
Abstract
Bioinformatics plays a crucial role in advancing genome editing techniques by utilizing computational tools and algorithms to analyze biological data. This chapter provides an overview of the role of bioinformatics in genome editing, focusing on key areas where it is applied. These areas include computational analysis of target sequences, prediction, and evaluation of off-target effects, designing and optimizing CRISPR systems, functional annotation of genomic variants, comparative genomics, homology analysis, and integration of multi-omics data. By leveraging bioinformatics, researchers can identify target sites for gene editing, predict and minimize off-target effects, enhance the efficiency of CRISPR systems, understand the functional consequences of genetic alterations, explore evolutionary relationships, and gain comprehensive insights into biological systems. Moreover, comparative genomics and homology analysis are discussed as vital approaches that leverage bioinformatics to understand evolutionary relationships and identify conserved elements across species. Integration of multi-omics data, such as genomics, transcriptomics, and proteomics, is highlighted as a powerful strategy for gaining comprehensive insights into biological systems. While challenges exist, such as accurate off-target prediction and data management, future directions involving machine learning and user-friendly tools hold promise. Bioinformatics continues to revolutionize genome editing, advancing precision medicine, agriculture, and biological research.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
8.1 Introduction to Genome Editing
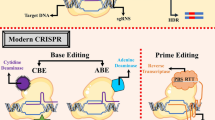
Genome editing is a revolutionary field in genetics that has opened up unprecedented opportunities for manipulating the DNA sequences of living organisms. It allows scientists to make precise modifications to the genetic material of cells, organisms, and even entire populations. This breakthrough technology has the potential to revolutionize various fields, including medicine, agriculture, and biotechnology (Khalil 2020). At its core, genome editing involves the deliberate alteration of DNA sequences within the genome. It enables scientists to add, delete, or replace specific genetic information, thereby modifying the characteristics of an organism (Zhang and Zhou 2014). This capability has far-reaching implications, as it can lead to the development of new treatments for genetic disorders, enhance crop productivity and nutritional value, and contribute to the understanding of fundamental biological processes. One of the most widely used and powerful genome editing techniques is CRISPR-Cas9, which stands for Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-associated protein 9. CRISPR-Cas9 utilizes a guide RNA molecule to target a specific DNA sequence and Cas9, a DNA-cutting enzyme, to introduce the desired modifications. The simplicity, versatility, and efficiency of CRISPR-Cas9 have revolutionized genome editing research and applications. However, genome editing is a complex process that requires careful planning and execution. This is where bioinformatics plays a crucial role. Bioinformatics is an interdisciplinary field that combines biology, computer science, and statistics to analyze and interpret biological data. In the context of genome editing, bioinformatics provides the necessary tools and computational resources to facilitate the design, analysis, and optimization of genome editing experiments.
Bioinformatics aids in the identification and selection of target sequences within the genome that can be modified using CRISPR-Cas9 or other genome editing tools. By analyzing the genomic context, bioinformatics algorithms can predict the potential off-target effects of genome editing and help researchers mitigate any unintended consequences (Akram et al. 2022). This computational analysis enables scientists to make informed decisions regarding target selection and minimize the risks associated with genome editing. Furthermore, bioinformatics assists in the annotation and functional characterization of genomic variants. It helps researchers understand the impact of specific genetic changes on gene function, protein structure, and biological pathways. By integrating multiple omics data, such as genomics, transcriptomics, and proteomics, bioinformatics enables comprehensive analysis of the effects of genome editing on various levels of biological regulation. Moreover, bioinformatics plays a crucial role in managing and integrating the vast amount of data generated during genome editing experiments. It provides the necessary tools and databases for storing, organizing, and analyzing genomic information. This data management infrastructure ensures that researchers can access and share their findings, promoting collaboration and accelerating scientific progress in the field of genome editing. Genome editing has emerged as a powerful tool for manipulating genetic information, with far-reaching implications across various domains. Bioinformatics serves as an essential component in the field of genome editing, providing computational tools and resources to facilitate target selection, optimize experimental design, predict off-target effects, annotate genomic variants, integrate multi-omics data, and manage the vast amount of generated information. By harnessing the power of bioinformatics, scientists can unlock the full potential of genome editing and contribute to advancements in medicine, agriculture, and biotechnology.
8.2 Overview of Bioinformatics
Bioinformatics is an interdisciplinary field that combines biology, computer science, and statistics to analyze and interpret biological data. It encompasses a wide range of techniques, tools, and methodologies aimed at extracting meaningful information from vast amounts of biological data. With the advancements in high-throughput technologies, such as next-generation sequencing, bioinformatics has become an indispensable discipline in modern biological research. At its core, bioinformatics focuses on developing computational algorithms and models to study biological systems. It involves the collection, storage, retrieval, and analysis of biological data, including genomic sequences, protein structures, gene expression profiles, and metabolic pathways. By leveraging computational techniques, bioinformatics enables researchers to gain valuable insights into the complex and intricate workings of living organisms. One of the primary areas of bioinformatics is genomics, which involves the study of an organism’s entire set of genes, known as its genome (Ratan et al. 2018). Bioinformatics tools and algorithms are extensively used to analyze and interpret genomic data. This includes tasks such as DNA sequence assembly, annotation of genes and regulatory elements, identification of genetic variations, and comparative genomics to understand the evolutionary relationships between species.
