and Kapsogeorgou 2010) and, in recent years, several lines of evidence have indicated that glandular epithelial cells in pSS lesions are aberrantly activated and may play an active role in the induction and perpetuation of the inflammatory processes (Lisi et al. 2012, 2013, 2014a, b; Manoussakis et al. 2007; Sisto et al. 2011). SGEC express immune-modulatory molecules implicated in innate and acquired immune responses, such as Toll-like receptors (TLRs), cytokines, chemokines, MHC molecules, adhesion and a plethora of co-stimulatory molecules (Tzioufas and Voulgarelis 2007). In addition, they have the potential to release autoantigens through autoantigen-loaded vesicles, contained in exosomes or apoptotic bodies (Tatouli and Tzioufas 2012). Furthermore, SGEC are able to mediate the recruitment of almost all types of immune cells, leading to their activation. It is now clear that while the infiltrating lymphocytes remain activated, the activated glandular epithelial cells undergo apoptotic cell death (Sisto et al. 2015).

Schematic representation of classification criteria recently adopted for the diagnosis of Sjögren’s syndrome
Amphiregulin
AREG is a heparin-binding molecule that binds EGFR (Cook et al.1991), originally isolated from conditioned medium of phorbol 12-myristate 13-acetate-stimulated MCF-7 breast cancer cells (Shoyab et al. 1988). The protein was termed AREG to reflect its bifunctional activity, interacting with the EGF/TGF-α receptor to stimulate the growth of normal fibroblasts and keratinocytes as well as tumor cells, and inhibitings the proliferation of several invasive carcer cell lines in culture (Shoyab et al. 1988). Structurally, the truncated form of AREG contains 78 amino acids (AA), whereas the larger form contains 84AA at the amino-terminus (Shoyab et al. 1989). The 84AA sequence has a lower binding affinity to EGFR and is the higher yield sequence, as compared to the 78AA sequence. Both the 78 and 84AA sequences have similar carboxyl-terminals, and both are biologically active (Shoyab et al. 1989). In humans the AREG gene is located on chromosome 4q13-21 and is transcribed as a 1.4 kb mRNA composed of six exons that code for the transmembrane polarized glycoprotein precursor (pro-AREG) of 252 aminoacids, also known as the transmembrane precursor or pro-form. Pro-AREG consists of a hydrophilic extracellular N-terminus (or ectodomain) containing an N-glycosylated heparin binding domain, a hydrophobic transmembrane domain and a hydrophilic cytoplasmic C-terminus that presents an EGF-like domain with a striking homology to EGF (38%) and TGF-α (32%) (Duffy et al. 2009; Luetteke et al. 1999). At the plasma membrane, proteolytic cleavage of the membrane-bound AREG precursor within its ectodomain leads to the release of the soluble form of AREG, and the 84 AA corresponded to the mature secreted form of AREG (Plowman et al. 1990). AREG was compared to other EGF-like growth factors and proteins, and like all family members, AREG’s bioactive domain is characterized by six essential cysteine 26 residues, spaced: CX7CX4CX10CX1CX8C, where C represents cysteines and X can be any AA (Shoyab et al.1989). The pro-AREG contains several cleavage sites and glycosylation motifs and needs to be cleaved to be active (Brown et al. 1998). Proteolytic processing of AREG requires TNF-α converting enzyme (TACE or also named ADAM17), a member of the disintegrins and metalloproteases family (Duffy et al. 2009).
AREG Exhibits Functional Differences Among EGFR Ligands
An essential first step to correlate a ligand’s chemotype with its pattern of functional modulation is to gain a proper understanding of the structural basis of ligand-specific EGFR interaction and determine the binding modes of different ligands to EGFR, thereby illuminating the pharmacology and biology of EGFR-mediated molecular signalling.
On the basis of experimental evidences, AREG has been shown to have a reduced binding affinity and intrinsic activity toward EGFR in comparison to others ligands such as EGF and TGF-α (Neelam et al. 1998; Shoyab et al. 1989). For example, in breast cancer cells, AREG or EGF interaction determines a markedly different pattern and intensity of EGFR tyrosine phosphorylation, suggesting that specific signal cascades emanating from the EGFR depend on the binding ligand (Gilmore et al. 2008). Moreover, ligand binding to the EGFR causes the formation of homo- and hetero-dimers, a process, which subsequently induces autophosphorylation that occurs through EGFR tyrosine kinase activity. Active EGFR undergoes internalization and endocytic trafficking. After endocytosis, it is well established that EGF, but not TGF-α, triggers efficient degradation of the EGF receptors (Johnson et al. 1993), while AREG does not induce significant EGFR degradation and, the relatively low binding affinity of AREG may cause the preferential dissociation and recycling of EGFR to the cell surface (Roepstorff et al. 2009; Stern et al. 2008).
Therefore, AREG does not target EGFR for lysosomal degradation but does induce fast as well as slow EGFR recycling; researchers have demonstrated that AREG stimulation leads to intense EGFR ubiquitination, but no receptor degradation. This is most likely because ubiquitination is rapidly lost, and EGFR is not targeted to lysosomal degradation, as occurs, for example, for TGF-α (Roepstorff et al. 2009). These separate effects appear to be linked to the AREG-reduced receptor-binding affinity correlated with a decreased EGFR phosphorylation and altered EGFR signalling (Riese et al. 1996). Recently, in fact, it was reported that AREG and EGF differ greatly in their induction of EGFR tyrosine phosphorylation (Gilmore et al. 2008). The reduced level of phosphorylation of EGFR tyrosine residues could provide a mechanistic explanation for ligand-specific EGFR signalling. The AREG-associated defects in EGFR degradation may be due to the failure to recruit Cbl, ubiquitin ligases involved in EGFR trafficking to lysosomes, which was closely correlated with a transient association of EGFR with c-Cbl, in particular, and transient EGFR ubiquitination (Baldys et al. 2009). Therefore, c-Cbl appears to be critical to drive EGFR away from the lysosomal degradative pathway and toward the recycling pathway (Baldys et al. 2009).
The differential trafficking fate of EGFR imposed by AREG in comparison to other EGFR ligands has a marked impact on signalling kinetics and downstream pathways; in fact AREG induces, increased DNA synthesis activity leading to cellular proliferation mainly dependent on extracellular signal-regulated kinase (ERK)1/2 mediated upregulation of cyclin D1 (note that upregulation of cyclin D1 protein is a major regulatory step in the G1 phase of cell cycle) (Shin et al. 2003).
Another relevant aspect distinguishing AREG from other EGFR ligands is that AREG has a heparan sulphate-binding domain unlike EGF or TGF-α (Billings and Pacifici 2015; Johnson and Wong 1994) and it has been amply demonstrated that only the EGF-members able to bind heparan sulphate proteoglycans (HSPGs) have a mitogenic activity (Mahtouk et al. 2006). Studies on colon carcinoma cell lines as well as on human keratinocytes suggested that autocrine and paracrine signalling by AR may require cellular HSPG, presumably as matrix or membrane proteoglycans (Cook et al. 1991; Li et al. 1992; Piepkorn et al. 1994).
AREG/EGFR Signalling
The activation of EGFR by AREG can occur through autocrine, paracrine, and juxtacrine signalling modes. Autocrine and paracrine mechanisms require a soluble ligand released through protease activity. AREG has been shown to be an autocrine factor in normal human keratinocytes (Cook et al. 1991), in normal human bronchial epithelial cells and normal urothelial cells (Tsao et al. 1996; Varley et al. 2005). In addition, an autocrine activity of AREG has been observed in cancer cells, including hepatocellular, colon, gastric, breast, and pancreatic cancer cells (Akagi et al. 1995; Castillo et al. 2006; Culouscou et al. 1992; Funatomi et al. 1997; Johnson et al. 1992; Willmarth and Ethier 2006). AREG was additionally demonstrated to be an autocrine growth factor as well as a mitogen for fibroblasts, astrocytes, Schwann cells, and immune cells [dendritic cells (DCs), neutrophils, mast cells and lymphocytes] (Zaiss et al. 2015). Dong et al. (1999) provided the first evidence that in the case of autocrine signalling through the EGFR, conversion of the membrane-anchored ligand to a soluble form is necessary to observe a significant biological activity; blocking AREG and TGF-α shedding using a metalloprotease inhibitor decreased the migration and proliferation of the human mammary epithelial cell line demonstrating the importance of proteolytic cleavage by metalloproteases. Confirming these conclusions, several other studies have suggested that cleavage of the AREG precursor to its mature form is required for activation of the EGFR (Kansra et al. 2004). The in vivo relevance of AREG shedding to activate EGFR-mediated signalling during morphogenesis had been demonstrated in earlier studies involving TNF-α converting enzyme (TACE)−/− mice. TACE, also known as ADAM17, regulates the shedding of TNF-α (Moss et al. 1997). Phenotypes of these mutant miceresembled those of the EGFR−/− and TGF-α−/− mice as they had defects in the morphogenesis of epithelial structures and, in addition, showed deficient TGF-α shedding (Peschon et al. 1998). Interestingly, TACE−/− mice have defects in lung branching morphogenesis, heart valves and in mammary gland ductal branching (similar to AREG−/− mice). These studies first identified TACE as a sheddase for TGF-α, but also for AREG (Jackson et al. 2003; Luetteke et al. 1999). Several authors have shown, in addition, that ADAM17-dependent AREG shedding is positively regulated by G-protein coupled receptors (GPCRs) (Gschwind et al. 2003). EGFR transactivation involves GPCR-induced AREG cleavage, as has been documented using GPCR agonists (Schafer et al. 2004) (Fig. 4).

Multifunctional role of EGFR signalling pathway. a EGFR signalling initiates by the binding of EGF family members such as EGF, TGF-α, and AREG to the extracellular domain of EGFR activating multiple important pathways that include the Ras/mitogen-activating protein (MAP) kinase pathway, phosphatidylinositide 3-kinases/protein kinase B (PI3K/AKT), v-Src avian sarcoma viral oncogene homolog (Src) family kinases, and signal transducers and activator of transcription (STAT) proteins. Activation of these pathways lead to cell proliferation, survival, adhesion, migration and organogenesis. b Schematic model that shows epithelial-mesenchymal crosstalk and ADAM17-AREG-EGFR signalling in mammary development
In mammary gland development, among the several candidate paracrine factors which have been suggested to regulate hormone-induced proliferation and morphogenesis, AREG appears to be the major paracrine mediator of estrogen-mediated ductal morphogenesis (Ciarloni et al. 2007). Unlike the other EGFR ligands, AREG is upregulated in the mammary gland during ductal elongation, rendering this factor a key mediator of hormone-driven epithelial proliferation (Wiesen et al. 1999). In addition, different key points of female reproduction are controlled by the EGFR signalling system (Ashkenazi et al. 2005). A recent study, exploring the expression of AREG in follicular fluid (Inoue et al. 2009) and revealing its presence, demonstrated that bioactive AREG accumulation is induced by human chorionic gonadotropin and that levels of AREG in follicular fluid support both oocyte maturation and cumulus expansion (Negishi et al. 2007). Interestingly, AREG has been identified as a key paracrine signal released by cumulus cells, driving the synthesis of specific proteins in oocytes to allow them to develop as embryos (Chen et al. 2013). AREG is also detected in the uterine epithelium during the early phases of pregnancy, progesterone regulates its expression and so AREG promotes blastocyst implantation (Das 1995) and trophoblast differentiation (Lysiak et al. 1995) (Fig. 4).
