
Abstract
Pathogenic variants in BRCA1 and BRCA2 (BRCA1/2) cause hereditary breast and ovarian cancer syndrome (HBOC). BRCA1/2 are involved in multiple cellular processes, including homologous recombination (HR), which maintain genome stability. The HR repair function of BRCA1/2 is thought to underpin their function as tumor suppressors. However, it is unclear how dysregulation of these proteins causes tissue-specific carcinogenesis. Previously, we found that cancer-derived variants and abnormal expression of BRCA1-associated proteins cause centrosome amplification in mammary tissue-derived cells, resulting in chromosome segregation errors.
HR-deficient cells are sensitive to poly (ADP-ribose) polymerase inhibitors and platinum agents. Recently, we developed an HR activity assay named Assay of Site-Specific HR Activity (ASHRA), which evaluates HR activity quantitively. Analyzing the HR activity of BRCA1 variants using ASHRA revealed that the assay can predict whether an individual has a moderate risk of breast and ovarian cancer, and their sensitivity to PARP inhibitors. Furthermore, we identified a novel mechanism underlying resistance to the PARP inhibitor olaparib and the platinum agent cisplatin, which is dependent on high expression of activating transcription factor 1 (ATF1) and the transactivation activity of BRCA1 with ATF1.
In this chapter, we describe the effects of BRCA1/2 impairment, which is thought to contribute to carcinogenesis, as well as regulation of centrosome number by BRCA1, which may play a role in tissue-specific carcinogenesis. Furthermore, we describe the mechanisms underlying resistance to PARP inhibitors and suggest a novel mechanism by which BRCA1/ATF1-mediated transcription leads to resistance to olaparib and cisplatin.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Hereditary breast and ovarian cancer syndrome (HBOC) is a BRCA1 and BRCA2 (BRCA1/2)-linked genetic disorder associated with a high risk of breast, ovarian, and other cancers [1,2,3]. A diagnosis of HBOC is made by genetic testing for BRCA1/2. Clinical management approaches include screening for early cancer detection, prophylactic surgery for healthy carriers, and chemotherapy for patients with cancer. Therefore, accurate diagnoses of pathogenic variants are critical for clinical decision-making and improved prognosis. The pathogenicity of BRCA1/2 variants is classified as benign, likely benign, variants of uncertain significance (VUS), likely pathogenic, or pathogenic. Most pathogenic variants are premature truncation variants generated by nonsense or frameshift mutations, whereas VUS are missense, small in-frame deletion or insertion, and splicing variants. The effects of these variants on function have not yet been determined.
BRCA1/2 functions in multiple cellular processes to maintain genome stability [4,5,6]. Homologous recombination (HR) repair is a critical error-free pathway for repairing DNA double-strand breaks; this pathway uses an intact sister chromatid as a template. BRCA1/2 function in HR repair is thought to underpin their role as tumor suppressors. Therefore, assays that evaluate HR activity have been used to estimate the pathogenicity of BRCA1/2 variants. Of these, the direct-repeat GFP (DR-GFP) assay is the most common [7,8,9,10,11,12,13].
On the other hand, it is unclear how dysregulation of BRCA1/2 causes tissue-specific carcinogenesis. Centrosomes, the major microtubule nucleation centers in animal cells, mediate formation of a bipolar spindle during mitosis [14]. Aberration of centrosome number and structure are common in various cancers [15] and are related to an invasive phenotype [16]. Centrosome amplification results in chromosome segregation errors, leading to chromosomal instability, which is in turn associated with carcinogenesis and cancer progression [17]. Recently, we identified BRCA1-interacting proteins that function to regulate centrosomes together with BRCA1. We found that abnormalities in these BRCA1-associated proteins in mammary tissue-derived cells cause centrosome amplification [18,19,20].
In addition to its role in carcinogenesis, HR repair activity in cells is important for predicting sensitivity to some anti-cancer agents, poly (ADP-ribose) polymerase (PARP) inhibitors, and platinum agents. PARP functions in various DNA damage repair pathways as well as the repair process for DNA single-stranded breaks (SSBs) [21]. PARP inhibitors impair the repair of DNA single-stranded breaks (SSBs), which results in the creation of DNA double-stranded breaks (DSBs), and trap PARP protein at DNA SSBs [22]. Furthermore, because PARP helps to restart stalled DNA replication forks, PARP inhibitors induce collapse of replication forks [23, 24]. Since HR contributes to repair of DSBs, PARP trapping, and collapse of replication forks [22], PARP inhibitors cause synthetic lethality in cells with HR deficiency caused by alteration of HR factors, including BRCA1/2 [25,26,27,28].
PARP inhibitors have been developed to treat various cancers, including breast, ovarian, pancreatic, and prostate cancers [29]; however, a number of resistance mechanisms have been reported [30,31,32]. Recently, we developed an assay to evaluate HR activity in cells and analyzed the HR activity of BRCA1 variants [33]. We found that this assay evaluates HR activity quantitively [34]. We also identified a novel mechanism underlying resistance via transactivation activity of BRCA1 [34].
Here, we describe the functions of BRCA1/2, whose impairment is involved in carcinogenesis. We also discuss our recent finding that BRCA1-interacting proteins regulate centrosomes. Furthermore, we explain the mechanisms underlying resistance to PARP inhibitors and describe a novel mechanism of resistance to PARP inhibitors and platinum agents that is dependent on the function of BRCA1 during transcription.