Another vital aspect of bioinformatics is proteomics, which focuses on the study of proteins, their structures, and functions. Bioinformatics plays a critical role in protein sequence analysis, predicting protein structures, and identifying protein–protein interactions (Fernandez-Recio et al. 2005). These insights are invaluable in understanding the complex mechanisms underlying various biological processes and diseases. Bioinformatics also contributes to the field of transcriptomics, which involves the study of gene expression patterns. By analyzing RNA sequencing data, bioinformatics can provide valuable information about which genes are active under specific conditions, identify alternative splicing events, and uncover regulatory networks governing gene expression. Furthermore, bioinformatics is essential in the field of metabolomics, which involves the study of small molecules called metabolites in biological systems. Bioinformatics tools assist in the identification and quantification of metabolites, as well as the integration of metabolomic data with other omics data to gain a comprehensive understanding of cellular processes. In addition to these specific areas, bioinformatics also encompasses other important applications such as systems biology, drug discovery, and precision medicine. It facilitates the integration of multiple data types, including genomics, proteomics, transcriptomics, and metabolomics, to gain a holistic view of biological systems and unravel their complexities (Kibar and Vingron 2023).
The field of bioinformatics continues to evolve rapidly, driven by advancements in computational technologies, machine learning, and artificial intelligence. These advancements enable the development of more sophisticated algorithms and models, allowing researchers to extract deeper insights from biological data. Moreover, bioinformatics promotes collaboration and data sharing through the development of databases, software tools, and public repositories, facilitating the dissemination of knowledge and fostering scientific advancements. Bioinformatics plays a vital role in modern biological research by leveraging computational techniques to analyze and interpret biological data. It encompasses various sub-disciplines, including genomics, proteomics, transcriptomics, and metabolomics, enabling researchers to gain valuable insights into the structure, function, and regulation of biological systems. With the continuous advancements in technology and computational methods, bioinformatics will continue to drive innovation and discovery in the field of life sciences.
8.3 Importance of Bioinformatics in Genome Editing
Bioinformatics plays a crucial role in the field of genome editing, providing valuable tools and resources that aid in the design, analysis, and optimization of genome editing experiments. The integration of bioinformatics with genome editing techniques enhances the precision, efficiency, and safety of genetic modifications. Let’s explore the importance of bioinformatics in genome editing. Firstly, bioinformatics enables the computational analysis of target sequences within the genome. By utilizing bioinformatics algorithms, researchers can identify and select optimal target sites for genome editing (Joshi et al. 2023). These algorithms take into account factors such as specificity, efficiency, and potential off-target effects, helping researchers make informed decisions in choosing suitable target sequences. This bioinformatics-guided target selection ensures precise and effective genome editing, minimizing unintended genetic alterations. Secondly, bioinformatics assists in the design and optimization of CRISPR systems. CRISPR-Cas9, the most widely used genome editing tool, requires the design of guide RNA molecules that can accurately target specific DNA sequences. Bioinformatics algorithms predict the efficiency and specificity of guide RNA sequences, facilitating the selection of optimal guides for successful genome editing. Additionally, bioinformatics aids in optimizing the delivery of CRISPR components into target cells, improving the overall efficiency of genome editing experiments.
Another vital role of bioinformatics in genome editing is the prediction and evaluation of off-target effects. Despite the remarkable specificity of CRISPR-Cas9, there is a possibility of unintended genetic modifications at off-target sites. Bioinformatics tools analyze the genomic context and sequence homology to predict potential off-target sites, allowing researchers to mitigate these risks. By identifying and evaluating off-target effects computationally, researchers can optimize their experimental designs and minimize unintended genetic alterations. Furthermore, bioinformatics provides essential resources for the functional annotation of genomic variants resulting from genome editing. Bioinformatics databases and tools assist in the identification and characterization of genetic alterations, enabling researchers to understand the impact of these modifications on gene function, regulatory elements, and protein structure. This functional annotation helps researchers assess the potential consequences of genome editing and aids in the interpretation of experimental results. Bioinformatics also facilitates comparative genomics and homology analysis, which are critical for studying the evolutionary relationships between species and identifying conserved regions within genomes. By comparing genomic sequences across different species, researchers can identify functional elements and target sites that are conserved, enhancing the effectiveness of genome editing strategies (Hatanaka et al. 2023).
Furthermore, bioinformatics enables the integration of multi-omics data generated during genome editing experiments. By integrating genomics, transcriptomics, proteomics, and metabolomics data, researchers can gain a comprehensive understanding of the effects of genome editing on various biological levels. Bioinformatics tools and algorithms assist in the analysis and interpretation of these multi-dimensional datasets, providing a holistic view of the genetic modifications and their impact on cellular processes. Lastly, bioinformatics plays a pivotal role in data management and integration. The vast amount of data generated during genome editing experiments requires efficient storage, organization, and retrieval. Bioinformatics provides the necessary infrastructure, including databases, software tools, and pipelines, to manage and integrate genomic information. This ensures that researchers can access and share their data, promoting collaboration and accelerating scientific progress in the field of genome editing. Bioinformatics is of paramount importance in the field of genome editing (Navaridas et al. 2023). It provides computational tools, resources, and algorithms that aid in target selection, guide RNA design, off-target prediction, functional annotation, comparative genomics, multi-omics integration, and data management. By leveraging bioinformatics, researchers can enhance the precision, efficiency, and safety of genome editing techniques, paving the way for advancements in medicine, agriculture, and biotechnology.
