
Abstract
Program death-1 (PD-1) is a co-inhibitory receptor inducibly expressed on activated T cells. PD-1 has been reported to be associated with the development of several autoimmune diseases including rheumatoid arthritis, but the precise cellular and molecular mechanisms have not been fully elucidated. To study the role of PD-1 in the pathogenesis of rheumatoid arthritis and the possible underlying mechanisms, we performed collagen-induced arthritis (CIA) in C57BL/6 mice. Here, we show that PD-1 deficiency leads to the development of severe CIA in mice. When analyzing T cells from CIA mice ex vivo, we noticed aberrant antigen-specific Th17 responses in mice lacking PD-1. This is possibly due to deregulated activation of PKC-θ and Akt. In support of this notion, treating Pdcd1 −/− mice with an inhibitor of PI3-kinase that is upstream of PKC-θ and Akt significantly suppressed the disease severity. Therefore, our data indicate that PD-1 dampens antigen-specific Th17 response, thus inhibiting the disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease of unknown etiology that primarily affects the synovial membranes of multiple joints (Koch 2007). The disease has been suspected to be autoimmune in nature and to be initiated by autoimmune CD4+ T cells (Wehrens et al. 2012). Collagen-induced arthritis (CIA) is the most widely used mouse model for RA, in which immunization of genetically susceptible strains of mice with type II collagen (CII) leads to the development of a severe polyarticular arthritis mediated by immune mechanisms (Brand et al. 2007; Li et al. 2013). Although CIA was originally considered as a Th1-mediated autoimmune disease, recent studies indicate that the newly identified Th17 subset that predominantly produces interleukin (IL)-17 is the major initiator of the disease (Lubberts et al. 2004; Miossec and Kolls 2012; Wehrens et al. 2012).
Program death-1 (PD-1) is encoded by the Pdcd1 gene on chromosome 1 in mice and chromosome 2 in humans. Its expression can be induced on T cells, B cells, NK cells, monocytes and dendritic cells upon activation (Keir et al. 2008). PD-1 is considered to play an important inhibitory role in immune responses as its deficiency causes lupus-like glomerulonephritis and proliferative arthritis in C57BL/6 (B6) mice and fatal autoimmune dilated cardiomyopathy in BALB/c mice (Okazaki and Honjo 2006). Single nucleotide polymorphisms on human PD-1 gene have been reported to be associated with RA (Lin et al. 2004). B7-H1, a major ligand of PD-1, has been documented to signal through PD-1 on T effector cells to prevent excessive activation and reduce autoimmune arthritis (Hamel et al. 2010). Blockade of PD-1-PDL interaction by PDL-Ig significantly ameliorated CIA as assessed by clinical arthritis score and histology in the joints (Wang et al. 2011). However, the direct intrinsic cellular and molecular mechanism(s) by which PD-1 participates in the pathogenesis of RA has not been well defined.
We previously showed that cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)-B7 interaction suppressed Th17 responses and then inhibited the induction of experimental autoimmune myocarditis (Ying et al. 2010), and that PD-1 may regulate T cell responses via an anergy-independent but inducible regulatory T cell-dependent mechanism (Qiao et al. 2012). However, the role of PD-1 in deregulated Th17 responses occurring during the development of CIA and possibly human RA is not fully characterized.
In this study, we show that PD-1 deficiency potentiates the development of CIA in B6 mice, with increased antigen-specific T cell proliferation and interleukin (IL)-17 production. Mechanistically, PD-1 may execute its role by suppressing Th17 responses possibly via inhibiting the activation of PI3K/Akt pathway.
Materials and Methods
Mice
Wild-type (WT) C57BL/6 (B6) mice were purchased from the National Cancer Institute (Fredrick, MD, USA) and Shanghai Experimental Animal Center. Pdcd1 −/− B6 mice were obtained from Dr. Tasuku Honjo (Kyoto University, Kyoto, Japan). All experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committees of the University of Chicago and Central South University.
