
Abstract
Acute myeloid leukaemia (AML) is an aggressive, heterogenous, and age-related haematological malignancy with dismal prognosis. Conventional therapy for AML consists of frontline induction therapy with cytarabine infusion for 7 days and administration of anthracyclines, most commonly daunorubicin, for 3 days (7 + 3), followed by subsequent consolidation with chemotherapy or allogeneic haematopoietic stem cell transplant (HSCT) for high-risk disease. However, the age-related nature of AML implies that a significant portion of patients are unfit for such intensive regimens and can only be put on palliative treatment. Increasing emphasis is being put on maximizing specificities and potencies of novel agents while minimizing treatment-related toxicities, entailing a future of personalized-therapy in AML. This chapter reviews recently approved agents and agents still in the pipeline for the treatment of AML both in the frontline and the relapsed/refractory setting.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
16.1 Introduction
Acute myeloid leukaemia (AML) is an aggressive, heterogenous, and age-related haematological malignancy with dismal prognosis. Conventional therapy for AML consists of frontline induction therapy with cytarabine infusion for 7 days and administration of anthracyclines, most commonly daunorubicin, for 3 days (7 + 3), followed by subsequent consolidation with chemotherapy or allogeneic haematopoietic stem cell transplant (HSCT) for high-risk disease. However, the age-related nature of AML implies that a significant portion of patients are unfit for such intensive regimens and can only be put on palliative treatment. Although treatment options for AML have remained stagnant for a long time, exciting progress has been made during recent years, with the U.S. Food and Drug Administration (FDA) approving nine novel agents indicated for this disease (Table 16.1). Increasing emphasis is being put on maximizing specificities and potencies of novel agents while minimizing treatment-related toxicities, entailing a future of personalized-therapy in AML.
16.2 Novel Chemotherapeutic Formulations
16.2.1 CPX-351
CPX-351 (Vyxeos) is an FDA-approved liposomal formulation of daunorubicin and cytarabine in a 5:1 molar ratio. While the combination of cytarabine and anthracyclines (7 + 3) has long been the conventional treatment for AML, their administration in the form of a liposomal capsule significantly prolongs their half-life and efficacy [1].
After encouraging results in a phase I study, subsequent phase II and III trials were carried out [2]. In a phase II study comparing CPX-351 with 7 + 3 in newly diagnosed AML patients, remarkable clinical benefit of CPX-351 was demonstrated, especially among patients with secondary AML, which is associated with poor prognosis [3]. In another phase II trial among relapsed or refractory (r/r) patients, CPX-351 induced superior responses when compared to standard salvage chemotherapy [4]. In a phase III trial, CPX-351 showed significantly prolonged survival compared to 7 + 3 induction [5]. These promising results were consistently replicated in subsequent trials [6, 7]. Side effects of CPX-351 are generally similar to those of 7 + 3, including myelosuppression, cardiotoxicity, with the exception of slower recoveries of neutrophil and platelet counts [3,4,5]. Combination of vyxeos with gemtuzumab ozogamicin (GO) and FLT3 inhibitors (quizartinib, midostaurin) also demonstrated clinical and preclinical efficacy respectively [8, 9].
Trials of CPX-351 as monotherapy or in combination with Ivosidenib, enasidenib, venetoclax, gilteritinib, midostaurin, quizartinib, palbociclib, glasdegib, GO, or fludarabine are underway (NCT04230239, NCT03988205, NCT03629171, NCT04668885, NCT04269213, NCT03555955, NCT04049539, NCT04493164, NCT03825796, NCT04075747, NCT04209725, NCT04038437, NCT03826992, NCT04293562, NCT04128748, NCT03844997, NCT04231851, NCT03878927, NCT03904251, NCT03672539, NCT02272478, NCT04425655). Studies comparing CPX-351 with other intensive chemotherapy regimens are also ongoing (NCT03897127, NCT04061239, NCT04293562, NCT04195945, NCT04802161).
16.3 Targeting Tyrosine Kinases
Tyrosine kinases regulate a wide range of cellular pathways and are crucial to signal transduction. Their aberrant activities can contribute to leukaemogenesis via promoting proliferation, impeding differentiation, and inhibiting apoptosis. Therefore, various agents have been developed against these kinases for the treatment of AML (Figs. 16.1, 16.2, and 16.3).

Downstream events of aberrant FLT3 signalling. AKT protein kinase B, ERK extracellular-signal-regulated kinase, FLT3-ITD FLT3 internal tandem duplication, FLT3-TKD FLT3 tyrosine kinase domain mutations, FLT3 Fms-like tyrosine kinase 3, MEK mitogen-activated protein kinase kinase, mTOR mammalian target of rapamycin complex, PI3K phosphoinositide 3-kinase, RAF rapidly accelerated fibrosarcoma, Ras rat sarcoma viral oncogene homolog, STAT5 signal transducer and activator of transcription 5

Actions of type I and type II FLT3 inhibitors. FL FLT3 ligand, FLT3 Fms-like tyrosine kinase 3

Agents targeting tyrosine kinases. AXL anexelekto, BTK Bruton tyrosine kinase, c-KIT cluster of differentiation 117, c-MET mesenchymal-epithelial transition factor, FL FLT3 ligand, FLT3 Fms-like tyrosine kinase 3, HGF hepatocyte growth factor, SCF stem cell factor, SFK Src family kinases, SYK spleen-associated tyrosine kinase
16.3.1 FLT3 Inhibitors
Figures 16.1, 16.2, and 16.3 and Table 16.2 summarize the role of FLT3 inhibitors and the major FLT3 inhibitors in development. Readers should refer to Chap. 12 of this title for further discussion.
16.3.2 c-KIT Inhibitors
c-KIT, also known as CD117, is an RTK expressed in haematopoietic cells for their normal development. Upon binding of stem cell factor (SCF), c-KIT dimerizes and undergoes autophosphorylation, which activates downstream PI3K/AKT/mTOR, JAK-STAT, and Ras/RAF/MAPK pathways, as well as Src family kinases (SFKs) [12, 13]. The expression of c-KIT is found in 60–80% of AML and its mutation is especially prevalent in core binding factor (CBF) AML [14]. Mutations in c-KIT mainly occur in exon 8 and exon 17, with the latter being associated with a more inferior clinical outcome [12]. Aberrant activation of c-KIT results in increased proliferation, reduced apoptosis, and subsequent leukaemogenesis [12].
Dasatinib and radotinib are multi-kinase inhibitors with potent activity against c-KIT. These agents induced apoptosis in c-KIT-positive AML cell lines and showed activity in downregulating other leukaemogenic pathways in various preclinical studies [15]. Dasatinib also showed synergistic efficacy with navitoclax against AML cells with NUP98-NSD1 and FLT3-ITD [16]. Addition of dasatinib to standard chemotherapy and its use as single agent maintenance therapy in patients with CBF AML showed favourable outcomes and a tolerable safety profile [17,18,19]. A phase III randomized controlled trial of chemotherapy with or without dasatinib in CBF AML patients is underway (NCT02013648). Other c-KIT inhibitors which are not actively evaluated for use in AML include imatinib, SU5416, and SU6668 [20, 21].
16.3.3 AXL Inhibitors
Anexelekto (AXL) is a member of the TYRO3, AXL, and MER (TAM) RTK family [22]. It is expressed on a multitude of cells and tissues and is crucial for the normal function of various haematopoietic cell types [22, 23]. Binding of Gas6 to AXL induces its dimerization and subsequent activation of PI3K, Ras, Src, and JAK/STAT pathways, resulting in cellular proliferation and migration [22]. In AML, AXL may be activated via mechanisms independent of Gas6 [22]. Aberrant signalling of AXL also acts as a key mediator of resistance against FLT3 inhibitors [23].
Similar to FLT3 inhibitors, AXL inhibitors are divided into two types. Type I AXL inhibitors bind to the ATP-binding site of the active AXL receptor [22]. Bemcentinib (BGB324) is a highly specific, potent, and safe small molecule type I inhibitor of AXL which showed efficacy against both FLT3-WT and FLT3-mutant AML cell lines [24, 25]. Due to promising results of its combination with LDAC in recent trials, bemcentinib has received fast track designation from the FDA [26, 27]. A phase II study regarding the use of bemcentinib in AML is currently underway (NCT03824080). Other type I inhibitors include gilteritinib and sunitinib. Type II inhibitors bind to the AXL receptor in its inactive form [22]. Among them, merestinib (LY2801653) is a potent and orally available inhibitor of AXL, FLT3, MNK, MET/RON, and other oncoproteins [28, 29]. It was proven to be safe in r/r AML patients in a phase I clinical trial [30]. Other novel AXL inhibitors with impressive preclinical efficacies against AML cell lines include the AXL/Mer dual inhibitors ONO-9330547 and ONO-7475, with ONO-7475 currently in a phase I/II trial as monotherapy or in combination with venetoclax (NCT03176277) [31,32,33].
16.3.4 c-MET Inhibitors
The MET RTK family consists of two members, c-Met and RON. Upon binding of their respective ligands (HGF for c-Met, MSP for RON), their tyrosine kinase domain activates and initiates signal transduction via PI3K, AKT, Β-catenin, Ras/MAPK, and JAK/STAT pathways [34]. Evidence of their expression in AML blasts led to studies evaluating their potential roles as therapeutic targets [34].
SU11274 is a c-Met inhibitor which demonstrated anti-leukaemic efficacy in preclinical studies [34,35,36]. Crizotinib also exhibited activity against AML cells, but seemed to induce resistance via a compensatory increase in HGF expression [35].
16.3.5 SYK Inhibitors
Spleen-associated tyrosine kinase (SYK) is a cytoplasmic tyrosine kinase with diverse biological activities, including roles in adaptive immune receptors signalling [37]. In AML, its increased expression is shown to be associated with inferior clinical outcomes [38]. Upon activation by FLT3-ITD or other upstream pathways, SYK undergoes phosphorylation and initiates a series of downstream signalling pathways, ultimately contributing to leukaemogenesis [39].
Preclinical studies with R406, an active metabolite of the SYK inhibitor fostamatinib, showed efficacy against AML cell lines by inducing differentiation and inhibiting proliferation [39]. Entospletinib (GS-9973) showed efficacy as monotherapy as well as in combination with chemotherapy in two early phase trials [40, 41]. TAK-659, a dual inhibitor of SYK and FLT3, also exhibited anti-leukemic activity in murine models and showed promising efficacy and safety profile in a phase Ib/II study in r/r AML patients [42, 43].
16.3.6 BTK Inhibitors
Bruton tyrosine kinase (BTK) is a member of the Tec family kinases [44]. This family of non-receptor, cytoplasmic kinases are mainly expressed on the surfaces of haematopoietic stem cells (HSCs) and other haematopoietic cells [44]. BTK, in particular, also plays a critical role in the development of B lymphocytes and is considered to be a key mediator in B-cell neoplasms [45, 46]. In AML, aberrant signalling of SFK, SYK, and PI3K leads to BTK activation and downstream activation of NFKB and other kinase pathways, resulting in leukaemogenesis. Emerging evidence of high BTK expression and constitutive activation in AML cells has led to interests on its potential role as a therapeutic target [47, 48]. In addition, FLT3-ITD may act as one of the upstream events leading to BTK autophosphorylation, implying the potential of BTK inhibitors for treating FLT3-ITD-positive AML [47].
Ibrutinib (CI-32765) is an irreversible inhibitor of BTK. In preclinical studies, it showed efficacy against AML cell lines by inhibiting downstream NFKB signalling, SDF1/CXCR4-mediated migration, and SDF1-induced activation of the AKT/MAPK pathway [48, 49]. Mutations in FLT3, NPM1, and DNMT3A were shown to be associated with increased sensitivity to ibrutinib [50]. In leukaemic blasts obtained from c-KIT-positive AML patients, ibrutinib also inhibited activation of BTK by c-KIT and their adhesion to bone marrow stromal cells [51]. Furthermore, specific inhibition of FLT3-ITD by ibrutinib in leukaemic cell lines has been reported, supporting the hypothesis that BTK inhibition may be efficacious against FLT3-ITD-positive AML [52]. Its combination with the recently approved Bcl-2 inhibitor, venetoclax, also showed promising results in preclinical studies [53]. However, a phase II clinical trial with ibrutinib monotherapy or in combination of azacitidine or cytarabine showed limited efficacy [54].
CG-806 is a dual FLT3/BTK inhibitor with remarkable activity and safety against AML cell lines and murine models [55]. This agent is currently evaluated in a phase I clinical trial (NCT04477291). Other novel BTK inhibitors with promising preclinical results include ARQ351 and abivertinib (AC0010) [56,57,58].
16.3.7 SFK Inhibitors
The non-receptor Src family of kinases include LYN, HCK, BLK, FGR, FYN, LCK, SRC, and YES [59]. In AML cells, FYN, LYN, HCK, and FGR are commonly expressed. Aberrant upstream signalling of FLT3, c-KIT, and other RTKs result in their activation, subsequently causing STAT5, Ras, and PI3K induction [59].
Bosutinib is an SFK inhibitor primarily used in the treatment of chronic myeloid leukaemia (CML). Recently, studies showed that its combination with all-trans retinoic acid (ATRA) enhances sensitivity of AML cell lines to ATRA, thus promoting differentiation of AML blasts [60]. It will be evaluated in a subsequent phase Ib trial in combination with glasdegib (NCT04655391).
RK-20449 is a selective HCK inhibitor which showed efficacy against chemotherapy-resistant AML cells in murine models [61]. An FGR inhibitor, TL02–59, also showed anti-leukaemic activity in a preclinical study [62]. Other SFK inhibitors with impressive preclinical evidences include PP2, dasatinib, ponatinib, PD180970, and SKI-606 [59, 63]. Although SAR103168 showed efficacy against AML cell lines in preclinical studies, results from a subsequent phase I trial were disappointing [64, 65].
16.4 Targeting the Hedgehog Pathway
The hedgehog (Hh) pathway is an essential mediator of embryonic development. In the canonical Hh pathway, Hh ligand binds to the transmembrane protein Patched (PTCH) to alleviate its inhibition on Smoothened (Smo), another transmembrane protein (Fig. 16.4). Smo then activates downstream glioma transcription factors (GLI) to stimulate gene transcription and proliferation [66]. In the non-canonical Hh pathway, activation of GLI is induced by other upstream pathways instead of Smo activation, such as PI3K/AKT/mTOR, RAS/RAF/MEK/ERK, protein kinase C (PKC), and many others [67]. In AML, the Hh pathway and oncogenic GLI activity may be constitutively activated, which is associated with radio- and chemo-resistance as well as poor prognosis [68,69,70]. Notably, crosstalk between the Hh pathway and FLT3-ITD has been discovered, prompting contemplations on its therapeutic role in FLT3-mutant AML [67].

Agents targeting the hedgehog pathway. AKT protein kinase B, ERK mitogen-activated protein kinase, GLI glioma transcription factors, Hh hedgehog, MEK mitogen-activated protein kinase kinase, mTOR mammalian target of rapamycin, PI3K phosphoinositide 3-kinase, PTCH patched, RAF rapidly accelerated fibrosarcoma, Ras rat sarcoma, Smo smoothened
16.4.1 Smo Inhibitors
Inhibition of Smo is the most widely studied among all potential therapeutic targets in the Hh pathway. Glasdegib (PF-04449913) is an FDA-approved selective Smo inhibitor. After encouraging results from preclinical studies, glasdegib was further studied in phase I clinical trials, where it was proven to be effective and tolerable in AML patients [71,72,73]. Subsequent trials investigating combinations of glasdegib with LDAC, decitabine, or standard chemotherapy all demonstrated clinical effectiveness [74,75,76,77]. Notably, addition of glasdegib to LDAC prolonged survival by nearly twofold compared to single agent LDAC, but did not increase toxicity in a multi-centre randomized phase II trial [76, 77]. Major side effects of glasdegib include febrile neutropenia, anaemia, and gastrointestinal disturbances [72,73,74,75,76,77]. Future trials include combination therapies with chemotherapy, LDAC, CPX-351, decitabine, azacitidine, GO, gilteritinib, ivosidenib, enasidenib, venetoclax, bosutinib, avelumab, and OX40 (NCT0341617, NCT02038777, NCT04231851, NCT04051996, NCT02367456, NCT04093505, NCT04655391, NCT03390296).
Sonidegib (LDE225) is another Smo inhibitor which demonstrated efficacy against doxorubicin-resistant AML cell lines and exhibited synergism with azacitidine in preclinical studies [78]. Its single agent therapy and combination with azacitidine or decitabine have been studied in phase I and phase II trials (NCT02129101, NCT01826214) [79].
Vismodegib (GDC-0449) also showed anti-leukaemic activity in preclinical studies, but had limited efficacy as monotherapy in a subsequent trial [80, 81]. Similarly, another trial of its use in combination with cytarabine was terminated due to the minimal responses observed among patients (NCT01880437).
16.4.2 GLI Inhibitors
Given that GLI activation can occur independently of Smo, direct inhibition of GLI is an attractive strategy against resistance to Smo inhibitors [67]. GANT61 is a GLI inhibitor which inhibited proliferation and induced apoptosis of AML in preclinical studies [82]. Its combination with sunitinib also prolonged survival of FLT3-mutant mice [83]. These optimistic results warrant clinical studies for GLI inhibitors in AML patients.
16.5 Targeting Apoptotic Pathways
16.5.1 BCL-2 Family Inhibitors
The anti-apoptotic B-cell lymphoma 2 (BCL-2) family prevents cellular apoptosis via the inhibition of proapoptotic proteins, such as BAX and BAK. Examples of members include Bcl-2 (B-cell lymphoma 2), myeloid cell leukaemia sequence 1 (MCL-1), and B-cell lymphoma-extra-large (Bcl-xL) [84]. Their actions are counteracted by the pro-apoptotic subfamily of BCL-2 (Fig. 16.5). In AML, their overexpression has been identified in multiple studies, which implies their influence on impairing apoptosis and promoting survival of leukemic cells [84, 85].