8.4 Computational Analysis of Target Sequences
Computational analysis of target sequences is a fundamental aspect of genome editing that relies on bioinformatics tools and algorithms to identify optimal sites for genetic modification. By utilizing computational techniques, researchers can select target sequences with high specificity and efficiency, maximizing the success of genome editing experiments. Let’s delve into the significance of computational analysis in identifying target sequences. The first step in computational analysis is the identification of potential target sites within the genome. This involves searching for specific DNA sequences that are amenable to modification using genome editing tools such as CRISPR-Cas9. Bioinformatics algorithms analyze the genomic sequence to identify regions that meet specific criteria, such as the presence of suitable protospacer adjacent motifs (PAMs) for CRISPR-Cas9 recognition (Cancellieri et al. 2023). These algorithms ensure the selection of target sequences that are compatible with the chosen genome editing tool. Furthermore, computational analysis assists in evaluating the uniqueness of target sequences. It is crucial to select target sites that are specific to the desired genomic region to minimize off-target effects. Bioinformatics algorithms compare the target sequence against the entire genome to assess its uniqueness and potential for off-target binding. This analysis helps researchers identify target sequences with minimal homology to non-intended regions, reducing the likelihood of unintended genetic modifications. Another aspect of computational analysis is the prediction of target site efficiency. Not all target sequences are equally efficient for genome editing. Bioinformatics tools predict the efficiency of target sites based on various factors, such as the accessibility of the DNA sequence, secondary structure formation, and nucleotide composition (Sharma et al. 2023). By evaluating these parameters, researchers can prioritize target sequences with high editing efficiency, increasing the success rate of genome editing experiments. Additionally, computational analysis aids in assessing potential limitations and challenges associated with target sequences. For instance, certain genomic regions may have high levels of repetitive elements or structural complexities, making them less amenable to genome editing. Bioinformatics algorithms can identify such regions and provide insights into potential challenges that may arise during the editing process. This information allows researchers to make informed decisions and adjust their experimental designs accordingly.
Moreover, computational analysis contributes to the identification of functional elements within target sequences. Bioinformatics algorithms scan the target sequence for important genomic features, such as coding regions, regulatory elements, and non-coding RNAs. This analysis helps researchers identify target sites that have the desired functional impact, such as modifying a specific gene or disrupting a regulatory element. By incorporating functional annotations, researchers can select target sequences that align with their experimental objectives. Furthermore, computational analysis facilitates the design of guide RNA molecules for CRISPR-Cas9-mediated genome editing (Table 8.1).
Guide RNAs guide the Cas9 enzyme to the target sequence for precise DNA cleavage. Bioinformatics algorithms predict and optimize guide RNA sequences to ensure their specificity and efficiency (Naeem and Alkhnbashi 2023). By analyzing factors such as off-target potential, secondary structure formation, and binding affinity, computational analysis helps design guide RNAs that can precisely target the desired genomic region. Computational analysis of target sequences is a critical step in genome editing, enabled by bioinformatics tools and algorithms. By leveraging computational techniques, researchers can identify optimal target sites with high specificity, efficiency, and functional impact. Computational analysis aids in target sequence identification, uniqueness assessment, efficiency prediction, identification of potential challenges, and guide RNA design. Through this computational approach, researchers can enhance the precision and success of genome editing experiments, driving advancements in various fields such as medicine, agriculture, and biotechnology.
Computational analysis of target sequences involves a series of steps to identify optimal sites for genetic modification. By leveraging bioinformatics tools and algorithms, researchers can perform an in-depth analysis of the genome to select target sequences with high specificity and efficiency (see Table 8.1). Here are the steps involved in computational analysis of target sequences:
-
Define the search criteria: Determine the specific requirements for the target sequence, such as the desired genomic region, sequence length, and any specific motifs or features to consider.
-
Retrieve the genomic sequence: Obtain the relevant genomic sequence from databases or sequencing experiments, ensuring it covers the region of interest.
-
Preprocess the genomic sequence: Perform necessary preprocessing steps, such as removing ambiguous characters, correcting sequencing errors, or handling variations in genome assembly.
-
Identify potential target sites: Utilize bioinformatics algorithms to scan the genomic sequence and identify potential target sites based on specific criteria. This may involve searching for suitable protospacer adjacent motifs (PAMs) for CRISPR-Cas9 or other recognition sequences for alternative genome editing tools.
-
Assess target site uniqueness: Compare the potential target sites against the entire genome to evaluate their uniqueness. Bioinformatics tools can help identify regions with homology to other non-intended genomic locations, minimizing the risk of off-target effects.
-
Predict target site efficiency: Utilize computational algorithms to predict the efficiency of target sites. Factors such as DNA accessibility, secondary structure formation, and nucleotide composition can be assessed to estimate the likelihood of successful genome editing.
-
Evaluate potential limitations: Analyze the target sequences for any limitations or challenges that may impact the editing process. This could include the presence of repetitive elements, structural complexities, or other known constraints. Identifying such limitations helps researchers anticipate potential difficulties and adjust their experimental design accordingly.
-
Consider functional elements: Scan the target sequences for important genomic features, such as coding regions, regulatory elements, or non-coding RNAs. This step allows researchers to select target sites that align with their specific experimental objectives and have the desired functional impact.