Induction of CIA and Assessment of Arthritis
Collagen-induced arthritis was induced as described previously with slight modification (Brand et al. 2007; Li et al. 2013). Briefly, chicken CII (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in 10 mM acetic acid and emulsified in equal volumes of complete Freund’s adjuvant (CFA; contains 5 mg/ml of heat-killed Mycobacterium tuberculosis). Mice age 12–14 weeks were injected intradermally at the base of the tail with 100 μg of CII in CFA on day 0 and 21; a dose of 3 mg zymosan (Sigma, St. Louis, MO, USA) in normal saline was given on day 26 after initial immunization (intraperitoneally: i.p.). The incidence and severity of arthritis were monitored for 8 weeks and then the mice were killed for further histological and immunological analysis. Each paw was scored for the degree of inflammation on a scale from 0 to 4:0, no evidence of erythema and swelling; 1, erythema and mild swelling confined to the midfoot (tarsals) or ankle joint; 2 erythema and mild swelling extending from the ankle to the midfoot; 3 erythema and mild swelling extending from the ankle to the metatarsal joints; 4 erythema and severe swelling encompassing the ankle, foot and digits (Brand et al. 2007). Hind paws were collected and fixed in 10 % formalin, and 5 μm sections were stained with H&E. Histologically, the joints were assessed on a scale of 0–5 based on the degree of inflammation, cartilage damage and bone erosion, as described previously (Buttgereit et al. 2009; Kondo et al. 2012). For in vivo inhibitor experiments, Pdcd1 −/− mice were administered (i.p.) with LY294002 (50 mg/kg) every other day from the time half of the mice had developed arthritis (day 30) till the end of the experiment (Hu et al. 2000; Skon et al. 2013; Toyama et al. 2010). Serum was collected for cytokine detection by ELISA (eBioscience, San Diego, CA, USA), and CD4+ T cells were isolated from lymph nodes using mouse CD4+ T cell isolation kits (R&D Systems, Minneapolis, MN, USA) when the mice were killed.
Ex vivo T Cell Recall Responses
At the end of CIA, the mice were killed and draining lymph nodes were collected. CD4+ T cells were isolated from lymph nodes using mouse CD4+ T cell isolation kits (R&D Systems, Minneapolis, MN, USA) and co-cultured with irradiated WT T cell-depleted splenocytes as feeder cells in the presence of different concentrations of CII. CII-specific T cell proliferation was determined by [3H]thymidine incorporation. For cytokine study, the supernatants were collected at 48 h and subjected to interferon (IFN)-γ, IL-4 and IL-17 detection by ELISA (eBioscience, San Diego, CA, USA).
Flow Cytometric Analysis
Draining lymph node cells were collected for further analysis at the end of CIA as described above. For regulatory T cell analysis, the cells were surface stained with antibodies against CD4 and CD25, followed by intracellular staining with anti-Foxp3 antibody (eBioscience; San Diego, CA, USA). For the Th1 and 17 cells analysis, the lymph node cells were stimulated with PMA (50 ng/ml; Sigma-Aldrich, St. Louis, MO, USA) plus ionomycin (1 μg/ml; Sigma-Aldrich, St. Louis, MO, USA) for 5 h in culture medium in the presence of GolgiStop (BD Biosciences, San Jose, CA, USA) and then surface stained with anti-CD4 antibody followed by intracellular staining with anti-IL-17 and anti-IFN-γ antibodies (eBioscience; San Diego, CA, USA). The cells were analyzed on an LSRII flow analyzer (BD Biosciences, San Jose, CA, USA) using FlowJo (Tree Star, Ashland, OR, USA) (Qiao et al. 2012; Ying et al. 2010).
In Vitro Stimulation and Immunoblotting
Naïve CD4+CD25− T cells from WT and Pdcd1 −/− mice were stimulated with anti-CD3 antibody in the presence or absence of anti-CD28 antibody for different time points and lysed. The cell lysates were blotted with monoclonal phospho-antibodies against Akt (Ser473), extracellular signal-regulated kinase (ERK; Thr202/Tyr204), p38 (Thr180/Tyr182), c-Jun N-terminal kinase (JNK; Thr183/Tyr185) and protein kinase C ((PKC)-θ, Thr538) (Cell Signaling, Boston, MA, USA). CD4+ T cells from CIA mice treated with LY294002 in vivo were lysed, and the cell lysates were blotted with monoclonal phospho-antibodies against Akt (Ser473) and PKC-θ (Thr538) (Cell Signaling, Boston, MA, USA). The membranes were reprobed with anti-β-actin.
Statistical Analysis
Data were expressed as the mean ± SD. Student’s t test to determine the differences between groups. The Mann–Whitney U test was used to determine the statistical significance in disease incidence and severity between WT and Pdcd1 −/− mice. The level of significance was set as p < 0.05.