Agents targeting apoptotic pathways. BAK Bcl-2-antagonist/killer 1, BAX apoptosis regulator BAX, Bcl-2, B-cell lymphoma 2, Bcl-xL B-cell lymphoma extra-large, BIM Bcl-2-like protein 11, BOK Bcl-2-related ovarian killer, DR4/5 death receptor 4 or 5, MCL-1 myeloid cell leukaemia sequence 1, NOXA Phorbol-12-myristate-13-acetate-induced protein 1, PUMA p53 upregulated modulator of apoptosis, tBID truncated BID, TRAIL TNF-related apoptosis-inducing ligand
16.5.2 Bcl-2 Inhibitors
Bcl-2 inhibitors exert anti-leukaemic activity by mimicking the BH3 domain of the pro-apoptotic BCL-2 proteins and freeing them from the anti-apoptotic BCL-2 protein, which induces apoptosis [85].
Despite unsatisfactory results in early trials with oblimersen and obatoclax, efforts on investigation of Bcl-2 inhibition were persistent, which led to the development of venetoclax [85]. Venetoclax (ABT-199) is an FDA-approved, potent, and selective Bcl-2 inhibitor. Venetoclax was proven to be effective and tolerable in preclinical and clinical studies, both as monotherapy and in combination with HMAs (azacitidine, decitabine) or cytarabine, both in newly diagnosed and r/r patients [86,87,88,89,90,91,92,93,94,95]. Notably, combination of venetoclax with HMAs induced remarkable responses in a wide range of patients, including those with high-risk cytogenetic features and mutant-TP53 [95, 96]. Preclinical studies also elucidated their efficacies in targeting LSCs via inhibition of complex 2 of the ETC [97].
Full approval of venetoclax by the FDA was prompted by the phase III randomized placebo-controlled VIALE-A and VIALE-C trials, which evaluated the use of venetoclax in combination with azacitidine and LDAC, respectively. Both trials illustrated improvements of survival outcomes and remission rates upon the addition of venetoclax, along with tolerable increases in haematological toxicities [98, 99]. However, it should be noted that these benefits did not reach statistical significance in the VIALE-C study. Major side effects of venetoclax include febrile neutropenia and thrombocytopenia [98, 99].
Finally, multiple novel combinations with venetoclax are also being studied to overcome resistance. Among them, agents downregulating activity of MCL-1 are intensively evaluated owing to associations between MCL-1 upregulation and venetoclax resistance [85]. Multiple trials of venetoclax as monotherapy or in combination with other agents are ongoing (Table 16.3).
Other Bcl-2 inhibitors currently engaged in clinical trials include VOB560, S 055746 (BCL201), S6548, and APG2575 (NCT04702425, NCT02920541, NCT03755154, NCT04501120).
16.5.2.1 Bcl-2/Bcl-xL Dual Inhibitors
ABT-737 demonstrated promising efficacy against AML cell lines in preclinical studies, but its clinical development has been limited by an unfavourable pharmacokinetic profile [85, 100]. A derivative of this agent, navitoclax (ABT-263), possesses superior pharmacokinetic properties, though its clinical investigation is still not of interest due to the major adverse effect of thrombocytopenia [85].
16.5.2.2 MCL-1 Inhibitors
MCL-1 is another attractive therapeutic target in AML due to its overexpression in AML and association with venetoclax-resistance. AZD5991 is an MCL-1 inhibitor which demonstrated synergistic actions with bortezomib against AML xenograft in a murine study and is currently evaluated in combination with venetoclax in r/r AML patients in a phase I/Ib/IIa trial (NCT03218683) [101]. AMG 176 and AM-8621 both showed single agent efficacy and synergistic activity with venetoclax, though only AMG 176 is selected for further clinical investigations as monotherapy and in combination with azacitidine or venetoclax given its superior pharmacokinetic profile (NCT02675452, NCT03797261) [102, 103]. Another agent, AMG 397, also showed favourable preclinical results and will be evaluated in r/r AML patients in a phase I trial (NCT03465540) [104]. In addition, S63845 demonstrated excellent anti-leukaemic efficacy as single agent and in combination with venetoclax, daunorubicin, or S55746 (Bcl-2 inhibitor) in preclinical studies [105,106,107]. A related agent, S64315, has been evaluated in AML patients in a phase I trial and will undergo further testing in combination with azacitidine, venetoclax, or VOB560 (NCT02979366, NCT04629443, NCT03672695, NCT04702425). Other MCL-1 inhibitors with preclinical efficacies against AML include Compound 42, VU661013, MIMI, and Cardone compound 9 [108,109,110,111].
16.5.3 TRAIL Inducers
TNF-related apoptosis-inducing ligand (TRAIL) induces p53-independent apoptosis upon binding to its cell surface receptors, namely death receptors (DR) 4 and 5 [112]. Imipridone compounds have been found to promote TRAIL transcription and expression, subsequently inducing apoptosis. Among them, ONC201 demonstrated potent anti-leukaemic effect against AML cells and LSCs, both as monotherapy and in combination with cytarabine or azacitidine [113,114,115]. Interestingly, its therapeutic activity relies on both the induction of TRAIL activity and stimulation of an integrated stress response (ISR) [113,114,115]. It is currently evaluated as monotherapy or in combination with LDAC, and as single agent post-HSCT maintenance in AML patients in phase I/II trials (NCT02392572, NCT03932643). ONC212, a more potent derivative of ONC201, exhibited single agent activity and synergism with venetoclax against AML cell lines and murine models [116, 117].
16.6 Targeting the TP53 Pathway
TP53 encodes the tumour suppressor p53 and is among the most commonly mutated genes in all human malignancies [118]. WT p53 promotes cell cycle arrest, inhibits proliferation, and induces cellular apoptosis upon cellular stress [119]. Its activity is counteracted by mouse double minute 2 (MDM2), an E3 ligase which induces proteasomal degradation of p53 with the aid of MDM4 (Fig. 16.6). In AML, TP53 mutations are associated with resistance to chemotherapeutic agents and dismal prognosis, which warrants the development of novel targeted therapies against this entity [120].

Agents targeting the TP53 pathway. ATO arsenic trioxide, GSH glutathione, MDM2 mouse double minute 2, MQ methylene quinuclidinone, mut-p53 mutant p53, ROS reactive oxygen species, WT wild type
16.6.1 Mutant TP53 Inhibitors
APR-246, a methylated analogue of p53 reactivation and induction of massive apoptosis (PRIMA-1), is a pro-drug of methylene quinuclidinone. Upon conversion into its active form, APR-246 restores the active conformation of p53 and its ability to induce apoptosis and cell cycle arrest in leukemic cells [120, 121]. APR0246 can also exert anti-tumour effect in a p53-independent manner via depletion of anti-oxidants and induction of oxidative stress [122]. Synergism with azacitidine in inducing G0/G1 cell cycle arrest, apoptosis, and downregulation of FLT3 signalling was also reported [123]. This agent demonstrated remarkable clinical efficacy in combination with azacitidine in TP53-mutant AML patients in an ongoing phase 1b/2 study [124] and is being further investigated in other trials (NCT03072043, NCT03931291).
Arsenic trioxide (ATO), an agent primarily used for the treatment of acute promyelocytic leukaemia, also demonstrated ability to induce proteasomal degradation of mutant p53 and restore normal function of WT p53 [125, 126]. Its combination with ascorbic acid selectively induced oxidative stress and apoptosis in TP53-mutant leukemic cells in a recent study [127]. In addition, this agent exhibited activity against NPM1-mutant AML cells by inducing mutant protein degradation in multiple studies [128,129,130]. The use of ATO as single agent and in combination with decitabine or all-trans-retinoic-acid (ATRA) is currently explored in patients with TP53 or NPM1 mutations in a number of clinical studies (NCT04689815, NCT03855371, NCT03031249).
16.6.2 MDM2 Inhibitors
Increased activity of MDM2 is associated with reduced p53 activity [118]. Therefore, inhibition of binding between MDM2 and p53 prevents degradation of p53 and restores its tumour suppressor functions [131]. Nutlins are the earliest selective inhibitors of MDM2 to be discovered, with nutlin 3 being widely used in preclinical studies investigating effects of MDM2 inhibition [131]. A small molecule MDM2 inhibitor, RG7112, demonstrated anti-leukaemic efficacy as monotherapy and in combination with cytarabine in AML patients [132, 133]. Another agent, idasanutlin (RG7388), is a potent, selective, and orally available second generation MDM2 inhibitor. Clinical studies of this agent as monotherapy and in combination with cytarabine had impressive responses. This agent was generally tolerable with gastrointestinal toxicity as a significant side effect [134]. In addition, idasanutlin exhibited synergistic activity with venetoclax in a preclinical study, which led to the initiation of a phase 1/1b trial with favourable results [135, 136]. Combination of idasanutlin with venetoclax or chemotherapy will be further evaluated in a phase 1/2 clinical trial (NCT04029688). Synergism between idasanutlin and XPO inhibitors (selinexor, eltanexor) was also discovered in a preclinical study [137].
Disappointingly, RO6839921, the pegylated prodrug of idasanutlin, showed inferior effectiveness compared to idasanutlin in a recent study and will not undergo further clinical development [138].
Another MDM2 inhibitor, AMG 232 (KRT232), showed modest clinical activity in combination with trametinib, a MEK inhibitor. This combination regimen was tolerable and common adverse effects include nausea, gastrointestinal disturbances, and poor appetite [139]. This agent will be tested in combination with cytarabine and venetoclax; cytarabine; decitabine; or with TL-895 (TKI) in subsequent trials (NCT04190550, NCT04113616, NCT04669067).
Siremadlin (HDM201) showed promising activity in a phase I trial with cytopenias and tumour lysis syndrome as the most significant side effects. It will undergo evaluation with midostaurin in r/r patients with TP53 and FLT3 mutations, as well as with MBG453 (TIM3 inhibitor) or venetoclax in AML patients (NCT04496999, NCT03940352) [140]. Another MDM2 inhibitor, Milademetan (DS-3032b), has been evaluated as monotherapy in a phase 1 trial and is currently evaluated in combination with azacitidine, or LDAC with or without venetoclax (NCT03671564, NCT02319369, NCT03634228). Finally, APG-115 is currently evaluated with azacitidine or cytarabine in a phase 1 trial (NCT04275518).
16.7 Targeting the PI3K/AKT/mTOR Pathway
The phosphoinositide 3-kinase (PI3K)-Protein kinase B (AKT)-mammalian target of rapamycin (mTOR) pathway is crucial to cellular metabolism and can be activated by a myriad of upstream pathways [141]. In AML, upregulation of this pathway supports leukaemic cell activities and can occur as a result of aberrant upstream tyrosine kinases signalling or constitutive activation [141]. Unfortunately, increased activity of this pathway seems to be associated with decreased survival [141]. Thus, pharmacological inhibition of this pathway is a logical and attractive novel strategy in AML (Fig. 16.7).

Agents targeting the PI3K/AKT/mTOR pathway. AKT protein kinase B, mTOR mammalian target of rapamycin, PDK 3-phosphoinositide-dependent protein kinase-1, PI3K phosphoinositide 3-kinase, TSC1 tuberous sclerosis complex 1, TSC2 tuberous sclerosis complex 2
Although PI3K/AKT/mTOR inhibition demonstrated anti-leukaemic efficacies in preclinical studies, these results did not translate into meaningful clinical benefits [141]. mTORC1 inhibitors, including sirolimus, everolimus (RAD001), deferolimus (AP23573, MK-8669), and temsirolimus (CCI-779), have been tested in multiple clinical trials as monotherapies or in combination with chemotherapy regimens among AML patients with mostly limited success [142,143,144,145,146,147]. Although dual inhibition of PI3K and mTORC1 was proposed as a mechanism against resistance to mTORC1 inhibitors [148], two dual PI3K/mTOR inhibitors, gedatolisib (PF-05212384) and BEZ235, did not improve patient survival as single-agent and as an adjunct to chemotherapy, respectively [149, 150]. Other strategies to overcome resistance, such as dual mTORC1/mTORC2 inhibition, are being explored for the treatment of AML [148].
16.8 Targeting Metabolic Pathways
Mitochondrial activity is fundamental to supporting cellular metabolisms of almost all types of body cells. This carries paramount significance for the treatment of AML due to the presence of mitochondrial abnormalities, which can be exploited for selective AML cells targeting [151] (Fig. 16.8). In addition, other aberrant metabolic pathways discovered in LSCs are also being explored as targets for LSC eradication [151].

Agents targeting metabolic pathways. 2-HG 2-hydroxyglutarate, Acetyl-CoA acetyl-coenzyme A, ADP adenosine diphosphate, ATP adenosine triphosphate, CPT1α carnitine palmitoyl transferase 1a, FAD flavin adenine dinucleotide, FADH flavin adenine dinucleotide hydrogen, IDH1 isocitrate dehydrogenase 1, IDH1m mutant IDH1, IDH2 isocitrate dehydrogenase 2, IDH2m mutant IDH2, NAD nicotinamide adenine dinucleotide, NADH nicotinamide adenine dinucleotide hydrogen, NADPH nicotinamide adenine dinucleotide phosphate hydrogen, α-KG α-ketoglutarate
16.8.1 IDH1/2 Inhibitors
This role of IDH1/2 inhibition and development of IDH1/2 inhibitors are further discussed in Chap. 11.
16.8.2 Oxidative Phosphorylation Inhibitors
Leukaemic stem cells (LSCs) reply on oxidative phosphorylation (OXPHOS) for their metabolism rather than anaerobic glycolysis, which is the predominant metabolic pathway in normal HSCs [152]. Since integrity of the mitochondrial electron transport chain (ETC) is essential for OXPHOS, its inhibition can disrupt metabolic activities of LSCs. IACS-010759, an inhibitor of complex 1 of the ETC, demonstrated selective anti-leukemic activity as monotherapy and synergism with venetoclax and vinorelbine, a microtubule destabilizer, against AML cells and xenograft models while sparing normal haematopoietic cells [153,154,155]. Compared to its predecessor BAY 87–2243, IACS-010759 also has a superior safety profile [152]. It is currently being studied in r/r AML patients in a phase I trial (NCT02882321). Another ETC complex 1 inhibitor, mubritinib (TAK-165), also exhibited activity against AML cells in a preclinical study [156].
16.8.3 Fatty Acid Oxidation Inhibitors
Fatty acid oxidation (FAO) generates acetyl coenzyme A (Acetyl-CoA) for the TCA cycle, and ultimately, OXPHOS [152]. The rate limiting step in FAO is catalysed by carnitine palmitoyl transferase 1a (CPT1a), thus, inhibition of this enzyme selectively impedes metabolism of leukaemic stem cells [152]. ST1326 is a CPT1a inhibitor which induced growth arrest, mitochondrial disruption, and apoptosis in various leukaemic cell lines, with the highest activity towards AML cells [157].
16.9 Targeting the Proteasome
The proteasome is a multimeric protein complex which mediates degradation of ubiquitinated proteins (Fig. 16.9). It controls a wide range of cellular activities, including cell cycle progression and survival [158]. Aberrant activities of the proteasome contribute to leukaemogenesis through various mechanisms, such as the activation of NF-κB signalling via degradation of its regulatory protein IκBα. Inhibition of the proteasome attenuates these pathways and induces autophagy of abnormal proteins, such as FLT3-ITD [158].

Agents targeting the proteasome. BAX apoptosis regulator BAX, BID BH3 interacting-domain death agonist, IκB α inhibitor of NF-κB alpha, NAE neural precursor cell expressed, developmentally downregulated 8 activating enzyme, NEDD8 neural precursor cell expressed, developmentally downregulated 8, Ub ubiquitin, UBC12 ubiquitin-conjugating enzyme 12
16.9.1 Proteasome Inhibitors
Bortezomib inhibits the 26S subunit of proteasome complex 2 [159]. This agent has been shown to exert anti-tumour activity via stabilization of p53, p27, IκBα, pro-apoptotic proteins BID and BAX, and other signalling proteins [159]. After demonstrating anti-leukaemic activity in preclinical studies, it was tested in AML patients in a number of clinical trials as monotherapy and in combination with other agents, including chemotherapy, hypomethylating agents, and HDAC inhibitors [158, 160]. Although it was minimally effective as a single agent, its combination regimens successfully induced remissions in varying portions of patients, with the highest response rates when added to intensive chemotherapy. Although bortezomib was generally tolerable, the risks of bortezomib-related peripheral neuropathy and potentially, pulmonary toxicity, are concerning [158, 160]. Other side effects of this agent include febrile neutropenia, nausea, and gastrointestinal disturbances [158, 160]. A phase 2 trial evaluating its role as a chemo-sensitizing agent is underway (NCT04173585).
16.9.2 NAE Inhibitors
Neural precursor cell expressed, developmentally downregulated 8 (NEDD8)-activating enzyme (NAE) promotes conjugation of NEDD8 to proteins, which results in their ubiquitination by Cullin-RING E3 ubiquitin ligase (CRL) and subsequent proteasomal degradation [161, 162].
Pevonedistat (MLN4924) is a first-in-class small molecule inhibitor of NAE. In preclinical studies, it downregulated NF-κB signalling, triggered oxidative stress, and caused apoptosis in AML cells [161, 162]. In view of its synergistic action with belinostat in inducing DNA SSBs and apoptosis in AML cells [163], this combination regimen will be tested in a phase I study in r/r AML patients (NCT03772925). The combination of pevonedistat and venetoclax also showed synergism in a preclinical model and yielded promising preliminary results in a phase I/II study [164, 165], prompting other phase I to III trials regarding this regimen (NCT04172844, NCT04266795, NCT03862157). Synergism between pevonedistat and LSD1 inhibitors was also demonstrated in another murine study [166]. These optimistic results paved way to phase I and randomized phase II trials evaluating the combination of pevonedistat and azacitidine, where it was effective and provided superior survival over azacitidine monotherapy along with a favourable safety profile [167, 168]. Common side effects of this agent include fever, peripheral edema, dyspnea, febrile neutropenia, nausea, gastrointestinal disturbances, and transaminitis. Pevonedistat will be evaluated in combination with LDAC (NCT03459859), cytarabine, and idarubicin (NCT03330821), HMAs (NCT04712942, NCT04090736, NCT03009240).
16.10 Targeting Nuclear Transport
16.10.1 XPO1 Inhibitors
Exportin 1 (XPO1), or chromosome maintenance protein 1 (CRM1), is a nuclear exporter responsible for the export of substances from the nucleus [169]. Aberrant activity of XPO1 contributes to the pathogenesis of AML via shuttling tumour suppressors, such as NPM1 and p53, into the cytoplasm, which perturbs their functions [169] (Fig. 16.10). Upregulation of XPO1 is also associated with FLT3 mutations and confers inferior prognosis in AML [169].