-
Optimize guide RNA design (if applicable): If using CRISPR-Cas9, design and optimize guide RNA molecules to guide the Cas9 enzyme to the target sequence. Computational analysis can predict off-target potential, assess secondary structure formation, and optimize binding affinity to ensure guide RNAs are specific and efficient.
-
Prioritize target sequences: Based on the results of the computational analysis, prioritize the identified target sequences according to their uniqueness, predicted efficiency, functional impact, and any other relevant criteria.
By following these steps, researchers can utilize computational analysis to identify and prioritize optimal target sequences for genome editing. This approach enhances the precision and success of genetic modifications, contributing to advancements in fields such as medicine, agriculture, and biotechnology.
8.5 Designing and Optimizing CRISPR Systems
Designing and optimizing CRISPR systems is a crucial step in genome editing, as it directly impacts the efficiency and precision of the gene editing process. CRISPR, or Clustered Regularly Interspaced Short Palindromic Repeats, is a revolutionary technology that allows researchers to precisely modify the DNA of organisms. The design and optimization of CRISPR systems involve several key considerations to ensure successful and accurate gene editing outcomes. The first step in designing a CRISPR system is the selection of the Cas9 protein or other nucleases that will be used to target the specific genomic region of interest. Cas9 is the most commonly used nuclease, but other nucleases such as Cpf1 are also employed. Factors such as the efficiency, specificity, and off-target effects of the nuclease need to be taken into account during the selection process. Once the nuclease is chosen, the next crucial step is designing the guide RNA (gRNA) that will guide the nuclease to the target DNA sequence (Tian et al. 2023). The gRNA is a short RNA molecule that binds to the target DNA and directs the nuclease to create a double-stranded break at the desired genomic location. Designing an effective gRNA involves identifying the protospacer adjacent motif (PAM) sequence, which is necessary for Cas9 binding, as well as optimizing the gRNA sequence to enhance its specificity and minimize off-target effects. Bioinformatics tools and algorithms play a vital role in the design and optimization of CRISPR systems. These tools help in identifying suitable target sites within the genome, predicting potential off-target effects, and optimizing the gRNA sequence for improved efficiency and specificity. Tools like CRISPR Design, E-CRISP, and CRISPRscan assist researchers in selecting optimal target sites and designing high-quality gRNAs.
Another important aspect of designing and optimizing CRISPR systems is the delivery method of the CRISPR components into the target cells or organisms. Different delivery methods, such as viral vectors, electroporation, or nanoparticle-mediated delivery, have varying efficiencies and capabilities to reach specific cell types or tissues. The choice of delivery method depends on factors such as the target organism, cell type, and intended application. Optimizing CRISPR systems also involves assessing and fine-tuning experimental parameters, such as the concentration of the CRISPR components, incubation time, and temperature. These parameters can significantly influence the editing efficiency and minimize potential off-target effects. Iterative optimization experiments are often performed to achieve the desired editing outcomes. Designing and optimizing CRISPR systems require careful consideration of various factors such as nuclease selection, gRNA design, delivery method, and experimental parameters. The use of bioinformatics tools and algorithms aids in efficient target site selection, gRNA design, and prediction of off-target effects. By optimizing these factors, researchers can enhance the efficiency, specificity, and accuracy of CRISPR-based genome editing, opening up new avenues for genetic research and potential therapeutic applications. The steps involved in designing and optimizing CRISPR systems:
-
Identify the target genomic region: Determine the specific region of the genome that needs to be edited or modified. This can be a gene, regulatory element, or other genomic features.
-
Select the appropriate nuclease: Choose the suitable nuclease for the intended application. Cas9 is commonly used, but other nucleases like Cpf1 or Cas12a can also be considered based on their specific properties.
-
Design the guide RNA (gRNA): Design a gRNA sequence that targets the desired genomic region. The gRNA should be complementary to the target DNA sequence and contain the necessary protospacer adjacent motif (PAM) sequence required for nuclease binding.
-
Evaluate potential off-target effects: Utilize bioinformatics tools to predict potential off-target sites where the gRNA may bind. Assess the specificity of the designed gRNA to minimize the risk of unintended modifications in other genomic regions.
-
Optimize the gRNA sequence: Fine-tune the gRNA sequence to enhance its efficiency and specificity. Consider parameters such as length, secondary structure, and GC content to improve the binding affinity and minimize off-target effects.
-
Determine the delivery method: Choose an appropriate delivery method for introducing the CRISPR components into the target cells or organisms. This can include viral vectors, electroporation, lipid-based transfection, or other specialized delivery systems.
-
Validate and optimize experimental parameters: Conduct preliminary experiments to optimize key parameters such as the concentration of CRISPR components, incubation time, temperature, and cell density. These parameters can significantly influence the efficiency and specificity of the editing process.
-
Assess editing efficiency: Evaluate the efficiency of the CRISPR system by analyzing the frequency and accuracy of desired edits in the target genomic region. Techniques like PCR, DNA sequencing, or reporter assays can be used for this purpose.
-
Iterate and refine: Based on the results obtained, refine the design and experimental parameters if necessary. Iterative optimization may involve modifying the gRNA sequence, adjusting nuclease concentrations, or exploring alternative delivery methods.