Results and Discussion
PD-1 Deficiency Potentiates CIA Induction
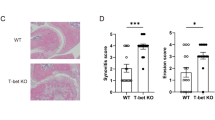
Although PD-1 has been reported to be involved in the development of CIA in mice (Hamel et al. 2010; Lin et al. 2004; Wang et al. 2011), the role of PD-1 in CIA has not been verified using mice deficient in PD-1. To provide the genetic evidence on PD-1 in CIA, WT and Pdcd1 −/− mice were immunized (subcutaneous) with type II chicken collagen emulsified in CFA and boosted on day 21. To enhance the incidence and severity of CIA, each mouse received zymosan injected (i.p.) on day 26 after initial immunization. The incidence and severity of arthritis were monitored. A standard scoring system, based upon swelling and redness of each paw, was used for the assessment of onset and arthritis severity. We found that although the arthritis incidence was comparable between WT and Pdcd1 −/− mice (both approximately 60 %), the severity was significantly higher in Pdcd1 −/− mice than WT mice (Fig. 1a). Erythema and swelling were more severe in Pdcd1 −/− mice than WT mice (Fig. 1b). There were severer bone and cartilage destruction and lymphocytes infiltration in Pdcd1 −/− mice than in WT mice (Fig. 1b). Therefore, we demonstrated that PD-1 suppresses the disease progression in CIA.

PD-1 deficiency potentiates CIA induction. Male WT and Pdcd1 −/− B6 mice (n = 10) at 10–12 weeks of age were immunized with chicken type II collagen (CII) and observed for 56 days as described in the “Materials and Methods”. a Arthritis incidence (left panel) and disease severity score (right panel) were determined by visual inspection of mouse paws. b Representative images of hind paws from WT and Pdcd1 −/− B6 mice 56 days after the first immunization with chicken CII. The joints were collected and fixed in 10 % formalin, and 5 mm sections were stained with H&E (×100). The values represent the mean ± SEM. The data shown are representative of two independent experiments (*p < 0.05, **p < 0.01)
PD-1 Deficiency Facilitated Antigen-Specific T Cell Proliferation and Th17 Differentiation
It has been reported that IL-17-positive cells were found in the synovium from RA patients and this synovium could produce functionally active IL-17 (Chabaud et al. 1999). Overexpression of IL-17 in the knee joint confers the induction of CIA and increased joint destruction (Lubberts et al. 2002), while deficiency of IL-17 or IL-17 receptor prevented the development of arthritis in animal models (Koenders et al. 2005; Nakae et al. 2003). Neutralization of IL-17 with specific antibody reduced the disease severity in CIA mice (Lubberts et al. 2004). However, whether inhibition of CIA development by PD-1 involves the attenuation of Th17 responses has not been established. To determine the possible cellular mechanisms involved in the pathogenesis of CIA, we detected the ex vivo CD4+ T cell recall response in response to chicken CII. As seen in Fig. 2a, CD4+ T cell from Pdcd1 −/− mice immunized with CII in CFA proliferated significantly higher than that from WT mice. Pdcd1 −/− CD4+ T cells produced more IL-17 than WT cells, while there was no significant difference in the production of IL-4 or IFN-γ between the two groups (Fig. 2b). Consistent with our previous data on natural regulatory T cells (Tregs) (Qiao et al. 2012), Tregs in draining lymph nodes were comparable between WT and Pdcd1 −/− mice with CIA. While Pdcd1 −/− CIA mice did show significantly expanded IL-17+ CD4+ T cells in the draining lymph nodes (Fig. 2c). These data indicate that in this CIA model, PD-1 may exert its protective role via inhibiting antigen-specific Th17 response (Fig. 2b, c), which is consistent with several recent reports (Iliopoulos et al. 2011; Wang et al. 2011).