Mechanism of actions of XPO1 inhibitors. IκB inhibitor of NF-κB, NPM1 nucleophosmin 1, p27 tumour protein 27, XPO1 exportin 1
Small molecule inhibitors of XPO1, known as selective inhibitors of nuclear export (SINE) or KPT-SINE, have diverse anti-leukaemic functions. These orally available agents irreversibly bind to the cysteine528 residue of XPO1 and alter its conformation, preventing export of tumour suppressors. They also induce differentiation via upregulation of the myeloid differentiation marker CD11b and downregulate WT and mutant FLT3 as well as c-KIT [169]. In addition, their strong activity against NPM1-mutant blasts is highlighted by a lower IC50 compared to NPM1-WT blasts [169]. An early KPT-SINE, KPT-185, demonstrated downregulation of FLT3 and induction of apoptosis in AML cell lines, while its analogue, KPT-276, prolonged survival in murine models [170].
Selinexor (KPT-330), a first generation SINE, demonstrated preclinical synergism with topoisomerase inhibitors (idarubicin, daunorubicin, mitoxantrone, etoposide), cytarabine, and sorafenib [171,172,173]. As monotherapy, Selinexor produced modest responses among patients in a phase I trial, but the subsequent randomized phase II Selinexor in Older Patients with Relapsed/Refractory AML (SOPRA) trial was terminated due to a failure of meeting the expected survival endpoint [174, 175]. Selinexor has been tested with multiple agents, including 7 + 3 induction (daunorubicin/idarubicin and cytarabine), fludarabine and cytarabine, cladribine, cytarabine, G-CSF (CLAG), high-dose cytarabine (HDAC) and mitoxantrone, and decitabine, where it induced excellent responses among patients [172, 176,177,178,179,180,181,182,183,184]. In combination with sorafenib, it also exhibited anti-leukemic efficacy in FLT3-mutant AML patients [185]. The use of selinexor as post-HSCT maintenance therapy has been explored with optimistic results in a phase I trial [186]. However, due to the CNS-penetrating properties of selinexor, its therapy is associated with dose-limiting toxicities such as cerebellar toxicity, anorexia, weight loss, and nausea [169, 174, 175]. Other major side effects include gastrointestinal disturbances, myelosuppression, and asymptomatic hyponatraemia [172, 174,175,176,177,178,179,180,181,182,183,184,185]. Preclinical studies also suggested that it may exert undesirable activity against normal haematopoietic cells [172]. Nevertheless, selinexor is currently studied as monotherapy in r/r paediatric AML, in combination with standard chemotherapy or with venetoclax in adult patients, and as post-transplant maintenance therapy (NCT02091245, NCT02403310, NCT02835222, NCT03955783, NCT02485535).
Eltanexor (KPT-8602) is a second-generation SINE with similar potency as selinexor. It is suggested to have an improved safety profile due to a lower degree of CNS penetration and reduced effect on normal haematopoiesis [187]. It exhibited potent single-agent anti-leukaemic effect and synergism with venetoclax in preclinical studies [187,188,189,190].
16.11 Targeting Epigenetic Pathways
Epigenetic regulators, such as DNMTs and HDACs, regulate transcription via controlling DNA methylation and acetylation [191] (Fig. 16.11). Aberrant activities of these pathways result in transcription of oncoproteins and/or transcriptional silencing of tumour suppressors, resulting in leukaemogenesis [191].

Agents targeting epigenetic pathways. 5hmC 5-hydroxymethylcytosine, 5mC 5-methylcytosine, Ac acetyl group, Arg arginine residue, BET bromodomain and extra-terminal domain, BRD bromodomain-containing protein 4, DNMT DNA methyltransferase, DOT1L disruptor of telomeric silencing 1-like, EED embryonic ectoderm development, EZH drosophila enhancer of zeste homolog, HDAC histone deacetylase, HMA hypomethylating agents, LSD1 lysine specific demethylase 1, Me methyl group, Me3 trimethyl group, MLL mixed-lineage leukemia, PRC2 polycomb repressive complex 2, PRMT protein arginine methyltransferase, SUZ12 suppressor of Zeste 12, TET ten-eleven-translocation
16.11.1 Hypomethylating Agents
Hypomethylating agents exert anti-leukemic activities by inhibition of DNA methyltransferases (DNMT), causing demethylation and reactivation of tumour suppressor genes [192]. Azacitidine and decitabine have been extensively studied and are widely used in AML patients. To enhance the ease of administration, an oral formulation of azacitidine (Onureg, CC-486) was developed and has recently been FDA-approved for the treatment of AML. In phase I trials, oral azacitidine demonstrated efficacy in DNA demethylation with a prolonged duration compared to subcutaneous azacitidine and a favourable safety profile, with common side effects being myelosuppression and gastrointestinal disturbances [193, 194]. In a subsequent randomized placebo-controlled phase III trial evaluating its use as maintenance therapy, oral azacitidine was significantly more effective at providing survival benefits [195, 196]. More randomized studies of oral azacitidine compared with placebos as maintenance therapies are ongoing (NCT04173533, NCT01757535).
Guadecitabine (SGI-110) is a deoxyguanosine analogue of decitabine with resistance to cytidine deaminase (CDA), thus prolonging its activity. Several trials of this agent in AML patients showed remarkable responses with tolerable toxicities, such as myelosuppression and infections [197,198,199]. However, subsequent phase III trials had disappointing results [200]. It is currently undergoing evaluation with talazoparib in r/r AML patients and with donor lymphocyte infusion (DLI) in post-HSCT patients (NCT02878785, NCT03454984, NCT02684162).
ASTX727, an oral formulation of decitabine with a cytidine deaminase inhibitor, cedazuridine, is currently compared with intravenous decitabine in a phase III randomized trial (NCT03306264). Its combinations with venetoclax, ivosidenib, enasidenib, and ASTX 660, a dual antagonist of cellular inhibitor of apoptosis protein (cIAP) 1 and X-linked inhibitor of apoptosis protein (XIAP), are also undergoing evaluation in clinical trials (NCT04657081, NCT04746235, NCT04774393, NCT04155580).
16.11.2 HDAC Inhibitors
Histone deacetylase (HDAC) and histone acetyltransferase (HATs) mediate deacetylation and acetylation of both histone and non-histone proteins. They are integral to the regulation of numerous cellular activities, such as gene transcription [201]. In AML, aberrant activation of HDAC by oncoproteins impairs the tumour suppressor function of p53, inhibits cellular differentiation, mediates aberrant signaling pathways (e.g. c-MYC), and induces abnormal proliferation [201]. Thus, the efficacies of multiple HDAC inhibitors have been studied in AML (Table 16.4) [201]. HDAC inhibitors can be classified into hydroxamines, benzamides, cyclic peptides, aliphatic acids, and electrophilic ketones according to their spectrum of activities and molecular structures [201]. Among them, vorinostat, panobinostat, and belinostat appear to be the most clinically promising. These agents are generally safe with only mild side effects, such as fatigue, nausea, and gastrointestinal disturbances.
16.11.3 LSD1 Inhibitors
Lysine specific demethylase 1 (LSD1) controls demethylation of H3K4 and can function both as a transcription activator and repressor [299]. Inhibition of LSD1 was shown to promote differentiation of AML cells [299]. Multiple agents targeting this enzyme have been studied as potential therapies for AML.
Tranylcypromine (TCP) is a selective LSD1 inhibitor which induced differentiation of AML cell lines and demonstrated synergistic effect with ATRA [300]. In a subsequent phase I/II trial, this combination was proven to be effective in AML patients [301]. This agent was tolerable, with hypotension, orthostatic dysregulation, vertigo, confusion, and cytopenias as its major adverse effects. Another trial regarding these two agents in AML is ongoing (NCT02717884).
Various analogues of TCP also demonstrated preclinical activities against AML cells [302,303,304,305,306,307,308,309,310,311,312,313]. Notably, iadademstat (ORY-1001) exhibited remarkable preclinical anti-leukemic efficacy and was effective and tolerable as monotherapy in AML patients in a phase I trial [314, 315]. A phase II trial regarding its combination with azacitidine is underway (EudraCT No.: 2018–000482-36). Another agent, GSK2879552, synergized with ATRA to exert anti-leukaemic efficacy in preclinical studies, but disappointing survival benefits from a phase I trial led to termination of the study (NCT02177812) [316]. Another LSD1 inhibitor, CC-90011, is also undergoing evaluation in combination with venetoclax and azacitidine (NCT0474884).
16.11.4 BET Inhibitors
Bromodomain and extra-terminal domain (BET) is a family of epigenetic readers responsible for regulating gene transcriptions [317]. Importantly, bromodomain-containing protein 4 (BRD4) is a member of this family which has been identified as a crucial mediator of various oncogenic pathways [299]. JQ-1 is a selective BRD4 inhibitor with potent preclinical anti-leukaemic efficacy as monotherapy and in combination with other agents, including cytarabine, ATRA, azacitidine, and ponatinib [318,319,320,321]. BI 894999 is another BRD inhibitor which also demonstrated marked single-agent anti-leukaemic activity and synergism with LDC000067, a CDK9 inhibitor in a preclinical study [322]. In addition, birabresib (OTX015/MK-8628) showed preclinical activity against AML cells as monotherapy and therapeutic synergy with either panobinostat or azacitidine [323]. It is now undergoing evaluation as monotherapy in a phase I/II trial (NCT02698189).
16.11.5 TET Inhibitors
Ten-eleven-translocation (TET) enzymes inhibit DNA methylation via oxidizing 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) [299]. In AML, mutant-TET causes hypermethylation of various gene loci, resulting in impaired differentiation and uncontrolled proliferation [299]. Ascorbic acid serves as a co-factor for TET2 to restore its normal activity and is frequently found to be deficient in AML patients. It showed anti-leukemic efficacy in preclinical studies and synergized with decitabine to prolong patient survival in a clinical trial [324,325,326]. A phase II trial of azacitidine in combination with ascorbic acid is currently underway (NCT03397173).
16.11.6 Menin-MLL Inhibitors
Mixed-lineage leukemia (MLL) is a lysine methyltransferase which methylates H3K4, while menin functions as its co-factor. MLL translocations result in generation of oncoproteins and are generally markers of poor prognosis. Small molecule inhibitors with preclinical efficacies against MLL complexes include MM-401, MI-503, MI-463, and MIV-6R [327,328,329,330]. Strikingly, these agents also exhibited potent activity against NPM1-mutant AML cell lines, possibly due to the reliance of mutant NPM1 on Menin-MLL1 interactions for its aberrant gene expression [331]. In particular, MI-503 and MI-3454 selectively targeted MLL1-rearranged and NPM1-mutant cells and prolonged survival in murine models [331, 332].
16.11.7 DOT1L Inhibitors
Disruptor of telomeric silencing 1-like (DOT1L) is a histone methyltransferase mediating the methylation of H3K79 [299]. Since its function is integral to the oncogenic activities of MLL fusion complexes, it can be used as a potential target against MLL-rearranged AML [299]. Pinometostat (EPZ5676) is a DOT1L inhibitor with remarkable preclinical efficacy against MLL-rearranged cell lines and showed modest single-agent clinical activity along with a favourable safety profile [333,334,335]. It is currently being tested in combination with standard chemotherapy in MLL-rearranged AML patients (NCT03724084). SYC-522 is another agent with preclinical efficacy against MLL-rearranged AML [336].
16.11.8 EZH Inhibitors
Drosophila enhancer of zeste homolog (EZH) is a subunit of polycomb repressive complex (PRC) 2, which regulates trimethylation of H3K27 and mediates gene transcription [299]. Interestingly, they can function both as a tumour suppressor and oncoprotein in AML [299]. The selective EZH2 inhibitor 3-Deazaneplanocin A (DZNep) and its analogue D9 both showed efficacy against MLL-rearranged AML cells [337,338,339]. UNC1999 is a dual inhibitor of EZH1 and 2 with preclinical efficacy against AML models with MLL gene rearrangement [340]. Finally, valemetostat (DS-3201) is another dual EZH1/2 inhibitor currently evaluated as monotherapy in a phase I trial (NCT03110354).
16.11.9 PRMT Inhibitors
Protein arginine methyltransferases (PRMTs) are mediators of arginine methylation of histone as well as non-histone proteins and their overexpression is frequently found in AML [299]. AMI-408 is a specific inhibitor of PRMT1 with growth suppressive effect on AML cell lines and murine models [341]. ERZ015666, an inhibitor of PRMT5, induced differentiation of AML cells and showed efficacy in murine models with MLL rearrangements [342]. GSK3326595 is another PRMT5 inhibitor currently undergoing evaluation in combination with azacitidine in a phase I trial (NCT03614728).
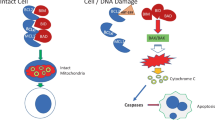
16.12 Targeting DNA Damage Response Pathways
DNA damage response (DDR) is essential for the maintenance of genomic stability via halting cell cycle progression for DNA repair [343]. In the case of substantial DNA damage beyond repair, the apoptotic cascade would be initiated [343]. Studies have shown that AML cells have defective DDR mechanisms and are thus more susceptible to combined inhibition of chemical and DDR pathways [344]. Importantly, IDH-mutant AML is proposed to be sensitive to further inhibition of DDR due to their intrinsic defects in homologous recombination (HR).
16.12.1 PARP Inhibitors
Poly (ADP-ribose) polymerases (PARP) are a superfamily of 18 enzymes responsible for DNA single strand breaks (SSBs) repair and survival of cells with DNA damage [345]. Some subtypes of AML, such as those with IDH1/2 and FLT3 mutations, are proposed to be more sensitive to the effects of PARP inhibitors [345]. PARP inhibitors are nicotinamide analogues which function via the inhibition of DNA SSB repair by PARP and induction of cytotoxic allosteric effects by trapping PARPs to damaged DNA [345, 346].
Olaparib is a potent and selective PARP inhibitor which showed excellent potency against AML cell lines and synergistic activity with two anti-CD33 antibody drug conjugates, GO and IMGN779, in preclinical studies [347, 348]. Olaparib is in a trial as monotherapy for r/r IDH-mutant AML (NCT03953898).
Other PARP inhibitors with promising preclinical activities against AML include veliparib, talazoparib (BMN-673), niraparib, rucaparib, and PJ34 [345, 349, 350]. These agents showed synergistic activity against AML cell lines in combination with IMGN632 (anti-CD123 antibody drug conjugate), MS275 (HDAC inhibitor), entinostat (MS275, HDAC inhibitor), and AZD1775 in preclinical studies [351,352,353,354]. Among them, results of veliparib as single agent or in combination with temazolomide (alkylating agent) or topotecan and carboplatin in r/r ALM patients were impressive [355, 356]. Two trials regarding the use of these two combinations in AML patients are ongoing (NCT00588991, NCT01139970). Talazoparib also demonstrated potent efficacy against IDH1-mutant AML cells [357]. Trials of talazoparib as monotherapy and in combination with decitabine are currently underway (NCT03974217, NCT02878785).
16.12.2 ATR Inhibitors
Ataxia telangiectasia and Rad3-related kinase (ATR) is responsible for detecting DNA SSBs. It subsequently activates downstream repair pathways or apoptotic cascades depending on the extent of DNA damage [343]. VX-970 and AZ20 are two ATR inhibitors which demonstrated single agent efficacy against AML cell lines [358, 359]. AZ20 also synergistically induced anti-leukemic activity with cytarabine in another preclinical study [360].
16.12.3 ATM Inhibitor
The function of ataxia telangiectasia mutated kinase (ATM) resembles that of ATR except for its detection of double strand breaks (DDBs) instead of SSBs in DNA [343]. AZD0156, an ATM inhibitor, prolonged survival of MLL-rearranged mice in a preclinical study [359]. In another study, KU-59403 also induced apoptosis in AML cell lines [361].
16.12.4 CHK Inhibitors
Checkpoint kinase (CHK) 1 and 2 inhibit CDK 1 and 2 and cause cell cycle arrest upon activation by ATR and ATM [343]. Their overexpression in AML is associated with inferior prognosis [362]. Prexasertib (LY2606368), MK-8776 (SCH900776), and rabusertib (LY2603618) are CHK inhibitors which synergistically induced apoptosis in combination with CPX-351 in TP53-WT and TP53-deleted AML cells [363]. Rabusertib also exhibited synergism with venetoclax against AML cells [364]. MK-8776 demonstrated activity at overcoming chemotherapeutic resistance and synergized with cytarabine and vorinostat [362, 365, 366]. However, the combination of MK-8776 with cytarabine did not provide survival benefit over single agent cytarabine in r/r AML patients in a subsequent trial [367]. A phase I trial of prexasertib in combination with cytarabine and fludarabine is underway (NCT02649764).
16.12.5 WEE1 Inhibitors
Wee1-like protein kinase (WEE1) is activated by CHK and induces cell cycle arrest by inhibition of CDK1 and 2 [343]. Adavosertib (AZD1775, MK-1775) exhibited synergism with panobinostat and olaparib, respectively, in AML cell lines [224, 354]. It also synergistically overcame cytarabine-resistance when combined with cytarabine in leukemic cells [368]. Unfortunately, a trial of adavosertib as monotherapy was terminated due to safety concerns and another trial of its combination with belinostat was terminated for unspecified reasons (NCT03718143, NCT02381548).
16.13 Targeting the Cell Cycle
The cell cycle is a 4-phased process and progression through each phase is under strict regulation by several mediators, including cyclin-dependent kinases (CDKs) and cell cycle checkpoints. Aberrant progression of the cell cycle results in uncontrolled proliferation and leukaemogenesis [151].
16.13.1 CDK Inhibitors
Cyclin-dependent kinases (CDKs) are regulators of cell cycle progression which are activated upon binding of cyclins. Among them, transcriptional CDKs (CDK7, 8, 9) are mainly responsible for regulating transcription (Fig. 16.12). Inhibition of CDKs can halt the cell cycle and inhibit aberrant gene expression, giving rise to anti-leukemic effects. Information regarding various CDK inhibitors is summarized in Table 16.5. Among them, palbociblib and alvocidib are the most widely studied in AML. These agents have an excellent safety profile with myelosuppression as a significant side effect.