-
Validate the desired edits: Confirm the desired genomic modifications through thorough analysis, such as targeted sequencing or functional assays. Validate the edited phenotype or functional outcome, depending on the specific objectives of the experiment.
8.6 Prediction and Evaluation of Off-Target Effects
Prediction and evaluation of off-target effects is a critical aspect of genome editing using CRISPR technology. While CRISPR systems offer remarkable precision, there is still a possibility of unintended modifications at genomic sites similar to the target sequence. Therefore, it is crucial to employ computational tools and experimental methods to predict and evaluate potential off-target effects. The first step in predicting off-target effects is the identification of potential off-target sites. This involves analyzing the genomic sequence for regions that share high similarity with the target sequence and the corresponding guide RNA (gRNA) (Spade 2023). Bioinformatics tools and algorithms have been developed to identify potential off-target sites based on sequence alignment and mismatch analysis. These tools search for sequences that possess similar protospacer adjacent motifs (PAMs) and exhibit only a few nucleotide mismatches with the target sequence. Once potential off-target sites are identified, the next step is to prioritize and evaluate their likelihood of being edited. Several factors come into play during this evaluation. One important consideration is the number and position of mismatches between the gRNA and the off-target site. Off-target sites with fewer mismatches and located near the PAM sequence are generally more prone to editing. Experimental validation is essential to confirm the presence and extent of off-target effects. Various techniques can be employed for this purpose, such as targeted sequencing, high-throughput sequencing, or genome-wide analyses. These approaches involve deep sequencing of the genomic regions surrounding the predicted off-target sites to detect any modifications or alterations.
Additionally, researchers can use control experiments to distinguish true off-target effects from potential artifacts. Control experiments involve comparing edited samples with appropriate negative controls, such as samples treated with an inactive nuclease or samples without any CRISPR components. This helps differentiate specific editing events from background noise or unintended modifications unrelated to CRISPR activity. Furthermore, advancements in CRISPR technology have led to the development of modified or engineered Cas proteins that exhibit improved specificity and reduced off-target effects. These modified nucleases, such as high-fidelity Cas9 variants or Cas9 fusion proteins offer enhanced targeting precision while minimizing unintended editing at off-target sites. Prediction and evaluation of off-target effects are crucial steps in CRISPR-based genome editing. Through the use of bioinformatics tools, computational analysis, and experimental validation, researchers can assess the likelihood and extent of off-target modifications. This knowledge enables the refinement of CRISPR designs and the development of strategies to minimize off-target effects, ultimately enhancing the specificity and accuracy of genome editing applications.
8.7 Functional Annotation of Genomic Variants
Functional annotation of genomic variants is a vital step in understanding the potential impact of genetic variations on gene function and disease susceptibility. With the advent of high-throughput sequencing technologies, numerous genomic variants can be identified in individuals, necessitating comprehensive annotation to interpret their functional significance. The process of functional annotation involves associating genomic variants with various functional elements in the genome. This includes identifying whether the variant falls within protein-coding regions, regulatory regions, non-coding RNA genes, or other important genomic features. Additionally, the annotation aims to determine the potential consequences of the variants, such as their impact on protein structure, gene expression, splicing, or regulatory interactions (Zhou et al. 2023).
Bioinformatics tools and databases play a crucial role in functional annotation. These resources provide comprehensive genomic annotations and integrate information from diverse data sources, including public databases, functional genomics experiments, evolutionary conservation analyses, and computational predictions. They assist in prioritizing variants for further investigation and provide insights into their potential functional consequences. Variant annotation typically involves the use of annotation tools that utilize reference genome sequences and incorporate variant calling data. These tools assign functional annotations based on known features, such as protein domains, DNA-binding motifs, and transcription factor binding sites. They can also predict the impact of variants on protein structure, function, and stability using algorithms and structural modeling approaches. Additionally, functional annotation often includes the analysis of allele frequencies in population databases. This information helps determine the prevalence of variants in different populations and it can be informative for studying genetic diversity, disease associations, or population-specific effects (Fig. 8.1).

Genomic data collection and its editing using bioinformatics tools to generate annotation of novel genetic functionality
Moreover, functional annotation extends beyond individual variants to consider their potential interactions within biological pathways or networks. Integration of variant data with functional pathway analysis allows for the identification of affected biological processes, enrichment of gene sets, and prioritization of pathways that may be dysregulated due to the presence of specific variants. Experimental validation is essential to confirm the functional impact of variants identified through annotation. Techniques such as functional assays, reporter assays, gene expression studies, or genome editing experiments can provide direct evidence of the effects of variants on gene function and cellular processes (Nagral et al. 2023). Functional annotation of genomic variants is a crucial step in interpreting their potential biological significance. By leveraging bioinformatics tools, databases, and experimental validations, researchers can gain insights into the functional consequences of genetic variations. This knowledge facilitates the understanding of disease mechanisms, identification of therapeutic targets, and personalized medicine approaches based on an individual’s genetic makeup.