2 Structure of BRCA1/2
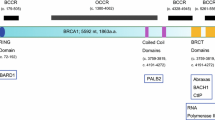
BRCA1 has a RING domain at its amino (N)-terminal region that binds directly to BARD1. There is a coiled-coil motif and two BRCT domains at the carboxy (C)-terminal region (Fig. 1a). The coiled-coil motif binds to PALB2, and the BRCT domains bind to BRIP1, CtIP, and ABRAXAS1. BARD1, PALB2, BRIP1, and ABRAXAS1 are also breast cancer susceptibility genes [6, 35].

Structure of BRCA1 and BRCA2. (a) BRCA1 has a RING domain in the N-terminal region, and a coiled-coil domain and two BRCT domains in the C-terminal region. BRCA1 binds to BARD1 via the RING domain. The coiled-coil domain of BRCA1 mediates complex formation with PALB2. The BRCT domains bind to BRIP1, CtIP, and ABRAXAS1. (b) BRCA2 has a PALB2 binding domain at the N-terminal region, eight BRC repeats in the middle, and a helical domain, three oligonucleotide/oligosaccharide binding (OB) folds, and a RAD51-binding domain, (C-terminal RAD51-binding; CTRB) at the C-terminal region. The middle portion, which includes eight BRC repeats, binds to RAD51. A helical domain and three OB folds in the C-terminal domain and the N-terminal DNA binding domain (NTD) are DNA binding domains
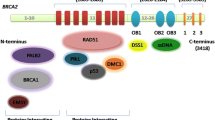
BRCA2 has eight BRC motifs in its middle segment. BRCA2 binds to the RAD51 recombinase (which plays a critical role in HR activity) via BRC motifs and its C-terminal region (Fig. 1b). The N-terminal region contains a PALB2 binding domain. The C-terminal region contains a helical domain and three oligonucleotide/oligosaccharide binding (OB) folds that bind to ssDNA. The N-terminal DNA binding domain (NTD) also has DNA binding activity.
3 Functions of BRCA1/2
3.1 HR
There are two major pathways that repair DNA DSBs: HR and non-homologous end joining (NHEJ). The HR is a pathway for error-free repair of DSBs uses the sister chromatid as a recombination template during the S/G2 phase of the cell cycle. By contrast, NHEJ repairs DSBs throughout the cell cycle by direct joining; however, it is error-prone and frequently causes deletion or insertion mutations in DNA [36].
The choice of which pathway is used to repair DSB is determined by DNA end resection, which is the processing of DNA ends to generate 3′ single strands. BRCA1/BARD1 competes with 53BP1 and promote end resection to proceed to HR pathway. DNA endoresection is initiated by the nuclease MRN complex, which comprises endonuclease MRE11, RAD50, and NBS1, to create short ssDNA (Fig. 2a). CtIP promotes end resection by MRE11. BRCA1/BARD1 plays a role in DNA end resection by interacting with the MRN complex and CtIP. ssDNA is occupied by replication protein A (RPA), which is then replaced by RAD51. Replacement of RPA by RAD51 is mediated by BRCA1-PALB2-BRCA2, thereby forming a RAD51-ssDNA nucleoprotein filament. The RAD51-ssDNA nucleoprotein filament functions in the homology search by invading duplex DNA molecules and facilitating base-pairing with complementary DNA sequences. BRCA1/BARD1 contributes to this homologous pairing. Therefore, BRCA1/BARD1 is involved in multiple steps of the HR repair pathway [4, 37, 38].

Functions of BRCA1 and BRCA2. (a) The HR repair pathway. BRCA1/BARD1 promotes end resection in competition with 53BP1. DNA end resection is initiated by the nuclease MRN complex with CtIP. BRCA1/BARD1 is implicated in DNA end resection via interaction with the MRN complex and CtIP. ssDNA is covered by RPA, which is then replaced by RAD51; this process is mediated BRCA1-PALB2-BRCA2. The RAD51 filament functions in the homology search by invading duplex DNA. (b) DNA replication fork protection. When DNA replication forks are stalled, nascent DNA strands pair with each other and the fork regresses (fork reversal). BRCA1/2 prevents MRE11-mediated degradation of the free DNA end stabilizing RAD51 filaments at the stalled forks. Alterations in BRCA1/2 restore MRE11-mediated degradation of free DNA ends, thereby collapsing the DNA replication fork. (c) Structure of the centrosome at G2 phase and the spindle poles at the mitotic phase. The centrosome comprises a pair of centrioles, mother centriole and daughter centriole, surrounded by pericentriolar material (PCM). (d) Model of the BRCA1/BARD1/OLA1/RACK1 complex. The N-terminal region of BRCA1 binds to the N-terminal region of BARD1. OLA1 binds to the N-terminal region of BRCA1, the C-terminal region of BARD1, and γ-tubulin. The middle portion of BRCA1 interacts with OLA1 via γ-tubulin. RACK1 binds to OLA1, the N-terminal region of BRCA1, and γ-tubulin. “N” indicates the N-terminal region. “C” indicates the C-terminal region. (e) Model of the conformational changes in the BRCA1/BARD1/OLA1/γ-tubulin complex induced by variants of BRCA1, BARD1, or OLA1. The I42V variant in BRCA1 and the E168Q variant in OLA1 impair binding of OLA1 to the N-terminal region of BRCA1. The C645R, V695L, and S761N variants in BARD1 and the S36A, S36C, F127A, and T325A variants in OLA1 impair binding of OLA1 to the C-terminal region of BARD1. These variants cause centrosome amplification. “N” indicates the N-terminal region. “C” indicates the C-terminal region
3.2 Stabilization of Replication Forks
When the DNA replication machinery encounters DNA lesions, or nucleotides become a limiting factor, DNA replication stalls. Newly synthesized DNA strands pair with each other, and the fork regresses (fork reversal), resulting a four-armed DNA structure that has free DNA end (Fig. 2b). BRCA1/2 prevents degradation of this free DNA end, which is mediated by the MRE11 nuclease, by stabilizing RAD51 filaments at the stalled forks. Therefore, BRCA1/2 prevent DNA damage. Alteration of BRCA1/2 results in the failure to protect replication forks, leading to collapse [37, 38].