PD-1 deficiency facilitates antigen-specific T cell proliferation and Th17 differentiation. a, b CD4+ T cell were isolated from draining lymph nodes of WT or Pdcd1 −/− mice with CIA as described in Fig. 1 and cultured in the presence of the indicated concentrations of CII with irradiated T-depleted WT splenocytes. a [3H]thymidine incorporation was measured for cell proliferation after 72 h. b The supernatants were collected after 48 h for the detection of IL-17, IFN-γ and IL-4 by ELISA. c Cells collected from draining lymph nodes of WT or Pdcd1 −/− mice with CIA were stained as described in “Materials and Methods”. Treg (upper panel), Th17 cells (lower panel) were analyzed with flow cytometry. The values represent mean ± SEM from triplicate (a, b). The data shown are representative of two independent experiments
PD-1 May Suppress Th17 Differentiation Through Inhibiting Akt/PKC-θ Phosphorylation
Previously, we have shown that heightened activation of Akt may be responsible for the defective inducible Tregs (iTregs) induction in vitro in the absence of PD-1. We also found more IL-17-producing cells in Pdcd1 −/− CD4+ T cells under Th17-biased condition (Qiao et al. 2012). We showed that no defect in phosphorylation of Stat3 or Smad2/3 was found in T cells in response to IL-6 or transforming growth factor β stimulation in the absence of PD-1 (data not shown). To further define the possible pathway(s) contributing to the regulation of Th17 differentiation by PD-1, we focused on the role of PD-1 in T cell receptor (TCR) signaling. Naïve CD4+ T cells from WT or Pdcd1 −/− mice were stimulated with anti-CD3 in the presence or absence of anti-CD28 in vitro for different times and lysed. T cell lysates were then blotted with different antibodies. As shown in Fig. 3, phosphorylation of ERK, JNK and p38 under TCR stimulation is comparable between WT and Pdcd1 −/− T cells. In addition to heightened phosphorylation of Akt, PKC-θ was also heavily phosphorylated at the Thr538 residue in Pdcd1 −/− CD4+ T cells upon TCR stimulation even in the absence of anti-CD28 costimulation (Fig. 3a), suggesting that both Akt and PKC-θ are negatively regulated by PD-1. Note that we found that the basal level of p-PKC-θ was higher in Pdcd1 −/− T cells than in WT T cells. Although the reason for this increase is currently unknown, this may be due to the effect of T cell intrinsic loss of PD-1 on TCR signaling, although PD-1 is not expressed in resting T cells. This finding indicates that PD-1 may regulate Th17 differentiation via controlling Akt and/or PKC-θ activation. Indeed, PKC-θ has been shown to inhibit iTregs in an Akt/Foxo1/3a-dependent manner (Ma et al. 2012). It has been demonstrated that the suppression of PI3K/Akt/mTORC1 axis impairs in vitro Th17 differentiation (Kurebayashi et al. 2012). Our previous data also showed that PD-1 deficiency increased Th17 differentiation in vitro (Qiao et al. 2012). These data indicate that PD-1 may suppress Th17 differentiation via inhibiting PI3K/Akt axis. To further verify this notion, we treated Pdcd1 −/− mice with a PI3K inhibitor LY294002 in vivo during CIA induction. LY294002 has proved useful to study the various functions of PI3K in vitro or in vivo. It is a classical reversible, ATP-competitive and pan-PI3K inhibitor. Treating the Pdcd1 −/− mice with the LY294002 did not change the incidence of CIA (data not shown), but it did decrease the disease severity as evaluated by erythema and swelling (Fig. 3b) and the phosphorylation of Akt and PKC-θ in CD4+ T cells (Fig. 3e). Histologically, treatment with LY294002 reduced cartilage destruction and lymphocyte infiltration within the synovial membrane (Fig. 3c). Inhibition of PI3K with LY294002 also decreased IL-17 production in the serum (Fig. 3d). Thus, PD-1 may suppress Th17 differentiation via inhibition of PI3K/Akt axis, eventually ameliorating the disease severity in CIA.

PD-1 inhibits the PI3K/Akt pathway, thus suppressing the development of CIA. a WT and Pdcd1 −/− CD4+CD25− T cells were stimulated with anti-CD3 (1 μg/ml) in the presence or absence of anti-CD28 (1 μg/ml). The cell lysates were blotted with phospho-antibodies against ERK, JNK, p38, Akt and PKC-θ. The membranes were stripped and reprobed with anti-actin. b–e Pdcd1 −/− mice were immunized with chicken type II collagen in CFA (n = 10) as described in Fig. 1. LY294002 was administered (i.p.) every other day when half of the mice had developed arthritis (day 30). Disease severity score was determined as described in Fig. 1 (b). The joints were collected and stained as described in Fig. 1 (×100) (c). IL-17 in the serum was detected via ELISA (d). CD4+ T cell lysates were detected for phosphorylation of Akt and PKC-θ (e). The data shown are representative of two independent experiments
In conclusion, we demonstrated that PD-1 is protective in the development of CIA, probably through inhibiting the induction of Th17 after exposure to the antigen. Mechanistically, PD-1 suppresses the activation of PI3K/Akt axis that eventually leads to the attenuation of Th17 differentiation. The identification of the potential mechanisms for PD-1 during the development of autoimmune arthritis will not only shed lights on further understanding of the pathogenesis of RA, but also lead to potential therapeutic approach to autoimmune arthritis and possibly RA in humans.