Physiologically, AREG plays a role in the modulation of mophogenesis of several others tissues such as the lung, prostate, kidney (Lee et al. 1999; Schuger et al. 1996; Tørring et al. 1998) pancreas, cardiac muscle, testis, colon, breast, and spleen (Berasain and Avila 2014). Furthermore, AREG is active during the proliferation of human normal cells, including fibroblasts, urothelial cells, keratinocytes and lung bronchial epithelial cells (Varley et al. 2005; Willmarth and Ethier 2006) and is also involved in physiological processes such as nerve regeneration (Nilsson et al. 2005). The EGFR and its ligands have been reported, also to play an important role in bone biology and in regulating the anabolic actions of parathyroid hormone (PTH) (Schneider et al. 2009); AREG, in particular, has been identified as a PTH target gene both in vitro and in vivo (Qin et al. 2005). Increased AREG mRNA levels were, in fact, detected in PTH-treated mouse cells and in rat primary osteoblastic cells, as well as in the femora of rats injected with PTH (Qin et al. 2005). Recently, it was shown that, during normal bone development, the PTH-mediated release of AREG from osteoblastic cells acts on the EGFRs expressed on mesenchymal progenitors to stimulate the Akt and p38 mitogen-activated protein kinase (MAPK) pathways, and subsequently promotes their migration in vitro to the bone surface (Zhu et al. 2012).
Particularly noteworthy was the marked expression of mRNA and protein levels of AREG in the healthy normal human and rodent liver (Berasain et al. 2005; Webber et al. 1993). Furthermore, hepatic AREG levels are markedly upregulated in the hepatic acute phase response during systemic inflammation and regeneration, performing important interactions with the immune and inflammatory responses (Pardo-Saganta et al. 2009).
AREG also plays a central role in skin homeostasis (Stoll et al. 2016), since the gene silencing of AREG markedly inhibits the expansion of human keratinocytes through mitotic failure and the accumulation of cells with a ≥4n DNA content (Stoll et al. 2016), AREG strongly stimulates keratinocyte proliferation in wound healing (Stoll et al. 2010a, b), and contributes to the expression of antimicrobial peptides (Johnston et al. 2011).
While autocrine and paracrine modes of signalling involve the secretion of chemicals outside the cell membrane to affect the behavior of the same cell or of the neighboring cells, juxtacrine stimulation was triggered by a non-diffusable cell-surface ligand. The ability of the EGFR ligands to signal via juxtacrine interactions is more controversial. AREG has been shown to activate EGFR in a juxtacrine fashion (Inui et al. 1997; Willmarth and Ethier 2006), and was found to interact with CD9; the CD9-AREG complex might cooperate with the inactive membrane-anchored-form of AREG to induce human keratinocyte growth in a juxtacrine manner (Inui et al. 1997). Interestingly, the ability of AREG to activate the EGFR in a juxtacrine fashion in breast cancer cells has been demonstrated, and that this activation could be blocked through the use of an AREG neutralizing antibody (Willmarth and Ethier 2006).
AREG-Mediated Pathways
AREG binding to phosphorylated EGFR transmits signals through a variety of intracellular substrates. The Ras/Raf/MEK/ERK pathway is involved in AREG-mediated activity in various cell types including pancreatic duct cells, normal human keratinocytes, bronchial epithelial cells, bone and human salivary gland (Blanchet et al. 2004; Kansra et al. 2004; Qin et al. 2005; Sisto et al. 2015; Wagner et al. 2002). AREG/EGFR activation and ERK signalling rapidly stimulates Elk1, c-fos and c-jun expression, required for the proliferation of a wide variety of cells (Oda et al. 2005; Wang et al. 2005; Wong et al. 1999). The sustained activation of the intracellular ERK/Elk1 signalling pathway determines the upregulation of cyclin D1 protein. Cyclin D1 expression, required for cell cycle G1/S transition, is regulated by AREG in pancreatic duct cells (Wagner et al. 2002), in vascular smooth muscle cells, known to proliferate actively during the progression of atherosclerosis (Shin et al. 2003), and during lung cancer progression associated with increased AKT and STAT3 activation (Hsu et al. 2011). Regards the involvement of AKT and STAT in the AREG/EGFR/ERK pathways, using molecular and pharmacological approaches, considerable evidence was found demonstrating that, in cancer cells, the activation of the downstream phosphatidylinositol 3-kinase (PI3K)/Akt pathway occurs through heterodimer formation of EGFR with HER3 (Yotsumoto et al. 2010). This cascade promotes cell survival through the up regulation of active NF-κB; the recruitment of NF-κB to the intercellular adhesion molecules (ICAM) and interleukins (IL) promoter, up-regulates ICAM and pro-inflammatory interleukin protein expression (Liu et al. 2015; Streicher et al. 2007). In addition, several authors demonstrated that the induction of IL-8 and ICAM-1, for example, occurs in a STAT-dependent manner. STAT are key components of the AREG/EGFR signalling, since AREG has been shown to induce STAT 1, 3, and 5, and transcriptional targets of STAT molecules are genes associated with cell proliferation, differentiation, motility and apoptosis (David et al. 1996; Liu et al. 2008). In addition to these mechanisms, recently So et al. (2014) demonstrated a pro-invasive potential of AREG and elucidated the underlying molecular mechanisms showing an important effect of AREG in down-regulating E-cadherin and promoting the cell invasion. AREG activates the ERK1/2 and PI3K/AKT pathways, which induce expression of the E-cadherin transcriptional repressor SLUG and the subsequent loss of E-cadherin (So et al. 2014). By this way, AREG stimulates ovarian cancer cell invasion by down-regulating E-cadherin. Similarly, AREG has been reported to reduce E-cadherin levels and adherence in keratinocytes (Chung et al. 2005a) Moreover, AREG promotes a reduction in membrane-localized E-cadherin and a motile morphology in MDCK cells (Chung et al. 2005b), suggesting that E-cadherin is a common mediator of AREG-stimulated cell motility.
Immunomodulatory effects of AREG needs to be revised in the context of inflammatory conditions; for example, in response to inflammatory stimuli, basophils (Qi et al. 2010), eosinophils (Matsumoto et al. 2009), mast cells (Okumura et al. 2005; Wang et al. 2005; Zaiss et al. 2013), lymphoid cells (Monticelli et al. 2011; Salimi et al. 2013), DCs (Bles et al. 2010) and CD4+ T cells (Qi et al. 2012; Zaiss et al. 2006) respond with an increased expression of AREG. In this regard, in conditioned knockout mice lacking AREG no morphologic or homeostatic abnormalities were displayed (Luetteke et al. 1999), but, in these mice, the immunological reactions were impaired in inflammatory conditions (Berasain et al. 2005; Meulenbroeks et al. 2015; Perugorria et al. 2008; Zaiss et al. 2006) supporting the idea that AREG plays a critical role in restoring tissue integrity following infection or injury. Furthermore, multiple immune mediators including prostaglandin E2 (Berasain et al. 2005; Qi et al. 2012; Shao et al. 2003), cAMP (Johansson et al. 2004), IGF-1 (Rodland et al. 2008) and TGF-β (Bennett et al. 1992; Zhou et al. 2012) can determine an increased AREG expression, suggesting that the protein has the capacity to modulate inflammation in multiple cell types. However, in situations of chronic inflammation, excessive collagen deposition can lead to fibrosis and impaired organ function (Allen and Wynn 2011; Gause et al. 2013). Therefore, deregulation of the interaction between the immune system and AREG signalling pathways can contribute to pathological fibrosis in the context of chronic inflammation. This has been demonstrated in chronic airway diseases (Enomoto et al. 2009; Hirota et al. 2012; Qi et al. 2010; Val et al. 2012; Zhou et al. 2012), fibrosis of the liver (Berasain et al. 2005; McKee et al. 2015) and autoimmune diseases (Davies et al. 2005; Ishii et al. 2005; Kawasaki et al. 2003; Lisi et al. 2010, 2013, 2014b; Sisto et al. 2010, 2015; Yamane et al. 2008).
Since AREG contributes to the inflammatory condition by triggering the release of pro-inflammatory cytokines, it has been postulated that AREG-mediated chronic inflammation is involved in lung, breast, and colon tumorigenesis (Berasain et al. 2009; Elinav et al. 2013; Houghton 2013; Tye and Jenkins 2013) suggesting a key role of AREG the regulation of immune responses during carcinogenesis. Hsu et al. (2011), have discovered that DCs associated to lung tumors secrete large amounts of AREG and play a determinant role in promoting lung cancer progression. The treatment of mice with anti-AREG antibodies decreased the incidence of lung tumors and increased survival rates (Hsu et al. 2011). ATP, released from tumor cells and present in the tumor microenvironment, might exert a tumorigenic action by stimulating the secretion of AREG from DCs and promoting tumor progression (Bles et al. 2010). Additionally, AREG is up-regulated in primary lesions of colorectal cancer and metastatic tumors of the liver, suggesting that AREG contributes to the metastatic process and could be considered an important predictive marker of liver metastasis. The majority of liver injuries activate acute inflammatory responses, inducing AREG expression that leads to liver fibrogenesis, a pre-cancerous state, and consequently to the development of hepatocellular carcinoma. Thereby, it establishes an autocrine loop that preserves the neoplastic phenotype of liver cancer cells (Castillo et al. 2006, 2009; Desbois-Mouthon et al. 2006). Furthermore, in a model of cancer associated to colitis, AREG has also been shown to promote the progression and development, via TLR-4, of inflammation-associated colorectal tumors (Fukata et al. 2007). Recently, it has been demonstrated that inflammatory breast cancer cells, constitutively overexpressed active EGFR and AREG protein; the proposed mechanism for AREG/EGFR-dependent cellular invasion seems to depend on the altered expression of MMPs (Baillo et al. 2011) and other factors involved in matrix degradation: uPA, EMMPRIN, and PAI-1 (Giusti et al. 2003; Silvy et al. 2001).
AREG Control of Mammary Gland Morphogenesis
Studies performed on mammary gland were useful to learn how AREG carries out its function during exocrine glands development (Ciarloni et al. 2007). The mammary gland, as well as the branching observed in other organs, undergoes most of its orchestrated morphogenesis during puberty, pregnancy, and lactation when the subsequent expansion in response to the reproductive hormones estrogen, progesterone, and prolactin gives rise to alveolar structures within the branches to be used for milk production (LaMarca and Rosen 2007).