3.3 Prevention of R-Loop Accumulation
R-loops, which comprise an RNA-DNA hybrid with a displaced ssDNA, occur on sites at which strong DNA secondary structures are formed due to perturbation of transcription or transcription-coupled processes such as mRNA splicing. Since an R-loop stalls the DNA replication machinery, cells avoid R-loop accumulation by preventing or removing them. BRCA1/BARD1 and BRCA2 prevent R-loop accumulation [37, 39].
3.4 Centrosome Regulation
Centrosomes regulate cell shape, polarity, and motility, in addition to formation of the mitotic spindle [14, 40]. The centrosome comprises a pair of centrioles, the mother and daughter centrioles, which are surrounded by the pericentriolar material (PCM) (Fig. 2c). Centrosomes are duplicated once per cell cycle, a process that is precisely controlled [41].
BRCA1 and BARD1, which localize to the centrosome throughout the cell cycle [42], function during centriole duplication [42,43,44]. BRCA1 interacts with major components of centrosomes: γ-tubulin [45] and mitotic kinases Aurora A and Polo-like kinase1 (PLK1) [46, 47]. BRCA1/BARD1 ubiquitinates centrosome proteins, including γ-tubulin [43]. We identified Obg-like ATPase 1 (OLA1) as a BARD1-interacting protein [48] and the receptor for activated C kinase 1 (RACK1) as an OLA1-interacting protein [49] (Fig. 2d). Aberrant expression of OLA1 and RACK1 occurs in many malignancies [50,51,52,53,54]. BRCA1 binds directly to OLA1 and RACK1, and functions to regulate centriole duplication together with these proteins. RACK1 regulates centriole duplication by controlling the centrosomal localization of BRCA1, as well as PLK1 phosphorylation by Aurora A [19, 20, 49].
A number of BRCA1 variants derived from familial breast cancers cause centrosome amplification in breast cancer cells [55]. Interestingly, some BRCA1 variants fail to regulate centrosome number; however, they are proficient in HR activity [8]. Cancer-derived variants of BRCA1, BARD1, OLA1, and RACK1 abolish their bindings each other. These variants and aberrant expression of these proteins cause centrosome amplification due to centriole overduplication only in mammary tissue-derived cells (Fig. 2e) [18, 19, 48, 49, 56]. These findings suggest that the BRCA1, together with these interacting proteins, regulates centrosomal numbers and contributes to tumor suppression.
Interestingly, the number of centrioles in cells with two γ-tubulin spots is higher in mammary tissue-derived cells than in cells derived from other tissues, suggesting that the efficiency of centriole duplication might be higher than that in cells derived from other tissues [49]. Thus, mammary cells might be sensitive to abnormality of centrosome proteins such as BRCA1-associated proteins, resulting in tissue-specific carcinogenesis.
BRCA2 also localizes to centrosomes and is involved in centriole duplication [57, 58]. Furthermore, BRCA2 interacts with a cytoskeletal cross-linker protein, plectin, and controls the position of the centrosome [59].
3.5 Other Functions
BRCA1/2 functions in multiple cellular processes in addition to the functions described above. The N-terminal region of BRCA1 has E3 ubiquitin ligase activity, which is enhanced significantly by forming a heterodimer with BARD1 [60]. The C-terminal region of BRCA1 interacts with RNA helicase A, a component of RNA polymerase II (Pol II) [61], and activates transcription. Since pathogenic variants abolish BRCA1-medicated transcriptional activation, functional assays were used to analyze a number of BRCA1 C-terminal missense variants to predict pathogenicity [6, 62, 63]. BRCA1 binds to several transcription factors, including p53, c-Myc, and GATA3. Furthermore, BRCA1 is involved in DNA damage checkpoint control, regulation of estrogen receptor α (ERα), apoptosis, and differentiation of luminal progenitor in breast tissues [6, 35, 62].
BRCA2 also regulates DNA damage checkpoint control [35]. In addition, BRCA2 is phosphorylated by PLK1, and regulates cytokinesis, a critical final step of cell division [64].
4 Evaluation of HR Activity
4.1 Evaluation of HR Activity in HBOC
Genetic alterations in HR factors in addition to BRCA1/2 cause hereditary cancers such as HBOC [35]. Therefore, evaluation of HR activity in cells is important to estimate the pathogenicity of HR factor variants. As described above, the DR-GFP assay has been used widely for this purpose. In addition, Ikegami et al. developed a functional assay to evaluate the HR activity of BRCA2 variants by assessing sensitivity of BRCA2-deficient cells to PARP inhibitor [65].
