
Abstract
Along with bacteria, other microorganisms are also key components of microbiota structure of freshwater, and those include archaea, microbial eukaryotes, and viruses that together build the diverse community of microorganisms. All those microorganisms play key ecological functions and can have important impacts on animal, plant, and human health. The traditional culture-based microbiology methods have been mostly replaced, or complemented, by molecular methods, due to their accuracy, fast response, and capability to detect microorganisms that are quite difficult to grow in culture media. This chapter addresses some of the current tendencies on microbial structure and its analysis. Those methods included the “omic-based” DNA studies that allow better and more comprehensive characterization of bacterial communities, leading to metagenomic studies that help to elucidate the population structure of microbiomes in aquatic environments. Strategies for detecting water-borne pathogens are focused on detecting bacteria, protozoa, yeast, and virus in different water sources, and these methods can also be complemented by molecular tools such as molecular marker (RAPD, Microsatellites, RFLPs, and AFLPs). Those methods also allow to detect pathogenic bacteria, fungus, and viruses of public health interest, such as the enterovirus and, more recently, the coronavirus. Water bodies can also be important reservoirs of antibiotic-resistant bacteria, and the group of ESKAPE bacteria represents an important study model for antibiotic resistance and its distribution. The acronym “ESKAPE” is associated with this group due to their ability to escape antimicrobial activity and develop high levels of resistance to multiple antibiotics (multidrug resistance). Water bodies play a central role on the selective pressure and is the most important cause of the dissemination and extension of resistance and has contributed to the genetic diversification of resistance genes also affecting ecological microbial structures.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1.1 Introduction
The biological role of freshwater microorganisms in the carbon cycle and its association with the uptake and emission of greenhouse gases is well documented as well as the impact that disturbance of microbial communities as a consequence of anthropogenic activities have on the climate and global change (Regnier et al. 2013; Premke et al. 2022).
Bacteria are prokaryote, unicellular, taxonomically diverse, and ubiquitous microorganisms. They are the most abundant organisms on earth and their habitats include virtually every ecosystem on the planet, even extreme aquatic environments such as hot springs and ice layers in the Arctic and Antarctic regions, under high pressures or extreme acidic environments (Takai et al. 2008; Dalmasso et al. 2016; Merino et al. 2019). The adaptation capabilities of bacteria to adverse environments explain their abundance within the different ecosystems on the earth; it is estimated that the average number of bacterial cells per gram of soil is around 40 million, whereas 1 million diverse bacterial cells can be found in 1 mL of environmental freshwater and it is even higher in estuaries and open ocean waters (Whitman et al. 1998; Anas et al. 2021).
In aquatic environments, the cross-feeding and metabolite interchange represent an important class of interactions, and thus, photosynthetically fixed dissolved carbon could produce chemotactic responses (Seymour et al. 2010). Early experimental evidence suggested the existence of strong positive and negative interactions among the photoautotroph and heterotroph microorganisms (Cole 1982). Besides bacteria, there are various microorganisms in fresh water, such as fungi, algae, and viruses, although they are not microorganisms. Their importance will be addressed in this chapter. The traditional culture-based microbiology methods have been mostly replaced, or complemented, by molecular methods, due to their accuracy and fast response. Here, some of the current tendencies on microbial structure and its analysis will be addressed.
1.2 Microbiome and DNA-Based Omic Studies
As we move forward to a better understanding at microorganic system level, complexity increases as the number of microorganisms interacting do. For this reason, it is important to develop pipelines to accurately characterize the diversity of bacteria and other microorganisms in specific niches and elucidate their role within the community and the ecosystem. Water is an essential natural resource for all living organisms, and the pollution consequence of human activities has a great impact ranging from environmental effects (sometime negative), animal and human health impact to important economic losses in food production. The causes of contamination of rivers, lakes, and ponds are diverse. Still, contamination with untreated or poorly treated sewage and residual waters containing heavy metals and other xenobiotics from industry, agriculture, and livestock production are among the main drivers that serve as disturbers of native bacteria communities. In the first case, sewage serves as an important source of enteric bacteria and viruses, whereas industrial residual waters modify the physical and chemical properties of the water itself, and these changes impact the bacterial population structure affecting the carbon flux between freshwater bodies and the environment among other important aspects (Schreiber et al. 2015).
Like other ecosystems, in freshwater bodies, bacteria and other microorganisms live in a complex multispecies structure. Begon et al. (1986) defined the microbial communities as the set of microorganisms coexisting in the same space and time. However, the characterization of bacterial communities was limited to microorganisms grown in culture media, and their study was focused mainly on morphological and biochemical profiles. In 1977, Woese and Fox proposed the 16S ribosomal RNA genes as a potential marker to establish phylogenetic relationships between bacteria, and it was only after the development of Sanger sequencing technology that the 16S ribosomal RNA genes analyses was used as the benchmark to fine-tune the classification system of microorganisms.
Later in 1998, Handelsman et al. proposed the term metagenomics in their research, which involved the isolation of total DNA from soil bacteria without previous culture on artificial media, and their cloning using BAC vectors in order to transform competent E. coli cells and then sequence the genomes and express genes detected, allowing a deep understanding of the bacterial population structures. The arrival of high-throughput next-generation sequencing (NGS) techniques in 2005 with the pyrosequencing method from Roche was a milestone in the microbial communities’ characterization because of its relative low cost and reliable methodology to rapid sequencing of short fragments of DNA. However, this platform is discontinued today by more efficient sequencing strategies such as synthesis sequencing, initially proposed from ion torrent, followed and enhanced then by Illumina and PacBio, all collectively called next-generation sequencing (NGS), that now represents the most accurate and used technique to characterize the metagenome of environmental samples at a different resolution of taxonomic levels; these sequencing technologies allow the rapid detection of perturbations in populations of native bacteria in freshwater deposits under different conditions that can be associated to ecological phenomena (Escobar-Zepeda et al. 2015). In the past few years, through NGS techniques, researchers worldwide have been able to elucidate the population structure of microbiomes in aquatic environments under different and extreme conditions (Table 1.1).
To this day, metagenomic characterization is mostly descriptive and without a careful approach design it does not provide insights into genes of interest or metabolic integration pathways in the analyzed environments. However, a comprehensive approach integrating every member of the microbial community transcriptome (metatranscriptomics) studies and the total set of proteins (metaproteomics) could improve the insight into the role played by every microbial member at an ecosystem level establishing accurate relationships between taxonomy and functionality profiles within microorganisms.
1.3 Current Strategies for Detection of Water-Borne Pathogens
1.3.1 Incidence of Bacterial Pathogens in Water
Water bodies, such as rivers, lakes, irrigation channels, damps, and hospital and industrial wastewaters, harbor a wide variety of microorganisms. The microorganisms commonly found in wastewater are enteric anaerobic bacteria, some yeasts and fungi, and some protists especially those resistant to harsh environments. Regarding the bacteria present in water bodies, it has been estimated that 90% of the genera and species of bacteria characterized belong to 4 phyla: Proteobacteria, Actinobacteria, Firmicutes, and Bacteroidetes. Some of the bacteria can be defined as pathogens such as Escherichia coli (Rice and Johnson 2000), Pseudomonas spp. (Pirnay et al. 2005), and Stenotrophomonas maltophilia (Brooke 2012). These bacteria, along with the emergence of antibiotic-resistant bacteria and its wide distribution in freshwater, is a concerning matter that will be addressed further in this chapter with the ESKAPE group.
1.3.2 Incidence of Yeast and Filamentous Fungi Pathogens in Water
The presence of these microorganisms in water has important ecological roles in organic matter transformation, and some have an additional impact, because these microorganisms can be pathogenic or display pathogenic properties and well as antifungal resistance (Monapathi et al. 2020). Some species of yeast that have been identified in water bodies include Candida, Clavispora, Cyberlindnera, Cryptococcus, Hanseniaspora, and Yarrowia. Yeast detected in cities water distribution systems and tap water include Candida, Clavispora, Cryptococcus, Debaromyces, Meyerozyma, and Pichia. Yeast isolates identified in sewage treatment plants are Candida, Cryptococcus, Debaryomyces, and Wickerhamonyces (Monapathi et al. 2020). Yeasts isolated from different water samples recognized as pathogenic include Candida spp. Cyberlindnera, and Cryptococcus. Fungal infections caused by Candida albicans and other Candida spp. are important complications in immunosuppressed patients, since frequently, candidiasis in mucosal tissues (oral, gastrointestinal, and vaginal) represent an early sign of immune system malfunction (Panizo and Reviákina 2001). Cryptococcus neoformans is an encapsulated yeast, and the cause of opportunistic infections, such as meningoencephalitis in immunocompromised patients (Pini et al. 2017).
Among filamentous fungi, some of the complications caused by some virulent species belonging to Aspergillus, Fusarium, and Alternaria may range from allergic to invasive syndromes. Immunocompetence facilitates clearance by initiating innate and adaptive host responses despite constant spore inhalation (Chotirmall et al. 2014). Regarding Aspergillus section Flavi includes 22 species, some of them with potentially pathogenic such as Aspergillus felis, A. fischeri, A. fumigatiaffinis, A. fumisynnematus, A. hiratsukae, A. laciniosus, A. lentulus, A. novofumigatus, A. parafelis, A. pseudofelis, A. pseudoviridinutans, A. spinosus, A. thermomutatus, and A. udagawae. The pathogenic species can display a wide range of differences in the frequency of production of toxins or toxic compounds (Tamayo-Ordóñez et al. 2021). Even though fewer pathogenic yeasts and molds have been reported in different water species compared to bacterial genera, their importance stands out, since their contact with humans with a compromised immune system could lead to serious complications and even cause death.