During embryogenesis, a rudimentary ductal system develops that grows isometrically leading to the formation of lactiferous ducts and their branches. These are already canalized by the end of prenatal life. At the time of birth only the main epithelial ducts are present, and these structures persist until puberty. At puberty, the distal end of the mammary ducts initiates branching morphogenesis, supported by growth hormone, estrogen, and IGF-1, to generate terminal end buds that fills the fat pad and create the typical branched duct system of the mature virgin gland. Terminal end-buds expand and increase greatly during this stage and, with consecutive menstrual cycles, the complexity of the milk duct system increases through the growth of lateral branches (Hinck and Silberstein 2005; Kenney et al. 1996). Side-branching is controlled by progesterone and intensifies during pregnancy (Shyamala 1999), when the alveoli become the sites of milk production, a process controlled by prolactin receptor signalling (Brisken 2002). Each phase of mammary branching, from embryonic through adolescent to adult, is regulated by specific hormones. Adolescent branching, for instance, requires the estrogen/estrogen receptor (ER)α receptor system, adult tertiary side-branching requires progesterone and its receptor (Cunha et al. 1997; Harris et al. 2003). Optimal mammary growth requires both estrogen and progesterone. Upon pregnancy, the combined actions of progesterone and prolactin are necessary to generate alveoli which secrete milk during lactation. Lack of demand for milk determines the involution of the gland, when the mammary epithelium regresses to its pre-pregnancy state (Macias and Hinck 2012).
These processes require numerous signalling pathways regulated by distinct enhancers that function at different stages of glandular development. AREG has been considered as a main paracrine regulator of ductal morphogenesis induced by estrogens (LaMarca and Rosen 2007) and the mitogenic effect of estrogens in the developing mammary gland is produced by an indirect mechanism due to the pattern of expression of the ER (Cunha et al. 1997; Woodworth et al. 1995). Current evidence supports the hypothesis that AREG expression is directly induced by estrogens acting as the key paracrine mediator of estrogen-stimulated pubertal ductal morphogenesis (McBryan et al. 2008). AREG is the only EGFR ligand whose expression has been demonstrated to increase strongly in the pubertal mammary gland paralleling ductal morphogenesis, an effect that is transcriptionally regulated by ovarian estrogen, and to decline during late pregnancy and lactation (McNally and Martin 2011; Schroeder and Lee 1998). To understand the role of AREG in normal mammary gland development, AREG gene knockout mice models have been generated and their mammary gland development has been studied (Ciarloni et al. 2007; Luetteke et al. 1999). Mice that lack AREG exhibit a slight ductal outgrowth and present only rudimentary ductal epithelial trees in comparison to wild-type glands of the same age, which are rich in terminal end bud structures (Ciarloni et al. 2007; Luetteke et al. 1999). These data suggested that although AREG is essential for pubertal ductal development, it is also dispensable for later stages of mammary gland development. Further transplantation experiments have been made to study ductal outgrowth induced by AREG, implanting the epithelium of one mouse group into the stroma of another. Wild-type epithelium grew, whereas AREG knockout epithelium showed a reduced growth in both wild-type and AREG knockout stromal compartments (Sternlicht et al. 2005). These experiments identify an essential requirement for AREG production in epithelial cells. Elaborate and elegant experiments of engrafted mammary glands by Ciarloni et al. (2007) have further suggested that AREG released from the AREG-positive cells acts locally to induce stromal growth factor(s) production, which in turn drives epithelial cell proliferation and contributes to all cell compartments of the ductal outgrowth (Sternlicht et al. 2005). Since AREG is the most strongly expressed EGFR ligand during pubertal mammary development, as a step toward understanding the requirements of the various EGFR ligands, intriguing experiments were made using EGF-or AREG-deficient mice and coupling to a TGF-α-null line, generating the various double and triple null mice lacking up to half of the EGFR ligand family. Analysis of these combination mutants confirms the importance of the EGFR system in both the developing and the differentiating mammary gland. Specifically, these data revealed a distinct and essential role for AREG in mammary ductal morphogenesis, and suggested the roles for EGF and TGF-α in lactogenesis (Luetteke et al. 1999). Therefore, during pubertal mammary gland development other members of the EGFR family of receptors may contribute to AREG action. In fact, a role for ErbB2 and ErbB3 has been demonstrated in ductal morphogenesis of the pubertal mammary gland. Transplantation studies, using day E13.5 mammary rudiments from ErbB2 gene knockout mice, showed significant delays in ductal elongation at puberty (Jackson-Fisher et al. 2004). In addition, ErbB3 deficient mice displayed low mammary ductal density and few branches (Qu et al. 2006). A plethora of molecular signals cooperate to execute mammary morphogenesis through a crosstalk between epithelial and stromal cells (Brisken and O’Malley 2010). Recent recombination studies have shown that ERα regulates the genetic program of growth in mammary glands in response to circulating ovarian hormones, and is required in the mammary stroma that subsequently exerts its effect on the epithelium through additional paracrine signalling events. Indeed, ERα knockout mice present a rudimental epithelial tree (Bocchinfuso and Korach 1997; Feng et al. 2007), whereas exogenous estrogen can rescue ductal development in ovariectomized mice (Gjorevski and Nelson 2011). ESR1 is the major intracellular estrogen receptors operating during ductal morphogenesis and knockout ESR1 mice show a hypoplastic development of the ductal system (Casimiro et al. 2013; Lubahn et al. 1993). AREG is currently the leading candidate for the factor that is released by ESR1-positive cells and signals cell proliferation. These observations were confirmed by conditioned targeted ablation of ESR1 in alveoli following ductal elongation, resulting in defective lobule alveolar development of mammary gland, and an insufficient milk supply (Feng et al. 2007). Particular interest was aroused by the identification of ATBF1, a candidate tumor suppressor that interacts with ER to inhibit the function of estrogen/ER signalling in gene regulation, cell proliferation, and one of the regulators controlling pubertal mammary gland development (Dong et al. 2010). During mammary gland development ATBF1 expression is dynamic, suggesting various roles in different stages of developing mammary gland; it, is likely more relevant to puberty and lactation. ATBF1 gene knockdown determined ductal elongation and bifurcation in pubertal mammary glands, which was indicated by extended ductal invasion, total ductal branching, and significantly upregulated the expression of AREG. Since AREG is a key mediator of the estrogen-driven epithelial cell proliferation and ductal elongation at puberty, it is possible that AREG mediates the acceleration of mammary gland branching and bifurcation upon the deletion of ATBF1 (Li et al. 2012).
Recently, authors have identified RIP140 as a co-factor that recruited together with ERα promoters a number of regulatory genes such as AREG, thereby stimulating their transcription and so regulating mammary gland development. For instance, when RIP140 expression is loss, there is lack of AREG expression leading to profound developmental defects and subsequently to altered mammary development (Nautiyal et al. 2013).
Recent interesting studies have provided evidence that cyclin D1, that regulates estrogen receptor ERα transactivation, participates in estrogen-regulated gene expression in vivo, governing growth factor and cytokine signalling. Growth factors induced by estrogens (E2) in a cyclin D1-dependent manner include AREG. Moreover, recent works have extended these findings and have demonstrated that the induction of AREG expression by E2 was abrogated in mice lacking cyclin D1. Endogenous cyclin D1 contributed to a 17-fold increase in E2-mediated AREG expression (Casimiro et al. 2013). In addition, recombination tissue experiments have shown that estrogen binds to ERα in the epithelium, thereby inducing the expression of AREG (Ciarloni et al. 2007; Coleman et al. 1988; Luetteke et al. 1999; Sebastian et al. 1998; Sternlicht et al. 2005), and its cleavage from the surface by the sheddase ADAM17 (Sternlicht et al. 2005). Cleaved AREG can influence the activity of stromal cells by binding to EGFR on the cell membrane (Fig. 4). The expression of EGFR is a key element in the stromal compartment (Wiesen et al. 1999); in fact, exogenous addition of EGFR ligands can rescue pubertal development of ovariectomized animals (Sternlicht 2006), which is consistent with the fact that EGFR initiates some common signalling pathways downstream of estrogen. Therefore, on the EGFR ligand side, only AREG-deficient mice showed significant defects in mammary gland development, confirming AREG as a central mediator of estrogen function (Luetteke et al. 1999). Links between the EGFR-activated pathways and mammary gland pathophysiology are further supported by the observation of a marked EGFR overexpression in breast cancer; transcripts for EGFR ligands such as EGF and AREG are frequently upregulated in human breast cancer biopsy samples and the expression of AREG has been associated with estrogen receptor positive breast cancer (Peterson et al. 2013). Little is known about intracellular signalling pathways triggered by AREG activity downstream of EGFR family activation. Interestingly, substantial evidence supports a critical role for AREG in the activation of the Ras/MAPK and PI3K/Akt pathways (Fig. 4). For instance, AREG has been shown to have an increased mitogenic potency on vascular smooth muscle cells in which it has been shown to activate a signal transduction pathway that includes the PI3K/Akt system, the p42/p44MAPK and the p38MAPK pathway (Kato et al. 2003). Furthermore, AREG is dispensable for the action of PTH that is able to increase EGFR phosphorylation in mesenchymal progenitors of osteoblasts. Zhu et al. (2012) have recently demonstrated that PTH increases the release of AREG from osteoblastic cells, which acts on the EGFRs expressed on mesenchymal progenitors to activate the Akt and p38MAPK pathways and subsequently promote their migration in vitro, suggesting a novel mechanism for the therapeutic effect of PTH on osteoporosis. It is interesting to note that similar pathways were documented in the pubertal mouse mammary gland, where high AREG levels were identified during epithelial cell proliferation of ductal morphogenesis, ErbB2 activation and Akt phosphorylation (LaRocca et al. 2011).
Signalling Moderation: the EGFR System in the Salivary Gland Disease Sjögren’s Syndrome
EGF and EGFR exert tropic effects on SGEC from SS patients, as reported by several authors (Gorgoulis et al. 1993; Nakamura et al. 2007) and resulted densely expressed in the epithelial duct cells, particularly in the areas of lymphocytic infiltration and tissue destruction. Nakamura et al. (2007) recently found a fundamental role of EGF in the anti-apoptotic and defence mechanisms activated in SGEC derived from SS patients. In fact, although SGEC apoptosis is increased in the SGs of SS patients (Fox et al. 2000), restoration mechanisms seem to be active in epithelial cells (Ohlsson et al. 2002). In this context, the defensive mechanism can be up-regulated by the augmented expression of the EGF/EGFR system, that is responsible for the activation of the classical anti-apoptogenic intracellular kinase cascades in which PI3K-Akt activates downstream IκB kinase leading to the phosphorylation and activation of the p65 subunit of NF-κB (Anest et al. 2004; Franke et al. 2003; Viatour et al. 2005) (Fig. 4).