8.8 Comparative Genomics and Homology Analysis
Comparative genomics and homology analysis are powerful approaches used to study the similarities and differences in genomic sequences among different organisms. These methods provide valuable insights into evolutionary relationships, functional conservation, and identification of important genomic elements across species. Comparative genomics involves the systematic comparison of genomic sequences from different organisms. By aligning and comparing DNA or protein sequences, researchers can identify regions of similarity and divergence. This analysis helps in understanding the evolutionary relationships between species and provides clues about the conservation of functional elements, such as protein-coding genes, regulatory sequences, or non-coding RNAs. Homology analysis is a fundamental aspect of comparative genomics. It aims to identify and characterize genes or genomic elements that have descended from a common ancestor. Through homology analysis, researchers can infer the presence of orthologous genes (genes in different species that have a common ancestor) or paralogous genes (genes that have arisen through gene duplication events within a species). Bioinformatics tools and algorithms are essential for conducting comparative genomics and homology analysis (Tao et al. 2023; Kaushik et al. 2022). These tools utilize sequence alignment algorithms, such as BLAST (Basic Local Alignment Search Tool), to compare sequences and identify regions of similarity. Multiple sequence alignment methods, such as ClustalW, MUSCLE, or MAFFT, are employed to align sequences from multiple organisms, allowing for the identification of conserved regions and the detection of evolutionary changes.
Comparative genomics and homology analysis have wide-ranging applications. They provide insights into gene function and regulation, identification of conserved non-coding elements, inference of gene regulatory networks, and discovery of candidate genes involved in specific biological processes or diseases. These approaches are particularly valuable for studying model organisms, as the knowledge gained from well-characterized species can be extrapolated to understand the biology of related organisms. Furthermore, comparative genomics and homology analysis have implications in fields such as evolutionary biology, phylogenetics, and drug discovery. By comparing genomes across species, researchers can trace the evolutionary history of genes and identify genetic variations that contribute to phenotypic differences or disease susceptibility. Comparative genomics and homology analysis provide valuable insights into the evolution and functional conservation of genomic sequences. Through the use of bioinformatics tools and algorithms, researchers can compare sequences, identify homologous genes, and unravel the relationships between different organisms. These approaches have broad applications in understanding gene function, unraveling evolutionary relationships, and advancing our knowledge of the biological processes underlying life.
8.9 Integration of Multi-Omics Data
Integration of multi-omics data has emerged as a powerful approach to unravel the complexities of biological systems by combining information from multiple molecular levels. Omics technologies, such as genomics, transcriptomics, proteomics, metabolomics, and epigenomics, generate vast amounts of data, providing a comprehensive view of cellular processes and their interconnections. Integration of these multi-omics datasets enables a deeper understanding of biological mechanisms, identification of biomarkers, and discovery of novel therapeutic targets. The integration of multi-omics data involves several key steps. First, data from different omics platforms need to be collected and preprocessed to ensure compatibility and quality. This includes data normalization, filtering, and transformation to account for technical variations and biases introduced during data generation. Next, bioinformatics methods and statistical algorithms are applied to integrate the multi-omics datasets (Cai et al. 2022). These methods aim to identify relationships, patterns, and associations between the different molecular layers. They can involve data fusion techniques, network analysis, machine learning algorithms, or statistical modeling approaches. The goal is to extract meaningful information and uncover molecular interactions, regulatory networks, and biological pathways that drive complex biological phenomena.
One of the main challenges in integrating multi-omics data is dealing with the high dimensionality and heterogeneity of the datasets. Various computational approaches have been developed to address these challenges, including dimensionality reduction techniques, feature selection methods, and data integration algorithms. These approaches help reduce noise, identify key features, and capture the underlying biological signals present in the data. Integration of multi-omics data has numerous applications across different fields of biology and medicine. In cancer research, for example, the integration of genomics, transcriptomics, and proteomics data can provide a comprehensive view of molecular alterations, identify driver mutations, and reveal potential therapeutic targets. In personalized medicine, integration of multi-omics data can aid in predicting treatment responses, stratifying patient populations, and guiding therapeutic decisions. Furthermore, the integration of multi-omics data has implications in systems biology and precision medicine. It enables the identification of biomarkers for early disease detection, understanding disease mechanisms, and discovering new drug targets (Zhang et al. 2022). By combining information from multiple molecular layers, researchers can gain a more comprehensive understanding of the complexity of biological systems and uncover novel insights that would be difficult to obtain from single-omics analyses. The integration of multi-omics data is a powerful approach that leverages the wealth of information provided by different omics technologies. By combining and analyzing data from genomics, transcriptomics, proteomics, metabolomics, and epigenomics, researchers can gain a deeper understanding of biological processes, identify molecular interactions, and discover new biomarkers and therapeutic targets. This integrative approach has the potential to revolutionize our understanding of complex diseases, drive precision medicine efforts, and advance our knowledge of the intricacies of living systems.
8.10 Data Management and Integration
Data management and integration play a crucial role in modern scientific research, especially in fields such as bioinformatics, genomics, and systems biology. As the volume and complexity of data generated from various sources continue to increase, effective data management and integration strategies are essential for organizing, storing, and analyzing large datasets and extracting meaningful insights. Data management involves the systematic organization and storage of data to ensure its accessibility, accuracy, and integrity. It encompasses various activities, including data acquisition, data cleaning, data storage, data documentation, and data sharing. Proper data management practices help researchers maintain data quality, enable reproducibility, and facilitate collaboration and data sharing within the scientific community (Yeo and Selvarajoo 2022). One of the key aspects of data management is data integration, which involves combining data from multiple sources or experiments to create a unified and comprehensive dataset. Integration enables researchers to merge diverse datasets, such as genomic data, clinical data, or environmental data, to gain a more holistic understanding of complex biological systems. It allows for the identification of patterns, correlations, and relationships that may not be apparent when analyzing individual datasets in isolation.