1.3.3 Identifying Virus in Water Samples
In raw wastewater, viruses may be ubiquitous and persistent and even after treatment, some viruses continue being distributed in treated wastewater that later arrive at water bodies. Adenovirus (HAdV), rotavirus (RoV), hepatitis A virus (HAV), and other enteric viruses, such as noroviruses (NoV), coxsackievirus, echovirus, reovirus, and astrovirus, are commonly detected in wastewater as well as some superficial water (rivers and irrigation channels) and are the principal human viral pathogens transmissible via water exposition (Corpuz et al. 2020). More dangerous viruses such as poliovirus can also be present in wastewaters. Enterovirus causing waterborne diseases such as diarrhea in children and adults are often associated with viral disease outbreaks, leading to several clinical manifestations (nausea, vomiting, and fever) and other gastrointestinal diseases, and some enteric viruses have also been related to respiratory, central nervous system (poliovirus), and other diseases (Corpuz et al. 2020).
Recently, a novel Coronavirus disease 2019 (COVID-19), which began in December 2019, that is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Guarner 2020) was reported. Since this virus can invade the gastrointestinal mucosa, it can be released in feces, and the presence of nucleic acid of SARS-CoV-2 has been reported in raw wastewater (Randazzo et al. 2020), sewage samples collected from hospitals (Wang et al. 2020b), and wastewater sample after secondary treatment (Randazzo et al. 2020). However, infection caused by exposition to water is not been documented, and its detection functions as an early warning system to indirectly detect SARS-Cov-2 infections in the population.
1.4 Molecular Markers for Genotyping and Identification of Bacteria in Water Bodies
1.4.1 Intraspecific and Interspecific Variability and Genotyping
Molecular markers are DNA fragments sequences associated with a physiological trait, and are used to identify a particular DNA sequence related to a function or a trait of interest (Langridge and Chalmers 2004). Different types of markers are distinguished by their ability to detect polymorphisms at single or multiple loci, or sequence patterns and can be of dominant or codominant type. Below we describe some of the molecular markers that have been most used to determine intra- and interspecific variability in bacterial genera isolated from different bodies of water.
RAPDs
RAPDs (randomly amplified DNA polymorphisms) are markers that randomly amplify segments of DNA in a wide variety of species. RAPDs are based on the statistical probability of complementary sites to the oligonucleotide of ten base pairs (bp) throughout the genome. The polymorphism of the bands between individuals is due to changes in the sequence of nucleotides at oligonucleotide coupling sites and by inserting or deleting fragments at these sites. These markers are dominant; they cannot discern the dominant homozygous or heterozygous for a particular segment, so allelic frequencies must be estimated indirectly, assuming Hardy Weinberg equilibrium. Among some of the applications of these molecular markers in bacteria, it has successfully demonstrated different RAPD profiles in multidrug-resistant coliform bacteria isolated from sewage samples of Ghaziabad city, India (Raj 2012). Since RAPD-derived markers include a genome-wide analysis, they can be a very good source of physiological markers that can be obtained even if little is known about the sequence of the microorganism being studied. Their application could be considered one of the first “genomic-wide” analysis performed before the massive sequencing technology was available; however, several problems with reproducibility limited its applications out of basic research.
Microsatellites
Simple repeat sequences (SSRs) or ISBPs (Insertion Site-Based Polymorphisms) are DNA sequences made up of 1–4 base pairs (mononucleotides (TT)n, dinucleotides (AT)n, or tetranucleotides (AAGG)n). These loci are found both in coding and noncoding regions of DNA, formed by breakage events that generate polymorphisms with values greater than 90%. Challagundla et al. (2018) analyzed 598 genome sequences of Staphylococcus aureus; this study showed that depending on the geographical area of the isolate, certain mutations in the CC5-MRSA marker are present allowing a deeply analysis of the epidemiology of these strains. A particular important application of simple repeat sequences was on M. tuberculosis research, because this allowed the design of a classification system of M. tuberculosis strains, based on a VNTR (variable number of tandem repeats) that was so specific to M. tuberculosis that later these were called mycobacterial interspersed repetitive units or MIRUs, allowing a great advance on mycobacterial isolates classifications, both from clinical and environmental sources. Dangerous strains such as the Beijing lineage were spotted with these markers and later with complementary genomic analyses on Beijing strains obtained from different clinical samples, waters, and substrates: this allowed us to know its biogeographic structure and evolutionary history of the Beijing lineage worldwide through the SNPs analysis of 4987 isolates from 99 countries (Merker et al. 2015).
RFLPs
This method allows the detection of specific DNA differences recognized by restriction enzymes. Each of the endonucleases (of bacterial origin) recognizes and cuts only a specific sequence in DNA, as long as they are not protected (methylated). Therefore, any DNA that is not methylated can be recognized and cut into fragments of defined length; any mutation within those sites could change the pattern of the fragment and allow an RFLP to be detected when comparing two or more genomes. Originally, RFLP were important tools to detect and related mutations and molecular marker to key physiological functions and to design the initial molecular diagnosis strategies, since sequencing was required only to confirm the design but not to apply the technique and had very good reproducibility results. RFLPs are still currently applied to understand bacterial community dynamics in wastewater treatment systems. Wang et al. (2010) investigated how bacterial communities change in treatment plants over a year using specific PCR followed by terminal restriction fragment length polymorphism (T-RFLP) of the 16S rRNA gene. The T-RFLP results indicated lack of stability in the bacterial community structures in 2 full-scale wastewater treatment systems, with 15 days average change rates observed in the 2 systems. On the other hand, through digestion of the 16S rRNA gene with the endonuclease Mse I, it was shown that distinctive patterns for Acrobacter species are observed, indicating variation in the population structure of the genera (Figueras et al. 2012).
AFLPs
AFLPs (amplified fragment length polymorphisms) is a technique that combines the digestion of two restriction enzymes, generally MseI and EcoRI, within a sequence, involving a selective amplification of restriction fragment lengths from a genome using PCR, and followed by gel electrophoresis to separate the amplified fragments and obtain a banding pattern, which can be used to identify genetic differences between individuals or populations, and can also be used for genetic mapping and phylogenetic analysis (Tamayo-Ordoñez et al. 2012).
This technique has been used to determine the biodiversity of the bacterium Pseudomonas aeruginosa in an aquatic environment in a study including 100 isolates of P. aeruginosa, where the oprL gene was analyzed, and a DNA-based fingerprint (AFLP) indicated a positive relationship between pollution and prevalence of P. aeruginosa. In the Woluwe River, P. aeruginosa community was almost as diverse as the global P. aeruginosa population. These findings illustrate the significance of river water as a stable reservoir and source for the distribution of potentially pathogenic P. aeruginosa strains (Pirnay et al. 2005).
In another recent study, antimicrobial tolerance to oxytetracycline and taxonomic diversity relationship in the culturable oxytetracycline-resistant (Otr) isolates of heterotrophic bacteria in two Belgian aquatic sites receiving wastewater was investigated. Profiles for ampicillin and kanamicin tolerance were detected from the taxonomic differences in the Otr bacteria detected at genera and subgenera level. In addition, Enterobacter sp., Stenotrophomonas maltophilia, and A. veronii resulted as potential indicator organisms to help to assess microbial tolerance in various compartments of the aquatic environment (Huys et al. 2001).
1.4.2 Identification of Species Through Sequencing of Molecular Markers
To date, depending on the purpose of each investigation, different genetic regions can be used to identify species from various samples. Among the most explored genes used for species identification are the 16S rRNA, 23S rRNA, rpoB, gyrB, dnaK, dsrAB, amoA, amoB, mip, horA, hitA, recA, ica, frc, oxc, 16S–23S rDNA ISR, and IS256.
Amplification and sequencing of 16S (V3–V4 of 16S rRNA) is commonly applied to study microbial communities with third-generation sequencers (MinION from Oxford Nanopore Technologies) and this made it possible to analyze the full length of the 16S rRNA gene, and this allows a deeper identification even to species level identification and at relative low cost. The analysis of nine indigenous bacteria that can be related to food poisoning and act as opportunistic infections was carried out with due diligence. Enterococcus faecalis and Enterococcus hirae were identified at the species level with an accuracy of 96.4–97.5%. Also, using these technologies, it is possible to evaluate the antibiotic sensitivities of multiple bacteria simultaneously. Kawai et al. (2022), using these technologies, allowed rapid evaluation of antibiotic activity spectrum at the species level containing a wide variety of bacteria, such as biofilm bacteria and gut microbiota. Even though the sequencing of these genes is a powerful tool to identify bacteria, it has some limitations when it comes to related bacterial complexes that can be difficult to distinguish.
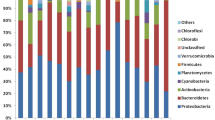
Since the sequencing and analysis of 16S rDNA ribosomal regions is the most used molecular marker, we decided to find out the abundance of bacteria identified in bodies of water and of these, which accessions have been reported in the Gen Bank from NCBI. It was possible to identify 103 sequences of 16S rDNA regions of bacteria present in different bodies of water (Fig. 1.1). After analysis, a total of 58 bacterial genera were identified.

Representation of the bacterial species identified in water bodies. The bacterial species were identified by searching in the sequences reported in the NCBI
Conclusively, we want to emphasize that the use of molecular markers allows to identify and genotype a wide variety of microorganisms from different water samples, highlighting the importance of molecular markers in the clinical and environmental areas.
1.4.3 Methods of Detection of Viruses in Waters
Viruses are important components of aquatic systems, since they are heavy regulators of bacteria populations, and some of them can be related to plants, animal, and human diseases, so their detection is crucial as indicated in numerous studies, including different water matrices such as surface water, treated wastewater, and irrigation water (Wang et al. 2020a; Ji et al. 2020; Rusiñol et al. 2020). Several methods to directly and indirectly detect and quantify viruses found in wastewater include epifluorescence microscopy, transmission electronic microscopy, pulsed-field gel electrophoresis, immunofluorescence assay, flow cytometry, traditional cell-culture, and molecular detection (Corpuz et al. 2020). Among the molecular techniques, PCR and especially real-time PCR are currently used.