4.2 Novel Assay to Evaluate HR Activity in Cells
To evaluate HR activity easily in cells, we developed the Assay for Site-specific HR Activity (ASHRA) (Fig. 3) [33]. In ASHRA, an expression vector for gRNA and Cas9 and a donor vector were co-transfected into the cells. The Cas9 endonuclease introduces a DSB specifically into the endogenous target locus of the genome. For our analysis, we chose the β-actin gene (ACTB), which is stably transcribed in cells, as a target gene. The donor vector contains a marker sequence flanked by two arms homologous to the target locus as a template for HR. Two days after transfection, genomic DNA was extracted. When the DSB is repaired by HR, the marker sequence is knocked-in to the target locus. HR activity is evaluated by quantifying the knock-in frequency by quantitative PCR. We confirmed that knockdown of BRCA1 or RAD51, but not that of non-HR factors, reduces the knock-in frequency [33].

Schematic of ASHRA. The Cas9 endonuclease creates double-strand breaks (DSBs) at the target site in the genome. When the DSBs are repaired by HR using a donor plasmid containing a marker sequence flanked by two arms homologous to the target site as a template, the marker sequence is knocked-in to the target site. HR activity in cells is evaluated by measuring the knock-in frequency of the marker sequence by quantitative PCR
4.3 A Novel HR Assay to Detect Intermediate HR Activity
Using ASHRA, we examined the HR activity of 30 BRCA1 missense variants at the N-terminal region that were previously analyzed using the DR-GFP assay [8, 66]. The DR-GFP assay categorized HR activity only as HR-proficient or HR-deficient, whereas ASHRA identified 10 BRCA1 variants as having intermediate HR activity, which were not distinguished by the DR-GFP assay [34]. Interestingly, the HR activity of these BRCA1 variants, as assessed by ASHRA, correlated significantly with the survival rates of cells expressing BRCA1 variants after exposure to the PARP inhibitor olaparib.
The BRCA1-R1699Q variant moderately elevates cancer risk [67, 68]. The BRCA1-V1736A variant increases the risk of ovarian cancer through biallelic variation [69]. To investigate the significance of intermediate HR activity determined by ASHRA, we analyzed the HR activity of these BRCA1 variants. ASHRA detected the intermediate HR activity of BRCA1-R1699Q and -V1736A; in addition, these variants showed intermediate sensitivity to olaparib. The DR-GFP assay categorized these variants as HR-deficient [9, 10]. These results suggest that HR activity determined by ASHRA can predict cancer risk and sensitivity to PARP inhibitors more accurately than the conventional DR-GFP assay.
4.4 Role of HR Activity in Cancer Treatment
Various sporadic cancers are HR-deficient [70]. The characteristics of tumors with germline BRCA1/2 mutations are referred to as BRCAness, which is observed in some sporadic tumors with somatic mutations or methylation of BRCA1/2, or inactivation of HR factors other than BRCA1/2 [71]. More than half of high-grade serous ovarian cancers are HR-deficient. Alterations of BRCA1/2 are found in more than half of HR-deficient high-grade serous ovarian cancers, whereas alterations in other HR factors account for the remainder [72].
Genetic testing for HR factors, as well as genomic scar assays, is used clinically to detect HR deficiency and to stratify patients in order to identify those likely to benefit from PARP inhibitors. However, a number of VUS of HR factors have been identified, and genomic scar assays do not detect resistance to PARP inhibitors mediated by revertant mutations, which restore HR activity [73,74,75].
4.5 Mechanisms Underlying Resistance to PARP Inhibitors
Various mechanisms underlie primary and acquired resistance to PARP inhibitors [30,31,32]. Two major mechanisms are restoration of HR activity and protection of replication forks. HR can be restored by acquired mutations in HR genes or via increased activity of effector proteins that mediate HR activity. Acquired mutations restore the reading frame and allow expression of the entire protein. Tumors in which the BRCA1 gene is silenced via promoter hypermethylation become resistant due to loss of hypermethylation and re-expression of BRCA1. Furthermore, restoration of HR activity is induced by suppression of the NHEJ pathway. 53BP1 regulates pathway choices, HR or NHEJ, in competition with BRCA1. Since 53BP1 collaborates with RIF1, REV7, and the Shieldin complex to prevents DNA end resection, these alterations trigger resistance by downregulating the NHEJ pathway. Protection of replication forks is caused by reduced recruitment of MRE11 and another DNA endonuclease, MUS81, leading to resistance to PARP inhibitors.
In addition, alterations in the PARP or PAR glycohydrolase proteins cause resistance to PARP inhibitors [31]. Similar to other anti-cancer agents, resistance is caused by increased drug efflux and reduced drug influx. Furthermore, Schlafen 11 (SFLN11) binds to replication forks in response to replication stress, thereby blocking further replication. SFLN11 sensitizes cells to a broad range of anti-cancer agents, including PARP inhibitors and platinum agents. Lack of SFLN11 expression in particular is involved in resistance to DNA-targeting anti-cancer agents [76, 77].
4.6 A Novel Mechanism of Resistance to PARP Inhibitors
Among the 30 BRCA1 missense variants analyzed, only the C61G variant shows significant discordance between HR activity and sensitivity to olaparib in HeLa cells, but not in MCF7 cells [34]. Interestingly, a similar C64G variant did not show these phenotypes. The C61G and C64G variants occur at the zinc-binding residues of the RING domain, resulting in defects in binding to BARD1, E3 ubiquitin ligase activity, and HR activity [8, 78, 79].