In agreement with the investigators focusing on the fundamental role of the EGF/EGFR system activation in SS, others authors have identified new EGF-mediated signalling pathways that are active in pSS. Sisto et al. (2015) recently reported that the activation of this system in pSS involves metalloproteinase ADAM17. The mechanism individuated, on the basis of experimental studies conducted in vitro on human SGEC derived from SS salivary gland biopsies, demonstrated that EGFR is a potent activator of the ERK1/ERK2, also known as MAPK3/MAPK1 pathway. Clarifying the signal transduction pathways, the authors reported that the EGFR-mediated activation of the downstream effectors ERK1/2 in pSS SGEC appeared to require ADAM17-dependent release of the endogenous EGFR ligand AREG and transactivation of the EGFR. Moreover, blockade of AREG bioactivity using a neutralizing antibody significantly reduced EGFR transactivation and ERK1/2 phosphorylation. In addition, pSS SGEC treated with the specific ADAM17 inhibitor TAPI-1 and with the EGFR inhibitor AG1478 exhibited deactivated AREG/EGFR/ERK signalling pathway and reduced pro-inflammatory cytokines released (Sisto et al. 2015) (Fig. 5). A review of the literature indicates that these assumptions are well supported by others studies in the autoimmunity field which suggest that an altered EGF/EGFR/ERK pathway is involved in the exacerbation of the chronic inflammatory condition characterizing autoimmune disorders, such as systemic lupus erythematosus and psoriasis (Mascia et al. 2003; Sawalha et al. 2008). Also in rheumatoid arthritis, investigators demonstrated that positive feedback loops of the ERK pathway, shedding of EGFR ligands and subsequent EGFR activation lead to cytokines production to boost the inflammatory response (Singh et al. 2009).

Schematic overview of the ADAM17/AREG/EGFR signalling pathway activated in Sjögren’s syndrome (SS) as reported in the text. The abbreviation SS Abs indicates serum autoantibodies characterizing Sjögren’s syndrome
Equally important is the discovery that the EGF/EGFR system cooperates in promoting nerve growth factor (NGF)-β release by pSS SGEC, since NGF-β acts as a pro-inflammatory neurokine in addition to its neurotrophic effect. This seems to occurs through ERK1/2 phosphorylation which leads to downstream Raf-1 and MEK activation (Lisi et al. 2014a). The Raf-1/MEK/ERK cascade is one of the major and best studied EGFR downstream pathways that couple signals from cell surface receptors with transcription factors, modulating genes expression involved in apoptosis or inflammatory reactions (Dunn et al. 2005; Yoon and Seger 2006). In vitro studies demonstrated that, when healthy SGEC cultures were exposed to additional TNF-α or IL-6 treatment, an enhanced NGF-β release was detected, while perturbing the Raf-1/MEK/ERK pathway using specific pharmacological MEK and Raf-1 inhibitors, a decrement in EGF-dependent NGF-β production was observed (Lisi et al. 2014a). This experimental hypothesis was tested also in pSS SGEC demonstrating that EGFR gene silencing inhibited ERK1/2 phosphorylation and NGF-β secretion in pSS SGEC, suggesting that pro-inflammatory cytokines release by infiltrating lymphocytes and pSS SGEC enhances NGF-β production via the EGFR/Raf-1/MEK/ERK pathway, and this, on the other hand, allows NGF to promote the release of inflammatory mediators (Fig. 5).
Most of the data supporting the relevance of the EGF/EGFR system in pSS derived from preliminary experiments conducted on pSS SGEC show a strong overexpression of AREG in salivary gland biopsies derived from pSS patients (Sisto et al. 2010) and a concomitant elevated expression of the active ADAM17 in the same samples (Lisi et al. 2010). The activation of AREG occurs through ADAM17-mediated shedding and active AREG modulates the ADAM17-mediated TNF-α signalling pathway, that has a significant role in the development of the pro-inflammatory state in pSS (Lisi et al. 2010). Cytokines are key molecules in systemic inflammation and contribute to the systemic complications of SS (Roescher et al. 2009) that result in cumulative damage to the SGs, impairing the secretory function (Lisi et al. 2010). The chronic inflammation observed in SS patients reflects an imbalance of cytokines expression both locally in the glands and systemically in the blood. From the works carried out by Sisto et al. (2010) it seems that the activation of the ADAM17/TNF-α/AREG axis is dependent on the presence in pSS serum of the anti-Ro/SSA autoantibodies (Abs) characterizing pSS. The gene silencing technique showed that TNF-α gene knockdown provokes a significant decrease of both pro-inflammatory cytokines production and AREG expression in healthy SGEC following anti-Ro/SSA Abs treatment (Lisi et al. 2010). Furthermore, AREG gene silencing determines a strong inhibitory effect on TNF-α-induced IL-6 and IL-8 secretion in healthy SGEC treated with anti-Ro/SSA Abs (Lisi et al. 2010) (Fig. 5).
Data obtained on AREG expression in pSS SGEC were supported by findings of Kawasaki et al. (2003) that demonstrated an up-regulated AREG gene expression in the conjunctival epithelium of patients with SS. The abnormal differentiation and keratinization that occur in the conjunctival epithelial cells of SS patients may be attributable to the up-regulation of AREG and/or c-fos through a mechanism involving increased interferon γ production, a critical factor in the pathogenesis of the ocular surface inflammation presented by patients with SS (Kawasaki et al. 2003). All these findings raise the possibility that specific EGF/EGFR/AREG inhibitors may be of therapeutic value for treating the chronic inflammation characterizing SS disease. The precise mechanisms by which EGFR-targeted treatments mediate their objective clinical responses in SS patients has remained, to date, unexplored, and further targeted research to determine the precise role of the immune system in the clinical successes of EGFR targeted treatments is needed. Our data have identified the ADAM17/AREG system and its dependent pathways as possible new targets in the prevention or treatment of SS.
Concluding Remarks
As can be inferred from the information collected in two decades of basic and clinical investigations, AREG is a very interesting molecule with a broad range of biological activities. In addition to its role in the development of specific organs such as the mammary gland, oocyte maturation and bone development, AREG resulted a key player in tissue repair, cell proliferation and inflammation. In this context, great strides have recently been made in our understanding of the regulation of AREG gene expression, AREG protein tissue distribution and AREG interaction with other signalling molecules involved in the EGFR-mediated molecular pathways. In particular, the role of AREG in the control of cell responses, and the molecular basis of ligand specificity in EGFR signalling have been clarified. It is now widely recognized that AREG’s major role is in the regulation of cell proliferation and it may function as a stimulatory or inhibitory growth factor depending on the phenotype of and environment surrounding a given cell, both normal and transformed. In addition, the identification of AREG as a new modulator of inflammation and autoimmunity opens new therapeutic avenues for the pharmacological modulation of immune responses and this, together with AREG involvement in tissue injury and cancer, could justify the development of AREG neutralizing strategies to disrupt AREG-mediated molecular pathways. So, AREG gene silencing by small interfering RNAs (siRNA), inhibition of AREG by neutralizing antibodies and the block of shedding of AREG using ADAM17 inhibitors or anti-ADAM17 siRNA are just some of the therapeutic options that are being explored in recent years. The clinical feasibility and relevance of such studies, although promising, remain, however, uncertain and future investigations will have to be carefully designed to explore the value of AREG as a biomarker in the identification of patients potentially responsive to targeted therapies.
References
Ahlgren U, Pfaff SL, Jessell TM et al (1997) Independent requirement for ISL1 in formation of pancreatic mesenchyme and islet cells. Nature 385:257–260
Akagi M, Yokozaki H, Kitadai Y et al (1995) Expression of amphiregulin in human gastric cancer cell lines. Cancer 75:1460–1466
Allen JE, Wynn TA (2011) Evolution of Th2 immunity: a rapid repair response to tissue destructive pathogens. PLoS Pathol 7:e1002003
Andl CD, Fargnoli BB, Okawa T et al (2006) Coordinated functions of E-cadherin and transforming growth factor beta receptor II in vitro and in vivo. Cancer Res 66:9878–9885
Andrew DJ, Ewald AJ (2010) Morphogenesis of epithelial tubes: insights into tube formation, elongation, and elaboration. Dev Biol 341:34–55
Anest V, Cogswell PC, Baldwin AS Jr (2004) IkappaB kinase alpha and p65/RelA contribute to optimal epidermal growth factor-induced c-fos gene expression independent of IkappaBalpha degradation. J Biol Chem 279:31183–31189
Ashkenazi H, Cao X, Motola S et al (2005) Epidermal growth factor family members: endogenous mediators of the ovulatory response. Endocrinology 146:77–84
Baillo A, Giroux C, Ethier SP (2011) Knock-down of amphiregulin inhibits cellular invasion in inflammatory breast cancer. J Cell Physiol 226:2691–2701
Baldys A, Göoz M, Morinelli TA et al (2009) Essential role of c-Cbl in amphiregulin-induced recycling and signaling of the endogenous epidermal growth factor receptor. Biochemistry 48:1462–1473
Bennett KL, Plowman GD, Buckley SD et al (1992) Regulation of amphiregulin mRNA by TGF-beta in the human lung adenocarcinoma cell line A549. Growth Factors 7:207–213
Berasain C, Avila MA (2010) AREG (amphiregulin (schwannoma-derived growth factor)). Atlas Genet Cytogenet Oncol Haematol 14:1–6
Berasain C, Avila MA (2014) Amphiregulin. Semin Cell Dev Biol 28:31–41
Berasain C, García-Trevijano ER, Castillo J et al (2005) Amphiregulin: an early trigger of liver regeneration in mice. Gastroenterology 128:424–432
Berasain C, Castillo J, Perugorría MJ (2009) Inflammation and liver cancer. Ann NY Acad Sci 1155:206–221
Biggs LC, Mikkola ML (2014) Early inductive events in ectodermal appendage morphogenesis. Semin Cell Dev Biol 25–26:11–21
Billings PC, Pacifici M (2015) Interactions of signaling proteins, growth factors and other proteins with heparan sulfate: mechanisms and mysteries. Connect Tissue Res 56:272–280