PCR is an in vitro enzymatic reaction that amplifies or generates millions of copies of a specific DNA sequence during several repeated cycles in which the target sequence is faithfully copied. This technique uses thermostable DNA polymerase to replicate DNA strands, for which alternate high and low temperature cycles are used to separate the newly formed DNA strands after each replication phase. Among the components used in this in vitro reaction are amplification template (DNA or complementary DNA, or RNA in the case of viruses), oligonucleotides, DNA polymerase (Taq polymerase), polymerase cofactor (MgCl2), PCR buffer, deoxyribonucleotide triphosphates, water, and equipment (thermocycler). Since viruses have DNA or RNA genomes, PCR is an important tool for detecting them in water samples. For the detection of RNA genome viruses, it is necessary to carry out an RT-PCR (Reverse Transcription-Polymerase Chain Reaction) that is a derived technique with a step where a RNA complementary DNA is generated that can be used directly in the PCR (Kadri 2020). An important derivative technique for virus study is the Real-time PCR or quantitative PCR that is a method that allows us to monitor the evolution (simultaneous amplification and quantification) of the PCR while it is being carried out. Cycle-by-cycle analysis of the changes in the accumulation of the PCR product, detected by a change in the fluorescent signal generated in the three steps of the PCR, is done with this technique and allows two types of determinations: the quantification absolute and quantitative comparison between samples. The quantification absolute determines the exact number of DNA or RNA molecules present in each sample. Applying this quantification method requires generating a standard curve and using the comparative Ct method. The other type of determination is the quantitative comparison of target nucleic acid, expressed in orders of magnitude concerning a calibrator.
Variants have been derived from both techniques, such as Multiplex PCR and ICC-PCR (integrated-cell culture PCR) (Corpuz et al. 2020). The application of multiplex PCR allows the simultaneous detection of multiple viruses present in a single sample using more than one set of primers in one reaction. Multiplex qPCR also enables the detection of different specific viruses using more than one fluorescent reporter. In the ICC-PCR method, the samples in which the viruses may be present are used to infect cell culture media and incubated for several days. Then the samples of cell culture are subjected to freezing to cause lysis of the cells. If viable viruses were present in the original sample and invaded the culture cells, those are from their host cells when lysis occurs. After this, PCR-based amplification is applied to the lysate (Corpuz et al. 2020), so the ICC-PCR is an effective enrichment method for virus detection.
1.4.3.1 Detection of Coronaviruses in Samples of Waters by Molecular Methods
There are extensive reviews in the literature describing the persistence of coronavirus in different types of wastewaters. The integrity of the coronavirus particle in water depends upon the wastewater characteristics, presence of suspended solids, organic matter, and temperature (Mandal et al. 2020). The methods of early detection of the virus on waters help to detect future immediate outbreaks, allow to implement measures to minimize the infections and deaths caused by COVID-19, and new molecular techniques are currently being developed that allow the effective detection of other viruses relevant for public health such as adenovirus and poliovirus,
1.5 ESKAPE Bacteria as a Model of Antibiotic-Resistant Bacteria Distribution in Water
1.5.1 Antibiotics and Drug Resistance
The use of antibiotic-producing microbes to prevent disease goes to back more than 2000 years ago from the use of moldy bread to treat open wounds (Hutchings et al. 2019). Still, it was not until the twentieth century that the microbiologist Alexander Fleming discovered the origin of Penicillin in 1928, a substance produced by mold (Penicillum notatum), that was lethal to several bacteria giving rise to the birth of the antibiotic era (Patel 2016), but also to the next milestone in history “antibiotic resistance” statement made by Alexander Fleming during his Nobel Prize acceptance speech (1945) (Podolsky 2018) predicting the extent and severity of antibiotic resistance over the next 10 years, statement that then became a reality, with hundreds of cases of resistance recorded throughout the world (Rosales Magallanes 2018).
Currently, antimicrobial resistance is one of the greatest threats to global health (Maillard et al. 2020). The widespread and inappropriate use of antibiotics has led to an increased prevalence of multidrug-resistant (MDR) bacteria in the environment. MDR infections led to 700,000 deaths globally in 2016 and are expected to rise to10 million by 2050 (Brooks et al. 2018). The ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeroginosa, and Enterobacter species) (Tigabu and Getaneh 2021) are a group of common opportunistic pathogens associated mainly with nosocomial infections (Santaniello et al. 2020), the acronym “ESKAPE” is associated to this group due to their ability to escape the antimicrobial activity and development of resistance to multiple antibiotics (multidrug-resistance) (Founou et al. 2018). The World Health Organization (WHO) listed in the critical priority list carbapenem-resistant A. baumannii, P. aeruginosa, K. pneumoniae, and Enterobacter spp., whereas vancomycin-resistant E. faecium (VRE) and methicillin/vancomycin-resistant S. aureus (MRSA or VRSA) in the list of high priority group (Mulani et al. 2019). The presence of the ESKAPE pathogens in the environment is likely due to contamination via sewage spills, hospital waste that has been discarded incorrectly; consequently, several antibiotic-resistant microorganisms and antibiotic-resistance genes (ARGs) have been detected in water bodies (Denissen et al. 2022). The water sources represent a reservoir of various chemical and microbiological pollutants such as chemical, heavy metal, organic pollutants, residues of pharmaceuticals (Voigt et al. 2020), including horizontal gene transfer of resistance genes, the misuse or overuse of antibiotics, and the contamination through livestock slurry and plant wastewater (Daniel et al. 2017) are factors that contribute to genetic selection pressure for the development of MDR bacterial infections in the community (Wang et al. 2018).
1.5.2 Enterococcus
Enterococci is a Gram-positive bacteria that commonly resides in the intestinal tracts of humans and animals but is also found in water, soil, and plants due to their high tolerance to different conditions (Cho et al. 2020), they can survive in a variety of environments, such as soil, water, food, plants, and animals (Castillo-Rojas et al. 2013), and they can survive in a wide range of temperatures (10–45 °C), pH (4.4–9.6), and in a hypersalty media with 6.5% NaCl (Ben Braïek and Smaoui 2019). Nowadays, Enterococcus is the leading cause of nosocomial infections, responsible for causing endocarditis, urinary tract infections, and bloodstream infections (Raza et al. 2018). There are more than 50 species of enterococci where E. faecium and E. faecalis are the most ecology and epidemiological relevant (Ramos et al. 2020). Furthermore, enterococci have intrinsic antimicrobial resistance to aminoglycosides, and cephalosporins antibiotic agents and can adapt to acquire resistance to antimicrobials such as β-lactams and vancomycin from the environment (Lee et al. 2019), by mutation and/or acquisition of genes through the plasmids and transposons (Grassotti et al. 2018). Vancomycin is an antibiotic of last resort used to treat MRSA. The vancomycin-resistance among clinical isolates is a growing health problem that involves the exchange of van genes between strains; where vanA and vanB are the most prevalent genotypes in E. faecium in clinical isolates (Melese et al. 2020), where the vanA gene encodes an enzyme that modifies the vancomycin binding site through the substitution of D-ala-D-ala by D-ala-D-lactate (Liu et al. 2021). Water ecosystems, in particular wastewater treatment plants, are considered one of the main hotspots of the evolution and spread of antibiotic-resistant bacteria (ARB) and ARGs into the natural environment that leads easily reach communities via aquatic environments (Ekwanzala et al. 2020). Enterococci and antimicrobials are commonly excreted in urine and feces, and most of this waste is treated in wastewater treatment plants where E. faecalis and E. faecium are dominant species identified before being discharged into surface water mainly during the dry season (Sanderson et al. 2020). E. faecalis and E. faecium are also used as an indicator of water quality criteria by fecal contamination in freshwater in the United States of America at geometric means of 33 and 35 CFU/100 mL (Wen et al. 2020) due to they are more resistant to stress than E. coli and other fecal coliform bacteria (Chidamba and Korsten 2018). Nevertheless, the distribution of resistant bacteria from the environment to humans may occur through contaminated food, manure, and contaminated surface water used for irrigation (Taučer-Kapteijn et al. 2016).
1.5.3 Staphylococcus
Staphylococcus aureus is a commensal opportunistic Gram-positive bacterium in humans (Pasachova et al. 2019) and are able to survive at a wide range of temperatures, dryness, dehydration, and low water activity (Silva et al. 2020). The genus Staphylococcus comprises more than 50 species, including coagulase-positive (S. aureus) and coagulase-negative staphylococci (E. epidermidis) (Kosecka-Strojek et al. 2019). S. aureus is one of the main pathogens in hospital and community infections associated with skin infections, pneumonia, and nosocomial bacteremia and represents public problem health (Cheung et al. 2021). The staphylococci became antibiotic resistant through genetic mutations, reducing outer membrane proteins, and acquiring resistance genes through HGT (Guo et al. 2020). The methicillin resistance in S. aureus (MRSA) is a significant global health concern determined by the expression of mecA gene, which encodes a penicillin-binding protein (PBP2a) with low affinity for β-lactam agents (Watkins et al. 2019). MRSA strains show a multidrug-resistant pattern with resistance to fluoroquinolones, macrolides, aminoglycosides, clindamycin, penicillins, cephalosporins, and carbapenem, making β-lactams ineffective (Gajdács 2019). MRSA is known as the major cause of hospital-/community-acquired infections, and morbidity infections are elevated worldwide. On the other hand, vancomycin has been one of the first-line drugs to treat MRSA infections; nowadays the intermediate and complete vancomycin-resistant S. aureus (VRSA) is a severe public health concern (Cong et al. 2020).