Agents targeting DNA damage responses and cell cycle. ATM ataxia telangiectasia mutated kinase, ATR ataxia telangiectasia and Rad3-related kinase, AURK aurora kinase, CDC25A cell division cycle 25 A, CDC25C cell division cycle 25 C, CDK1 cyclin-dependent kinase 1, CDK2 cyclin-dependent kinase 2, CDK4/6 cyclin-dependent kinase 4/6, CHK1 checkpoint kinase 1, CHK2 checkpoint kinase 2, DSB double strand breaks, P phosphate group, PARP poly (ADP-ribose) polymerases, PLK1 polo-like kinases 1, RSK p90 ribosomal S6 kinase, SSB single strand breaks, WEE1 Wee1-like protein kinase
16.13.2 Aurora Kinase Inhibitors
The aurora kinase (AURK) family consists of three members, AURK A, B, and C. These enzymes are responsible for entry into the M phase and normal progression of mitosis [393]. In AML, their overexpression has been observed and is associated with poor-risk cytogenetics. Alisertib (MLN8237) is an AURKA inhibitor with oral bioavailability. Investigational use of this agent in a phase II study in combination with induction chemotherapy among poor-risk AML patients illustrated its clinical effectiveness and safety [394]. Barasertib (AZD1152) is an AURKB inhibitor which demonstrated anti-leukaemic efficacy along with a less desirable safety profile in a phase I/II study in AML patients, with major side effects being febrile neutropenia and oral mucositis [395]. In another trial, it was tested in combination with LDAC and showed favourable outcomes and tolerability [396]. A trial of barasertib as monotherapy or in combination with venetoclax and/or azacitidine is underway (NCT03217838).
16.13.3 PLK Inhibitors
Polo-like kinases (PLKs) promote cell cycle progression via inducing degradation of WEE1 and activating CDK1 [397, 398]. It also inhibits apoptosis by activating Bcl-xL [398]. In AML, its overexpression is frequently observed [399].
Rigosertib (ON01910) is a dual inhibitor of PLK1 and PI3K. In addition to exhibiting preclinical anti-leukemic efficacy, it was effective and tolerable as monotherapy and in combination with azacitidine in clinical trials [400,401,402]. Major adverse events were gastrointestinal disturbances, myelosuppression, and pneumonia. A phase II study of oral rigosertib in combination with azacitidine is underway (NCT01926587).
Volasertib (BI6727) is a selective PLK1/2/3 inhibitor. Encouraging results from preclinical studies in AML models paved way for subsequent clinical trials in AML patients [403]. In summary, volasertib was safe and effective as monotherapy and demonstrated synergism in combination with LDAC and decitabine, respectively, among AML patients in phase I and II trials [404,405,406,407]. However, responses of its combination with LDAC did not meet expectations in the randomized phase III POLO-AML-2 trial [408]. Significant side effects of volasertib include myelosuppression and fatigue. It is currently undergoing evaluation as monotherapy or in combination with cytarabine in several trials (NCT00804856, NCT01721876). Another oral PLK1 inhibitor, onvansertib (NMS-1286937), demonstrated impressive efficacy and safety in combination with decitabine, but limited activity with LDAC in a phase Ib study [409].
BI2536 also exhibited anti-leukemic effect in a preclinical study and had modest single agent activity in AML as reported in a phase I/Ib trial [410,411,412]. Other PLK1 inhibitors with preclinical efficacies against AML include TAK-960 and NMS-P937 [413, 414].
16.13.4 CDC25 Inhibitors
Cell division cycle 25 (CDC25) is a protein phosphatase which modulates cell cycle progression via dephosphorylation of CDKs [415]. In a preclinical study, several CDC25 inhibitors, namely NSC95397, ALX1, ALX2, ALX3, and ALX4, inhibited proliferation of AML cells, but did not demonstrate cytotoxic effects [415].
16.13.5 RSK Inhibitor
p90 Ribosomal S6 Kinase (RSK) is a downstream mediator of the Ras/MAPK/ERK pathway and controls a wide range of cellular pathways, including the promotion of cell cycle progression via activation of CDC25 and CDK1 [416]. In AML, upregulation of RSK has been discovered in patient samples and is indicative of poor prognosis. BI-D1870 is an RSK inhibitor which exerts potent anti-leukemic activity via S phase cell cycle arrest, impeding mitotic exit, and induction of DNA damage [416, 417]. It was effective as monotherapy and showed synergism with vincristine in AML cell lines [416, 417].
16.14 Targeting the Bone Marrow Microenvironment
The bone marrow microenvironment (BMM) plays crucial roles for the normal development of HSCs and other haematopoietic cells. In AML, the complex interactions between leukaemic cells and the BMM are integral to their development and disease progression [418]. With overwhelming evidence suggesting the substantial abnormalities in the BMM of AML patients, multiple therapeutic strategies to target these aberrant pathways are being explored (Fig. 16.13) [418].

Agents targeting the bone marrow microenvironment. CD44 cluster of differentiation 44, CXCR4 C-X-C chemokine receptor type 4, SDF1 stromal-derived factor 1
16.14.1 SDF1/CXCR4 Inhibitors
C-X-C chemokine receptor (CXCR) type 4 is a HSC surface G-protein-coupled chemokine receptor for stromal-derived factor 1 (SDF1), also known as CXCR12, which is produced by mesenchymal stromal cells. Their interactions promote survival, quiescence, and marrow homing of HSCs. Leukaemic cells exploit this mechanism by upregulating their expressions of CXCR4, which grants them chemoresistance due to protection by marrow stromal cells.
Plerixafor (AMD3100) is a small molecule inhibitor of CXCR4 commonly used as an off-label stem cell mobilizing agent. Promising results from preclinical studies prompted several trials of plerixafor in combination with chemotherapies [419, 420], decitabine [421], as well as with G-CSF with or without sorafenib [422]. These studies all showed impressive survival outcomes and demonstrated the remarkable potential of plerixafor as a chemo-sensitizing and AML blast mobilizing agent. It will be tested as a chemo-sensitizing agent prior to pre-transplant conditioning in a phase II trial (NCT02605460).
16.14.2 E-Selectin Inhibitors
E-selectins are molecules expressed by vascular endothelial cells which mediate cellular adhesion. Leukaemic cells express CD44, the ligand for E-selectins, to promote their engraftment in the bone marrow. Uproleselan (GMI-1271) is an inhibitor of e-selectin which showed preclinical efficacy in overcoming chemoresistance and synergism with chemotherapeutic agents [423]. Its use in several clinical trials in combination with chemotherapy yielded profound response rates, excellent tolerability, and even reduction in risks of mucositis [424,425,426]. Phase III trials evaluating comparing responses to chemotherapy with or without uproleselan are underway (NCT03616470, NCT03701308).
16.15 Immunotherapy
Immunotherapy represents a new era of therapies in AML and has been intensively studied in recent years. Broadly, these strategies can be classified into antibody-based or T/NK-cell-based depending on their mechanism of actions. The former involves targeting cell surface antigens of leukemic cells, while the latter relies on activation of immune responses against leukaemic cells. Compared to conventional chemotherapy, they are generally more tolerable due to reduced toxicity on normal cells.
16.15.1 Antibody-Based Immunotherapies
16.15.1.1 Antibody-Drug Conjugates
Antibody-drug conjugates (ADJs) are synthesized via the conjugation of cytotoxic agents to antibodies against various cell surface antigens of AML cells or LSCs. Upon cell surface receptor binding, they are endocytosed and release their cytotoxic moieties to induce leukaemic cell death (Fig. 16.14).

Mechanism of actions of antibody-based immunotherapies. 131I Iodine-131, 225Ac Actinium-225, 90Yt Yttrium-90, CD123 cluster of differentiation 123, CD33 cluster of differentiation 33, CD45 cluster of differentiation 45, DNA deoxyribonucleic acid
16.15.1.1.1 Anti-CD33 ADJs
CD3 is expressed primarily on LSCs and not in normal haematopoietic cells [152]. Thus, targeting this cell surface antigen allows selective eradiation of LSC while sparing normal haematopoietic cells [152]. Gemtuzumab ozogamicin (GO; Mylotarg) is an FDA-approved anti-CD33 ADJ with the cytotoxic agent calicheamicin as a conjugate. GO was first FDA-approved for the treatment of AML in 2000, but was withdrawn in 2010 in view of its non-superior survival benefit compared to standard 7 + 3 induction and high risks of toxicities, such as veno-occlusive diseases (VODs), and hepatotoxicity [427]. Despite these discouraging events, GO was continually studied at fractionated and lower doses with optimistic results. Notably, the phase III randomized ALFA-0701 study showed that the addition of GO to standard induction chemotherapy provided marked survival benefits with only a slight increase in risks of VODs [428, 429]. Another randomized phase III trial (AML-19) also reported that GO improved patient survival to a larger extent than best supportive care [430]. Following these encouraging results, GO was re-approved by the FDA for newly diagnosed and relapsed AML patients. However, it should be noted that subsequent controlled trials still reported higher risks of VODs and early mortality with GO therapy than in control groups [431]. Apart from VODs, other toxicities of GO include haemorrhage, infections, gastrointestinal disturbances, febrile neutropenia, and myelosuppression.
Further trials of GO include its use as monotherapy (NCT03737955) or in combination with pracinostat (NCT03848754) and venetoclax (NCT04070768); talozoparib (NCT04207190); OX40, venetoclax, avelumab, glasdegib, and azacitidine (NCT03390296); mitoxantrone and etoposide (NCT03839446), CPX-351 (NCT03904251, NCT03878927, NCT03672539), midostaurin and standard induction therapy (NCT03900949, NCT04385290), CLAG (NCT04050280), CLAG, and mitoxantrone (CLAG-M) (NCT03531918); fludrabine, cytarabine, G-CSF, idarubicin (NCT00801489); cytarabine, daunorubicin, erwinase, and etoposide (NCT04326439). It will also be tested as induction therapy followed by glasdegib (NCT04093505) or non-engraftment donor leukocyte infusion (NCT03374332),
Vadastuximab talirine (SGN33A) is another anti-CD33 ADJ linked to a pyrrolobenzodiazepine dimer. In preclinical studies, it exhibited remarkable anti-leukaemic activity in a diverse panel of cell lines, including those with TP53-mutant and multi-drug-resistant phenotypes [432]. It induced remarkable responses as monotherapy in AML patients in a number of trials, with some achieving MRD negativity [433, 434]. Although it also showed potent efficacy and induced MRD-negativity in combination with azacitidine in a phase I trial [435, 436], the subsequent randomized-phase III CASCADE trial was discontinued due to increased mortality in the experimental arm [437]. Treatment-related deaths were attributed to severe infections rather than VODs [437].
IMGN779 also targets CD33 and is conjugated to DGN462, an alkylating agent. It showed anti-leukaemic activity against multiple AML cell lines and murine models, with the highest activity in cells harbouring FLT3-ITD [438, 439]. Its use in a phase I trial yielded impressive response rates and tolerability [440].
16.15.1.1.2 Anti-CD123 ADJs
CD123 functions as an interleukin (IL)-3 receptor and mediates downstream proliferation induced by IL-3 [152]. It was also found to be highly expressed on LSCs and is an attractive target for eliminating leukaemic colony forming activities [152].
Tagraxofusp (SL-401) consists of an anti-CD33 antibody conjugated to part of the diphteria toxin [152]. It will be evaluated as monotherapy or in combination with venetoclax with or without azacitidine in AML patients (NCT04342962, NCT03113643).
IMGN632 is conjugated to DNA mono-alkylating portion of the indolinobenzodiazepine pseudodimer. It showed encouraging activity and favourable safety profile in a phase I trial [441]. Since it demonstrated synergism with azacitidine and venetoclax in a preclinical study [442], its combination with venetoclax, azacitidine, or both agents will be tested in a phase I/II trial (NCT04086264). Its use as monotherapy in AML patients will also be tested (NCT03386513).
16.15.1.2 Radioimmunotherapy
Radioimmunotherapy (RIT) involves the use of monoclonal antibodies linked with radionuclides, which then bind to leukaemic cell surface antigens and continually release ionizing radiation, resulting in selective anti-leukaemic effects (Fig. 16.14) [443]. In AML, RITs mainly utilize Iodine(I)-131 and Yttrium(Yt)-90 and are usually studied as pre-transplant conditioning regimens.
131I-anti-CD45 RIT markedly improve post-transplant outcomes in various clinical trials in combination with various conditioning regimens, including total body irradiation (TBI), busulfan and cyclophosphamide, as well as fludarabine and low-dose TBI [444,445,446,447]. The use of 90Y-anti-CD45 RIT also resulted in remarkable survival outcomes and prolonged donor engraftment [448, 449].
While the above two agents emit β-radiation, 225Actinium(Ac)-lintuzumab (225Ac-anti-CD33) emits short-ranged α-radiation. Although it induced blast reduction in patients, no remissions were seen in a phase I trial combining this agent with LDAC in newly diagnosed patients [450]. 225Ac-lintuzumab and 211Astatine(At)-anti-CD45 will undergo further testing either as therapy for r/r patients or as part of conditioning regimens in multiple clinical trials (NCT03867682, NCT03441048, NCT03670966, NCT03128034).
16.15.2 T-Cell-Based Immunotherapies
T cells are integral to the normal functioning of the adaptive immune system. In particular, cytotoxic T cells are responsible for the elimination of cells carrying abnormal antigens, including leukaemic cells. However, these activities often impaired in AML, giving rise to immune evasion of leukaemic blasts. Thus, intensifying the anti-tumour responses of T cells is an attractive strategy against AML.
16.15.2.1 Immune-Related Adverse Events
Although immune-cell-based immunotherapies are generally considered to be more tolerable than conventional therapies, their resulting alterations in immune responses cause a distinct group of side effects termed “immune-related adverse events”. They can present in a multitude of ways, including as skin rash, pneumonitis, and colitis, among others [451]. Fortunately, the majority of these events are tolerable and not lethal. In addition, cytokine release syndrome (CRS) is especially common with the use of multivalent antibodies and CAR-T, with presentations ranging from mild flu-like symptoms to severe multi-organ failures and encephalopathy [452]. Cautious monitoring and proper management of CRS are keys to preventing significant morbidities and mortality.
16.15.2.2 Immune Checkpoint Inhibitors
Immune checkpoints (ICs) inhibit aberrant T-cell responses against normal body cells and are paramount to self-tolerance. However, leukemic cells can also express checkpoint ligands, which cause anergy of T-cells upon their binding, resulting in immune evasion and uncontrolled proliferation. Therefore, inhibition of these pathways allows reactivation of immune responses against leukemic cells (Fig. 16.15).

Mechanisms of actions of immune checkpoint inhibitors and OX40 agonists. CD47 cluster of differentiation 47, CD80/86 cluster of differentiation 80 or 86, CTLA4 cytotoxic T-lymphocyte-associated protein 4, MHC I major histocompatibility complex class I, OX40 tumor necrosis factor receptor superfamily, member 4, PD-1 programmed death 1, PD-L1 programmed death ligand 1, SIRPα signal regulatory protein alpha, TIM3 T cell immunoglobulin and mucin domain-containing protein 3
16.15.2.2.1 PD-1/PD-L1 Inhibitors
Programmed death 1 (PD-1) is expressed on the surface of T cells while its ligand, Programmed death ligand 1 (PD-L1), is expressed on leukaemic cells [453]. Nivolumab, an anti-PD-1 antibody, induced impressive responses in combination with azacitidine in older r/r AML patients in a phase II trial [454]. Addition of ipilimumab to this regimen further improved survival outcomes at the cost of increased toxicities and immune-related adverse events [455]. The above study is still currently ongoing (NCT02397720). Another trial of nivolumab with cytarabine or idarubicin showed remarkable remission rates with measurable residual disease (MRD) negativity in more than half of the cohort [451]. Nivolumab is generally tolerable with mostly immune-related adverse events, such as skin rash, transaminitis, and nephritis. However, another trial of nivolumab as post-transplant therapy demonstrated minimal efficacy and unacceptable adverse effects [456]. In view of these encouraging results, it will be further evaluated in multiple trials, including combination with azacitidine in r/r paediatric AML patients, as monotherapy in post-transplant relapsed patients, and as post-chemotherapy or post-HSCT maintenance as monotherapy or in combination with ipilimumab (NCT03825367, NCT01822509, NCT02275533, NCT02532231, NCT03600155, NCT02846376). Its combination with NY-ESO-1 vaccination and decitabine will also be tested in a clinical trial (NCT03358719).
Pembrolizumab is another anti-PD-1 antibody which has been tested in combination with azacitidine, decitabine, and following HDAC in AML patients; their respective trials all showed optimistic outcomes and tolerable adverse effects which were mostly immune-related [457,458,459]. Further trials will evaluate this agent in combination with decitabine, galinpepimut-S, azacitidine, venetoclax, with intensive chemotherapy as frontline therapy, with azacitidine in NPM1-mutant AML patients with molecular relapse, and as monotherapy in patients with post-HSCT relapse (NCT03969446, NCT03761914, NCT04284787, NCT04284787, NCT03769532, NCT02981914, NCT03286114).
Avelumab, an anti-PD-L1 mAb, showed anti-leukaemic efficacy and tolerability in combination with decitabine in a phase I trial and is currently involved in a phase I/II trial with venetoclax, PF-04518600, glasdegib, GO, and azacitidine (NCT03390296) [460].
However, durvalumab (MEDI-4736), another anti-PD-L1 antibody, did not provide additional survival benefits when added to azacitidine in a randomized phase II study [461].
16.15.2.2.2 CTLA-4 Inhibitors
Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is expressed on the surface of T cells. It competitively binds to CD80 or CD86 expressed by leukaemic cells with a higher affinity than CD28 [453], inducing T cell anergy. Ipilimumab is an anti-CTLA-4 antibody which induced responses in AML patients who experienced relapse after HSCT, with graft-versus-host disease (GVHD) as the dose limiting toxicity in some patients [462]. It is undergoing evaluation in combination with decitabine, as monotherapy in patients with post-transplant relapse, as post-HSCT maintenance either as monotherapy, in combination with nivolumab, or in combination with donor lymphocyte infusion (NCT02890329, NCT01822509, NCT03600155, NCT02846376, NCT03912064).
16.15.2.2.3 TIM-3 Inhibitors
T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3) receptors on T cells are activated by the binding of galectin-9 on leukemic cell surface [453]. Sabatolimab (MBG453) is an anti-TIM3 antibody. In a phase Ib trial, its combination with either azacitidine or decitabine showed promising anti-leukaemic activity [463]. This agent was tolerable, with myelosuppression and immune-related adverse effects being major side effects. Trials will further explore its combination with azacitidine and venetoclax, HDM201, and decitabine (NCT04150029, NCT03940352, NCT03066648). Its use in MRD-positive post-transplant patients will also be tested (NCT04623216).
16.15.2.2.4 CD47 Inhibitors
CD47 functions as an immune checkpoint by binding to Signal regulatory protein alpha (SIRPα) receptors on macrophage and preventing phagocytosis of CD47-positive cells [464]. The anti-CD47 mAb magrolimab is currently undergoing clinical evaluation in combination with azacitidine with optimistic preliminary results from a phase Ib trial [465]. It has been granted fast track designation by the FDA and will be tested in combination with azacitidine and venetoclax in a phase I/II trial (NCT04435691). A phase III trial comparing magrolimab combined with azacitidine against standard therapy is also underway (NCT04778397).
16.15.2.3 Targeting Co-Stimulatory Pathways
16.15.2.3.1 OX40 Agonists
OX40 is a cell surface receptor predominantly expressed by activated T cells, while its ligand, OX40L, is widely expressed by activated antigen presenting cells. The binding of OX40L to OX40 provides a co-stimulatory signal necessary for further T cell activation, clonal expansion, and anti-leukaemic immune responses [466] (Fig. 16.15). (PF-8600) is an anti-OX40 agonist monoclonal antibody currently investigated in combination with venetoclax, avelumab, glasdegib, GO, and azacitidine in a phase I/II trial (NCT03390296).
16.15.2.4 Multivalent Antibody Therapies
Multivalent antibody therapies facilitate interactions between immune cells and leukaemic cells. These recombinant antibodies are constructed by the combination of antibodies targeting these two types of cells and thus carry specificity against multiple antigens. This allows them to bring immune cells to the proximity of leukaemic cells for exerting anti-tumour effects (Fig. 16.16). Broadly, they can be divided into non-IgG-like and IgG-like, where only IgG-like multivalent antibodies retain the Fc region to promote additional immune pathways, such as antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) [467]. Currently, bivalent antibodies are the most widely studied in AML.