Blanchet S, Ramgolam K, Baulig A et al (2004) Fine particulate matter induces amphiregulin secretion by bronchial epithelial cells. Am J Respir Cell Mol Biol 30:421–427
Bles N, Di Pietrantonio L, Boeynaems JM et al (2010) ATP confers tumori-genic properties to dendritic cells by inducing amphiregulin secretion. Blood 116:3219–3226
Bocchinfuso WP, Korach KS (1997) Mammary gland development and tumorigenesis in estrogen receptor knockout mice. J Mammary Gland Biol Neoplasia 2:323–334
Brantley DM, Chen CL, Muraoka RS et al (2001) Nuclear factor-kappaB (NF-kappaB) regulates proliferation and branching in mouse mammary epithelium. Mol Biol Cell 12:1445–1455
Brisken C (2002) Hormonal control of alveolar development and its implications for breast carcinogenesis. J Mammary Gland Biol Neoplasia 7:39–48
Brisken C, O’Malley B (2010) Hormone action in the mammary gland. Cold Spring Harb Perspect Biol 2:a003178
Brown CL, Meise KS, Plowman GD et al (1998) Cell surface ectodomain cleavage of human amphiregulin precursor is sensitive to a metalloprotease inhibitor. Release of a predominant N-glycosylated 43-kDa soluble form. J Biol Chem 273:17258–17268
Busser B, Coll JL, Hurbin A (2009) The increasing role of amphiregulin in non-small cell lung cancer. Pathol Biol 57:511–512
Busser B, Sancey L, Brambilla E et al (2011) The multiple roles of amphiregulin in human cancer. Biochim Biophys Acta 1816:119–131
Casimiro MC, Wang C, Li Z et al (2011) Cyclin D1 determines estrogen signaling in the mammary gland in vivo. Mol Endocrinol 27:1415–1428
Castillo J, Erroba E, Perugorría MJ et al (2006) Amphiregulin contributes to the transformed phenotype of human hepato-cellular carcinoma cells. Cancer Res 66:6129–6138
Castillo J, Goni S, Latasa MU et al (2009) Amphiregulin induces the alternative splicing of p73 into its oncogenicisoform DeltaEx2p73 in human hepatocellular tumors. Gastroenterology 137:1805–1815
Chen J, Diacovo TG, Grenache DG et al (2002) The a2 integrin subunit-deficient mouse: a multifaceted phenotype including defects of branching morphogenesis and hemostasis. Am J Pathol 161:337–344
Chen J, Torcia S, Xie S et al (2013) Somatic cells regulate maternal mRNA translation and developmental competence of mouse oocytes. Nat Cell Biol 15:1415–1423
Chulam TC, Noronha Francisco AL, Goncalves Filho J et al (2013) Warthin’s tumour of the parotid gland: our experience. Acta Otorhinolaryngol Ital 33:393–397
Chung E, Cook PW, Parkos CA et al (2005a) Amphiregulin causes functional down regulation of adherens junctions in psoriasis. J Invest Dermatol 124:1134–1140
Chung E, Graves-Deal R, Franklin JL et al (2005b) Differential effects of amphiregulin and TGF-alpha on the morphology of MDCK cells. Exp Cell Res 309:149–160
Ciarloni L, Mallepell S, Brisken C (2007) Amphiregulin is an essential mediator of estrogen receptor alpha function in mammary gland development. Proc Natl Acad Sci USA 104:5455–5460
Coleman S, Silberstein GB, Daniel CW (1988) Ductal morphogenesis in the mouse mammary gland: evidence supporting a role for epidermal growth factor. Dev Biol 127:304–315
Cook PW, Mattox PA, Keeble WW et al (1991) A heparin sulfate-regulated human keratinocyte autocrine factor is similar or identical to amphiregulin. Mol Cell Biol 11:2547–2557
Cowin P, Wysolmerski J (2010) Molecular mechanisms guiding embryonic mammary gland development. Cold Spring Harb Perspect Biol 2:a003251
Culouscou JM, Remacle-Bonnet M, Carlton GW et al (1992) Colorectum cell-derived growth factor (CRDGF) is homologous to amphiregulin, a member of the epidermal growth factor family. Growth Factors 7:195–205
Cunha GR, Young P, Hom YK et al (1997) Elucidation of a role for stromal steroid hormone receptors in mammary gland growth and development using tissue recombinants. J Mammary Gland Biol Neoplasia 2:393–402
Daniel CW, Strickland P, Friedmann Y (1995) Expression and functional role of E- and P-cadherins in mouse mammary ductal morphogenesis and growth. Dev Biol 169:511–519
Das SK (1995) Amphiregulin is an implantation-specific and progesterone-regulated gene in the mouse uterus. Mol Endocrinol 9:691–705
Davenport TG, Jerome-Majewska LA, Papaioannou VE (2003) Mammary gland, limb and yolk sac defects inmice lacking Tbx3, the gene mutated in human ulnar mammary syndrome. Development 130:2263–2273
David M, Wong L, Flavell R et al (1996) STAT activation by epidermal growth factor (EGF) and amphiregulin. Requirement for the EGF receptor kinase but not for tyrosine phosphorylation sites or JAK1. J Biol Chem 271:9185–9188
Davidson EH, Rast JP, Oliveri P et al (2002) A genomic regulatory network for development. Science 295:1669–1678
Davies JA (2002) Do different branching epithelia use a conserved developmental mechanism? BioEssays 24:937–948
Davies MR, Harding CJ, Raines S et al (2005) Nurr1 dependent regulation of pro-inflammatory mediators in immortalised synovial fibroblasts. J Inflamm 25:2–15
de la Cuadra-Blanco C, Peces-Peña MD, Mérida-Velasco JR (2003) Morphogenesis of the human lacrimal gland. J Anat 203:531–536
De Moerlooze L, Spencer-Dene B, Revest JM et al (2000) An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signaling during mouse organogenesis. Development 127:483–492
Delaleu N, Jonsson R, Koller MM (2005) Sjogren’s syndrome. Eur J Oral Sci 113:101–113
Desbois-Mouthon C, Cacheux W, Blivet-Van Eggelpoël MJ et al (2006) Impact of IGF-1R/EGFR cross-talks on hepatoma cell sensitivity to gefitinib. Int J Cancer 119:2557–2566
Dong C, Hemminki K (2001) Second primary neoplasms among 53 159 haematolymphoproliferative malignancy patients in Sweden, 1958–1996: a search for common mechanisms. Br J Cancer 85:997–1005
Dong J, Opresko LK, Dempsey PJ et al (1999) Metalloprotease-mediated ligand release regulates autocrine signaling through the epidermal growth factor receptor. Proc Natl Acad Sci USA 96:6235–6240
Dong XY, Sun X, Guo P et al (2010) ATBF1 inhibits estrogen receptor (ER) function by selectively competing with AIB1 for binding to the ER in ER-positive breast cancer cells. J Biol Chem 285:32801–32809
Duffy MJ, McKiernan E, O’Donovan N et al (2009) Role of ADAMs in cancer formation and progression. Clin Cancer Res 15:1140–1144
Dunn KL, Espino PS, Drobic B et al (2005) The Ras-MAPK signal transduction pathway, cancer and chromatin remodeling. Biochem Cell Biol 83:1–14
Elinav E, Nowarski R, Thaiss CA et al (2013) Inflammation-inducedcancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 13:759–771
Enomoto Y, Orihara K, Takamasu T et al (2009) Tissue remodeling induced by hypersecreted epidermal growth factor and amphiregulin in the airway after an acute asthma attack. J Allergy Clin Immunol 124:913–920
Faraci-Orf E, McFadden C, Vogel WF (2006) DDR1 signaling is essential to sustain Stat5 function during lactogenesis. J Cell Biochem 97:109–121
Fata JE, Werb Z, Bissell MJ (2004) Regulation of mammary gland branching morphogenesis by the extracellular matrix and its remodeling enzymes. Breast Cancer Res 6:1–11
Feng Y, Manka D, Wagner KU et al (2007) Estrogen receptor-α expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc Natl Acad Sci USA 104:14718–14723
Fischer AH, Jacobson KA, Rose J et al (2008) Hematoxylin and eosin staining of tissue and cell sections. CSH Protoc 2008:pdb.prot4986
Fonseca FP, Carvalho Mde V, de Almeida OP et al (2012) Clinicopathologic analysis of 493 cases of salivary gland tumors in a Southern Brazilian population. Oral Surg Oral Med Oral Pathol Oral Radiol 114:230–239
Fox RI, Stern M, Michelson P (2000) Update in Sjögren’s syndrome. Curr Opin Rheumtol 12:391–398
Franke TF, Hornik CP, Segev L et al (2003) PI3K/Akt and apoptosis: size matters. Oncogene 22:8983–8998
Fukata M, Chen A, Vamadevan AS et al (2007) Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 133:1869–1881
Funatomi H, Itakura J, Ishiwata T et al (1997) Amphiregulin antisense oligonucleotide inhibits the growth of T3M4 human pancreatic cancer cells and sensitizes the cells to EGF receptor-targeted therapy. Int J Cancer 72:512–517
Gardner TS, di Bernardo D, Lorenz D et al (2003) Inferring genetic networks and identifying compound mode of action via expression profiling. Science 301:102–105
Gause WC, Wynn TA, Allen JE et al (2013) Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat Rev Immunol 13:607–614
Gilmore JL, Scott JA, Bouizar Z et al (2008) Amphiregulin-EGFR signaling regulates PTHrP gene expression in breast cancer cells. Breast Cancer Res Treat 110:493–505
Giusti C, Desruisseau S, Ma L et al (2003) Transforming growth factor beta-1 and amphiregulin act in synergy to increase the production of urokinase-type plasminogen activator in transformed breast epithelial cells. Int J Cancer 105:769–778
Gjorevski N, Nelson CM (2011) Integrated morphodynamic signalling of the mammary gland. Nat Rev Mol Cell Biol 12:581–593
Gonzalez S, Aguilera S, Urzua U et al (2011) Mechanotransduction and epigenetic control in autoimmune diseases. Autoimmun Rev 10:175–179
Gorgoulis V, Giatromanolaki A, Iliopoulos A et al (1993) EGF and EGF-R immunoexpression in Sjögren’s syndrome secondary to rheumatoid arthritis. Correlation with EBV expression? Clin Exp Rheumatol 11:623–627
Gresik EW, Koyama N, Hayashi T et al (2009) Branching morphogenesis in the fetal mouse submandibular gland is codependent on growth factors and extracellular matrix. J Med Invest 56(Suppl):228–233
Grobstein C (1953) Morphogenetic interaction between embryonic mouse tissues separated by a membrane filter. Nature 172:869–870
Gschwind A, Hart S, Fischer OM et al (2003) TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO J 22:2411–2421
Gu G, Brown JR, Melton DA (2003) Direct lineage tracing reveals theontogeny of pancreatic cell fates during mouse embryogenesis. Mech Dev 120:35–43
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation cell. Cell 144:646–674
Harris RC, Chung E, Coffey RJ (2003) EGF receptor ligands. Exp Cell Res 284:2–13
Harunaga J, Hsu JC, Yamada KM (2011) Dynamics of salivary gland morphogenesis. J Dent Res 90:1070–1077
Hauser BR, Hoffman MP (2015) Regulatory mechanisms driving salivary gland organogenesis. Curr Top Dev Biol 115:111–130
Headington JT, Batsakis JG, Beals TF et al (1977) Membranous basal cell adenoma of parotid gland, dermal cylindromas and trichoepitheliomas. Comparative histochemistry and ultrastructure. Cancer 39:2460–2469
Hennighausen L, Robinson GW (2001) Signaling pathways in mammary gland development. Dev Cell 1:467–475