Environmental factors such as pH, temperature, nutrient content, salinity, and dissolved oxygen play important roles in biofilm development, influencing their persistence in water, especially in piping due to inefficient water treatment (Silva et al. 2022). Despite this, wastewater treatment reduces the prevalence of S. aureus, thus reducing its presence in surface water, a fact attributable to the fact that the effluents are diluted in the river water (Zieliński et al. 2020). Although Staphylococci are less frequent in surface water than E. coli, Enterococcus, and other ESKAPE pathogens, they are indicators of antimicrobial resistance in the environment mainly by the presence of mecC gen associated with S. aureus (Silva et al. 2021).
1.5.4 Klebsiella pneumoniae
Klebsiella spp. are Gram-negative and nonmotile bacteria resident in the environment (surface water, sewage, soil, and plants), and on mucosal surfaces such as humans and animals (Bengoechea and Sa Pessoa 2019). In humans, K. pneumoniae is a commensal and opportunistic pathogen found in gastrointestinal and respiratory tracts and skin of healthy people, and is responsible for causing community-acquired and nosocomial infections (urinary, respiratory tract infections, and infections of wounds and soft tissue) (Herridge et al. 2020). In contrast to other nosocomial pathogens, K. pneumoniae clones are divided into two phenotypic groups characterized by multidrug resistance and hypervirulence (Gonzalez-Ferrer et al. 2021). K. pneumoniae shows high resistance to a broad spectrum of antibiotics such as ESBL, carbapenem, quinolones, polymyxin, tigecycline, fluoroquinolones, and aminoglycosides. Recently, the WHO recognized extended-spectrum β-lactam (ESBL) producing and carbapenem-resistant K. pneumoniae as a critical public health threat (Wyres et al. 2020).
β-lactam antibiotics are the first-line option for the treatment of infections, but ESBL enzymes hydrolyze β-lactams (penicillins, monobactams, carbapenems, and cephalosporins) (Pillai 2022); on the other hand, the carbapenems are β-lactam antibiotics of last resort with the broadest spectrum of activity, that, in less than a decade after their use, become a global public health crisis (Cherak et al. 2021), mainly because infections caused by the ESBL/carbapenemase-producing Enterobacteriaceae are associated with increased mortality, length stay, and health-care costs (De angelis et al. 2020).
The environment is an important reservoir for disseminating ARB, antibiotic-resistance genes, and mobile genetic elements that interact and spread to other parts or to human and animal hosts (Samreen et al. 2021). Carbapenemase and ESBL-producing K. pneumoniae have been reported in aquatic environments, such as rivers, hospital effluents, and posthospital wastewater (Furlan et al. 2020). Some human health concerns associated with antibiotic resistance in the environment are the alteration of the human microbiome toward the emergence of antibiotic resistance and the potential hazard of selective pressure to create an antibiotic resistance environment (Ben et al. 2019). Nevertheless, measurements of the amounts and types of hazards (ARBs and ARGs) associated with environmental sources (water, air, soil, and food) that are related to the acquisition of resistant infections in humans are essential to establish suitable mitigation strategies (Vikesland et al. 2017).
1.5.5 Acinetobacter baumannii
The genus Acinetobacter comprises a group of Gram-negative, nonfermenting, strictly aerobic, catalase-positive and oxidase-negative coccobacillus (Torres et al. 2010). Acinetobacter species are widely distributed in the environment, making it possible to recover Acinetobacter isolates in almost 100% of the soil and water samples, as these are considered their main ecological niches (Salazar de Vegasa and Nieves 2005). Among the 32 varieties of species of the genus Acinetobacter, the most representative species is Acinetobacter baumanii, whose, while being distributed in nature, prevalence is strongly associated with the clinical-hospital environment, where some strains can survive in wet or dry inanimate objects in the hospital environment for weeks or months; in addition, it is a microorganism that is part of the normal flora of human skin and is distinguished as the microorganism that is most frequently carried persistently on the skin by hospital personnel (Phillips 2014).
Thus, all these factors increase the chances that patients will be colonized, in addition to promoting contamination of medical equipment, thus playing a determining role in the spread of infection and prolonged hospital outbreaks. The risk factors for Acinetobacter baumannii infection will depend on the type of infection (intrahospital, extrahospital) and its drug resistance profile, the latter being the most important. Community-acquired Acinetobacter baumannii infections, unlike those acquired in the hospital environment, are reported less frequently, because other risk factors are involved, such as residence in a tropical developing country, alcoholism, smoking, or diseases such as diabetes mellitus and chronic lung disease, conditions that easily allow infection to result in bacteremia or pneumonia.
It is worth mentioning that despite being rarely detected, this type of infection is of greater concern once it is acquired in the community, since it is a more severe infection than nosocomial infection and, in most cases, it is fulminant, dying in 60% of patients (Antunes et al. 2014). The A. baumannii genome contains several genes grouped into resistance islands, whose structure, in addition to providing high intrinsic resistance to antimicrobials, facilitates the acquisition of resistance mechanisms from other species of bacteria present in its environment, such as Pseudomonas spp. through mobile genetic elements (plasmids, transposons and integrons) (Agodi et al. 2013; Fournier et al. 2006), thus giving rise to an Acinetobacter isolate with a multidrug-resistant (MDR) phenotype with the capacity to produce extended-spectrum beta-lactamases, which confers resistance to most of the known antimicrobials, including carbapenems and colistin (Towner 2009). Therapeutic difficulties, added to the great capacity that it must survive for a long time in the hospital environment, together with the potential to develop in an acidic pH and at low temperatures, which allows it to increase its ability to invade devitalized tissues, together with the development of a biofilm (“biofilm”) on human surfaces and cells, constitute this microorganism as an emerging public health problem (Gaddy and Actis 2009).
1.5.6 Pseudomonas aeruginosa
Bacteria of the genus Pseudomonas are Gram-negative microorganisms that can be classified as facultative anaerobes and are characterized as being ubiquitous and preferring humid environments (Luján Roca 2014). Specifically, Pseudomonas aeruginosa is a bacterium that successfully colonizes an enormous diversity of niches, as a consequence of its great nutritional versatility, it can survive in extreme environmental conditions; however, naturally, we can find it widely distributed in nature, its reservoir is the moist soil, water, wastewater, vegetation, humans and animals, humans and animals being the main hosts (CDC 2016). P. aeruginosa is an opportunistic pathogen responsible for a wide range of infections, mainly nosocomial, which is why it is considered highly prevalent in infections associated with health-care infections (HAIs), since it has a high adaptation to repeated changes in the microenvironment, nutrient availability, resistance mechanisms, and capacities to form biofilms (Villanueva-Ramos et al. 2019).
In humans, the species most at risk from infection is Pseudomonas aeruginosa, but infections by other species such as P. paucimobilis, P. putida, P. fluorescens, or P. acidovorans may also occur (Dumaru et al. 2019). Invasive infections by P. aeruginosa occur mainly in patients with underlying diseases that cause immunosuppression and mostly occur in the hospital setting. Community-acquired infection may occasionally occur in previously healthy patients. These infections are usually localized at the skin level (folliculitis, cellulitis, or ecthyma gangrenosum) without associated bacteremia, or present as severe clinical forms (Brady 2004; De almeida et al. 2002;). In the case of community-acquired infections, such as sepsis due to Pseudomonas aeruginosa in previously healthy patients, it is an infrequent clinical entity that must be considered when ecthyma gangrenosum, neutropenia, and gastrointestinal manifestations are present.
However, there is a possibility that P. aeruginosa can colonize parts of the human body; however, the prevalence of this colonization in healthy people is low (Rossolini and Mantengoli 2005). In case of acquiring an infection caused by P. aeruginosa, the vast majority of these will be related to the hospital environment, constituting a severe clinical problem, since, in most cases, there is a compromise of the host’s defenses (Lyczak et al. 2000). This is because once the infection is established, P. aeruginosa produces a series of toxic compounds that cause not only extensive tissue damage but also interfere with the functioning of the immune system. Among the proteins involved in P. aeruginosa infection, we find toxins, as well as hydrolytic enzymes that degrade the membranes and connective tissue of various organs. This situation is aggravated by the difficulty in treating P. aeruginosa infections, since this bacterium is intrinsically resistant to multiple classes of antibiotics that are not structurally related to each other (Strateva and Yordanov 2009) due to decreased permeability of its outer membrane, the constitutive expression of several efflux pumps, and the production of enzymes that inactivate antibiotics. In addition, it can acquire new resistance mechanisms via mutations (Mesaros et al. 2007). Furthermore, in aqueous environments, this bacterium adheres to surfaces, producing a kind of aggregate called a biofilm. The formation of these accumulations of bacteria and extracellular material represents a health problem, since it contaminates devices implanted inside the body, such as intrauterine devices, catheters, or heart valves. Biofilms also represent a problem in the production process of various industries as they cause clogging and corrosion of connections and filters (Villanueva-Ramos et al. 2019). It is estimated that 65% of bacterial infections are due to the formation of biofilm and its mechanisms of tolerance to antibiotics, which represents a problem in the health field; the appearance of biofilms is strongly associated with recurrent chronic and delayed wound healing (Romeo 2020). When growing in a biofilm state, Pseudomonas aeruginosa increases tolerance to antibiotics up to a thousand times more than when it grows in its free form (Torres et al. 2010) and is mainly associated with the following mechanisms: quorum sensing (QS) signal, porins, efflux pumps, gene expression, membrane vesicles, extracellular DNA, and enzymes (Bolívar-Vargas et al. 2021). P. aeruginosa appears as an exceptional bacterium; the wide variety of virulence factors, the breadth of infections it causes, and its multiple mechanisms of resistance to antibiotics make it stand out among the pathogenic microorganisms for humans.