Bioinformatics tools and databases play a vital role in data management and integration. These resources provide platforms for data storage, retrieval, and analysis, as well as standardized formats and protocols for data exchange. Researchers can leverage these tools and databases to manage and integrate various types of biological data, including DNA sequences, gene expression profiles, protein structures, and functional annotations. Furthermore, data management and integration often involve the use of data integration frameworks and computational algorithms. These methods facilitate the seamless integration of diverse datasets by addressing issues such as data heterogeneity, data format conversion, and data mapping. Data integration frameworks enable researchers to merge datasets with different structures, ontologies, or data models, ensuring compatibility and consistency across the integrated dataset. Effective data management and integration have numerous benefits in scientific research. They enable researchers to uncover hidden insights, generate new hypotheses, and make data-driven decisions. Integration of diverse datasets enhances the power and robustness of analyses, allowing for a more comprehensive understanding of complex biological phenomena. Furthermore, proper data management practices ensure the long-term preservation and availability of valuable research data, promoting transparency and reproducibility. Data management and integration are essential components of scientific research in the era of big data. By implementing effective data management strategies, researchers can ensure data quality, accessibility, and reproducibility. Integration of diverse datasets enables researchers to extract meaningful insights and gain a deeper understanding of complex biological systems. Embracing proper data management and integration practices is crucial for advancing scientific knowledge, facilitating collaboration, and driving discoveries across various disciplines.
8.11 Challenges and Future Directions in Bioinformatics for Genome Editing
Bioinformatics has played a crucial role in advancing genome editing technologies, such as CRISPR-Cas9, and has greatly facilitated the design, analysis, and optimization of gene-editing experiments. However, several challenges remain, and future directions in bioinformatics are poised to address these challenges and further enhance the efficiency and precision of genome editing techniques. One of the primary challenges in bioinformatics for genome editing is the accurate prediction of off-target effects. While considerable progress has been made in developing computational tools and algorithms to predict potential off-target sites, there is still room for improvement. Enhancing the specificity and accuracy of off-target prediction algorithms will be crucial in minimizing unintended modifications and ensuring the safety of genome editing applications. Another challenge lies in the prediction of on-target editing efficiency. While bioinformatics tools can identify potential target sites for genome editing, accurately estimating the editing efficiency at these sites remains a challenge (Han et al. 2022). Factors such as chromatin accessibility, DNA structure, and epigenetic modifications can influence the editing outcomes. Integrating these factors into computational models will improve the prediction of on-target editing efficiency and aid in selecting optimal target sites. Furthermore, the analysis and interpretation of large-scale genomics and multi-omics datasets pose significant challenges. The integration of genomics, transcriptomics, proteomics, and other omics data requires sophisticated algorithms and computational methods. Developing comprehensive and scalable bioinformatics pipelines that can handle the vast amounts of data generated by high-throughput sequencing technologies will be crucial for leveraging these datasets in genome editing research.
Additionally, the interpretation of functional consequences resulting from genomic modifications is an ongoing challenge. While bioinformatics tools can predict the impact of genetic variants and editing events on protein function and gene regulation, accurately understanding the functional implications in complex biological systems remains complex. Integrating experimental validation, functional assays, and advanced computational approaches will be instrumental in unraveling the intricate relationship between genomic alterations and phenotypic outcomes. As for future directions, advancements in machine learning and artificial intelligence hold great promise for bioinformatics in genome editing. Deep learning algorithms and neural networks can potentially enhance the accuracy and efficiency of off-target prediction, on-target editing efficiency prediction, and functional annotation of genomic variants. Integrating these advanced computational techniques into existing bioinformatics pipelines will contribute to more precise and reliable genome editing outcomes. Another future direction lies in the development of user-friendly bioinformatics tools and software platforms. Simplifying the accessibility and usability of bioinformatics tools will democratize their use and enable a broader community of researchers to employ these powerful techniques in their genome editing experiments. User-friendly interfaces, intuitive workflows, and comprehensive documentation will enhance the adoption and impact of bioinformatics in the field. Bioinformatics plays a pivotal role in genome editing, but challenges persist. Addressing these challenges and embracing future directions will propel the field forward. By improving off-target prediction, on-target editing efficiency estimation, multi-omics data analysis, and functional interpretation, bioinformatics will continue to revolutionize genome editing technologies, opening new avenues for precision medicine, agriculture, and fundamental biological research.
8.12 Conclusion
Bioinformatics has emerged as a vital discipline in the field of genome editing, facilitating various aspects of the gene-editing process. Through computational analysis of target sequences, researchers can identify suitable target sites for genome editing and assess their potential impact. Designing and optimizing CRISPR systems using bioinformatics tools enables the development of more efficient and precise gene-editing tools. Prediction and evaluation of off-target effects using computational algorithms aid in minimizing unintended modifications and ensuring the safety of genome editing applications. Functional annotation of genomic variants provides insights into the functional consequences of genetic alterations, guiding researchers in understanding their impact on protein function and gene regulation. Comparative genomics and homology analysis help unravel evolutionary relationships and identify conserved elements across species, contributing to our understanding of gene function and evolution. Integration of multi-omics data allows for a comprehensive view of biological systems, enabling the identification of molecular interactions and the discovery of novel biomarkers and therapeutic targets. Challenges, such as accurate prediction of off-target effects, on-target editing efficiency, data management, and functional interpretation, exist in the field of bioinformatics for genome editing. However, these challenges present opportunities for future advancements, including the integration of machine learning and artificial intelligence, development of user-friendly tools, and enhanced data analysis techniques. By addressing these challenges and embracing future directions, bioinformatics will continue to revolutionize genome editing technologies, empowering researchers with powerful tools for precision medicine, agriculture, and biological research.