Hens JR, Wysolmerski JJ (2005) Key stages of mammary gland development: Molecular mechanisms involved in the formation of the embryonic mammary gland. Breast Cancer Res 7:220–224
Hinck L, Silberstein GB (2005) Key stages in mammary gland development: the mammary end bud as a motile organ. Breast Cancer Res 7:245–251
Hiremath M, Wysolmerski J (2013) Parathyroid hormone-related protein specifies the mammary mesenchyme and regulates embryonic mammary development. J Mammary Gland Biol Neoplasia 18:171–177
Hirota N, Risse PA, Novali M et al (2012) Histamine may induce airway remodeling through release of epidermal growth factor receptor ligands from bronchial epithelial cells. FASEB J 26:1704–1716
Hoffman MP, Kidder BL, Steinberg ZL et al (2002) Gene expression profiles of mouse submandibular gland development: FGFR1 regulates branching morphogenesis in vitro through BMP- and FGF-dependent mechanisms. Development 129:5767–5778
Hogan BL (1999) Morphogenesis. Cell 96:225–233
Houghton AM (2013) Mechanistic links between COPD and lung cancer. Nat Rev Cancer 13:233–345
Hsu YL, Huang MS, Cheng DE et al (2011) Lung tumor-associated dendritic cell-derived amphiregulin increased cancer progression. J Immunol 187:1733–1744
Hynes NE, Watson CJ (2010) Mammary gland growth factors: roles in normal development and in cancer. Cold Spring Harb Perspect Biol 2:a003186
Iber D, Menshykau D (2013) The control of branching morphogenesis. Open Biol 3:130088
Iezzoni JC, Gaffey MJ, Weiss LM (1995) The role of Epstein-Barr virus in lymphoepithelioma-like carcinomas. Am J Clin Pathol 103:308–315
Inoue Y, Miyamoto S, Fukami T et al (2009) Amphiregulin is much more abundantly expressed than transforming growth factor-alpha and epidermal growth factor in human follicular fluid obtained from patients undergoing in vitro fertilization-embryo transfer. Fertil Steril 91:1035–1041
Inui S, Higashiyama S, Hashimoto K et al (1997) Possible role of coexpression of CD9 with membrane-anchored heparin-binding EGF-like growth factor and amphiregulin in cultured human keratinocyte growth. J Cell Physiol 171:291–298
Ishii T, Onda H, Tanigawa A et al (2005) Isolationand expression profiling of genes upregulated in the peripheral blood cells of systemic lupus erythematosus patients. DNA Res 12:429–439
Jackson LF, Qiu TH, Sunnarborg SW et al (2003) Defective valvulogenesis in HB-EGF and TACE-null mice is associated with aberrant BMP signaling. EMBO J 22:2704–2716
Jackson-Fisher AJ, Bellinger G, Ramabhadran R et al (2004) ErbB2 is required for ductal morphogenesis of the mammary gland. Proc Natl Acad Sci USA 101:17138–17143
Jaskoll T, Melnick M (1999) Submandibular gland morphogenesis: stage-specific expression of TGF-alpha, EGF, IGF, TGF-beta, TNF and IL-6 signal transduction in normal mice and the phenotypic effects of TGF-beta2, TGF-beta3, and EGF-R null mutations. Anat Rec 256:252–268
Jaskoll T, Zhou YM, Chai Y et al (2002) Embryonic submandibular gland morphogenesis: stage-specific protein localization of FGFs, BMPs, Pax 6 and Pax 9 and abnormal SMG phenotypes in Fgf/R2-IIIc+/Δ, BMP7−/− and Pax6−/− mice. Cells Tissues Organs 170:83–98
Jaskoll T, Witcher D, Toreno L et al (2004) FGF8 dose-dependent regulation of embryonic submandibular salivary gland morphogenesis. Dev Biol 268:457–469
Jeffers L, Webster-Cyriaque JY (2011) Viruses and salivary gland disease (SGD): lessons from HIV SGD. Adv Dent Res 23:79–83
Johansson CC, Yndestad A, Enserink JM et al (2004) The epidermal growth factor-like growth factor amphiregulin is strongly induced by the adenosine 3′,5′-monophosphate pathway in various cell types. Endocrinology 145:5177–5184
Johnson GR, Wong L (1994) Heparan sulfate is essential to amphiregulin-induced mitogenic signaling by the epidermal growth factor receptor. J Biol Chem 269:27149–27154
Johnson GR, Saeki T, Gordon AW et al (1992) Autocrine action of amphiregulin in a colon carcinoma cell line and immunocytochemical localization of amphiregulin in human colon. J Cell Biol 118:741–751
Johnson GR, Kannan B, Shoyab M (1993) Amphiregulin induces tyrosine phosphorylation of the epidermal growth factor receptor and p185 erbB2. Evidence that amphiregulin acts exclusively through the epidermal growth factor receptor at the surface of human epithelial cells. J Biol Chem 268:2924–2931
Johnston JE, Gudjonsson A, Aphale AM et al (2011) EGFR and IL-1 signaling synergistically promote keratinocyte antimicrobial defenses in a differentiation-dependent manner. J Invest Dermatol 131:329–337
Kadoya Y, Yamashina S (2005) Salivary gland morphogenesis and basement membranes. Anat Sci Int 80:71–79
Kansra S, Stoll SW, Johnson JL et al (2004) Autocrine extracellular signal-regulated kinase (ERK) activation in normal human keratinocytes: metalloproteinase-mediated release of amphiregulin triggers signaling from ErbB1 to ERK. Mol Biol Cell 15:4299–4309
Kashimata M, Gresik EW (1997) Epidermal growth factor system is a physiological regulator of development of the mouse fetal submandibular gland and regulates expression of the alpha6-integrin subunit. Dev Dyn 208:149–161
Kashimata MW, Sakagami HW, Gresik EW (2000a) Intracellular signaling cascades activated by the EGF receptor and/or integrins, with potential relevance for branching morphogenesis of the fetal mouse submandibular gland. Eur J Morphol 38:269–275
Kashimata M, Sayeed S, Ka A et al (2000b) The ERK-1/2 signaling pathway is involved in the stimulation of branching morphogenesis of fetal mouse submandibular glands by EGF. Dev Biol 220:183–196
Kato M, Inazu T, Kawai Y et al (2003) Amphiregulin is a potent mitogen for the vascular smooth muscle cell line, A7r5. Biochem Biophys Res Commun 301:1109–1115
Kawasaki S, Kawamoto S, Yokoi N et al (2003) Up-regulated gene expression in the conjunctival epithelium of patients with Sjögren’s syndrome. Exp Eye Res 77:17–26
Keibel F, Elze C (1908) Tabla De Desarrollo Cronológico Del Ser Humano. Verlag von Gustav Fischer, Jena
Kenney NJ, Smith GH, Rosenberg K et al (1996) Induction of ductal morphogenesis and lobular hyperplasia by amphiregulin in the mouse mammary gland. Cell Growth Differ 7:1769–1781
Kere J, Srivastava AK, Montonen O et al (1996) X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet 13:409–416
Knudsen KA, Sauer C, Johnson KR et al (2005) Effect of N-cadherin misexpression by the mammary epithelium in mice. J Cell Biochem 95:1093–1107
Kotwall CA (1992) Smoking as an etiologic factor in the development of Warthin’s tumor of the parotid gland. Am J Surg 164:646–647
Kovacs L, Szodoray P, Kiss E (2010) Secondary tumours in Sjogren’s syndrome. Autoimmun Rev 9:203–206
LaMarca HL, Rosen JM (2007) Estrogen regulation of mammary gland development and breast cancer: amphiregulin takes center stage. Breast Cancer Res 9:304
LaRocca J, Pietruska J, Hixon M (2011) Akt1 is essential for postnatal mammary gland development, function, and the expression of Btn1a1. PLoS One 6:e24432
Larsen M, Yamada KM, Musselmann K (2010) Systems analysis of salivary gland development and disease. Wiley Interdiscip Rev Syst Biol Med 2:670–682
Lee SB, Lee SB, Huang K et al (1999) The Wilmstumor suppressor WT1 encodes a transcriptional activator of amphiregulin. Cell 98:663–673
Lee BH, Tudares MA, Nguyen CQ (2009) Sjogren’s syndrome: an old tale with a new twist. Arch Immunol Ther Exp 57:57–66
Li S, Plowman GD, Buckley SD et al (1992) Heparin inhibition of autonomous growth implicates amphiregulin as an autocrine growth factor for normal human mammary epithelial cells. J Cell Physiol 153:103–111
Li M, Fu X, Ma G et al (2012) Atbf1 regulates pubertal mammary gland development likely by inhibiting the pro-proliferative function of estrogen-ER signaling. PLoS One 7:e51283
Lisi S, Sisto M, Lofrumento DD et al (2010) Pro-inflammatory role of Anti-Ro/SSA autoantibodies through the activation of Furin-TACE-amphiregulin axis. J Autoimmun 35:160–170
Lisi S, Sisto M, Lofrumento DD et al (2012) Altered IκBα expression promotes NF-κB activation inmonocytes from primary Sjögren’s syndrome patients. Pathology 44:557–561
Lisi S, Sisto M, D’Amore M et al (2013) Emerging avenues linking inflammation, angiogenesis and Sjögren’s syndrome. Cytokine 61:693–703
Lisi S, Sisto M, Ribatti D et al (2014a) Chronic inflammation enhances NGF-β/TrkA system expression viaEGFR/MEK/ERK pathway activation in Sjögren’s syndrome. J Mol Med 92:523–537
Lisi S, D’Amore M, Sisto M (2014b) ADAM17 at the interface between inflammation and autoimmunity. Immunol Lett 162:159–169
Liu K, Gualano RC, Hibbs ML et al (2008) Epidermal growth factor receptor signaling to Erk1/2 and STATs control the intensity of the epithelial inflammatory responses to rhinovirus infection. J Biol Chem 283:9977–9985
Liu JF, Tsao YT, Hou CH (2015) Amphiregulin enhances intercellular adhesion molecule-1 expression and promotes tumor metastasis in human osteosarcoma. Oncotarget 6:40880–40895
Lovicu FJ, Kao WW, Overbeek PA (1999) Ectopic gland induction by lens-specific expression of keratinocyte growth factor (FGF-7) in transgenic mice. Mech Dev 88:43–53
Lu P, Werb Z (2008) Patterning mechanisms of branched organs. Science 322:1506–1509
Lu P, Sternlicht MD, Werb Z (2006) Comparative mechanisms of branching morphogenesis in diverse systems. J Mammary Gland Biol Neoplasia 11:213–228
Lubahn DB, Moyer JS, Golding TS et al (1993) Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci USA 90:11162–11166
Luetteke NC, Qiu TH, Fenton SE et al (1999) Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development 126:2739–2750
Lysiak JJ, Johnson GR, Lala PK (1995) Localization of amphiregulin in the human placenta and decidua throughout gestation: role in trophoblast growth. Placenta 16:359–366
Macias H, Hinck L (2012) Mammary gland development. Wiley Interdiscip Rev Dev Biol 1:533–557
Mahtouk K, Cremer FW, Rème T et al (2006) Heparan sulphate proteoglycans are essential for the myeloma cell growth activity of EGF-family ligands in multiple myeloma. Oncogene 25:7180–7191
Makarenkova HP, Ito M, Govindarajan V (2000) FGF10 is an inducer and Pax6 a competence factor for lacrimal gland development. Development 127:2563–2572
Manoussakis MN, Kapsogeorgou EK (2010) The role of intrinsic epithelial activation in the pathogenesis of Sjögren’s syndrome. J Autoimmun 35:219–224
Manoussakis MN, Boiu S, Korkolopoulou P et al (2007) Rates of infiltration by macrophages and dendritic cells and expression of interleukin-18 and interleukin-12 in the chronic inflammatory lesions of Sjögren’s syndrome: correlation with certain features of immune hyperactivity and factors associated with high risk of lymphoma development. Arthritis Rheum 56:3977–3988