Mechanism of actions of multivalent antibodies. BiTE bispecific T cell engager, CD3 cluster of differentiation 3, CD123 cluster of differentiation 123, DART dual-affinity retargeting antibodies, Fab antigen-binding fragment, Fc fragment crystallizable, FcRn neonatal Fc receptor, NK natural killer, scFv single chain variable fragment
16.15.2.4.1 Non-IgG like Multivalent Antibodies
16.15.2.4.1.1 BiTE
Bispecific T-cell engagers (BiTEs) contain both the heavy and light chain variable domains (VH and VL) of the single chain variable fragments (scFv) from two antibodies targeting T cells (e.g. CD3) and leukaemic cells (e.g. CD33, CD123), respectively.
AMG330 is an anti-CD33 x anti-CD3 BiTE with encouraging preclinical efficacy and is now currently evaluated in a trial with r/r or MRD-positive AML patients (NCT02520427) [468, 469]. AMG673 is another anti-CD33 x anti-CD3 BiTE. Preliminary results from an ongoing first-in-human phase I study revealed anti-leukaemic efficacy (NCT03224819) [470]. Manageable adverse effects, such as CRS and myelosuppression, were observed. AMV564 has the same antigen specificity as the above BiTEs, but is tetravalent, i.e. it contains two VH and VL chains from each types of antibody, which further promotes target binding. Favourable preclinical data promoted a phase I trial, which showed promising preliminary results (NCT03144245) [471, 472].
16.15.2.4.1.2 Dart
Dual-affinity retargeting (DART) antibodies are similar in principle to BiTEs, except the VH and VL chains from the two antibodies are cross-linked to further increase efficiency [467]. Flotetuzumab (MGD-006) is anti-CD3 x anti-CD123 DART which induced T-cell activation against CD123-positive leukaemic blasts in preclinical studies. Preliminary results from the subsequent in-human trial showed potent anti-leukaemic activity and manageable side effects, which mainly included CRS [473].
16.15.2.4.2 IgG-Like Multivalent Antibodies
XmAb14045 is an anti-CD123 x anti-CD3 multivalent antibody. Its long serum half-life of 6.2 days can be attributed to the binding of its bispecific Fc domain to neonatal Fc receptor (FcRn), which prevents its degradation [452, 467]. This agent demonstrated preclinical anti-leukaemic efficacy and induced T-cell activation. Although a phase 1 study in AML patients was suspended following occurrences of patient mortalities and major toxicities, including CRS and pulmonary edema, the partial suspension was shortly lifted by the FDA [474, 475]. A subsequent phase I study reported encouraging efficacy with manageable adverse events, such as CRS (NCT02730312) [476, 477]. The clinical activity and tolerability of XmAb14045 will be further elucidated in another trial (NCT02730312).
16.15.2.5 Chimeric Antigen Receptor T Cells Therapy
Chimeric antigen receptor T (CAR-T) cells therapy involves the use of genetically engineered T cells expressing chimeric antigen receptors (CAR) against leukaemic cell surface antigens. After infusion of CAR-T cells, binding of CAR to leukaemic cells triggers cytotoxic responses and leukaemic cell death (Fig. 16.17) [427].

Mechanism of action of chimeric antigen receptor (CAR) T cells. NKG2D natural killer group 2D, NKG2D-L NKG2D ligand
CYAD-01 is a CAR-T cell product expressing the natural killer group 2D (NKG2D) fused to a CD3ζ signalling domain. NKG2D is normally expressed by natural killer cells, CD8+ T cells and NK-T cells. It is activated upon binding to NKG2D ligand (NKG2D-L) expressed by leukaemic cells, while a co-stimulatory signal is provided by DNAX-activating protein 10 (DAP10) [478]. Astonishingly, CYAD-01 is capable of inducing a co-stimulatory signal via DAP10-independent pathways. In a phase I trial, it was determined to possess anti-leukaemic activity. However, frequent adverse effects, including CRS and pneumonitis, were observed [479]. Another phase I trial with the use of NKG2D CAR-Tx cells in AML patients is underway (NCT04658004).
CAR-T cell products can also be engineered to target more than one leukaemic antigens to further improve potency. Notably, a compound CAR-T (cCAR-T) product constructed to target C-type lectin-like molecule-1 (CLL1) and CD33 was evaluated in a phase I study with remarkable results, with seven out of nine patients achieving remission and MRD-negativity [480, 481].
Other CAR-T products with preclinical successes in AML include c-Kit-targeting and FLT3 scFv-targeting CAR-Ts [482, 483]. CAR-T is undergoing intensive studies in numerous clinical trials, including novel CAR-T strategies such as donor-derived CAR-T (NCT04766840), CD123/CLL1 CAR-T (NCT03631576), CD38-targeted CAR-T (NCT04351022), and IL3 CAR-T (NCT04599543), among many others.
16.15.3 NK Cell-Based Immunotherapies
NK cells are paramount effectors of anti-tumour immune responses. After binding of an antibody to a cell surface antigen, the Fc receptors of NK cells bind to the Fc region of the antibody, resulting in activation of NK cells and release of cytotoxic materials for target cell killing. This process is known as antigen-dependent cellular cytotoxicity (ADCC). NK cell-based immunotherapies aim at harnessing the cytotoxic activity of intrinsic or foreign NK cells against leukaemic cells (Fig. 16.18).

Summary of NK cell-based immunotherapies. CAR chimeric antigen receptor, NK natural killer
16.15.3.1 Unconjugated Antibodies
Unlike ADJs, unconjugated antibodies exert cytotoxicity by stimulating ADCC as well as CDC activated by their Fc domains. Daratumumab is an anti-CD38 mAb which demonstrated anti-leukaemic activity against AML cell lines via induction of ADCC and CDC [484]. Interestingly, it also targets leukaemic blasts via perturbing cellular metabolism [485]. It is currently studied as monotherapy and in combination with FT538 (NCT04714372, NCT03067571). A study of daratumumab in combination with DLI for patients who relapsed post-HSCT is also underway (NCT03537599).
Isatuximab is another anti-CD38 mAb with potent anti-leukaemic activity in a preclinical study [486]. It will undergo further evaluation in combination with chemotherapy in r/r paediatric AML patients (NCT03860844).
Talacotuzumab (CSL362) is a CD123 which demonstrated high potency against CD123 in preclinical studies [487]. However, its uses as monotherapy or in combination with decitabine were only minimally effective in multiple clinical trials and caused high incidences of treatment termination [488, 489].
16.15.3.2 CAR-NK Cells Therapy
CAR-NK cells are engineered to express receptors which enhance ADCC and are administered in conjunction to unconjugated antibodies [490]. FT538 is a CAR-NK cell product expressing an IL-5 receptor alpha fusion protein and high affinity non-cleavable CD16 [490]. It demonstrated promising preclinical efficacy against multiple myeloma cells and will undergo further testing in a phase I trial with daratumumab in r/r AML patients (NCT03067571) [491].
16.15.4 Vaccination
Vaccination of tumour-associated antigens is a strategy created to induce antigen presentation of dendritic cells to T cells, which then generate anti-leukaemic immune responses and prolonged immunological memory against leukaemic cells (Fig. 16.19) [427]. They can potentially prevent future relapses by inducing eradication of all remaining abnormal blasts in the haematopoietic system. Given the dismal prognosis of r/r AML, their development carries substantial significance for patients.