A literature search to identify different phenotypes of the C61G and C64G variants revealed that the BRCA1-C61G variant (but not C64G variant) functions to coactivate transcription by activating transcription factor 1 (ATF1) (similar to wild-type BRCA1) [80]. ATF1, which belongs to the c-AMP response element-binding protein/activating transcription factor (CREB/ATF) family, activates gene transcription to regulate cell proliferation and survival [81]. We found that BRCA1-C61G (but not the C64G variant) binds to ATF1 and activates transcription of NRAS and BIRC2, which are involved in cell proliferation and survival, respectively, similar to wild-type BRCA1 [34] (Fig. 4a). ATF1 was expressed at markedly higher levels in HeLa cells than in MCF7 cells. Furthermore, we found MCF10A is another ATF1-low cell line, and BT-549 is another ATF1-high cell line. Exogenous expression of ATF1 caused olaparib resistance in BRCA1-C61G–expressing ATF1-low cells. By contrast, knockdown of ATF1 increased the sensitivity of BRCA1-C61G–expressing ATF1-high cells to olaparib. The level of ATF1 expression did not affect HR activity. These data suggest that high expression of the ATF1 protein confers resistance to olaparib in cells expressing BRCA1-C61G, independent of HR activity (Fig. 4b).

Schematic illustrating ATF1-dependent sensitivity to PARP inhibitors and cisplatin. (a) Wild-type BRCA1 and the BRCA1-C61G variant bind to ATF1 and activates transcription of NRAS and BIRC2. By contrast, BRCA1-C64G fails binds to ATF1 and so does not activate the transcription of NRAS and BIRC2. (b) When treated with PARP inhibitors or platinum agents, HR-proficient cells repair DNA and survive. HR-deficient cells cannot repair DNA damage by HR. However, in cells proficient in BRCA1/ATF1-mediated transcription, cell survival is dependent on the expression level of ATF1. High ATF1-expressing cells proficient in BRCA1/ATF1-mediated transcription survive, but low-expressing cells die. HR-deficient cells harboring BRCA1-C61G or altered non-BRCA1 HR factors such as BRCA2, but possessing wild-type BRCA1, activate ATF1-mediated transcription and survive. Therefore, ATF1 might be a good biomarker for drug resistance in tumor cells. (c) In HR-deficient tumors, genes downstream of BRCA1/ATF1 transactivation might be good biomarkers for sensitivity to PARP inhibitors and platinum agents, regardless of BRCA1/ATF1 transactivation
4.7 BRCA1/ATF1-Mediated Transactivation Confers Resistance to Cisplatin
BRCA1-C61G is HR-deficient, but functions as a coactivator of ATF1-regulated transcription, which causes resistance to olaparib in the presence of high ATF1 expression. Thus, we speculated that BRCA1/ATF1-mediated transcription confers resistance to olaparib in HR-deficient cells due to alterations in other HR factors. As expected, we found that ATF1 overexpression induced olaparib resistance in BRCA2- or RAD51-knockdown MCF7 cells, but not in BRCA1-knockdown cells. Similar results were obtained for cells treated with cisplatin [34]. These data suggest that BRCA1/ATF1-mediated transcription induces the resistance to olaparib and cisplatin upon knockdown of BRCA2 or RAD51. Therefore, the level of ATF1 expression could be a biomarker for the efficacy of PARP inhibitors and platinum agents against tumors that are HR-deficient, but proficient in BRCA1/ATF1-mediated transcription (e.g., BRCA2-deficient tumors) (Fig. 4b).
Tian et al. identified ATF1-target genes [81]. We found that there are two types of ATF1-target genes, BRCA1-dependent (NRAS and BIRC2) and BRCA1-independent (BRAF and MYC) [34]. Although we did not examine whether NRAS and BIRC2 are involved in resistance to PARP inhibitors and platinum agents, their expression levels might be a biomarker for the efficacy of PARP inhibitors and platinum agents against HR-deficient cells (Fig. 4c). However, it is possible that other genes upregulated by BRCA1/ATF1 might be responsible for resistance. Identification of the responsible genes might make it possible to develop therapies that overcome resistance to PARP inhibitors and platinum agents.
BRCA1-C61G is an important founder mutation in the Polish population; therefore, it is analyzed in panel tests for HBOC diagnosis and for cancer treatment [82, 83]. Therefore, resistance to PARP inhibitors and platinum agents induced by BRCA1/ATF1-mediated transcription might be considered in the Polish population.
Overexpression of ATF1, NRAS, and BIRC2 occurs in some malignancies, including breast and ovarian cancers (COSMIC, the Catalogue Of Somatic Mutations In Cancer (https://cancer.sanger.ac.uk/cosmic)). ATF1 forms a fusion gene with EWSR1 or FUS in sarcomas such as clear cell sarcoma and angiomatoid fibrous histiocytoma [84, 85]. Therefore, ATF1-fusion gene products might be involved in resistance of these sarcomas to chemotherapy.
5 Conclusions
HR activity is important for the tumor suppressor functions of BRCA1/2. We used the CRISPR/Cas9 system to develop ASHRA, an assay designed to evaluate HR activity. ASHRA can measure HR activity quantitatively; the results show that HR activity correlates with cancer risk and sensitivity to PARP inhibitors. Furthermore, we identified a novel mechanism involving BRCA1/ATF1-mediated transcription that underlies resistance to olaparib and cisplatin. This assay will contribute to classification of VUS of HR factors that might be involved in cancer risk and sensitivity to PARP inhibitors and platinum agents. It will also be useful for identifying novel resistance mechanisms and target molecules to facilitate development of effective cancer therapies.