References
Akram F, Haq IU, Sahreen S, Nasir N, Naseem W, Imitaz M, Aqeel A (2022) CRISPR/Cas9: a revolutionary genome editing tool for human cancers treatment. Technol Cancer Res Treat 21:15330338221132078
Cai Z, Poulos RC, Liu J, Zhong Q (2022) Machine learning for multi-omics data integration in cancer. Iscience 25:103798
Cancellieri S, Zeng J, Lin LY, Tognon M, Nguyen MA, Lin J, Pinello L (2023) Human genetic diversity alters off-target outcomes of therapeutic gene editing. Nat Genet 55(1):34–43
Fernandez-Recio J, Totrov M, Skorodumov C, Abagyan R (2005) Optimal docking area: a new method for predicting protein–protein interaction sites. Proteins 58(1):134–143
Han P, Teo WZ, Yew WS (2022) Biologically engineered microbes for bioremediation of electronic waste: Wayposts, challenges and future directions. Eng Biol 6(1):23–34
Hatanaka F, Suzuki K, Shojima K, Yu J, Takahashi Y, Sakamoto A, Belmonte JCI (2023) Therapeutic strategy for spinal muscular atrophy by combining gene supplementation and genome editing. bioRxiv 2023:535786
Joshi A, Song HG, Yang SY, Lee JH (2023) Integrated molecular and bioinformatics approaches for disease-related genes in plants. Plants 12(13):2454
Kaushik V, Jain P, Akhtar N, Joshi A, Gupta LR, Grewal RK, Chawla M (2022) Immunoinformatics-aided design and in vivo validation of a peptide-based multiepitope vaccine targeting canine circovirus. ACS Pharmacol Transl Sci 5(8):679–691
Khalil AM (2020) The genome editing revolution. J Genet Eng Biotechnol 18(1):1–16
Kibar G, Vingron M (2023) Prediction of protein–protein interactions using sequences of intrinsically disordered regions. Proteins 91:980
Naeem M, Alkhnbashi OS (2023) Current bioinformatics tools to optimize CRISPR/Cas9 experiments to reduce off-target effects. Int J Mol Sci 24(7):6261
Nagral A, Mallakmir S, Garg N, Tiwari K, Masih S, Nagral N, Aggarwal R (2023) Genomic variations in ATP7B gene in Indian patients with Wilson disease. Indian J Pediatr 90(3):240–248
Navaridas R, Vidal-Sabanés M, Ruiz-Mitjana A, Perramon-Güell A, Megino-Luque C, Llobet-Navas D, Dolcet X (2023) Transient and DNA-free in vivo CRISPR/Cas9 genome editing for flexible modeling of endometrial carcinogenesis. Cancer Commun 43(5):620
Ratan ZA, Son YJ, Haidere MF, Uddin BMM, Yusuf MA, Zaman SB, Cho JY (2018) CRISPR-Cas9: a promising genetic engineering approach in cancer research. Ther Adv Med Oncol 10:1758834018755089
Sharma P, Dahiya S, Kaur P, Kapil A (2023) Computational biology: role and scope in taming antimicrobial resistance. Indian J Med Microbiol 41:33–38
Spade G (2023) The revolutionary genome editor: CRISPR-Cas9 systems
Tao X, Xu T, Lin X, Xu S, Fan Y, Guo B, Yue H (2023) Genomic profiling reveals the variant landscape of sporadic parathyroid adenomas in Chinese population. J Clin Endocrinol Metabol 108(7):1768–1775
Tian M, Zhang R, Li J (2023) Emergence of CRISPR/Cas9-mediated bioimaging: a new dawn of in-situ detection. Biosens Bioelectron 232:115302
Yeo HC, Selvarajoo K (2022) Machine learning alternative to systems biology should not solely depend on data. Brief Bioinform 23(6):bbac436
Zhang L, Zhou Q (2014) CRISPR/Cas technology: a revolutionary approach for genome engineering. Sci China Life Sci 57:639–640
Zhang C, Chen Y, Zeng T, Zhang C, Chen L (2022) Deep latent space fusion for adaptive representation of heterogeneous multi-omics data. Brief Bioinform 23(2):bbab600
Zhou H, Arapoglou T, Li X, Li Z, Zheng X, Moore J, Lin X (2023) FAVOR: functional annotation of variants online resource and annotator for variation across the human genome. Nucleic Acids Res 51(D1):D1300–D1311
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2024 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Joshi, A., Kumar, A., Kaushik, V., Kumar, P., Dubey, S. (2024). Role of Bioinformatics in Genome Editing. In: Singh, V., Kumar, A. (eds) Advances in Bioinformatics. Springer, Singapore. https://doi.org/10.1007/978-981-99-8401-5_8
Download citation
DOI: https://doi.org/10.1007/978-981-99-8401-5_8
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-8400-8
Online ISBN: 978-981-99-8401-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)