1.5.7 Enterobacter spp.
The Enterobacter genus is a member of the ESKAPE group, which contains the main resistant bacterial pathogens (Rice 2010). It consists of fermentative, facultative anaerobic, Gram-negative bacterial species belonging to the Enterobacteriaceae family. This genus is associated with a variety of environmental habitats, their presence in the intestinal tract as natural commensals of animal and human intestinal microbiota resulting in their wide distribution in soil, plants, water, and wastewater (Singh et al. 2018). Among this genus of bacteria, only certain subspecies/species have been associated with hospital-acquired infections and outbreaks. In humans, multiple Enterobacter species are known to act as opportunistic pathogens (disease-causing organisms): species, such as E. cloacae, E. aerogenes, E. gergoviae, and E. agglomerans, are associated with hospital-acquired infections and outbreaks (Akbari et al. 2016). These bacteria cause a variety of conditions, including eye and skin infections, meningitis, bacteremia (bacterial infection blood), pneumonia, and urinary tract infections.
The Enterobacter aerogenes, E. cloacae, and E. hormaechei species represent the most frequently described isolated species in clinical infections, especially in immunocompromised patients hospitalized in an intensive care unit (ICU), due to the adaptability of these species to antimicrobial agents and their behavior as opportunistic pathogens. These pathogens are frequently associated with a multidrug resistance (MDR) phenotype, mainly due to their adaptation to the hospital setting and their ability to readily acquire numerous mobile genetic elements containing resistance and virulence genes. These species have intrinsic resistance to ampicillin, amoxicillin, first-generation cephalosporins, and cefoxitin due to the expression of a constitutive AmpC β-lactamase. In addition, the production of extended-spectrum β-lactamases has been reported in these bacteria, making their treatment difficult (Davin-Regli et al. 2016). Antibiotic resistance, the regulation of resistance genes, and the expression of extended-spectrum beta-lactamases and carbapenemases significantly reduce therapeutic options, creating a particularly worrisome scenario with this microorganism as the protagonist of a global crisis of multiresistance to antibiotics.
Recent studies show that carbapenem-resistant enterobacteriaceae (CRE) can contaminate aquatic environments such as marine surface waters, rivers, estuaries, and contaminated drinking water (Mahon et al. 2019). CRE can reach aquatic bodies as a consequence of organic contamination from multiple sources (Mathys et al. 2019), including hospital effluents, wastewater treatment plants (PTARs), discharges from livestock and agricultural farms, water seepage, and others. Once mixed with the aquatic body, these effluents can introduce not only resistant microorganisms but also high doses of antibiotics, likely triggering the spread of resistance (Aydin et al. 2019). However, evaluating the link between the clinical and aquatic epidemiology of CRE is often challenging. While some studies have shown that clinical strains can be found in aquatic bodies, others have yet to prove such a link (Piedra-Carrasco et al. 2017).
1.5.8 Current Situation of Resistance
During the last 20 years, an increase in resistance has been observed in the 6 bacteria of the ESKAPE group; this represents a new crisis and a global public health problem (WHO 2015). Specifically, in Latin America, more than 50% of infections acquired in intensive care units (ICU) are caused by ESKAPE bacteria, with a growing tendency toward extreme drug resistance (resistance to all families of antimicrobials except 2 or 1 of them) and pan drug resistance (resistance to all families of antimicrobials) (WHO 2015).
Selective pressure is the most important cause of the dissemination and extension of resistance; in the last 70 years, the indiscriminate use of antibiotics had contributed to the genetic diversification of resistance genes, as can be seen in the current number of TEM beta-lactamases, where to date, there are at least 187 described, when in 1982 before third-generation cephalosporins were introduced into the clinic, only TEM-1 and TEM-2 were known (Corvec et al. 2013).
Antimicrobial resistance is favored by the inappropriate use of antimicrobials in human medicine, veterinary medicine, agriculture, and aquaculture. Insufficient prevention and control measures for infections associated with hospital care, incomplete treatment by patients, and lack of hygiene and sanitation, are the other factors that complicate global efforts to contain it (Bolívar-Vargas et al. 2021). The prevalence of resistance has not only affected the efforts made to hold it but has also complicated the interpretation of the phenotypic profile and addressing the appropriate treatment, since the association of different resistance mechanisms for the same is increasingly frequent.
Furthermore, the prevalence of resistance has not only increased in infection-causing bacteria. Intestinal colonization of healthy people by ESBL-producing Enterobacteriaceae has reached pandemic levels worldwide in just a few years, and it is estimated that there are 1753 million colonized people worldwide (Phillips 2014; Woerther et al. 2013). Other factors that contribute to the extension and dissemination of resistance are a consequence of the current situation in the world, where the hyperconnection among borders, for example, the food and animal trade, tourism, health and business trips, emigration, and refugees, among other events, allows resistant strains to reach anywhere (Rogers et al. 2011). In addition, wild animals can also act as a reservoir and a potential source of dissemination. An example of this is the presence of bacteria resistant to antibiotics in migratory birds, a situation that could undoubtedly favor the spread of resistance over long distances (Simoes et al. 2010). In all of the above examples, water bodies do play an important role as reservoirs and distribution nodes of antibiotic-resistant bacteria, especially those of the ESKAPE group, which, besides commonly being multidrug resistant, have an enhanced ability to transfer antibiotic-resistance genes to related bacteria present in the water bodies.
References
Agodi A, Auxilia F, Barchitta M, Brusaferro S, D’Alessandro D, Grillo OC et al (2013) Trends, risk factors and outcomes of health-care-associated infections within the Italian network SPIN-UTI. J Hosp Infect 84:52–58
Akbari M, Bakhshi B, Najar PS (2016) Particular distribution of Enterobacter cloacae strains isolated from urinary tract infection within clonal complexes. Iran Biomed J 20:49–55
Anas A, Tharakan B, Jasmin C, Chandran C, Vipindas PV, Narayanan S, Jaleel A (2021) Microbial community shifts along an estuary to open ocean continuum. Reg Stud Mar Sci 41:101587
Antunes LC, Visca P, Toener DJ (2014) Acinetobacter baumannii: evolution of a global pathogen. Pathogens Dis 71:292–301
Aydin S, Aydin ME, Ulvi A, Kilic H (2019) Antibiotics in hospital effluents: occurrence, contribution to urban wastewater, removal in a wastewater treatment plant, and environmental risk assessment. Environ Sci Pollut Res Int 26(1):544–558
Begon M, Harper JL, Townsend CR (1986) Ecology: individuals, populations and communities. Blackwell Scientific Publications, Oxford
Ben Braïek O, Smaoui S (2019) Enterococci: between emerging pathogens and potential probiotics. Biomed Res Int 2019:5938210. https://doi.org/10.1155/2019/5938210
Ben Y, Fu C, Hu M, Liu L, Wong MH, Zheng C (2019) Human health risk assessment of antibiotic resistance associated with antibiotic residues in the environment: a review. Environ Res 169:483–493. https://doi.org/10.1016/j.envres.2018.11.040
Bengoechea JA, Sa Pessoa J (2019) Klebsiella pneumoniae infection biology: living to counteract host defenses. FEMS Microbiol Rev 43:123–144. https://doi.org/10.1093/femsre/fuy043
Bolívar-Vargas AF, Torres-Caycedo MI, Sánchez-Neira Y (2021) Biofilms de Pseudomonas aeruginosa como mecanismos de resistencia y tolerancia a antibióticos. Revisión narrativa. Revista de la Facultad de Ciencias de la Salud de la Universidad del Cauca 23(2):47–57. https://doi.org/10.47373/rfcs.2021.v232.1780
Brady MT (2004) Pseudomonas and related genera. In: Feigin RD, Cherry JD, Demmier GJ, Kaplan SL (eds) Textbook of pediatric infectious diseases, 5th edn. Saunders, Philadelphia, pp 1557–1573
Brooke JS (2012) Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clin Microbiol Rev 25:2–41. https://doi.org/10.1128/CMR.00019-11
Brooks LE, Ul-Hasan S, Chan BK, Sistrom MJ (2018) Quantifying the evolutionary conservation of genes encoding multidrug efflux pumps in the ESKAPE pathogens to identify antimicrobial drug targets. MSystems 3:e00024-18. https://doi.org/10.1128/msystems.00024-18