Mansouri A, Stoykova A, Gruss P (1994) Pax genes in development. J Cell Sci 18(Suppl):35–42
Mascia F, Mariani V, Girolomoni G et al (2003) Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol 163:303–312
Maślińska M, Przygodzka M, Kwiatkowska B et al (2015) Sjögren’s syndrome: still not fully understood disease. Rheumatol Int 35:233–241
Mathews SA, Kurien BT, Scofield RH (2008) Oral manifestations of Sjogren’s syndrome. J Dent Res 87:308–318
Matsumoto K, Fukuda S, Nakamura Y et al (2009) Amphiregulin production by human eosinophils. Int Arch Allergy Immunol 149(Suppl 1):39–44
McBryan J, Howlin J, Napoletano S et al (2008) Amphiregulin: role in mammary gland development and breast cancer. J Mammary Gland Biol Neoplasia 13:159–169
McKee C, Sigala B, Soeda J et al (2015) Amphiregulin activates human hepatic stellate cells and is upregulated in non alcoholic steatohepatitis. Sci Rep 5:8812
McNally S, Martin F (2011) Molecular regulators of pubertal mammary gland development. Ann Med 43:212–234
Melnick M, Jaskoll T (2000) Mouse submandibular gland morphogenesis: a paradigm for embryonic signal processing. Crit Rev Oral Biol Med 11:199–215
Melnick M, Chen H, Min Zhou Y et al (2001a) The functional genomic response of developing embryonic submandibular glands to NF-kappa B inhibition. BMC Dev Biol 1:15
Melnick M, Chen H, Zhou Y et al (2001b) An alternatively spliced Muc10 glycoprotein ligand for putative L-selectin binding during mouse embryonic submandibular gland morphogenesis. Arch Oral Biol 46:745–747
Melnick M, Chen H, Zhou Y et al (2001c) Embryonic mouse submandibular salivary gland morphogenesis and the TNF/TNF-R1 signal transduction pathway. Anat Rec 262:318–330
Melnick M, Chen H, Zhou Y et al (2001d) Interleukin-6 signaling and embryonic mouse submandibular salivary gland morphogenesis. Cells Tissues Organs 168:233–245
Melnick M, Phair RD, Lapidot SA et al (2009) Salivary gland branching morphogenesis: a quantitative systems analysis of the Eda/Edar/NFkappaB paradigm. BMC Dev Biol 9:32
Mendelsohn J, Baselga J (2006) Epidermal growth factor receptor targeting in cancer. Semin Oncol 33:369–385
Meulenbroeks C, van Weelden H, Schwartz C et al (2015) Basophil-derived amphiregulin is essential for UVB irradiation-induced immune suppression. J Invest Dermatol 135:222–228
Mikkola ML (2008) TNF superfamily in skin appendage development. Cytokine Growth Factor Rev 19:219–230
Monreal AW, Zonana J, Ferguson B (1998) Identification of a new splice form of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am J Hum Genet 63:380–389
Monticelli LA, Sonnenberg GF, Abt MC et al (2011) Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol 12:1045–1054
Moss ML, Jin SL, Milla ME et al (1997) Cloning of a disintegrin metalloproteinase factor processes precursor tumour-necrosis factor-alpha. Nature 385:733–736
Moutsopoulos HM (1994) Sjögren’s syndrome: autoimmune epithelitis. Clin Immunol Immunopathol 72:162–165
Nakamura H, Kawakami A, Ida H et al (2007) Epidermal growth factor inhibits Fas-mediated apoptosis in salivary epithelial cells of patients with primary Sjögren’s syndrome. Clin Exp Rheumatol 25:831–837
Napenas JJ, Brennan MT, Fox PC (2009) Diagnosis and treatment of xerostomia (dry mouth). Odontology 97:76–83
Nautiyal J, Steel JH, Mane MR et al (2013) The transcriptional co-factor RIP140 regulates mammary gland development by promoting the generation of key mitogenic signals. Development 140:1079–1089
Neelam B, Richter A, Chamberlin SC et al (1998) Structure-function studies of ligand-induced epidermal growth factor receptor dimerization. Biochemistry 37:4884–4891
Negishi H, Ikeda C, Nagai Y et al (2007) Regulation of amphiregulin, EGFR-like factor expression by hCG in cultured human granulosa cells. Acta Obstet Gynecol Scand 86:706–710
Nilsson A, Moller K, Dahlin L et al (2005) Early changes in gene expression in the dorsal root ganglia after transection of the sciatic nerve; effects of amphiregulin and PAI-1 on regeneration. Brain Res Mol Brain Res 136:65–74
Oakes SR, Hilton HN, Ormandy CJ (2006) The alveolar switch: coordinating the proliferative cues and cell fate decisions that drive the formation of lobuloalveoli from ductal epithelium. Breast Cancer Res 8:207
Oda K, Matsuoka Y, Funahashi A et al (2005) A comprehensive pathway map of epidermal growth factor receptor signaling. Mol Syst Biol 1(2005):0010
Ohlsson M, Szodoray P, Loro LL et al (2002) CD40, CD154, Bax and Bcl-2 expression in Sjögren’s syndrome salivary glands: a putative anti-apoptotic role during its effector phases. Scand J Immunol 56:561–571
Ohuchi H, Hori Y, Yamasaki M et al (2000) FGF10 acts as a major ligand for FGF receptor 2 IIIb in mouse multi-organ development. Biochem Biophys Res Commun 277:643–649
Okumura S, Sagara H, Fukuda T et al (2005) FcεRI-mediated amphiregulin production by human mast cells increases mucin gene expression in epithelial cells. J Allergy Clin Immunol 115:272–279
Okumura K, Shinohara M, Endo F (2012) Capability of tissue stem cells to organize into salivary rudiments. Stem Cells Int 2012:502136
Ornitz DM, Itoh N (2001) Fibroblast growth factors. Genome Biol 2:REVIEWS3005
Pardo-Saganta A, Latasa MU, Castillo J et al (2009) The epidermal growth factor receptor ligand amphiregulin is a negative regulator of hepatic acute-phase gene expression. J Hepatol 51:1010–1020
Parkin NT, Kitajewski J, Varmus HE (1993) Activity of Wnt-1 as a transmembrane protein. Genes Dev 7:2181–2193
Patel VN, Hoffman MP (2014) Salivary gland development: a template for regeneration. Semin Cell Dev Biol 25–26:52–60
Patel VN, Rebustini IT, Hoffman MP (2006) Salivary gland branching morphogenesis. Differentiation 74:349–364
Perugorria MJ, Latasa MU, Nicou A et al (2008) The epidermal growth factor receptor ligand amphiregulin participates in the development of mouse liver fibrosis. Hepatology 48:1251–1261
Peschon JJ, Slack JL, Reddy P et al (1998) An essential role for ectodomain shedding in mammalian development. Science 282:1281–1284
Peterson EA, Pectasides E, Shabbeer S et al (2013) Evaluation of serum Amphiregulin levels in breast cancer patients and cancer-free controls. Exp Hematol Oncol 2:25
Phippard DJ, Weber-Hall SJ, Sharpe PT et al (1996) Regulation of Msx-1, Msx-2, Bmp-2 and Bmp-4 during foetal and postnatal mammary gland development. Development 122:2729–2737
Piepkorn M, Lo C, Plowman G (1994) Amphiregulin-dependent proliferation of cultured human keratinocytes: autocrine growth, the effects of exogenous recombinant cytokine, and apparent requirement for heparin-like glycosaminoglycans. J Cell Physiol 159:114–120
Plowman GD, Whitney GS, Neubauer MG et al (1990) Molecular cloning and expression of an additional epidermal growth factor receptor-related gene. Proc Natl Acad Sci USA 87:4905–4909
Pozzi A, Zent R (2011) Extracellular matrix receptors in branched organs. Curr Opin Cell Biol 23:547–553
Prasannan L, Pu A, Hoff P (1999) Parotid carcinoma as a second malignancy after treatment of childhood acute lymphoblastic leukemia. J Pediatr Hematol Oncol 21:535–538
Proctor GB, Carpenter GH (2007) Regulation of salivary gland function by autonomic nerves. Auton Neurosci 133:3–18
Qi Y, Operario DJ, Oberholzer CM et al (2010) Human basophils express amphiregulin in response to T cell-derived IL-3. J Allergy Clin Immunol 126:1260–1266
Qi Y, Operario DJ, Georas SN et al (2012) The acute environment, rather than T cell subset pre-commitment, regulates expression of the human T cell cytokine amphiregulin. PLoS One 7:e39072
Qin L, Tamasi J, Raggatt L et al (2005) Amphiregulin is a novel growth factor involved in normal bone development and in the cellular response to parathyroid hormone stimulation. J Biol Chem 280:3974–3981
Qu S, Rinehart C, Wu HH et al (2006) Gene targeting of ErbB3 using a Cre-mediated unidirectional DNA inversion strategy. Genesis 44:477–486
Rao PK, Shetty SR, Hegde D (2012) Ectopic pleomorphic adenoma. N Am J Med Sci 4:190–192
Riese DJ, Kim ED, Elenius K et al (1996) The epidermal growth factor receptor couples transforming growth factor-α, heparin-binding epidermal growth factor-like factor, and amphiregulin to Neu, ErbB-3, and ErbB-4. J Biol Chem 271:20047–20052
Robinson GW, Hennighausen L, Johnson PF (2000) Side-branching in the mammary gland: the progesterone-Wnt connection. Genes Dev 14:889–894
Rodland KD, Bollinger N, Ippolito D et al (2008) Multiple mechanisms are responsible for transactivation of the epidermal growth factor receptor in mammary epithelial cells. J Biol Chem 283:31477–31487
Roepstorff K, Grandal MV, Henriksen L et al (2009) Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 10:1115–1127
Roescher N, Tak PP, Illei GG (2009) Cytokines in Sjögren’s syndrome. Oral Dis 15:519–526
Sakakura T (1987) Embryogenesis. In: Neville MC, Daniel CW (eds) Development, regulation and function. Plenum, New York, pp 37–65
Salimi M, Barlow JL, Saunders SP et al (2013) A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med 210:2939–2950
Sawalha AH, Jeffries M, Webb R et al (2008) Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun 9:368–378
Schafer B, Marg B, Gschwind A et al (2004) Distinct ADAM metalloproteinases regulate G protein-coupled receptor-induced cell proliferation and survival. J Biol Chem 279:47929–47933
Schneider MR, Sibilia M, Erben RG (2009) The EGFR network in bone biology and pathology. Trends Endocrinol Metab 20:517–524
Schroeder JA, Lee DC (1998) Dynamic expression and activation of ERBB receptors in the developing mouse mammary gland. Cell Growth Differ 9:451–464
Schuger L, Johnson GR, Gilbride K et al (1996) Amphiregulin in lung branching morphogenesis: interaction with heparan sulfate proteoglycan modulates cell proliferation. Development 122:1759–1767
Sebastian J, Richards RG, Walker MP et al (1998) Activation and function of the epidermal growth factor receptor and erbB-2 during mammary gland morphogenesis. Cell Growth Differ 9:777–785
Segatto O, Anastasi S, Alemà S (2011) Regulation of epidermal growth factor receptor signaling by inducible feedback inhibitors. J Cell Sci 124:1785–1793
Sequeira SJ, Larsen M, DeVine T (2010) Extracellular matrix and growth factors in salivary gland development. Front Oral Biol 14:48–77
Shams I, Rohmann E, Eswarakumar VP et al (2007) Lacrimo-auriculo-dento-digital syndrome is caused by reduced activity of the fibroblast growth factor 10 (FGF10)-FGF receptor 2 signaling pathway. Mol Cell Biol 27:6903–6912