Mechanism of action of vaccination for the treatment of AML. DC dendritic cell, WT-1 Wilm’s tumour 1
Wilm’s tumour 1 (WT-1) antigens are attractive targets for peptide vaccination due to their high expression in leukaemic cells [427]. Galinpepimut-S and OCV-501 are examples of WT-1 vaccines which showed potency at inducing immunological responses and improving survival in AML patients in CR1/2 in phase I or II trials [492,493,494]. The combination of galinpepimut-S with pembrolizumab is currently under evaluation (NCT03761914). Ombipepimut-S (DSP-7888) is another WT-1 peptide vaccine evaluated in AML (NCT04747002).
Owing to evidence suggesting that allogeneic dendritic cells (DCs) induce stronger immune responses compared to autologous ones, an allogeneic DC vaccine, DCP-001, was manufactured and examined in a phase I trial with optimistic outcomes and tolerability [495]. This vaccine is under clinical investigation in a phase II trial among AML patients in remission (NCT03697707).
In addition to the above agents, NY-SEO-1 vaccination is currently studied in a phase I trial in combination with decitabine and nivolumab (NCT03358719). This vaccine formulation comprises three components: (1) a mAb against DEC-2015 (CD205), a dendritic cell surface receptor which promotes antigen presentation; (2) NY-SEO-1, a leukaemic cell surface antigen; and (3) polyinosinic-polycytidylic acid complexed with poly-L-lysine and carboxymethylcellulose (Poly-ICLC), a double-stranded mRNA complex which serves as an immune stimulant [496, 497]. No preliminary results are available at the moment.
16.16 Conclusion
Given the plethora of aberrant pathways in AML, the aforementioned novel strategies only provide a glimpse of the endless therapeutic options against this aggressive haematological malignancy. Although the current prognosis of AML remains suboptimal, intensive efforts on the development of novel agents may soon bring about unprecedented pharmacological breakthroughs.
References
Alfayez M, Kantarjian H, Kadia T, Ravandi-Kashani F, Daver N. CPX-351 (vyxeos) in AML. Leuk Lymphoma. 2020;61(2):288–97.
Feldman EJ, Lancet JE, Kolitz JE, Ritchie EK, Roboz GJ, List AF, et al. First-in-man study of CPX-351: a liposomal carrier containing cytarabine and daunorubicin in a fixed 5:1 molar ratio for the treatment of relapsed and refractory acute myeloid leukemia. J Clin Oncol. 2011;29(8):979–85.
Lancet JE, Cortes JE, Hogge DE, Tallman MS, Kovacsovics TJ, Damon LE, et al. Phase 2 trial of CPX-351, a fixed 5:1 molar ratio of cytarabine/daunorubicin, vs. cytarabine/daunorubicin in older adults with untreated AML. Blood. 2014;123(21):3239–46.
Cortes JE, Goldberg SL, Feldman EJ, Rizzeri DA, Hogge DE, Larson M, et al. Phase II, multicenter, randomized trial of CPX-351 (cytarabine:daunorubicin) liposome injection versus intensive salvage therapy in adults with first relapse AML. Cancer. 2015;121(2):234–42.
Lancet JE, Uy GL, Cortes JE, Newell LF, Lin TL, Ritchie EK, et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J Clin Oncol. 2018;36(26):2684–92.
Guolo F, Fianchi L, Minetto P, Clavio M, Gottardi M, Galimberti S, et al. CPX-351 treatment in secondary acute myeloblastic leukemia is effective and improves the feasibility of allogeneic stem cell transplantation: results of the Italian compassionate use program. Blood Cancer J. 2020;10(10):96.
Chiche E, Rahmé R, Bertoli S, Dumas P-Y, Micol J-B, Hicheri Y, et al. Real-life experience with CPX-351 and impact on the outcome of high-risk AML patients: a multicentric French cohort. Blood Adv. 2021;5(1):176–84.
Ramos Perez JM, Kadia TM, Montalban-Bravo G, Benton CB, Faderl S, Sasaki K, et al. Liposomal cytarabine and daunorubicin (CPX-351) in combination with gemtuzumab ozogamicin (GO) in relapsed refractory (R/R) patients with acute myeloid leukemia (AML) and post-hypomethylating agent (post-HMA) failure high-risk myelodysplastic syndrome (HR-MDS). Blood. 2019;134(Suppl_1):2642.
Edwards DKV, Javidi-Sharifi N, Rofelty A, Rosenfeld C, Roth-Carter R, Tardi P, et al. Effective combination of CPX-351 with FLT3 inhibitors in AML blasts harboring the FLT3-ITD mutation. Blood. 2016;128(22):5124.
Weis TM, Marini BL, Bixby DL, Perissinotti AJ. Clinical considerations for the use of FLT3 inhibitors in acute myeloid leukemia. Crit Rev Oncol Hematol. 2019;141:125–38.
Scholl S, Fleischmann M, Schnetzke U, Heidel FH. Molecular mechanisms of resistance to FLT3 inhibitors in acute myeloid leukemia: ongoing challenges and future treatments. Cells. 2020;9(11):2493.
Liang J, Wu YL, Chen BJ, Zhang W, Tanaka Y, Sugiyama H. The C-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9(5):435–43.
Linnekin D. Early signaling pathways activated by c-kit in hematopoietic cells. Int J Biochem Cell Biol. 1999;31(10):1053–74.
Malaise M, Steinbach D, Corbacioglu S. Clinical implications of c-kit mutations in acute myelogenous leukemia. Curr Hematol Malig Rep. 2009;4(2):77–82.
Heo S-K, Noh E-K, Kim JY, Jeong YK, Jo J-C, Choi Y, et al. Targeting c-KIT (CD117) by dasatinib and radotinib promotes acute myeloid leukemia cell death. Sci Rep. 2017;7(1):15278.
Kivioja JL, Thanasopoulou A, Kumar A, Kontro M, Yadav B, Majumder MM, et al. Dasatinib and navitoclax act synergistically to target NUP98-NSD1+/FLT3-ITD+ acute myeloid leukemia. Leukemia. 2019;33(6):1360–72.
Nicolas B, Aline R, Thibaut L, Pascale Cornillet L, Christian R, Thibaud L, et al. Dasatinib in high-risk core binding factor acute myeloid leukemia in first complete remission: a French acute myeloid leukemia intergroup trial. Haematologica. 2015;100(6):780–5.
Paschka P, Schlenk RF, Weber D, Benner A, Bullinger L, Heuser M, et al. Adding dasatinib to intensive treatment in core-binding factor acute myeloid leukemia-results of the AMLSG 11-08 trial. Leukemia. 2018;32(7):1621–30.
Marcucci G, Geyer S, Laumann K, Zhao W, Bucci D, Uy GL, et al. Combination of dasatinib with chemotherapy in previously untreated core binding factor acute myeloid leukemia: CALGB 10801. Blood Adv. 2020;4(4):696–705.
Kindler T, Breitenbuecher F, Marx A, Beck J, Hess G, Weinkauf B, et al. Efficacy and safety of imatinib in adult patients with c-kit-positive acute myeloid leukemia. Blood. 2004;103(10):3644–54.
Smolich BD, Yuen HA, West KA, Giles FJ, Albitar M, Cherrington JM. The antiangiogenic protein kinase inhibitors SU5416 and SU6668 inhibit the SCF receptor (c-kit) in a human myeloid leukemia cell line and in acute myeloid leukemia blasts. Blood. 2001;97(5):1413–21.
Zhu C, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer. 2019;18(1):153.
Park IK, Mundy-Bosse B, Whitman SP, Zhang X, Warner SL, Bearss DJ, et al. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia. 2015;29(12):2382–9.
Ben-Batalla I, Schultze A, Wroblewski M, Erdmann R, Heuser M, Waizenegger JS, et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood. 2013;122(14):2443–52.
Loges S, Heuser M, Chromik J, Vigil CE, Paschka P, Re F, et al. Durable responses observed in elderly AML patients unfit for intensive chemotherapy with first-in class selective AXL inhibitor bemcentinib (BGB324) in combination with LDAC: phase II open-label study. Blood. 2019;134(Suppl_1):3943.
BerGenBio. BerGenBio presents preliminary phase II clinical data at EHA 24: bemcentinib in combination with low dose chemotherapy yields durable responses in AML patients unfit for intensive chemotherapy. 2019. https://www.bergenbio.com/bergenbio-to-present-preliminary-phase-ii-clinical-data-showing-bemcentinib-in-combination-with-low-dose-chemotherapy-yields-durable-responses-in-aml-patients-unfit-for-intensive-chemotherapy-at-the-2/.
BerGenBio. BerGenBio receives FDA approval of fast track designation for bemcentiniB. 2019.
Yan SB, Peek VL, Ajamie R, Buchanan SG, Graff JR, Heidler SA, et al. LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Investig New Drugs. 2013;31(4):833–44.
Kosciuczuk EM, Saleiro D, Kroczynska B, Beauchamp EM, Eckerdt F, Blyth GT, et al. Merestinib blocks Mnk kinase activity in acute myeloid leukemia progenitors and exhibits antileukemic effects in vitro and in vivo. Blood. 2016;128(3):410–4.
Garcia JS, Gandler HI, Fell G, Fiore AJ, Neuberg DS, Anderson A, et al. Targeting MET and FGFR in relapsed or refractory acute myeloid leukemia: preclinical, clinical, and correlative studies. Blood. 2019;134(Suppl_1):2549.
Yasuhiro T, Yoshizawa T, Fujikawa R, Tanaka K, Koike T, Kawabata K. Development of an Axl/Mer dual inhibitor, ONO-9330547: promising single agent activity in an acute myeloid leukemia (AML) model. Blood. 2014;124(21):999.
Gilmour M, Scholtz A, Ottmann OG, Hills RK, Knapper S, Zabkiewicz J. Axl/Mer inhibitor ONO-9330547 as a novel therapeutic agent in a stromal co-culture model of primary acute myeloid Leukaemia (AML). Blood. 2016;128(22):2754.
Ruvolo PP, Ma H, Ruvolo VR, Zhang X, Mu H, Schober W, et al. Anexelekto/MER tyrosine kinase inhibitor ONO-7475 arrests growth and kills FMS-like tyrosine kinase 3-internal tandem duplication mutant acute myeloid leukemia cells by diverse mechanisms. Haematologica. 2017;102(12):2048–57.
Fialin C, Larrue C, Vergez F, Sarry JE, Bertoli S, Mansat-De Mas V, et al. The short form of RON is expressed in acute myeloid leukemia and sensitizes leukemic cells to cMET inhibitors. Leukemia. 2013;27(2):325–35.
Kentsis A, Reed C, Rice KL, Sanda T, Rodig SJ, Tholouli E, et al. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat Med. 2012;18(7):1118–22.
Mulgrew NM, Kettyle LMJ, Ramsey JM, Cull S, Smyth LJ, Mervyn DM, et al. C-met inhibition in a HOXA9/Meis1 model of CN-AML. Dev Dyn. 2014;243(1):172–81.
Mócsai A, Ruland J, Tybulewicz VLJ. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 2010;10(6):387–402.
Boros K, Puissant A, Back M, Alexe G, Bassil CF, Sinha P, et al. Increased SYK activity is associated with unfavorable outcome among patients with acute myeloid leukemia. Oncotarget. 2015;6(28):25575–87.
Polak A, Bialopiotrowicz E, Krzymieniewska B, Wozniak J, Stojak M, Cybulska M, et al. SYK inhibition targets acute myeloid leukemia stem cells by blocking their oxidative metabolism. Cell Death Dis. 2020;11(11):956.
Walker AR, Bhatnagar B, Marcondes AMQ, DiPaolo J, Vasu S, Mims AS, et al. Interim results of a Phase 1b/2 study of entospletinib (GS-9973) monotherapy and in combination with chemotherapy in patients with acute myeloid leukemia. Blood. 2016;128(22):2831.
Walker AR, Byrd JC, Blum W, Lin T, Crosswell HE, Zhang D, et al. Abstract 819: high response rates with entospletinib in patients with t(v;11q23.3);KMT2A rearranged acute myeloid leukemia and acute lymphoblastic leukemia. Cancer Res. 2018;78(13 Suppl):819.
Yu J, Huck J, Theisen M, He H, Tirrell S, Zhang M, et al. Anti-tumor activity of TAK-659, a dual inhibitor of SYK and FLT-3 kinases, in AML models. J Clin Oncol. 2016;(34, 15_suppl):e14091.
Pratz K, Levis MJ, Morris JC, Wise-Draper T, Levy M, Bixby DL, et al. A phase (ph) 1b/2 study of TAK-659, an investigational dual FLT-3 and SYK inhibitor, in patients (Pts) with relapsed or refractory acute myelogenous leukemia (R/R AML). Blood. 2017;130(Suppl 1):2622.
Mohamed AJ, Yu L, Bäckesjö C-M, Vargas L, Faryal R, Aints A, et al. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev. 2009;228(1):58–73.
Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol Cancer. 2018;17(1):57.
Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol. 2012;31(2):119–32.
Pillinger G, Abdul-Aziz A, Zaitseva L, Lawes M, MacEwan DJ, Bowles KM, et al. Targeting BTK for the treatment of FLT3-ITD mutated acute myeloid leukemia. Sci Rep. 2015;5:12949.
Rushworth SA, Murray MY, Zaitseva L, Bowles KM, MacEwan DJ. Identification of Bruton’s tyrosine kinase as a therapeutic target in acute myeloid leukemia. Blood. 2014;123(8):1229–38.
Zaitseva L, Murray MY, Shafat MS, Lawes MJ, MacEwan DJ, Bowles KM, et al. Ibrutinib inhibits SDF1/CXCR4 mediated migration in AML. Oncotarget. 2014;5(20):9930–8.
Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562(7728):526–31.
Rushworth SA, Pillinger G, Abdul-Aziz A, Piddock R, Shafat MS, Murray MY, et al. Activity of Bruton’s tyrosine-kinase inhibitor ibrutinib in patients with CD117-positive acute myeloid leukaemia: a mechanistic study using patient-derived blast cells. Lancet Haematol. 2015;2(5):e204–11.
Wu H, Hu C, Wang A, Weisberg EL, Wang W, Chen C, et al. Ibrutinib selectively targets FLT3-ITD in mutant FLT3-positive AML. Leukemia. 2016;30(3):754–7.
Eide CA, Kurtz SE, Kaempf A, Long N, Agarwal A, Tognon CE, et al. Simultaneous kinase inhibition with ibrutinib and BCL2 inhibition with venetoclax offers a therapeutic strategy for acute myeloid leukemia. Leukemia. 2020;34(9):2342–53.
Cortes JE, Jonas BA, Graef T, Luan Y, Stein AS. Clinical experience with ibrutinib alone or in combination with either cytarabine or azacitidine in patients with acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2019;19(8):509–15.e1.
Goldberg AD, Ohanian M, Koller P, Altman JK, Cherry M, Tomlinson B, Chandhok N, Zhang H, Rastgoo N, Benbatoul K, Jin Y. A phase 1a/b dose escalation study of the mutation agnostic FLT3/BTK inhibitor luxeptinib (CG-806) in patients with relapsed or refractory acute myeloid leukemia. Blood. 2021;138:1272.
Debora S, Stefania O, Samantha R, Paola M, Claudia M, Luca A, et al. The new small tyrosine kinase inhibitor ARQ531 targets acute myeloid leukemia cells by disrupting multiple tumor-addicted programs. Haematologica. 2019;105(10):2420–31.
Elgamal OA, Mehmood A, Jeon JY, Carmichael B, Lehman A, Orwick SJ, et al. Preclinical efficacy for a novel tyrosine kinase inhibitor, ArQule 531 against acute myeloid leukemia. J Hematol Oncol. 2020;13(1):8.
Huang S, Pan J, Jin J, Li C, Li X, Huang J, et al. Abivertinib, a novel BTK inhibitor: anti-leukemia effects and synergistic efficacy with homoharringtonine in acute myeloid leukemia. Cancer Lett. 2019;461:132–43.
Voisset E, Brenet F, Lopez S, de Sepulveda P. SRC-family kinases in acute myeloid leukaemia and mastocytosis. Cancers (Basel). 2020;12(7):1996.
MacDonald RJ, Bunaciu RP, Ip V, Dai D, Tran D, Varner JD, et al. Src family kinase inhibitor bosutinib enhances retinoic acid-induced differentiation of HL-60 leukemia cells. Leuk Lymphoma. 2018;59(12):2941–51.
Saito Y, Yuki H, Kuratani M, Hashizume Y, Takagi S, Honma T, et al. A pyrrolo-pyrimidine derivative targets human primary AML stem cells in vivo. Sci Transl Med. 2013;5(181):181ra52.
Weir MC, Shu ST, Patel RK, Hellwig S, Chen L, Tan L, et al. Selective inhibition of the myeloid Src-family kinase Fgr potently suppresses AML cell growth in vitro and in vivo. ACS Chem Biol. 2018;13(6):1551–9.
Ozawa Y, Williams AH, Estes ML, Matsushita N, Boschelli F, Jove R, et al. Src family kinases promote AML cell survival through activation of signal transducers and activators of transcription (STAT). Leuk Res. 2008;32(6):893–903.
Bourrié B, Brassard DL, Cosnier-Pucheu S, Zilberstein A, Yu K, Levit M, et al. SAR103168: a tyrosine kinase inhibitor with therapeutic potential in myeloid leukemias. Leuk Lymphoma. 2013;54(7):1488–99.
Roboz GJ, Khoury HJ, Jabbour E, Session W, Ritchie EK, Miao H, et al. Phase I trial of SAR103168, a novel multi-kinase inhibitor, in patients with refractory/relapsed acute leukemia or high-risk myelodysplastic syndrome. Leuk Lymphoma. 2015;56(2):395–400.
Terao T, Minami Y. Targeting hedgehog (Hh) pathway for the acute myeloid leukemia treatment. Cells. 2019;8(4):312.
Aberger F, Hutterer E, Sternberg C, Del Burgo PJ, Hartmann TN. Acute myeloid leukemia—strategies and challenges for targeting oncogenic hedgehog/GLI signaling. Cell Commun Signal. 2017;15(1):8.
Wellbrock J, Latuske E, Köhler J, Wagner K, Stamm H, Vettorazzi E, et al. Expression of hedgehog pathway mediator GLI represents a negative prognostic marker in human acute myeloid leukemia and its inhibition exerts antileukemic effects. Clin Cancer Res. 2015;21(10):2388–98.
Zahreddine HA, Culjkovic-Kraljacic B, Assouline S, Gendron P, Romeo AA, Morris SJ, et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature. 2014;511(7507):90–3.
Li X, Chen F, Zhu Q, Ding B, Zhong Q, Huang K, et al. Gli-1/PI3K/AKT/NF-kB pathway mediates resistance to radiation and is a target for reversion of responses in refractory acute myeloid leukemia cells. Oncotarget. 2016;7(22):33004–15.
Fukushima N, Minami Y, Kakiuchi S, Kuwatsuka Y, Hayakawa F, Jamieson C, et al. Small-molecule hedgehog inhibitor attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Sci. 2016;107(10):1422–9.
Minami Y, Minami H, Miyamoto T, Yoshimoto G, Kobayashi Y, Munakata W, et al. Phase I study of glasdegib (PF-04449913), an oral smoothened inhibitor, in Japanese patients with select hematologic malignancies. Cancer Sci. 2017;108(8):1628–33.
Martinelli G, Oehler VG, Papayannidis C, Courtney R, Shaik MN, Zhang X, et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: a phase 1 safety and pharmacokinetics study. Lancet Haematol. 2015;2(8):e339–46.
Savona MR, Pollyea DA, Stock W, Oehler VG, Schroeder MA, Lancet J, et al. Phase Ib study of glasdegib, a hedgehog pathway inhibitor, in combination with standard chemotherapy in patients with AML or high-risk MDS. Clin Cancer Res. 2018;24(10):2294–303.
Cortes JE, Douglas Smith B, Wang ES, Merchant A, Oehler VG, Arellano M, et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high-risk MDS: Phase 2 study results. Am J Hematol. 2018;93(11):1301–10.
Cortes JE, Heidel FH, Hellmann A, Fiedler W, Smith BD, Robak T, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia. 2019;33(2):379–89.
Heuser M, Robak T, Montesinos P, Leber B, Fiedler WM, Pollyea DA, et al. Glasdegib (GLAS) plus low-dose cytarabine (LDAC) in AML or MDS: BRIGHT AML 1003 final report and four-year overall survival (OS) follow-up. J Clin Oncol. 2020;38(15_suppl):7509.
Huang K, Ding B, Zhong Q, Jiang X, Li X, Wang Z, et al. Hh/IGF-1R/PI3K/Akt/MRP1 pathway induce refractory acute myeloid leukemia and its targeting therapy. Blood. 2014;124(21):3612.
Tibes R, Kosiorek HE, Dueck A, Palmer J, Slack JL, Knight EA, et al. Phase I/IB study of azacitidine and hedgehog pathway inhibition with sonidegib (LDE225) in myeloid malignancies. Blood. 2017;130(Suppl 1):2629.
Shallis RM, Bewersdorf JP, Boddu PC, Zeidan AM. Hedgehog pathway inhibition as a therapeutic target in acute myeloid leukemia. Expert Rev Anticancer Ther. 2019;19(8):717–29.