Cabello-Yeves PJ, Zemskaya TI, Rosselli R, Coutinho FH, Zakharenko AS, Blinov VV, Rodriguez-Valera F (2017) Genomes of novel microbial lineages assembled from the sub-ice waters of Lake Baikal. Appl Environ Microbiol 84(1):e02132-17. https://doi.org/10.1128/AEM.02132-17
Castillo-Rojas G, Mazari-Hiríart M, Ponce de León S, Amieva-Fernández RI, Agis-Juárez RA, Huebner J, López-Vidal Y (2013) Comparison of Enterococcus faecium and Enterococcus faecalis strains isolated from water and clinical samples: antimicrobial susceptibility and genetic relationships. PLoS ONE 8(4). https://doi.org/10.1371/journal.pone.0059491
Centers for Disease Control and Prevention (CDC) (2016) Healthy Swimming. Available online: https://www.cdc.gov/healthywater/swimming. Accessed on 4 April 2022
Chakraborty J, Sapkale V, Shah M, Rajput V, Mehetre G, Agawane S, Kamble S, Dharne M (2020) Metagenome sequencing to unveil microbial community composition and prevalence of antibiotic and metal resistance genes in hypersaline and hyperalkaline Lonar Lake, India. Ecol Indicators 110:105827. https://doi.org/10.1016/j.ecolind.2019.105827
Challagundla L, Reyes J, Rafiqullah I, Sordelli DO, Echaniz-Aviles G, Velazquez-Meza ME, Robinson DA (2018) Phylogenomic classification and the evolution of clonal complex 5 methicillin-resistant Staphylococcus aureus in the Western hemisphere. Front Microbiol 9:1901. https://doi.org/10.3389/fmicb.2018.01901
Chen H, Jing L, Yao Z, Meng F, Teng Y (2019) Prevalence, source and risk of antibiotic resistance genes in the sediments of Lake Tai (China) deciphered by metagenomic assembly: a comparison with other global lakes. Environ Int 127:267–275. https://doi.org/10.1016/j.envint.2019.03.048
Cherak Z, Loucif L, Moussi A, Rolain JM (2021) Carbapenemase-producing gram-negative bacteria in aquatic environments: a review. J Glob Antimicrob Resist 25:287–309. https://doi.org/10.1016/j.jgar.2021.03.024
Cheung GYC, Bae JS, Otto M (2021) Pathogenicity and virulence of Staphylococcus aureus. Virulence 12:547–569. https://doi.org/10.1080/21505594.2021.1878688
Chidamba L, Korsten L (2018) Relative proportions of E. coli and Enterococcus spp. may be a good indicator of potential health risks associated with the use of roof harvested rainwater stored in tanks. Environ Monit Assess 190:177. https://doi.org/10.1007/s10661-018-6554-1
Cho S, Jackson CR, Frye JG (2020) The prevalence and antimicrobial resistance phenotypes of salmonella, Escherichia coli and enterococcus sp. in surface water. Lett Appl Microbiol 71:3–25. https://doi.org/10.1111/lam.13301
Chotirmall SH, Mirkovic B, Lavelle GM, McElvaney NG (2014) Immunoevasive Aspergillus virulence factors. Mycopathologia 178:363–370. https://doi.org/10.1007/s11046-014-9768-y
Cole JJ (1982) Interactions between bacteria and algae in aquatic ecosystems. Annu Rev Ecol Syst 13:291–314
Cong Y, Yang S, Rao X (2020) Vancomycin resistant Staphylococcus aureus infections: a review of case updating and clinical features. J Adv Res 21:169–176. https://doi.org/10.1016/j.jare.2019.10.005
Corpuz MVA, Buonerba A, Vigliotta G, Zarra T, Ballesteros F Jr, Campiglia P, Naddeo V (2020) Viruses in wastewater: occurrence, abundance and detection methods. Sci Total Environ 745:140910. https://doi.org/10.1016/j.scitotenv.2020.140910
Corvec S, Beyrouthy R, Cremet L, Aubin GG, Robin F, Bonnet R et al (2013) TEM-187, a new extended-spectrum beta-lactamase with weak activity in a Proteus mirabilis clinical strain. Antimicrob Agents Chemother 57:2410–2412
Dalmasso C, Oger P, Selva G, Courtine D, L’Haridon S, Garlaschelli A et al (2016) Thermococcus piezophilus sp. nov., a novel hyperthermophilic and piezophilic archaeon with a broad pressure range for growth, isolated from a deepest hydrothermal vent at the Mid-Cayman Rise. Syst Appl Microbiol 39:440–444. https://doi.org/10.1016/j.syapm.2016.08.003
Daniel DS, Lee SM, Gan HM, Dykes GA, Rahman S (2017) Genetic diversity of enterococcus faecalis isolated from environmental, animal and clinical sources in Malaysia. J Infect Public Health 10:617–623. https://doi.org/10.1016/j.jiph.2017.02.006
Davin-Regli A, Masi M, Bialek S, Nicolas-Chanoine MH, Pagès JM (2016) Antimicrobial resistance and drug efflux pumps in Enterobacter and Klebsiella. In: Li X-Z, Elkins CA, Zgurskaya HI (eds) Efflux-mediated drug resistance in bacteria: mechanisms, regulation and clinical implications. Springer International Publishing, Basel, pp 281–306
De Almeida JF, Sztajnbok J, Trostser EJ, Vaz FA, Case report. (2002) Pseudomonas aeruginosa septic shock associated with ecthyma gangrenosum in an infant with agammaglobulinemia. Rev Inst Med Trop S Paulo 44:167–169
De Angelis G, Del GP, Posteraro B, Sanguinetti M, Tumbarello M (2020) Molecular mechanisms, epidemiology, and clinical importance of β-lactam resistance in enterobacteriaceae. Int J Mol Sci 21:1–22. https://doi.org/10.3390/ijms21145090
Denissen J, Reyneke B, Waso-Reyneke M, Havenga B, Barnard T, Khan S et al (2022) Prevalence of ESKAPE pathogens in the environment: antibiotic resistance status, community-acquired infection and risk to human health. Int J Hyg Environ Health 244:114006. https://doi.org/10.1016/j.ijheh.2022.114006
Dumaru R, Baral R, Shrestha LB (2019) Study of biofilm formation and antibiotic resistance pattern of gram-negative Bacilli among the clinical isolates at BPKIHS, Dharan. BMC Res Notes 12(1):38
Ekwanzala MD, Dewar JB, Kamika I, Momba MNB (2020) Comparative genomics of vancomycin-resistant enterococcus spp. revealed common resistome determinants from hospital wastewater to aquatic environments. Sci Total Environ 719:137275. https://doi.org/10.1016/j.scitotenv.2020.137275
Escobar-Zepeda A, Vera-Ponce de León A, Sanchez-Flores A (2015) The road to metagenomics: from microbiology to DNA sequencing technologies and bioinformatics. Front Genet 6:348. https://doi.org/10.3389/fgene.2015.00348
Figueras MJ, Levican A, Collado L (2012) Updated 16S rRNA-RFLP method for the identification of all currently characterised Arcobacterspp. BMC Microbiol 12:1–7. https://doi.org/10.1186/1471-2180-12-292
Founou RC, Founou LL, Essack SY (2018) Extended spectrum beta-lactamase mediated resistance in carriage and clinical gram-negative ESKAPE bacteria: a comparative study between a district and tertiary hospital in South Africa. Antimicrob Resist Infect Control 7:1–11. https://doi.org/10.1186/s13756-018-0423-0
Fournier PE, Vallenet D, Barbe V (2006) Comparative genomics of multidrug resistance in Acinetobacter baumannii. PLoS Genet 2:e7
Furlan JPR, Savazzi EA, Stehling EG (2020) Genomic insights into multidrug-resistant and hypervirulent Klebsiella pneumoniae co-harboring metal resistance genes in aquatic environments. Ecotoxicol Environ Saf 201:110782. https://doi.org/10.1016/j.ecoenv.2020.110782
Gaddy JA, Actis LA (2009) Regulation of Acinetobacter baumannii biofilm formation. Future Microbiol 4:273–278
Gajdács M (2019) The continuing threat of methicillin-resistant Staphylococcus aureus. Antibiotics 8:52. https://doi.org/10.3390/antibiotics8020052
Gonzalez-Ferrer S, Peñaloza HF, Budnick JA, Bain WG, Nordstrom HR, Lee JS et al (2021) Finding order in the chaos: outstanding questions in Klebsiella pneumoniae pathogenesis. Infect Immun 89:e00693-20. https://doi.org/10.1128/IAI.00693-20
Grassotti TT, De Angelis ZD, Da Fontoura Xavier Costa L, De Araújo AJG, Pereira RI, Soares RO et al (2018) Antimicrobial resistance profiles in enterococcus spp. isolates from fecal samples of wild and captive black capuchin monkeys (Sapajus nigritus) in South Brazil. Front Microbiol 9:2366. https://doi.org/10.3389/fmicb.2018.02366
Guarner J (2020) Three emerging coronaviruses in two decades: the story of SARS, MERS, and now COVID-19. Am J Clin Pathol 153:420–421. https://doi.org/10.1093/ajcp/aqaa029
Guo Y, Song G, Sun M, Wang J, Wang Y (2020) Prevalence and therapies of antibiotic-resistance in Staphylococcus aureus. Front Cell Infect Microbiol 10:1–11. https://doi.org/10.3389/fcimb.2020.00107
Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM (1998) Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol 5:R245–R249. https://doi.org/10.1016/s1074-5521(98)90108-9
Herridge WP, Shibu P, O’Shea J, Brook TC, Hoyles L (2020) Bacteriophages of Klebsiella spp., their diversity and potential therapeutic uses. J Med Microbiol 69:176–194. https://doi.org/10.1099/jmm.0.001141
Hutchings M, Truman A, Wilkinson B (2019) Antibiotics: past, present and future. Curr Opin Microbiol 51:72–80. https://doi.org/10.1016/j.mib.2019.10.008
Huys G, Gevers D, Temmerman R, Cnockaert M, Denys R, Rhodes G, Swings J (2001) Comparison of the antimicrobial tolerance of oxytetracycline-resistant heterotrophic bacteria isolated from hospital sewage and freshwater fishfarm water in Belgium. Syst Appl Microbiol 24:122–130. https://doi.org/10.1078/0723-2020-00008