Shao J, Lee SB, Guo H et al (2003) Prostaglandin E2 stimulates the growth of colon cancer cells via induction of amphiregulin. Cancer Res 63:5218–5223
Shiboski SC, Shiboski CH, Criswell L et al (2012) American College of Rheumatology classification criteria for Sjögren’s syndrome: a data-driven, expert consensus approach in the Sjögren’s International Collaborative Clinical Alliance cohort. Arthritis Care Res 64:475–487
Shin HS, Lee HJ, Nishida M et al (2003) Betacellulin and amphiregulin induce upregulation of cyclin D1 and DNA synthesis activity through differential signaling pathways in vascular smooth muscle cells. Circ Res 93:302–310
Shoyab M, McDonald VL, Bradley JG et al (1988) Amphiregulin: a bifunctional growth-modulating glycoprotein produced by the phorbol 12-myristate 13-acetate-treated human breast adenocarcinoma cell line MCF-7. Proc Natl Acad Sci USA 85:6528–6532
Shoyab M, Plowman GD, McDonald VL et al (1989) Structure and function of human amphiregulin: a member of the epidermal growth factor family. Science 243(4894 Pt 1):1074–1076
Shyamala G (1999) Progesterone signaling and mammary gland morphogenesis. J Mammary Gland Biol Neoplasia 4:89–104
Silvy M, Giusti C, Martin PM et al (2001) Differential regulation of cell proliferation and protease secretion by epidermal growth factor and amphiregulin in tumoral versus normal breast epithelial cells. Br J Cancer 84:936–945
Simian M, Hirai Y, Navre M et al (2001) The interplay of matrix metalloproteinases, morphogens and growth factors is necessary for branching of mammary epithelial cells. Development 128:3117–3131
Singh K, Deshpande P, Pryshchep S et al (2009) ERK-dependent T cell receptor threshold calibration in rheumatoid arthritis. J Immunol 183:8258–8267
Sisto M, Lisi S, Lofrumento DD et al (2010) Expression of pro-inflammatory TACE-TNF-α-amphiregulin axis in Sjögren’s syndrome salivary glands. Histochem Cell Biol 134:345–353
Sisto M, Lisi S, Lofrumento DD et al (2011) A failure of TNFAIP3 negative regulation maintains sustained NF-κB activation in Sjögren’s syndrome. Histochem Cell Biol 135:615–625
Sisto M, Lisi S, D’Amore M et al (2015) The metalloproteinase ADAM17 and the epidermal growth factor receptor (EGFR) signaling drive the inflammatory epithelial response in Sjögren’s syndrome. Clin Exp Med 15:215–225
So WK, Fan Q, Lau MT et al (2014) Amphiregulin induces human ovarian cancer cell invasion by down-regulating E-cadherin expression. FEBS Lett 588:3998–4007
Speight PM, Barrett AW (2002) Salivary gland tumours. Oral Dis 8:229–240
Srivastava AK, Pispa J, Hartung AJ et al (1997) The Tabby phenotype is caused by mutation in a mouse homologue of the EDA gene that reveals novel mouse and human exons and encodes a protein (ectodysplasin-A) with collagenous domains. Proc Natl Acad Sci USA 94:13069–13074
Steinberg Z, Myers C, Heim VM et al (2005) FGFR2b signaling regulates ex vivo submandibular gland epithelial cell proliferation and branching morphogenesis. Development 132:1223–1234
Stern KA, Place TL, Lill NL (2008) EGF and amphiregulin differentially regulate Cbl recruitment to endosomes and EGF receptor fate. Biochem J 410:585–594
Sternlicht MD (2006) Key stages in mammary gland development: the cues that regulate ductal branching morphogenesis. Breast Cancer Res 8:201
Sternlicht MD, Sunnarborg SW (2008) The ADAM17-amphiregulin-EGFR axis in mammary development and cancer. J Mammary Gland Biol Neoplasia 13:181–194
Sternlicht MD, Sunnarborg SW, Kouros-Mehr H et al (2005) Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulin. Development 132:3923–3933
Stoll SW, Johnson JL, Li Y et al (2010a) Amphiregulin carboxy-terminal domain is required for autocrine keratinocyte growth. J Invest Dermatol 130:2031–2040
Stoll SW, Johnson JL, Bhasin A et al (2010b) Metalloproteinase-mediated, context-dependent function of amphiregulin and HB-EGF in human keratinocytes and skin. J Invest Dermatol 130:295–304
Stoll SW, Stuart PE, Swindell WR et al (2016) The EGF receptor ligand amphiregulin controls cell division via FoxM1. Oncogene 35:2075–2086
Streicher KL, Willmarth NE, Garcia J et al (2007) Activation of a nuclear factor kappaB/interleukin-1 positive feedback loop by amphiregulin in human breast cancer cells. Mol Cancer Res 5:847–861
Swanson GM, Burns PB (1997) Cancers of the salivary gland: workplace risks among women and men. Ann Epidemiol 7:369–374
Sympson CJ, Talhouk RS, Alexander CM et al (1994) Targeted expression of stromelysin-1 in mammarygland provides evidence for a role of proteinases in branchingmorphogenesis and the requirement for an intact basement membranefor tissue-specific gene expression. J Cell Biol 125:681–693
Tatouli IP, Tzioufas AG (2012) Pathogenetic aspects of humoral autoimmunity in Sjögren’s syndrome. Lupus 21:1151–1154
Tørring N, Sørensen BS, Bösch ST et al (1998) Amphiregulin is expressed in primary cultures of prostate myofibroblasts, fibroblasts, epithelial cells, and in prostate tissue. Prostate Cancer Prostatic Dis 1:262–267
Tripathi BJ, Tripathi RC (1990) Evidence for the neuroectodermal origin of the human lacrimal gland. Invest Ophthalmol Vis Sci 31:393–395
Tsao MS, Zhu H, Viallet J (1996) Autocrine growth loop of the epidermal growth factor receptor in normal and immortalized human bronchial epithelial cells. Exp Cell Res 223:268–273
Tucker AS (2007) Salivary gland development. Semin Cell Dev Biol 18:237–244
Tye H, Jenkins BJ (2013) Tying the knot between cytokine and toll-like receptor signaling in gastrointestinal tract cancers. Cancer Sci 104:1139–1145
Tzioufas AG, Voulgarelis M (2007) Update on Sjogren’s syndrome autoimmune epithelitis: from classification to increased neoplasias. Best Pract Res Clin Rheumatol 21:989–1010
Val S, Belade E, George I et al (2012) Fine PM induce airway MUC5AC expression through the autocrine effect of amphiregulin. Arch Toxicol 86:1851–1859
van Genderen C, Okamura RM, Fariñas I et al (1994) Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in LEF-1-deficientmice. Genes Dev 8:2691–2703
Varley C, Hill G, Pellegrin S et al (2005) Autocrine regulation of human urothelial cell proliferation and migration during regenerative responses in vitro. Exp Cell Res 306:216–229
Viatour P, Merville MP, Bours V et al (2005) Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci 30:43–52
Vitali C (2003) Classification criteria for Sjögren’s syndrome. Ann Rheum Dis 62:94–95
Wagner M, Weber CK, Bressau F et al (2002) Transgenic overexpression of amphiregulin induces a mitogenic response selectively in pancreatic duct cells. Gastroenterology 122:1898–1912
Wang J, Laurie GW (2004) Organogenesis of the exocrine gland. Dev Biol 273:1–22
Wang Z, Juttermann R, Soloway PD (2000) TIMP-2 is required for efficient activation of proMMP-2 in vivo. J Biol Chem 275:26411–26415
Wang SW, Oh CK, Cho SH et al (2005) Amphiregulin expression in human mast cells and its effect on the primary human lung fibroblasts. J Allergy Clin Immunol 115:287–294
Webber EM, Fitzgerald MJ, Brown PI et al (1993) Transforming growth factor-α expression during liver regeneration after partial hepatectomy and toxic injury and potential interactions between transforming growth factor-α and hepatocyte growth factor. Hepatology 18:1422–1431
Wieduwilt MJ, Moasser MM (2008) The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci 65:1566–1584
Wiesen JF, Young P, Werb Z et al (1999) Signaling through the stromal epidermal growth factor receptor is necessary for mammary ductal development. Development 126:335–344
Williams MF (1999) Sialolithiasis. Otolaryngol Clin North Am 32:819–834
Willmarth NE, Ethier SP (2006) Autocrine and juxtacrine effects of amphiregulin on the proliferative, invasive, and migratory properties of normal and neoplastic human mammary epithelial cells. J Biol Chem 281:37728–37737
Wilson KF, Meier JD, Ward PD (2014) Salivary gland disorders. Am Fam Physician 89:882–888
Witty JP, Wright JH, Matrisian LM (1995) Matrix metalloproteinasesare expressed during ductal and alveolar mammary morphogenesis, andmisregulation of stromelysin-1 in transgenic mice induces un scheduled alveolar development. Mol Biol Cell 6:1287–1303
Wong L, Deb TB, Thompson SA et al (1999) A differential requirement for the COOH-terminal region of the epidermal growth factor (EGF) receptor in amphiregulin and EGF mitogenic signaling. J Biol Chem 274:8900–8909
Woodworth CD, McMullin E, Iglesias M et al (1995) Interleukin 1 alpha and tumor necrosis factor alpha stimulate autocrine amphiregulin expression and proliferation of human papillomavirus-immortalized and carcinoma-derived cervical epithelial cells. Proc Natl Acad Sci USA 92:2840–2844
Yamane S, Ishida S, Hanamoto Y et al (2008) Proinflammatory role of amphiregulin, an epidermal growth factor family member whose expression is augmented in rheumatoid arthritis patients. J Inflamm 5:5
Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signaling network. Nat Rev Mol Cell Biol 2:127–137
Yoon S, Seger R (2006) The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24:21–24
Yotsumoto F, Fukami T, Yagi H et al (2010) Amphiregulin regulates the activation of ERK and Akt through epidermal growth factor receptor and HER3 signals involved in the progression of pancreatic cancer. Cancer Sci 101:2351–2360
Zaiss DM, Yang L, Shah PR et al (2006) Amphiregulin, a TH2 cytokine enhancing resistance to nematodes. Science 314:1746
Zaiss DM, van Loosdregt J, Gorlani A et al (2013) Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity 38:275–284
Zaiss DM, Gause WC, Osborne LC et al (2015) Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 42:216–226
Zhou Y, Lee JY, Lee CM et al (2012) Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-beta-induced pulmonary fibrosis. J Biol Chem 287:41991–42000
Zhu J, Siclari VA, Liu F et al (2012) Amphiregulin-EGFR signaling mediates the migration of bone marrow mesenchymal progenitors toward PTH-stimulated osteoblasts and osteocytes. PLoS One 7:e50099
Acknowledgements
We are grateful to M.V.C. Pragnell, B.A., a professional scientific text editor, for the revision of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
About this article

Cite this article
Sisto, M., Lorusso, L., Ingravallo, G. et al. Exocrine Gland Morphogenesis: Insights into the Role of Amphiregulin from Development to Disease. Arch. Immunol. Ther. Exp. 65, 477–499 (2017). https://doi.org/10.1007/s00005-017-0478-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-017-0478-2