BRCA1/2 plays role in multiple cellular processes. However, the role of BRCA1/2 alterations in mechanisms of underlying tissue-specific carcinogenesis remains unclear. The roles played by BRCA1/2 alterations in cancer risk and sensitivity to anti-cancer agents seem to be different. Thus, functional assays to evaluate the effects of BRCA1/2 variants should be changed dependent on the purpose of the analysis: prediction of cancer risk or sensitivity to anti-cancer agents.
References
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71.
Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378(6559):789–92.
Momozawa Y, Sasai R, Usui Y, Shiraishi K, Iwasaki Y, Taniyama Y, et al. Expansion of cancer risk profile for BRCA1 and BRCA2 pathogenic variants. JAMA. Oncologia. 2022;8:871.
Zhao W, Wiese C, Kwon Y, Hromas R, Sung P. The BRCA tumor suppressor network in chromosome damage repair by homologous recombination. Annu Rev Biochem. 2019;88:221–45.
Gorodetska I, Kozeretska I, Dubrovska A. BRCA genes: the role in genome stability, cancer Stemness and therapy resistance. J Cancer. 2019;10(9):2109–27.
Takaoka M, Miki Y. BRCA1 gene: function and deficiency. Int J Clin Oncol. 2018;23(1):36–44.
Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15(24):3237–42.
Ransburgh DJ, Chiba N, Ishioka C, Toland AE, Parvin JD. Identification of breast tumor mutations in BRCA1 that abolish its function in homologous DNA recombination. Cancer Res. 2010;70(3):988–95.
Anantha RW, Simhadri S, Foo TK, Miao S, Liu J, Shen Z, et al. Functional and mutational landscapes of BRCA1 for homology-directed repair and therapy resistance. elife. 2017;6:6.
Bouwman P, van der Gulden H, van der Heijden I, Drost R, Klijn CN, Prasetyanti P, et al. A high-throughput functional complementation assay for classification of BRCA1 missense variants. Cancer Discov. 2013;3(10):1142–55.
Guidugli L, Carreira A, Caputo SM, Ehlen A, Galli A, Monteiro AN, et al. Functional assays for analysis of variants of uncertain significance in BRCA2. Hum Mutat. 2014;35(2):151–64.
Guidugli L, Pankratz VS, Singh N, Thompson J, Erding CA, Engel C, et al. A classification model for BRCA2 DNA binding domain missense variants based on homology-directed repair activity. Cancer Res. 2013;73(1):265–75.
Guidugli L, Shimelis H, Masica DL, Pankratz VS, Lipton GB, Singh N, et al. Assessment of the clinical relevance of BRCA2 missense variants by functional and computational approaches. Am J Hum Genet. 2018;102(2):233–48.
Nigg EA, Holland AJ. Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat Rev Mol Cell Biol. 2018;19(5):297–312.
Nigg EA, Schnerch D, Ganier O. Impact of centrosome aberrations on chromosome segregation and tissue architecture in cancer. Cold Spring Harb Symp Quant Biol. 2017;82:137–44.
LoMastro GM, Holland AJ. The emerging link between centrosome aberrations and metastasis. Dev Cell. 2019;49(3):325–31.
Gonczy P. Centrosomes and cancer: revisiting a long-standing relationship. Nat Rev Cancer. 2015;15(11):639–52.
Otsuka K, Yoshino Y, Qi H, Chiba N. The function of BARD1 in centrosome regulation in cooperation with BRCA1/OLA1/RACK1. Genes (Basel). 2020;11(8):842.
Yoshino Y, Fang Z, Qi H, Kobayashi A, Chiba N. Dysregulation of the centrosome induced by BRCA1 deficiency contributes to tissue-specific carcinogenesis. Cancer Sci. 2021;112:1679.
Yoshino Y, Chiba N. Roles of RACK1 in centrosome regulation and carcinogenesis. Cell Signal. 2022;90:110207.
Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol. 2017;18(10):610–21.
Pommier Y, O’Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016;8(362):362ps17.
Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, et al. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol. 2012;19(4):417–23.
Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, Bartek J. High speed of fork progression induces DNA replication stress and genomic instability. Nature. 2018;559(7713):279–84.
Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105(44):17079–84.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7.
McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109–15.
Pilie PG, Gay CM, Byers LA, O’Connor MJ, Yap TA. PARP inhibitors: extending benefit beyond BRCA-mutant cancers. Clin Cancer Res. 2019;25(13):3759–71.
D’Andrea AD. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst). 2018;71:172–6.
Zheng F, Zhang Y, Chen S, Weng X, Rao Y, Fang H. Mechanism and current progress of poly ADP-ribose polymerase (PARP) inhibitors in the treatment of ovarian cancer. Biomed Pharmacother. 2020;123:109661.
Reilly NM, Novara L, Di Nicolantonio F, Bardelli A. Exploiting DNA repair defects in colorectal cancer. Mol Oncol. 2019;13(4):681–700.
Yoshino Y, Endo S, Chen Z, Qi H, Watanabe G, Chiba N. Evaluation of site-specific homologous recombination activity of BRCA1 by direct quantitation of gene editing efficiency. Sci Rep. 2019;9(1):1644.