Ji P, Aw TG, Van Bonn W, Rose JB (2020) Evaluation of a portable nanopore-based sequencer for detection of viruses in water. J Virol Methods 278:113805. https://doi.org/10.1016/j.jviromet.2019.113805
Kadri K (2020) Polymerase chain reaction (PCR): principle and applications. In: Nagpal ML, Boldura OM, Balta C, Enany S (eds) Synthetic biology—new interdisciplinary science. IntechOpen, London. https://doi.org/10.5772/intechopen.86491
Kawai Y, Ozawa N, Fukuda T, Suzuki N, Mikata K (2022) Development of an efficient antimicrobial susceptibility testing method with species identification by nanopore sequencing of 16S rRNA amplicons. PLoS One 17:e0262912. https://doi.org/10.1371/journal.pone.0262912
Koo H, Hakim JA, Morrow CD, Crowley MR, Andersen DT, Bej AK (2018) Metagenomic analysis of microbial community compositions and cold-responsive stress genes in selected Antarctic lacustrine and soil ecosystems. Life 8:29. https://doi.org/10.3390/life8030029
Kosecka-Strojek M, Sabat AJ, Akkerboom V, Becker K, van Zanten E, Wisselink G et al (2019) Development and validation of a reference data set for assigning Staphylococcus species based on next-generation sequencing of the 16S-23S rRNA region. Front Cell Infect Microbiol 9:1–19. https://doi.org/10.3389/fcimb.2019.00278
Langridge P, Chalmers K (2004) The principle: identification and application of molecular markers. In: Molecular marker systems in plant breeding and crop improvement. Springer, Berlin, pp 3–22. https://doi.org/10.1007/3-540-26538-4_1
Lee T, Pang S, Abraham S, Coombs GW (2019) Antimicrobial-resistant CC17 Enterococcus faecium: the past, the present and the future. J Glob Antimicrob Resist 16:36–47. https://doi.org/10.1016/j.jgar.2018.08.016
Liu WT, Chen EZ, Yang L, Peng C, Wang Q, Xu Z et al (2021) Emerging resistance mechanisms for 4 types of common anti-MRSA antibiotics in Staphylococcus aureus: a comprehensive review. Microb Pathog 156:104915. https://doi.org/10.1016/j.micpath.2021.104915
Luján Roca DA (2014) Pseudomonas aeruginosa: un adversario peligroso. Acta bioquím. clín. latinoam, vol 48, no 4. La Plata dic
Lyczak JB, Cannon CL, Pier GB (2000) Stablishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microb Infect 2(9):1051–1060
Mahon BM, Brehony C, Cahill N, McGrath E, O’Connor L, Varley A et al (2019) Detection of OXA-48-like-producing Enterobacterales in Irish recreational water. Sci Total Environ 10(690):1–6
Maillard JY, Bloomfield SF, Courvalin P, Essack SY, Gandra S, Gerba CP et al (2020) Reducing antibiotic prescribing and addressing the global problem of antibiotic resistance by targeted hygiene in the home and everyday life settings: a position paper. Am J Infect Control 48:1090–1099. https://doi.org/10.1016/j.ajic.2020.04.011
Mandal P, Gupta AK, Dubey BK (2020) A review on presence, survival, disinfection/removal methods of coronavirus in wastewater and progress of wastewater-based epidemiology. J Environ Chem Eng 8:104317. https://doi.org/10.1016/j.jece.2020.104317
Mathys DA, Mollenkopf DF, Feicht SM, Adams RJ, Albers AL, Stuever DM et al (2019) Carbapenemase-producing Enterobacteriaceae and Aeromonas spp present in wastewater treatment plant effluent and nearby surface waters in the US. PLoS One 14(6):e0218650
Melese A, Genet C, Andualem T (2020) Prevalence of vancomycin resistant enterococci (VRE) in Ethiopia: a systematic review and meta-analysis. BMC Infect Dis 20:124. https://doi.org/10.1186/s12879-020-4833-2
Merino N, Aronson HS, Bojanova DP, Feyhl-Buska J, Wong ML, Zhang S, Giovannelli D (2019) Living at the extremes: extremophiles and the limits of life in a planetary context. Front Microbiol 10:780. https://doi.org/10.3389/fmicb.2019.00780
Merker M, Blin C, Mona S, Duforet-Frebourg N, Lecher S, Willery E, Wirth T (2015) Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat Genet 47:242–249. https://doi.org/10.1038/ng.3195
Mesaros N, Nordmann P, Plésiat P, Roussel-Delvallez M, Van Eldere J, Glupczynski Y et al (2007) Pseudomonas aeruginosa: résistance et options thérapeutiques à l’aube du deuxième millénaire. Antibiotiques 9(3):189–198
Monapathi ME, Bezuidenhout CC, James-Rhode OH (2020) Aquatic yeasts: diversity, characteristics and potential health implications. J Water Health 18:91–105. https://doi.org/10.2166/wh.2020.270
Mulani MS, Kamble EE, Kumkar SN, Tawre MS, Pardesi KR (2019) Emerging strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: a review. Front Microbiol 10:539. https://doi.org/10.3389/fmicb.2019.00539
Nakatsu CH, Byappanahalli MN, Nevers MB (2019) Bacterial community 16S rRNA gene sequencing characterizes riverine microbial impact on Lake Michigan. Front Microbiol 10:996. https://doi.org/10.3389/fmicb.2019.00996
Panizo MM, Reviákina V (2001) Candida albicans y su efecto patógeno sobre las mucosas. Rev Soc Venez Microbiol 21:38–45
Pasachova J, Ramirez S, Muñoz L (2019) Staphylococcus aureus: generalidades, mecanismos de patogenicidad y colonización celular. Ther Nova 17:25–38
Patel B (2016) Antibiotics: a savior in need of saving. City Tech Writer 11:46–49
Peña-Ocaña BA, Ovando-Ovando CI, Puente-Sánchez F, Tamames J, Servín-Garcidueñas LE, González-Toril E, Gutiérrez-Sarmiento W, Jasso-Chávez R, Ruíz-Valdiviezo VM (2022) Metagenomic and metabolic analyses of poly-extreme microbiome from an active crater volcano lake. Environ Res 203:111862. https://doi.org/10.1016/j.envres.2021.111862
Phillips M (2014) Acinetobacter species. In: Mandell GL, Dolin R, Blaser MJ (eds) Principles and practices of infectious diseases, vol 2, 8th edn. Imprint Elsevier Inc., Philadelphia, pp 2551–2558
Piedra-Carrasco N, Fabrega A, Calero-Caceres W, Cornejo-Sanchez T, Brown-Jaque M, Mir-Cros A et al (2017) Carbapenemase-producing enterobacteriaceae recovered from a Spanish river ecosystem. PLoS One 12(4):e0175246
Pillai D (2022) Genetic diversity and prevalence of extended spectrum beta-lactamase producing Escherichia coli and Klebsiella pneumoniae in aquatic environment receiving untreated hospital effluents. Res Sq:1–19
Pirnay JP, Matthijs S, Colak H, Chablain P, Bilocq F, Eldere V, Cornelis P (2005) Global Pseudomonas aeruginosa biodiversity as reflected in a Belgian river. Environ Microbiol 7:969–980. https://doi.org/10.1111/j.1462-2920.2005.00776.x
Pini G, Faggi E, Bravetti E (2017) Molecular typing of clinical and environmental Cryptococcus neoformans strains isolated in Italy. Open J Med Microbiol 7(04):77. https://doi.org/10.4236/ojmm.2017.74007
Podolsky SH (2018) The evolving response to antibiotic resistance (1945–2018). Palgrave Commun 4:124. https://doi.org/10.1057/s41599-018-0181-x
Premke K, Wurzbacher C, Felsmann K, Fabian J, Taube R, Bodmer P, Attermeyer K, Nitzsche KN, Schroer S, Koschorreck M, Hübner E, Mahmoudinejad T, Kyba C, Monaghan MT, Hölker F (2022) Large-scale sampling of the freshwater microbiome suggests pollution-driven ecosystem changes. Environ Pollut 308:119627. Proc Natl Acad Sci U.S.A. vol 95, p 12
Raj A (2012) Antibiotic resistance, plasmid and RAPD profiles of multidrug-resistant coliform bacteria isolated from sewage samples of Ghaziabad City, India. UJERT 2:318–324
Ramos S, Silva V, de Lurdes Enes Dapkevicius M, Igrejas G, Poeta P (2020) Enterococci, from harmless bacteria to a pathogen. Microorganisms 8:1118. https://doi.org/10.3390/microorganisms8081118
Randazzo W, Truchado P, Cuevas-Ferrando E, Simón P, Allende A, Sánchez G (2020) SARS-CoV-2 RNA in wastewater anticipated COVID-19 occurrence in a low prevalence area. Water Res 181:115942. https://doi.org/10.1016/j.watres.2020.115942
Raza T, Ullah SR, Mehmood K, Andleeb S (2018) Vancomycin resistant enterococci: a brief review. J Pak Med Assoc 68:768–772
Regnier P, Friedlingstein P, Ciais P et al (2013) Anthropogenic perturbation of the carbon fluxes from land to ocean. Nat Geosci 6:597–607. https://doi.org/10.1038/ngeo1830
Rice LB (2010) Progress and challenges in implementing the research on ESKAPE pathogens. Infect Control Hosp Epidemiol 31:7–10
Rice EW, Johnson CH (2000) Survival of Escherichia coli O157: H7 in dairy cattle drinking water. J Dairy Sci 83:2021–2023. https://doi.org/10.3168/jds.S0022-0302(00)75081-8
Rogers BA, Aminzadeh Z, Hayashi Y, Paterson DL (2011) Country-to-country transfer of patients and the risk of multi-resistant bacterial infection. Clin Infect Dis 53:49–56
Romeo A (2020) Biofilm y Resistencia antimicrobiana. Arch Med camagüey 24(4):1–4
Rosales Magallanes GF (2018) The end of the antibiotic era, 1. https://doi.org/10.15761/cmp.1000103
Rossolini GM, Mantengoli E (2005) Treatment and control of severe infections caused by multiresistant Pseudomonas aeruginosa. Clin Microbiol Infect 11(Suppl 4):17–32