Bixby D, Noppeney R, Lin TL, Cortes J, Krauter J, Yee K, et al. Safety and efficacy of vismodegib in relapsed/refractory acute myeloid leukaemia: results of a phase Ib trial. Br J Haematol. 2019;185(3):595–8.
Masetti R, Bertuccio SN, Astolfi A, Chiarini F, Lonetti A, Indio V, et al. Hh/Gli antagonist in acute myeloid leukemia with CBFA2T3-GLIS2 fusion gene. J Hematol Oncol. 2017;10(1):26.
Latuske EM, Stamm H, Klokow M, Vohwinkel G, Muschhammer J, Bokemeyer C, et al. Combined inhibition of GLI and FLT3 signaling leads to effective anti-leukemic effects in human acute myeloid leukemia. Oncotarget. 2017;8(17):29187–201.
Wei AH, Roberts AW, Spencer A, Rosenberg AS, Siegel D, Walter RB, et al. Targeting MCL-1 in hematologic malignancies: rationale and progress. Blood Rev. 2020;44:100672.
Wei Y, Cao Y, Sun R, Cheng L, Xiong X, Jin X, et al. Targeting Bcl-2 proteins in acute myeloid leukemia. Front Oncol. 2020;10(2137):584974.
Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014;4(3):362–75.
Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6(10):1106–17.
Bogenberger JM, Kornblau SM, Pierceall WE, Lena R, Chow D, Shi CX, et al. BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia. 2014;28(8):1657–65.
Bogenberger JM, Delman D, Hansen N, Valdez R, Fauble V, Mesa RA, et al. Ex vivo activity of BCL-2 family inhibitors ABT-199 and ABT-737 combined with 5-azacytidine in myeloid malignancies. Leuk Lymphoma. 2015;56(1):226–9.
DiNardo CD, Pratz KW, Letai A, Jonas BA, Wei AH, Thirman M, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018;19(2):216–28.
DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17.
Winters AC, Gutman JA, Purev E, Nakic M, Tobin J, Chase S, et al. Real-world experience of venetoclax with azacitidine for untreated patients with acute myeloid leukemia. Blood Adv. 2019;3(20):2911–9.
Aldoss I, Yang D, Aribi A, Ali H, Sandhu K, Al Malki MM, et al. Efficacy of the combination of venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Haematologica. 2018;103(9):e404–e7.
Wei AH, Strickland SA Jr, Hou JZ, Fiedler W, Lin TL, Walter RB, et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a Phase Ib/II study. J Clin Oncol. 2019;37(15):1277–84.
DiNardo CD, Rausch CR, Benton C, Kadia T, Jain N, Pemmaraju N, et al. Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am J Hematol. 2018;93(3):401–7.
Aldoss I, Yang D, Pillai R, Sanchez JF, Mei M, Aribi A, et al. Association of leukemia genetics with response to venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Am J Hematol. 2019;94(10):E253–e5.
Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. 2018;24(12):1859–66.
DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, et al. Azacitidine and Venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617–29.
Wei AH, Montesinos P, Ivanov V, DiNardo CD, Novak J, Laribi K, et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo-controlled trial. Blood. 2020;135(24):2137–45.
Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10(5):375–88.
Tron AE, Belmonte MA, Adam A, Aquila BM, Boise LH, Chiarparin E, et al. Discovery of mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun. 2018;9(1):5341.
Caenepeel S, Brown SP, Belmontes B, Moody G, Keegan KS, Chui D, et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov. 2018;8(12):1582–97.
Caenepeel SR, Belmontes B, Sun J, Coxon A, Moody G, Hughes PE. Abstract 2027: preclinical evaluation of AMG 176, a novel, potent and selective mcl-1 inhibitor with robust anti-tumor activity in mcl-1 dependent cancer models. Cancer Res. 2017;77(13 Suppl):2027.
Caenepeel S, Karen R, Belmontes B, Verlinsky A, Tan H, Yang Y, et al. Abstract 6218: discovery and preclinical evaluation of AMG 397, a potent, selective and orally bioavailable MCL1 inhibitor. Cancer Res. 2020;80(16 Suppl):6218.
Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538(7626):477–82.
Moujalled DM, Pomilio G, Ghiurau C, Ivey A, Salmon J, Rijal S, et al. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia. 2019;33(4):905–17.
Anstee NS, Bilardi RA, Ng AP, Xu Z, Robati M, Vandenberg CJ, et al. Impact of elevated anti-apoptotic MCL-1 and BCL-2 on the development and treatment of MLL-AF9 AML in mice. Cell Death Differ. 2019;26(7):1316–31.
Lee T, Christov PP, Shaw S, Tarr JC, Zhao B, Veerasamy N, et al. Discovery of potent myeloid cell Leukemia-1 (mcl-1) inhibitors that demonstrate in vivo activity in mouse xenograft models of human cancer. J Med Chem. 2019;62(8):3971–88.
Ramsey HE, Fischer MA, Lee T, Gorska AE, Arrate MP, Fuller L, et al. A novel MCL1 inhibitor combined with venetoclax rescues venetoclax-resistant acute myelogenous leukemia. Cancer Discov. 2018;8(12):1566–81.
Cohen NA, Stewart ML, Gavathiotis E, Tepper JL, Bruekner SR, Koss B, et al. A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chem Biol. 2012;19(9):1175–86.
Richard DJ, Lena R, Bannister T, Blake N, Pierceall WE, Carlson NE, et al. Hydroxyquinoline-derived compounds and analoguing of selective mcl-1 inhibitors using a functional biomarker. Bioorg Med Chem. 2013;21(21):6642–9.
Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22(53):8628–33.
Prabhu VV, Talekar MK, Lulla AR, Kline CLB, Zhou L, Hall J, et al. Single agent and synergistic combinatorial efficacy of first-in-class small molecule imipridone ONC201 in hematological malignancies. Cell Cycle. 2018;17(4):468–78.
Edwards H, Ge Y. ONC201 shows promise in AML treatment. Cell Cycle. 2018;17(3):277.
Ishizawa J, Kojima K, Chachad D, Ruvolo P, Ruvolo V, Jacamo RO, et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci Signal. 2016;9(415):ra17.
Wagner J, Kline CL, Ralff MD, Lev A, Lulla A, Zhou L, et al. Preclinical evaluation of the imipridone family, analogs of clinical stage anti-cancer small molecule ONC201, reveals potent anti-cancer effects of ONC212. Cell Cycle. 2017;16(19):1790–9.
Nii T, Prabhu VV, Ruvolo V, Madhukar N, Zhao R, Mu H, et al. Imipridone ONC212 activates orphan G protein-coupled receptor GPR132 and integrated stress response in acute myeloid leukemia. Leukemia. 2019;33(12):2805–16.
Konopleva M, Martinelli G, Daver N, Papayannidis C, Wei A, Higgins B, et al. MDM2 inhibition: an important step forward in cancer therapy. Leukemia. 2020;34(11):2858–74.
Loizou E, Banito A, Livshits G, Ho Y-J, Koche RP, Sánchez-Rivera FJ, et al. A gain-of-function p53-mutant oncogene promotes cell fate plasticity and myeloid leukemia through the pluripotency factor FOXH1. Cancer Discov. 2019;9(7):962.
Barbosa K, Li S, Adams PD, Deshpande AJ. The role of TP53 in acute myeloid leukemia: challenges and opportunities. Genes Chromosom Cancer. 2019;58(12):875–88.
Tiong IS, Wei AH. New drugs creating new challenges in acute myeloid leukemia. Genes Chromosom Cancer. 2019;58(12):903–14.
Sallman DA. To target the untargetable: elucidation of synergy of APR-246 and azacitidine in TP53 mutant myelodysplastic syndromes and acute myeloid leukemia. Haematologica. 2020;105(6):1470–2.
Maslah N, Salomao N, Drevon L, Verger E, Partouche N, Ly P, et al. Synergistic effects of PRIMA-1(met) (APR-246) and 5-azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica. 2020;105(6):1539–51.
Sallman DA, DeZern AE, Steensma DP, Sweet KL, Cluzeau T, Sekeres MA, et al. Phase 1b/2 combination study of APR-246 and azacitidine (AZA) in patients with TP53 mutant myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). Blood. 2018;132(Suppl 1):3091.
Yan W, Jung YS, Zhang Y, Chen X. Arsenic trioxide reactivates proteasome-dependent degradation of mutant p53 protein in cancer cells in part via enhanced expression of Pirh2 E3 ligase. PLoS One. 2014;9(8):e103497.
Yan W, Zhang Y, Zhang J, Liu S, Cho SJ, Chen X. Mutant p53 protein is targeted by arsenic for degradation and plays a role in arsenic-mediated growth suppression. J Biol Chem. 2011;286(20):17478–86.
Noguera NI, Pelosi E, Angelini DF, Piredda ML, Guerrera G, Piras E, et al. High-dose ascorbate and arsenic trioxide selectively kill acute myeloid leukemia and acute promyelocytic leukemia blasts in vitro. Oncotarget. 2017;8(20):32550–65.
Schlenk RF, Döhner K, Kneba M, Götze K, Hartmann F, Del Valle F, et al. Gene mutations and response to treatment with all-trans retinoic acid in elderly patients with acute myeloid leukemia. Results from the AMLSG trial AML HD98B. Haematologica. 2009;94(1):54–60.
Martelli MP, Gionfriddo I, Mezzasoma F, Milano F, Pierangeli S, Mulas F, et al. Arsenic trioxide and all-trans retinoic acid target NPM1 mutant oncoprotein levels and induce apoptosis in NPM1-mutated AML cells. Blood. 2015;125(22):3455–65.
El Hajj H, Dassouki Z, Berthier C, Raffoux E, Ades L, Legrand O, et al. Retinoic acid and arsenic trioxide trigger degradation of mutated NPM1, resulting in apoptosis of AML cells. Blood. 2015;125(22):3447–54.
Khurana A, Shafer DA. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: perspectives on the therapeutic potential of idasanutlin (RG7388). Onco Targets Ther. 2019;12:2903–10.
Yee K, Martinelli G, Assouline S, Kasner M, Vey N, Kelly KR, et al. Phase 1b study of the MDM2 antagonist RG7112 in combination with 2 doses/schedules of cytarabine. Blood. 2013;122(21):498.
Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, et al. Results of the Phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Cancer Res. 2016;22(4):868–76.
Yee K, Martinelli G, Vey N, Dickinson MJ, Seiter K, Assouline S, et al. Phase 1/1b study of RG7388, a potent MDM2 antagonist, in acute Myelogenous leukemia (AML) patients (Pts). Blood. 2014;124(21):116.
Dangl M, Chien Y, Lehmann C, Friess T. Abstract 5505: synergistic anticancer activity of clinical stage, non-genotoxic apoptosis inducing agents RG7388 (MDM2 antagonist) and ABT-199 (GDC-0199, BCL2 inhibitor) in p53 wild-type AML tumor models. Cancer Res. 2014;74(19 Suppl):5505.
Daver NG, Garcia JS, Jonas BA, Kelly KR, Assouline S, Brandwein JM, et al. Updated results from the venetoclax (Ven) in combination with idasanutlin (Idasa) arm of a phase 1b trial in elderly patients (Pts) with relapsed or refractory (R/R) AML ineligible for cytotoxic chemotherapy. Blood. 2019;134(Suppl_1):229.
Nishida Y, Ishizawa J, Ruvolo V, Kojima K, Montoya RH, Daver NG, et al. Dual inhibition of MDM2 and XPO1 synergizes to induce apoptosis in acute myeloid leukemia progenitor cells with wild-type TP53 through nuclear accumulation of p53 and suppression of c-Myc. Blood. 2019;134(Suppl_1):2556.
Abdul Razak AR, Miller WH Jr, Uy GL, Blotner S, Young AM, Higgins B, et al. A phase 1 study of the MDM2 antagonist RO6839921, a pegylated prodrug of idasanutlin, in patients with advanced solid tumors. Investig New Drugs. 2020;38(4):1156–65.
Erba HP, Becker PS, Shami PJ, Grunwald MR, Flesher DL, Zhu M, et al. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019;3(13):1939–49.
ASH Clinical News. Early-phase trials of HDM201 show promise in leukemias. 2017. https://www.ashclinicalnews.org/meeting-news/early-phase-trials-hdm201-show-promise-leukemias/.
Nepstad I, Hatfield KJ, Grønningsæter IS, Reikvam H. The PI3K-Akt-mTOR signaling pathway in human acute myeloid leukemia (AML) cells. Int J Mol Sci. 2020;21(8):2907.
Perl AE, Kasner MT, Tsai DE, Vogl DT, Loren AW, Schuster SJ, et al. A phase I study of the mammalian target of rapamycin inhibitor sirolimus and MEC chemotherapy in relapsed and refractory acute myelogenous leukemia. Clin Cancer Res. 2009;15(21):6732–9.
Park S, Chapuis N, Saint Marcoux F, Recher C, Prebet T, Chevallier P, et al. A phase Ib GOELAMS study of the mTOR inhibitor RAD001 in association with chemotherapy for AML patients in first relapse. Leukemia. 2013;27(7):1479–86.
Yee KW, Zeng Z, Konopleva M, Verstovsek S, Ravandi F, Ferrajoli A, et al. Phase I/II study of the mammalian target of rapamycin inhibitor everolimus (RAD001) in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res. 2006;12(17):5165–73.
Alan KB, Emma Das G, Steve K, Asim K, Marion S, Lars K, et al. Addition of the mammalian target of rapamycin inhibitor, everolimus, to consolidation therapy in acute myeloid leukemia: experience from the UK NCRI AML17 trial. Haematologica. 2018;103(10):1654–61.
Amadori S, Stasi R, Martelli AM, Venditti A, Meloni G, Pane F, et al. Temsirolimus, an mTOR inhibitor, in combination with lower-dose clofarabine as salvage therapy for older patients with acute myeloid leukaemia: results of a phase II GIMEMA study (AML-1107). Br J Haematol. 2012;156(2):205–12.
Rizzieri DA, Feldman E, Dipersio JF, Gabrail N, Stock W, Strair R, et al. A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res. 2008;14(9):2756–62.
Herschbein L, Liesveld JL. Dueling for dual inhibition: means to enhance effectiveness of PI3K/Akt/mTOR inhibitors in AML. Blood Rev. 2018;32(3):235–48.
Vargaftig J, Farhat H, Ades L, Briaux A, Benoist C, Turbiez I, et al. Phase 2 trial of single agent Gedatolisib (PF-05212384), a dual PI3K/mTOR inhibitor, for adverse prognosis and relapse/refractory AML: clinical and Transcriptomic results. Blood. 2018;132(Suppl 1):5233.
Lang F, Wunderle L, Badura S, Schleyer E, Brüggemann M, Serve H, et al. A phase I study of a dual PI3-kinase/mTOR inhibitor BEZ235 in adult patients with relapsed or refractory acute leukemia. BMC Pharmacol Toxicol. 2020;21(1):70.
Abou Zahr A, Borthakur G. Emerging cell cycle inhibitors for acute myeloid leukemia. Expert Opin Emerg Drugs. 2017;22(2):137–48.
Carter JL, Hege K, Yang J, Kalpage HA, Su Y, Edwards H, et al. Targeting multiple signaling pathways: the new approach to acute myeloid leukemia therapy. Signal Transduct Target Ther. 2020;5(1):288.
Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. 2018;24(7):1036–46.
Liu F, Kalpage HA, Wang D, Edwards H, Hüttemann M, Ma J, et al. Cotargeting of mitochondrial complex I and Bcl-2 shows antileukemic activity against acute myeloid leukemia cells reliant on oxidative phosphorylation. Cancers (Basel). 2020;12(9):2400.
Panina SB, Pei J, Baran N, Konopleva M, Kirienko NV. Utilizing synergistic potential of mitochondria-targeting drugs for leukemia therapy. Front Oncol. 2020;10:435.
Baccelli I, Gareau Y, Lehnertz B, Gingras S, Spinella J-F, Corneau S, et al. Mubritinib targets the electron transport chain complex i and reveals the landscape of OXPHOS Dependency in acute myeloid leukemia. Cancer Cell. 2019;36(1):84–99.e8.
Ricciardi MR, Mirabilii S, Allegretti M, Licchetta R, Calarco A, Torrisi MR, et al. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood. 2015;126(16):1925–9.
Cloos J, Roeten MS, Franke NE, van Meerloo J, Zweegman S, Kaspers GJ, et al. (Immuno)proteasomes as therapeutic target in acute leukemia. Cancer Metastasis Rev. 2017;36(4):599–615.
Enrique C, Stela Á-F, Patricia M, Jesús M-S, Maria Belén V, Mercedes G, et al. The effect of the proteasome inhibitor bortezomib on acute myeloid leukemia cells and drug resistance associated with the CD34+ immature phenotype. Haematologica. 2008;93(1):57–66.
Tomlinson BK, Tuscano JM, Abedi M, Welborn J, Arora M, O’Donnell RT, et al. A phase II study of bortezomib in combination with pegylated liposomal doxorubicin for acute myeloid leukemia. Am J Hematol. 2019;94(11):E291–E4.
Swords RT, Kelly KR, Smith PG, Garnsey JJ, Mahalingam D, Medina E, et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. 2010;115(18):3796–800.
Sen S, De Leon JP, Smith PG, Roboz GJ, Guzman ML. Investigational NEDD8-activating enzyme (NAE) inhibitor, MLN4924, demonstrates activity against primary AML blast, progenitor and stem cell populations. Blood. 2011;118(21):1414.
Zhou L, Chen S, Zhang Y, Kmieciak M, Leng Y, Li L, et al. The NAE inhibitor pevonedistat interacts with the HDAC inhibitor belinostat to target AML cells by disrupting the DDR. Blood. 2016;127(18):2219–30.
Knorr KL, Schneider PA, Meng XW, Dai H, Smith BD, Hess AD, et al. MLN4924 induces Noxa upregulation in acute myelogenous leukemia and synergizes with Bcl-2 inhibitors. Cell Death Differ. 2015;22(12):2133–42.
Short NPB, Dinardo C, Garcia-Manero G, Muftuoglu M, Alaniz Z, Patel K, Montalban-Bravo G, Jain N, Alvarado Y, Jabbour E, Andreeff M, Delumpa R, Kantarjian H, Cortes J. Preliminary results of a phase I/II study of azacitidine, venetoclax and pevonedistat in patients with secondary acute myeloid leukemia who are unfit for intensive chemotherapy. 2020. https://library.ehaweb.org/eha/2020/eha25th/294475/nicholas.short.preliminary.results.of.a.phase.i.ii.study.of.azacitidine.html?f=listing%3D0%2Abrowseby%3D8%2Asortby%3D2%2Asearch%3Dblast.
Ishikawa Y, Nakayama K, Morimoto M, Mizutani A, Nakayama A, Toyoshima K, et al. Synergistic anti-AML effects of the LSD1 inhibitor T-3775440 and the NEDD8-activating enzyme inhibitor pevonedistat via transdifferentiation and DNA rereplication. Oncogenesis. 2017;6(9):e377.
Swords RT, Coutre S, Maris MB, Zeidner JF, Foran JM, Cruz J, et al. Pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood. 2018;131(13):1415–24.
Sekeres MA, Watts J, Radinoff A, Sangerman MA, Cerrano M, Lopez PF, et al. Randomized phase 2 trial of pevonedistat plus azacitidine versus azacitidine for higher-risk MDS/CMML or low-blast AML. Leukemia. 2021;
Talati C, Sweet KL. Nuclear transport inhibition in acute myeloid leukemia: recent advances and future perspectives. Int J Hematol Oncol. 2018;7(3):Ijh04.
Ranganathan P, Yu X, Na C, Santhanam R, Shacham S, Kauffman M, et al. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood. 2012;120(9):1765–73.
Ranganathan P, Kashyap T, Yu X, Meng X, Lai T-H, McNeil B, et al. XPO1 inhibition using selinexor synergizes with chemotherapy in acute myeloid leukemia by targeting DNA repair and restoring topoisomerase IIα to the nucleus. Clin Cancer Res. 2016;22(24):6142–52.
Ramzi A, Ezhilarasi C, Michael PR, Kathryn MT, Peter AR, Camille NA, et al. Selinexor combined with cladribine, cytarabine, and filgrastim in relapsed or refractory acute myeloid leukemia. Haematologica. 2020;105(8):e404–e7.
Zhang W, Ly C, Ishizawa J, Mu H, Ruvolo V, Shacham S, et al. Combinatorial targeting of XPO1 and FLT3 exerts synergistic anti-leukemia effects through induction of differentiation and apoptosis in FLT3-mutated acute myeloid leukemias: from concept to clinical trial. Haematologica. 2018;103(10):1642–53.
Garzon R, Savona M, Baz R, Andreeff M, Gabrail N, Gutierrez M, et al. A phase 1 clinical trial of single-agent selinexor in acute myeloid leukemia. Blood. 2017;129(24):3165–74.