Endo S, Yoshino Y, Shirota M, Watanabe G, Chiba N. BRCA1/ATF1-mediated transactivation is involved in resistance to PARP inhibitors and cisplatin. Cancer Research Communications. 2021;1(2):90–105.
Nielsen FC, van Overeem HT, Sorensen CS. Hereditary breast and ovarian cancer: new genes in confined pathways. Nat Rev Cancer. 2016;16(9):599–612.
Scully R, Panday A, Elango R, Willis NA. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 2019;20(11):698–714.
Tarsounas M, Sung P. The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nat Rev Mol Cell Biol. 2020;21(5):284–99.
Chen CC, Feng W, Lim PX, Kass EM, Jasin M. Homology-directed repair and the role of BRCA1, BRCA2, and related proteins in genome integrity and cancer. Annu Rev Cancer Biol. 2018;2:313–36.
Santos-Pereira JM, Aguilera A. R loops: new modulators of genome dynamics and function. Nat Rev Genet. 2015;16(10):583–97.
Conduit PT, Wainman A, Raff JW. Centrosome function and assembly in animal cells. Nat Rev Mol Cell Biol. 2015;16(10):611–24.
Fujita H, Yoshino Y, Chiba N. Regulation of the centrosome cycle. Mol Cell Oncol. 2016;3(2):e1075643.
Sankaran S, Starita LM, Groen AC, Ko MJ, Parvin JD. Centrosomal microtubule nucleation activity is inhibited by BRCA1-dependent ubiquitination. Mol Cell Biol. 2005;25(19):8656–68.
Starita LM, Machida Y, Sankaran S, Elias JE, Griffin K, Schlegel BP, et al. BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol Cell Biol. 2004;24(19):8457–66.
Ko MJ, Murata K, Hwang DS, Parvin JD. Inhibition of BRCA1 in breast cell lines causes the centrosome duplication cycle to be disconnected from the cell cycle. Oncogene. 2006;25(2):298–303.
Hsu LC, Doan TP, White RL. Identification of a gamma-tubulin-binding domain in BRCA1. Cancer Res. 2001;61(21):7713–8.
Ertych N, Stolz A, Stenzinger A, Weichert W, Kaulfuss S, Burfeind P, et al. Increased microtubule assembly rates influence chromosomal instability in colorectal cancer cells. Nat Cell Biol. 2014;16(8):779–91.
Zou J, Rezvani K, Wang H, Lee KS, Zhang D. BRCA1 downregulates the kinase activity of polo-like kinase 1 in response to replication stress. Cell Cycle. 2013;12(14):2255–65.
Matsuzawa A, Kanno S, Nakayama M, Mochiduki H, Wei L, Shimaoka T, et al. The BRCA1/BARD1-interacting protein OLA1 functions in centrosome regulation. Mol Cell. 2014;53(1):101–14.
Yoshino Y, Qi H, Kanazawa R, Sugamata M, Suzuki K, Kobayashi A, et al. RACK1 regulates centriole duplication by controlling localization of BRCA1 to the centrosome in mammary tissue-derived cells. Oncogene. 2019;38(16):3077–92.
Sun H, Luo X, Montalbano J, Jin W, Shi J, Sheikh MS, et al. DOC45, a novel DNA damage-regulated nucleocytoplasmic ATPase that is overexpressed in multiple human malignancies. Mol Cancer Res. 2010;8(1):57–66.
Duff D, Long A. Roles for RACK1 in cancer cell migration and invasion. Cell Signal. 2017;35:250–5.
Cao XX, Xu JD, Liu XL, Xu JW, Wang WJ, Li QQ, et al. RACK1: a superior independent predictor for poor clinical outcome in breast cancer. Int J Cancer. 2010;127(5):1172–9.
Cao XX, Xu JD, Xu JW, Liu XL, Cheng YY, Wang WJ, et al. RACK1 promotes breast carcinoma proliferation and invasion/metastasis in vitro and in vivo. Breast Cancer Res Treat. 2010;123(2):375–86.
Al-Reefy S, Mokbel K. The role of RACK1 as an independent prognostic indicator in human breast cancer. Breast Cancer Res Treat. 2010;123(3):911; author reply 2
Kais Z, Chiba N, Ishioka C, Parvin JD. Functional differences among BRCA1 missense mutations in the control of centrosome duplication. Oncogene. 2012;31(6):799–804.
Yoshino Y, Qi H, Fujita H, Shirota M, Abe S, Komiyama Y, et al. BRCA1-interacting protein OLA1 requires interaction with BARD1 to regulate centrosome number. Mol Cancer Res. 2018;16:1499–511.
Nakanishi A, Han X, Saito H, Taguchi K, Ohta Y, Imajoh-Ohmi S, et al. Interference with BRCA2, which localizes to the centrosome during S and early M phase, leads to abnormal nuclear division. Biochem Biophys Res Commun. 2007;355(1):34–40.
Han X, Saito H, Miki Y, Nakanishi A. A CRM1-mediated nuclear export signal governs cytoplasmic localization of BRCA2 and is essential for centrosomal localization of BRCA2. Oncogene. 2008;27(21):2969–77.
Niwa T, Saito H, Imajoh-ohmi S, Kaminishi M, Seto Y, Miki Y, et al. BRCA2 interacts with the cytoskeletal linker protein plectin to form a complex controlling centrosome localization. Cancer Sci. 2009;100(11):2115–25.
Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, et al. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem. 2001;276(18):14537–40.
Anderson SF, Schlegel BP, Nakajima T, Wolpin ES, Parvin JD. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat Genet. 1998;19(3):254–6.
Zhang X, Li R. BRCA1-dependent transcriptional regulation: implication in tissue-specific tumor suppression. Cancers (Basel). 2018;10(12):513.
Fernandes VC, Golubeva VA, Di Pietro G, Shields C, Amankwah K, Nepomuceno TC, et al. Impact of amino acid substitutions at secondary structures in the BRCT domains of the tumor suppressor BRCA1: implications for clinical annotation. J Biol Chem. 2019;294(15):5980–92.
Takaoka M, Saito H, Takenaka K, Miki Y, Nakanishi A. BRCA2 phosphorylated by PLK1 moves to the midbody to regulate cytokinesis mediated by nonmuscle myosin IIC. Cancer Res. 2014;74(5):1518–28.
Ikegami M, Kohsaka S, Ueno T, Momozawa Y, Inoue S, Tamura K, et al. High-throughput functional evaluation of BRCA2 variants of unknown significance. Nat Commun. 2020;11(1):2573.
Towler WI, Zhang J, Ransburgh DJ, Toland AE, Ishioka C, Chiba N, et al. Analysis of BRCA1 variants in double-strand break repair by homologous recombination and single-strand annealing. Hum Mutat. 2013;34(3):439–45.
Moghadasi S, Meeks HD, Vreeswijk MP, Janssen LA, Borg A, Ehrencrona H, et al. The BRCA1 c. 5096G>A p.Arg1699Gln (R1699Q) intermediate risk variant: breast and ovarian cancer risk estimation and recommendations for clinical management from the ENIGMA consortium. J Med Genet. 2018;55(1):15–20.
Spurdle AB, Whiley PJ, Thompson B, Feng B, Healey S, Brown MA, et al. BRCA1 R1699Q variant displaying ambiguous functional abrogation confers intermediate breast and ovarian cancer risk. J Med Genet. 2012;49(8):525–32.
Domchek SM, Tang J, Stopfer J, Lilli DR, Hamel N, Tischkowitz M, et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov. 2013;3(4):399–405.
Riaz N, Blecua P, Lim RS, Shen R, Higginson DS, Weinhold N, et al. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat Commun. 2017;8(1):857.
Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–9.
Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15.
Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013;229(3):422–9.
Dhillon KK, Swisher EM, Taniguchi T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci. 2011;102(4):663–9.
Kondrashova O, Nguyen M, Shield-Artin K, Tinker AV, Teng NNH, Harrell MI, et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor Rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2017;7(9):984–98.
Murai J, Zhang H, Pongor L, Tang SW, Jo U, Moribe F, et al. Chromatin remodeling and immediate early gene activation by SLFN11 in response to replication stress. Cell Rep. 2020;30(12):4137–51 e6.
Murai J, Thomas A, Miettinen M, Pommier Y. Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies. Pharmacol Ther. 2019;201:94–102.
Wu LC, Wang ZW, Tsan JT, Spillman MA, Phung A, Xu XL, et al. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet. 1996;14(4):430–40.
Brzovic PS, Keeffe JR, Nishikawa H, Miyamoto K, Fox D 3rd, Fukuda M, et al. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc Natl Acad Sci U S A. 2003;100(10):5646–51.
Houvras Y, Benezra M, Zhang H, Manfredi JJ, Weber BL, Licht JD. BRCA1 physically and functionally interacts with ATF1. J Biol Chem. 2000;275(46):36230–7.
Tian J, Chang J, Gong J, Lou J, Fu M, Li J, et al. Systematic functional interrogation of genes in GWAS loci identified ATF1 as a key driver in colorectal cancer modulated by a promoter-enhancer interaction. Am J Hum Genet. 2019;105(1):29–47.
Szwiec M, Jakubowska A, Gorski B, Huzarski T, Tomiczek-Szwiec J, Gronwald J, et al. Recurrent mutations of BRCA1 and BRCA2 in Poland: an update. Clin Genet. 2015;87(3):288–92.
Menkiszak J, Chudecka-Glaz A, Gronwald J, Bedner R, Cymbaluk-Ploska A, Wezowska M, et al. Characteristics of selected clinical features in BRCA1 mutation carriers affected with breast cancer undergoing preventive female genital tract surgeries. Ginekol Pol. 2013;84(9):758–64.
Hocar O, Le Cesne A, Berissi S, Terrier P, Bonvalot S, Vanel D, et al. Clear cell sarcoma (malignant melanoma) of soft parts: a clinicopathologic study of 52 cases. Dermatol Res Pract. 2012;2012:984096.
Thway K, Fisher C. Angiomatoid fibrous histiocytoma: the current status of pathology and genetics. Arch Pathol Lab Med. 2015;139(5):674–82.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Fang, Z., Yoshino, Y., Chiba, N. (2023). New Functions of BRCA1/2 in Regulating Carcinogenesis and Drug Sensitivity. In: Aoki, D., Nakamura, S., Miki, Y. (eds) Practical Guide to Hereditary Breast and Ovarian Cancer. Springer, Singapore. https://doi.org/10.1007/978-981-99-5231-1_7
Download citation
DOI: https://doi.org/10.1007/978-981-99-5231-1_7
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-5230-4
Online ISBN: 978-981-99-5231-1
eBook Packages: MedicineMedicine (R0)