References
Brand DD, Latham KA, Rosloniec EF (2007) Collagen-induced arthritis. Nat Protoc 2:1269–1275
Buttgereit F, Zhou H, Kalak R et al (2009) Transgenic disruption of glucocorticoid signaling in mature osteoblasts and osteocytes attenuates K/BxN mouse serum-induced arthritis in vivo. Arthritis Rheum 60:1998–2007
Chabaud M, Durand JM, Buchs N et al (1999) Human interleukin-17: a T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum 42:963–970
Hamel KM, Cao Y, Wang Y et al (2010) B7-H1 expression on non-B and non-T cells promotes distinct effects on T- and B-cell responses in autoimmune arthritis. Eur J Immunol 40:3117–3127
Hu L, Zaloudek C, Mills GB et al (2000) In vivo and in vitro ovarian carcinoma growth inhibition by a phosphatidylinositol 3-kinase inhibitor (LY294002). Clin Cancer Res 6:880–886
Iliopoulos D, Kavousanaki M, Ioannou M et al (2011) The negative costimulatory molecule PD-1 modulates the balance between immunity and tolerance via miR-21. Eur J Immunol 41:1754–1763
Keir ME, Butte MJ, Freeman GJ et al (2008) PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 26:677–704
Koch AE (2007) The pathogenesis of rheumatoid arthritis. Am J Orthop 36(7 suppl):5–8
Koenders MI, Kolls JK, Oppers-Walgreen B et al (2005) Interleukin-17 receptor deficiency results in impaired synovial expression of interleukin-1 and matrix metalloproteinases 3, 9, and 13 and prevents cartilage destruction during chronic reactivated streptococcal cell wall-induced arthritis. Arthritis Rheum 52:3239–3247
Kondo Y, Iizuka M, Wakamatsu E et al (2012) Overexpression of T-bet gene regulates murine autoimmune arthritis. Arthritis Rheum 64:162–172
Kurebayashi Y, Nagai S, Ikejiri A et al (2012) PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating gfi1 expression and nuclear translocation of RORγ. Cell Rep 1:360–373
Li J, Wei B, Guo A et al (2013) Deficiency of β-arrestin1 ameliorates collagen-induced arthritis with impaired TH17 cell differentiation. Proc Natl Acad Sci USA 110:7395–7400
Lin SC, Yen JH, Tsai JJ et al (2004) Association of a programmed death 1 gene polymorphism with the development of rheumatoid arthritis, but not systemic lupus erythematosus. Arthritis Rheum 50:770–775
Lubberts E, Joosten LA, van de Loo FA et al (2002) Overexpression of IL-17 in the knee joint of collagen type II immunized mice promotes collagen arthritis and aggravates joint destruction. Inflamm Res 51:102–104
Lubberts E, Koenders MI, Oppers-Walgreen B et al (2004) Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum 50:650–659
Ma J, Ding Y, Fang X et al (2012) Protein kinase C-θ inhibits inducible regulatory T cell differentiation via an AKT-Foxo1/3a-dependent pathway. J Immunol 188:5337–5347
Miossec P, Kolls JK (2012) Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov 11:763–776
Nakae S, Nambu A, Sudo K et al (2003) Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol 171:6173–6177
Okazaki T, Honjo T (2006) The PD-1-PD-L pathway in immunological tolerance. Trends Immunol 27:195–201
Qiao G, Yang L, Li Z et al (2012) Program death-1 regulates peripheral T cell tolerance via an anergy-independent mechanism. Clin Immunol 143:128–133
Skon CN, Lee JY, Anderson KG et al (2013) Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol 14:1285–1293
Toyama S, Tamura N, Haruta K et al (2010) Inhibitory effects of ZSTK474, a novel phosphoinositide 3-kinase inhibitor, on osteoclasts and collagen-induced arthritis in mice. Arthritis Res Ther 12:R92
Wang G, Hu P, Yang J et al (2011) The effects of PDL-Ig on collagen-induced arthritis. Rheumatol Int 31:513–519
Wehrens EJ, Prakken BJ, van Wijk F (2012) T cells out of control—impaired immune regulation in the inflamed joint. Nat Rev Rheumatol 9:34–42
Ying H, Yang L, Qiao G et al (2010) Cutting edge: CTLA-4–B7 interaction suppresses Th17 cell differentiation. J Immunol 185:1375–1378
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NNSF 81201371, to L. Yang), Young Researcher Culturing Plan from Xiangya Hospital, Young Teacher Exploring Plan from Central South University (to L. Yang) and NIH R01 AR049775 and AI090901 (to J. Zhang).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
About this article

Cite this article
Yang, L., Qiao, G., Hassan, Y. et al. Program Death-1 Suppresses Autoimmune Arthritis by Inhibiting Th17 Response. Arch. Immunol. Ther. Exp. 64, 417–423 (2016). https://doi.org/10.1007/s00005-016-0404-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-016-0404-z