Rusiñol M, Martínez-Puchol S, Timoneda N, Fernández-Cassi X, Pérez-Cataluña A, Fernández-Bravo A, Moreno-Mesonero L, Moreno Y, Alonso JL, Figueras MJ, Abril JF, Bofill-Mas S, Girones R (2020) Metagenomic analysis of viruses, bacteria and protozoa in irrigation water. Int J Hyg Environ Health 224:113440. https://doi.org/10.1016/j.ijheh.2019.113440
Salazar de Vegasa EZ, Nieves B (2005) Acinetobacter spp: Aspectos microbiológicos, clínicos y epidemiológicos. Rev Soc Venez Microbiol 25(2):64–71
Samreen AI, Malak HA, Abulreesh HH (2021) Environmental antimicrobial resistance and its drivers: a potential threat to public health. J Glob Antimicrob Resist 27:101–111. https://doi.org/10.1016/j.jgar.2021.08.001
Sanderson H, Ortega-Polo R, Zaheer R, Goji N, Amoako KK, Brown RS et al (2020) Comparative genomics of multidrug-resistant Enterococcus spp. isolated from wastewater treatment plants. BMC Microbiol 20:20. https://doi.org/10.1186/s12866-019-1683-4
Santaniello A, Sansone M, Fioretti A, Menna LF (2020) Systematic review and meta-analysis of the occurrence of eskape bacteria group in dogs, and the related zoonotic risk in animal-assisted therapy, and in animal-assisted activity in the health context. Int J Environ Res Public Health 17:3278. https://doi.org/10.3390/ijerph17093278
Schreiber C, Rechenburg A, Rind E, Kistemann T (2015) The impact of land use on microbial surface water pollution. Int J Hyg Environ Health 218(2):181–187. https://doi.org/10.1016/j.ijheh.2014.09.006
Seymour JR, Ahmed T, Durham WM, Stocker R (2010) Chemotactic response of marine bacteria to the extracellular products of Synechococcus and Prochlorococcus. Aquat Microb Ecol 59(2):161–168
Silva V, Caniça M, Capelo JL, Igrejas G, Poeta P (2020) Diversity and genetic lineages of environmental staphylococci: a surface water overview. FEMS Microbiol Ecol 96:1–13. https://doi.org/10.1093/femsec/fiaa191
Silva V, Ferreira E, Manageiro V, Reis L, Tejedor-Junco MT, Sampaio A et al (2021) Distribution and clonal diversity of Staphylococcus aureus and other staphylococci in surface waters: detection of ST425-t742 and ST130-t843 mecC-positive MRSA strains. Antibiotics 10:1416. https://doi.org/10.3390/antibiotics10111416
Silva V, Pereira E, Igrejas G (2022) Influence of environmental factors on biofilm formation of Staphylococci isolated from wastewater and surface water. Pathogens 11:1069
Simoes RR, Poirel L, da Costa PM, Nordmann P (2010) Seagulls and beaches as reservoirs for multidrug-resistant Escherichia coli. Emerg Infect Dis 16:110–112
Singh NK, Bezdan D, Checinska Sielaff A, Wheeler K, Mason CE, Venkateswaran K (2018) Multi-drug resistant Enterobacter bugandensis species isolated from the International Space Station and comparative genomic analyses with human pathogenic strains. BMC Microbiol 18:175. https://doi.org/10.1186/s12866-018-1325-2
Strateva T, Yordanov D (2009) Pseudomonas aeruginosa - a phenomenon of bacterial resistance. J Med Microbiol 58(9):1133–1148
Takai K, Nakamura K, Toki T, Tsunogai U, Miyazaki M, Miyazaki J et al (2008) Cell proliferation at 122 C and isotopically heavy CH4 production by a hyperthermophilic methanogen under high-pressure cultivation. Proc Natl Acad Sci U S A 105:10949–10954. https://doi.org/10.1073/pnas.0712334105
Tamayo-Ordoñez M, Huijara-Vasconselos J, Quiroz-Moreno A, Ortíz-García M, Sánchez-Teyer LF (2012) Plant tissue culture and molecular markers. In: Plant cell culture protocols. Humana Press, Totowa, pp 343–356. https://doi.org/10.1007/978-1-61779-818-4_26
Tamayo-Ordóñez MC, Contreras-Esquivel JC, Ayil-Gutiérrez BA, De la Cruz-Arguijo EA, Tamayo-Ordóñez FA, Ríos-González LJ, Tamayo-Ordóñez YJ (2021) Interspecific evolutionary relationships of alpha-glucuronidase in the genus Aspergillus. Fungal Biol 125:560–575. https://doi.org/10.1016/j.funbio.2021.02.005
Taučer-Kapteijn M, Hoogenboezem W, Heiliegers L, de Bolster D, Medema G (2016) Screening municipal wastewater effluent and surface water used for drinking water production for the presence of ampicillin and vancomycin resistant enterococci. Int J Hyg Environ Health 219:437–442. https://doi.org/10.1016/j.ijheh.2016.04.007
Tigabu A, Getaneh A (2021) Staphylococcus aureus, ESKAPE bacteria challenging current health care and community settings: a literature review. Clin Lab 67:1539–1549. https://doi.org/10.7754/Clin.Lab.2020.200930
Torres HA, Vázquez EG, Yagüe G, Gómez JG. (2010) Multidrug resistant Acinetobacter baumannii: clinical update and new highlights. Rev Esp Quimioter 23(1):12–19
Towner KJ (2009) Acinetobacter: an old friend, but a new enemy. J Hosp Infect 73:355–363
Tran PQ, Bachand SC, McIntyre PB et al (2021) Depth-discrete metagenomics reveals the roles of microbes in biogeochemical cycling in the tropical freshwater Lake Tanganyika. ISME J 15:1971–1986. https://doi.org/10.1038/s41396-021-00898-x
Tseng CH, Chiang PW, Shiah FK et al (2013) Microbial and viral metagenomes of a subtropical freshwater reservoir subject to climatic disturbances. ISME J 7:2374–2386. https://doi.org/10.1038/ismej.2013.118
Vikesland PJ, Pruden A, Alvarez PJJ, Aga D, Bürgmann H, Li XD et al (2017) Toward a comprehensive strategy to mitigate dissemination of environmental sources of antibiotic resistance. Environ Sci Technol 51:13061–13069. https://doi.org/10.1021/acs.est.7b03623
Villanueva-Ramos NB, De la Mora-Fernández AR, Ríos-Burgueño ER, de Peraza-Garay FJ (2019) Detección de biopelículas en tejido de amígdalas y adenoides en pacientes con procesos infecciosos crónicos y obstructivos. Anales de Otorrinolaringología Mexicana 64:1–17
Voigt AM, Ciorba P, Döhla M, Exner M, Felder C, Lenz-Plet F et al (2020) The investigation of antibiotic residues, antibiotic resistance genes and antibiotic-resistant organisms in a drinking water reservoir system in Germany. Int J Hyg Environ Health 224:113449. https://doi.org/10.1016/j.ijheh.2020.113449
Wang X, Wen X, Criddle C, Yan H, Zhang Y, Ding K (2010) Bacterial community dynamics in two full-scale wastewater treatment systems with functional stability. J Appl Microbiol 109:1218–1226. https://doi.org/10.1111/j.1365-2672.2010.04742.x
Wang W, Arshad MI, Khurshid M, Rasool MH, Nisar MA, Aslam MA et al (2018) Antibiotic resistance: a rundown of a global crisis. Infect Drug Resist 11:1645–1658
Wang H, Kjellberg I, Sikora P, Rydberg H, Lindh M, Bergstedt O, Norder H (2020a) Hepatitis E virus genotype 3 strains and a plethora of other viruses detected in raw and still in tap water. Water Res 168:115141. https://doi.org/10.1016/j.watres.2019.115141
Wang J, Feng H, Zhang S, Ni Z, Ni L, Chen Y, Qu T (2020b) SARS-CoV-2 RNA detection of hospital isolation wards hygiene monitoring during the coronavirus disease 2019 outbreak in a Chinese hospital. Int J Infect Dis 94:103–106. https://doi.org/10.1016/j.ijid.2020.04.024
Watkins RR, Holubar M, David MZ (2019) Antimicrobial resistance in methicillin-resistant Staphylococcus aureus to newer antimicrobial agents. Antimicrob Agents Chemother 63:e01216-19. https://doi.org/10.1128/AAC.01216-19
Wen X, Chen F, Lin Y, Zhu H, Yuan F, Kuang D et al (2020) Microbial indicators and their use for monitoring drinkingwater quality—a review. Sustainability 12:1–14. https://doi.org/10.3390/su12062249
Whitman WB, Coleman DC, Wiebe WJ (1998) Prokaryotes: the unseen majority. Proc Natl Acad Sci U S A 95(12):6578–6583. https://doi.org/10.1073/pnas.95.12.6578. PMID: 9618454; PMCID: PMC33863
Woerther PL, Burdet C, Chachaty E, Andremont A (2013) Trends ins human fecal carriage of extended-spectrum beta-lactamases in the community: toward the globalization of CTX-M. Clin Microbiol Rev 26:744–758
Woese CR, Fox GE (1977) Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A 74:5088–5090. https://doi.org/10.1073/pnas.74.11.5088
World Health Organization (2015) Worldwide country situation analysis: response to antimicrobial resistance: summary. World Health Organization. https://apps.who.int/iris/handle/10665/163473
Wyres KL, Lam MMC, Holt KE (2020) Population genomics of Klebsiella pneumoniae. Nat Rev Microbiol 18:344–359. https://doi.org/10.1038/s41579-019-0315-1
Zieliński W, Korzeniewska E, Harnisz M, Hubeny J, Buta M, Rolbiecki D (2020) The prevalence of drug-resistant and virulent Staphylococcus spp. in a municipal wastewater treatment plant and their spread in the environment. Environ Int 143:143. https://doi.org/10.1016/j.envint.2020.105914
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Bocanegra-García, V. et al. (2023). Freshwater Microbiology: Recent Updates and Prospects. In: Soni, R., Suyal, D.C., Morales-Oyervides, L., Sungh Chauhan, J. (eds) Current Status of Fresh Water Microbiology. Springer, Singapore. https://doi.org/10.1007/978-981-99-5018-8_1
Download citation
DOI: https://doi.org/10.1007/978-981-99-5018-8_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-5017-1
Online ISBN: 978-981-99-5018-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)