Karyopharm Press Release. Karyopharm annouces results from interim analysis of phase II OPRA study evaluating selinexor in relapsed/refractory acute myeloid leukemia. 2017. https://www.globenewswire.com/news-release/2017/03/02/930523/0/en/Karyopharm-Announces-Results-from-Interim-Analysis-of-Phase-2-SOPRA-Study-Evaluating-Selinexor-in-Relapsed-Refractory-Acute-Myeloid-Leukemia.html.
Pardee TS, Pladna KM, Lyerly S, Dralle S, Manuel M, Ellis LR, Howard DS, Bhave R, Powell BL. Frontline selinexor and chemotherapy is highly active in older adults with acute myeloid leukemia (AML). Blood. 2020;136(Suppl 1):24–5.
Sweet K, Komrokji R, Padron E, Cubitt CL, Turner JG, Zhou J, et al. Phase I clinical trial of selinexor in combination with daunorubicin and cytarabine in previously untreated poor-risk acute myeloid leukemia. Clin Cancer Res. 2020;26(1):54–60.
Fiedler W, Heuser M, Chromik J, Thol F, Bokemeyer C, Theile S, et al. Phase II results of Ara-C and Idarubicin in combination with the selective inhibitor of nuclear export (SINE) compound Selinexor (KPT-330) in patients with relapsed or refractory AML. Blood. 2016;128(22):341.
Fiedler W, Chromik J, Amberg S, Kebenko M, Thol F, Schlipfenbacher V, et al. A Phase II study of selinexor plus cytarabine and idarubicin in patients with relapsed/refractory acute myeloid leukaemia. Br J Haematol. 2020;190(3):e169–e73.
Alexander TB, Lacayo NJ, Choi JK, Ribeiro RC, Pui CH, Rubnitz JE. Phase I study of selinexor, a selective inhibitor of nuclear export, in combination with fludarabine and cytarabine, in pediatric relapsed or refractory acute leukemia. J Clin Oncol. 2016;34(34):4094–101.
Uy GL, Rettig MP, Fletcher T, Riedell PA, Stockerl-Goldstein KE, Ghobadi A, et al. Selinexor in combination with cladribine, cytarabine and G-CSF for relapsed or refractory AML. Blood. 2017;130(Suppl 1):816.
Wang AY, Weiner HL, Green M, Larson RA, Odenike O, Artz A, et al. Combination of selinexor with high-dose cytarabine (HiDAC) and mitoxantrone (Mito) for remission induction in acute myeloid leukemia (AML) is feasible and tolerable. Blood. 2016;128(22):212.
Wang AY, Weiner H, Green M, Chang H, Fulton N, Larson RA, et al. A phase I study of selinexor in combination with high-dose cytarabine and mitoxantrone for remission induction in patients with acute myeloid leukemia. J Hematol Oncol. 2018;11(1):4.
Bhatnagar B, Zhao Q, Mims AS, Vasu S, Behbehani GK, Larkin K, et al. Selinexor in combination with decitabine in patients with acute myeloid leukemia: results from a phase 1 study. Leuk Lymphoma. 2020;61(2):387–96.
Daver NG, Assi R, Kantarjian HM, Cortes JE, Ravandi F, Konopleva MY, et al. Final results of Phase I/II study of selinexor (SEL) with sorafenib in patients (pts) with relapsed and/or refractory (R/R) FLT3 mutated acute myeloid leukemia (AML). Blood. 2018;132(Suppl 1):1441.
Cooperrider JH, Fulton N, Artz AS, Larson RA, Stock W, Kosuri S, et al. Phase I trial of maintenance selinexor after allogeneic hematopoietic stem cell transplantation for patients with acute myeloid leukemia and myelodysplastic syndrome. Bone Marrow Transplant. 2020;55(11):2204–6.
Hing ZA, Fung HY, Ranganathan P, Mitchell S, El-Gamal D, Woyach JA, et al. Next-generation XPO1 inhibitor shows improved efficacy and in vivo tolerability in hematological malignancies. Leukemia. 2016;30(12):2364–72.
Etchin J, Berezovskaya A, Conway AS, Galinsky IA, Stone RM, Baloglu E, et al. KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia. 2017;31(1):143–50.
Fischer MA, Arrate PM, Chang H, Gorska AE, Fuller LS, Ramsey HE, et al. Abstract 1877: selective inhibitor of nuclear export (SINE) compound, eltanexor (KPT-8602), synergizes with venetoclax (ABT-199) to eliminate leukemia cells and extend survival in an in vivo model of acute myeloid leukemia. Cancer Res. 2018;78(13 Suppl):1877.
Fischer MA, Friedlander SY, Arrate MP, Chang H, Gorska AE, Fuller LD, et al. Venetoclax response is enhanced by selective inhibitor of nuclear export compounds in hematologic malignancies. Blood Adv. 2020;4(3):586–98.
Fennell KA, Bell CC, Dawson MA. Epigenetic therapies in acute myeloid leukemia: where to from here? Blood. 2019;134(22):1891–901.
Duchmann M, Itzykson R. Clinical update on hypomethylating agents. Int J Hematol. 2019;110(2):161–9.
Garcia-Manero G, Gore SD, Cogle C, Ward R, Shi T, Macbeth KJ, et al. Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J Clin Oncol. 2011;29(18):2521–7.
Savona MR, Kolibaba K, Conkling P, Kingsley EC, Becerra C, Morris JC, et al. Extended dosing with CC-486 (oral azacitidine) in patients with myeloid malignancies. Am J Hematol. 2018;93(10):1199–206.
Roboz GJ, Montesinos P, Selleslag D, Wei A, Jang JH, Falantes J, et al. Design of the randomized, phase III, QUAZAR AML maintenance trial of CC-486 (oral azacitidine) maintenance therapy in acute myeloid leukemia. Future Oncol. 2016;12(3):293–302.
Wei AH, Döhner H, Pocock C, Montesinos P, Afanasyev B, Dombret H, et al. The QUAZAR AML-001 maintenance trial: results of a phase III international, randomized, double-blind, placebo-controlled study of CC-486 (oral formulation of azacitidine) in patients with acute myeloid leukemia (AML) in first remission. Blood. 2019;134(Suppl_2):LBA-3.
Kantarjian HM, Roboz GJ, Kropf PL, Yee KWL, O’Connell CL, Tibes R, et al. Guadecitabine (SGI-110) in treatment-naive patients with acute myeloid leukaemia: phase 2 results from a multicentre, randomised, phase 1/2 trial. Lancet Oncol. 2017;18(10):1317–26.
Issa GC, Kantarjian HM, Xiao L, Ning J, Alvarado Y, Borthakur G, et al. Phase II trial of CPX-351 in patients with acute myeloid leukemia at high risk for induction mortality. Leukemia. 2020;34(11):2914–24.
Roboz GJ, Kantarjian HM, Yee KWL, Kropf PL, O’Connell CL, Griffiths EA, et al. Dose, schedule, safety, and efficacy of guadecitabine in relapsed or refractory acute myeloid leukemia. Cancer. 2018;124(2):325–34.
Astex Pharmaceuticals. Astex and Otsuka announce results of phase 3 ASTRAL-2 and ASTRAL-3 studies of guadecitabine (SGI-110) in patients with previously treated acute myeloid leukemia (AML) and myelodysplastic syndromes or chronic myelomonocytic leukemia (MDS/CMML). Pleasanton, CA: Astex Pharmaceuticals; 2020.
San José-Enériz E, Gimenez-Camino N, Agirre X, Prosper F. HDAC inhibitors in acute myeloid leukemia. Cancers (Basel). 2019;11(11):1794.
Fiskus W, Wang Y, Joshi R, Rao R, Yang Y, Chen J, et al. Cotreatment with vorinostat enhances activity of MK-0457 (VX-680) against acute and chronic myelogenous leukemia cells. Clin Cancer Res. 2008;14(19):6106–15.
Miller CP, Rudra S, Keating MJ, Wierda WG, Palladino M, Chandra J. Caspase-8 dependent histone acetylation by a novel proteasome inhibitor, NPI-0052: a mechanism for synergy in leukemia cells. Blood. 2009;113(18):4289–99.
Shiozawa K, Nakanishi T, Tan M, Fang HB, Wang WC, Edelman MJ, et al. Preclinical studies of vorinostat (suberoylanilide hydroxamic acid) combined with cytosine arabinoside and etoposide for treatment of acute leukemias. Clin Cancer Res. 2009;15(5):1698–707.
Wei Y, Kadia T, Tong W, Zhang M, Jia Y, Yang H, et al. The combination of a histone deacetylase inhibitor with the BH3-mimetic GX15-070 has synergistic antileukemia activity by activating both apoptosis and autophagy. Autophagy. 2010;6(7):976–8.
Zhou L, Zhang Y, Chen S, Kmieciak M, Leng Y, Lin H, et al. A regimen combining the Wee1 inhibitor AZD1775 with HDAC inhibitors targets human acute myeloid leukemia cells harboring various genetic mutations. Leukemia. 2015;29(4):807–18.
Lin WH, Yeh TK, Jiaang WT, Yen KJ, Chen CH, Huang CT, et al. Evaluation of the antitumor effects of BPR1J-340, a potent and selective FLT3 inhibitor, alone or in combination with an HDAC inhibitor, vorinostat, in AML cancer. PLoS One. 2014;9(1):e83160.
Schaefer EW, Loaiza-Bonilla A, Juckett M, DiPersio JF, Roy V, Slack J, et al. A phase 2 study of vorinostat in acute myeloid leukemia. Haematologica. 2009;94(10):1375–82.
Kadia TM, Yang H, Ferrajoli A, Maddipotti S, Schroeder C, Madden TL, et al. A phase I study of vorinostat in combination with idarubicin in relapsed or refractory leukaemia. Br J Haematol. 2010;150(1):72–82.
Garcia-Manero G, Tambaro FP, Bekele NB, Yang H, Ravandi F, Jabbour E, et al. Phase II trial of vorinostat with idarubicin and cytarabine for patients with newly diagnosed acute myelogenous leukemia or myelodysplastic syndrome. J Clin Oncol. 2012;30(18):2204–10.
Walter RB, Medeiros BC, Gardner KM, Orlowski KF, Gallegos L, Scott BL, et al. Gemtuzumab ozogamicin in combination with vorinostat and azacitidine in older patients with relapsed or refractory acute myeloid leukemia: a phase I/II study. Haematologica. 2014;99(1):54–9.
Walter RB, Medeiros BC, Powell BL, Schiffer CA, Appelbaum FR, Estey EH. Phase II trial of vorinostat and gemtuzumab ozogamicin as induction and post-remission therapy in older adults with previously untreated acute myeloid leukemia. Haematologica. 2012;97(5):739–42.
Kirschbaum M, Gojo I, Goldberg SL, Bredeson C, Kujawski LA, Yang A, et al. A phase 1 clinical trial of vorinostat in combination with decitabine in patients with acute myeloid leukaemia or myelodysplastic syndrome. Br J Haematol. 2014;167(2):185–93.
How J, Minden MD, Brian L, Chen EX, Brandwein J, Schuh AC, et al. A phase I trial of two sequence-specific schedules of decitabine and vorinostat in patients with acute myeloid leukemia. Leuk Lymphoma. 2015;56(10):2793–802.
Mims AS, Mishra A, Orwick S, Blachly J, Klisovic RB, Garzon R, et al. A novel regimen for relapsed/refractory adult acute myeloid leukemia using a KMT2A partial tandem duplication targeted therapy: results of phase 1 study NCI 8485. Haematologica. 2018;103(6):982–7.
Sayar H, Cripe LD, Saliba AN, Abu Zaid M, Konig H, Boswell HS. Combination of sorafenib, vorinostat and bortezomib for the treatment of poor-risk AML: report of two consecutive clinical trials. Leuk Res. 2019;77:30–3.
Craddock CF, Houlton AE, Quek LS, Ferguson P, Gbandi E, Roberts C, et al. Outcome of azacitidine therapy in acute myeloid leukemia is not improved by concurrent vorinostat therapy but is predicted by a diagnostic molecular signature. Clin Cancer Res. 2017;23(21):6430–40.
Holkova B, Supko JG, Ames MM, Reid JM, Shapiro GI, Perkins EB, et al. A phase I trial of vorinostat and alvocidib in patients with relapsed, refractory, or poor prognosis acute leukemia, or refractory anemia with excess blasts-2. Clin Cancer Res. 2013;19(7):1873–83.
Anne M, Sammartino D, Barginear MF, Budman D. Profile of panobinostat and its potential for treatment in solid tumors: an update. Onco Targets Ther. 2013;6:1613–24.
Blagitko-Dorfs N, Schlosser P, Greve G, Pfeifer D, Meier R, Baude A, et al. Combination treatment of acute myeloid leukemia cells with DNMT and HDAC inhibitors: predominant synergistic gene downregulation associated with gene body demethylation. Leukemia. 2019;33(4):945–56.
Fiskus W, Buckley K, Rao R, Mandawat A, Yang Y, Joshi R, et al. Panobinostat treatment depletes EZH2 and DNMT1 levels and enhances decitabine mediated de-repression of JunB and loss of survival of human acute leukemia cells. Cancer Biol Ther. 2009;8(10):939–50.
Gopalakrishnapillai A, Kolb EA, McCahan SM, Barwe SP. Epigenetic drug combination induces remission in mouse xenograft models of pediatric acute myeloid leukemia. Leuk Res. 2017;58:91–7.
Schwartz J, Niu X, Walton E, Hurley L, Lin H, Edwards H, et al. Synergistic anti-leukemic interactions between ABT-199 and panobinostat in acute myeloid leukemia ex vivo. Am J Transl Res. 2016;8(9):3893–902.
Qi W, Zhang W, Edwards H, Chu R, Madlambayan GJ, Taub JW, et al. Synergistic anti-leukemic interactions between panobinostat and MK-1775 in acute myeloid leukemia ex vivo. Cancer Biol Ther. 2015;16(12):1784–93.
Fiskus W, Sharma S, Saha S, Shah B, Devaraj SG, Sun B, et al. Pre-clinical efficacy of combined therapy with novel β-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia. 2015;29(6):1267–78.
Fiskus W, Sharma S, Shah B, Portier BP, Devaraj SG, Liu K, et al. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia. 2014;28(11):2155–64.
Fiskus W, Sharma S, Qi J, Valenta JA, Schaub LJ, Shah B, et al. Highly active combination of BRD4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol Cancer Ther. 2014;13(5):1142–54.
Pietschmann K, Bolck HA, Buchwald M, Spielberg S, Polzer H, Spiekermann K, et al. Breakdown of the FLT3-ITD/STAT5 axis and synergistic apoptosis induction by the histone deacetylase inhibitor panobinostat and FLT3-specific inhibitors. Mol Cancer Ther. 2012;11(11):2373–83.
Jiang XJ, Huang KK, Yang M, Qiao L, Wang Q, Ye JY, et al. Synergistic effect of panobinostat and bortezomib on chemoresistant acute myelogenous leukemia cells via AKT and NF-κB pathways. Cancer Lett. 2012;326(2):135–42.
Mandawat A, Fiskus W, Buckley KM, Robbins K, Rao R, Balusu R, et al. Pan-histone deacetylase inhibitor panobinostat depletes CXCR4 levels and signaling and exerts synergistic antimyeloid activity in combination with CXCR4 antagonists. Blood. 2010;116(24):5306–15.
Maiso P, Colado E, Ocio EM, Garayoa M, Martín J, Atadja P, et al. The synergy of panobinostat plus doxorubicin in acute myeloid leukemia suggests a role for HDAC inhibitors in the control of DNA repair. Leukemia. 2009;23(12):2265–74.
Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin a and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114(13):2733–43.
Ocio EM, Herrera P, Olave MT, Castro N, Pérez-Simón JA, Brunet S, et al. Panobinostat as part of induction and maintenance for elderly patients with newly diagnosed acute myeloid leukemia: phase Ib/II panobidara study. Haematologica. 2015;100(10):1294–300.
Wieduwilt MJ, Pawlowska N, Thomas S, Olin R, Logan AC, Damon LE, et al. Histone deacetylase inhibition with panobinostat combined with intensive induction chemotherapy in older patients with acute myeloid leukemia: Phase I study results. Clin Cancer Res. 2019;25(16):4917–23.
Garcia-Manero G, Sekeres MA, Egyed M, Breccia M, Graux C, Cavenagh JD, et al. A phase 1b/2b multicenter study of oral panobinostat plus azacitidine in adults with MDS, CMML or AML with ⩽30% blasts. Leukemia. 2017;31(12):2799–806.
Schlenk RF, Krauter J, Raffoux E, Kreuzer KA, Schaich M, Noens L, et al. Panobinostat monotherapy and combination therapy in patients with acute myeloid leukemia: results from two clinical trials. Haematologica. 2018;103(1):e25–e8.
Dai Y, Chen S, Wang L, Pei XY, Kramer LB, Dent P, et al. Bortezomib interacts synergistically with belinostat in human acute myeloid leukaemia and acute lymphoblastic leukaemia cells in association with perturbations in NF-κB and Bim. Br J Haematol. 2011;153(2):222–35.
Kirschbaum MH, Foon KA, Frankel P, Ruel C, Pulone B, Tuscano JM, et al. A phase 2 study of belinostat (PXD101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: a California cancer consortium study. Leuk Lymphoma. 2014;55(10):2301–4.
Holkova B, Tombes MB, Shrader E, Cooke SS, Wan W, Sankala H, et al. Phase I trial of belinostat and bortezomib in patients with relapsed or refractory acute leukemia, myelodysplastic syndrome, or chronic myelogenous leukemia in blast crisis. Blood. 2011;118(21):2598.
Holkova B, Bose P, Tombes MB, Shrader E, Wan W, Weir-Wiggins C, et al. Phase I trial of belinostat and bortezomib in patients with relapsed or refractory acute leukemia, myelodysplastic syndrome, or chronic myelogenous leukemia in blast crisis—one year update. Blood. 2012;120(21):3588.
Abaza YM, Kadia TM, Jabbour EJ, Konopleva MY, Borthakur G, Ferrajoli A, et al. Phase 1 dose escalation multicenter trial of pracinostat alone and in combination with azacitidine in patients with advanced hematologic malignancies. Cancer. 2017;123(24):4851–9.
ClinicalTrials.gov. An efficacy and safety study of pracinostat in combination with azacitidine in adults with acute myeloid leukemia. 2020. https://clinicaltrials.gov/ct2/show/NCT03151408.
Barbetti V, Gozzini A, Rovida E, Morandi A, Spinelli E, Fossati G, et al. Selective anti-leukaemic activity of low-dose histone deacetylase inhibitor ITF2357 on AML1/ETO-positive cells. Oncogene. 2008;27(12):1767–78.
Golay J, Cuppini L, Leoni F, Micò C, Barbui V, Domenghini M, et al. The histone deacetylase inhibitor ITF2357 has anti-leukemic activity in vitro and in vivo and inhibits IL-6 and VEGF production by stromal cells. Leukemia. 2007;21(9):1892–900.
Zabkiewicz J, Gilmour M, Hills R, Vyas P, Bone E, Davidson A, et al. The targeted histone deacetylase inhibitor tefinostat (CHR-2845) shows selective in vitro efficacy in monocytoid-lineage leukaemias. Oncotarget. 2016;7(13):16650–62.
Vey N, Prebet T, Thalamas C, Charbonnier A, Rey J, Kloos I, et al. Phase 1 dose-escalation study of oral abexinostat for the treatment of patients with relapsed/refractory higher-risk myelodysplastic syndromes, acute myeloid leukemia, or acute lymphoblastic leukemia. Leuk Lymphoma. 2017;58(8):1880–6.
Li Y, Chen K, Zhou Y, Xiao Y, Deng M, Jiang Z, et al. A new strategy to target acute myeloid leukemia stem and progenitor cells using Chidamide, a histone Deacetylase inhibitor. Curr Cancer Drug Targets. 2015;15(6):493–503.
Lin L, Que Y, Lu P, Li H, Xiao M, Zhu X, et al. Chidamide inhibits acute myeloid leukemia cell proliferation by lncRNA VPS9D1-AS1 Downregulation via MEK/ERK signaling pathway. Front Pharmacol. 2020;11:569651.
Mao J, Li S, Zhao H, Zhu Y, Hong M, Zhu H, et al. Effects of chidamide and its combination with decitabine on proliferation and apoptosis of leukemia cell lines. Am J Transl Res. 2018;10(8):2567–78.
Li Q, Huang JC, Liao DY, Wu Y. Chidamide plus decitabine synergistically induces apoptosis of acute myeloid leukemia cells by upregulating PERP. Am J Transl Res. 2020;12(7):3461–75.
Li X, Yan X, Guo W, Huang X, Huang J, Yu M, et al. Chidamide in FLT3-ITD positive acute myeloid leukemia and the synergistic effect in combination with cytarabine. Biomed Pharmacother. 2017;90:699–704.
Huang H, Wenbing Y, Dong A, He Z, Yao R, Guo W. Chidamide enhances the cytotoxicity of cytarabine and sorafenib in acute myeloid leukemia cells by modulating H3K9me3 and autophagy levels. Front Oncol. 2019;9:1276.
Wang H, Liu YC, Zhu CY, Yan F, Wang MZ, Chen XS, et al. Chidamide increases the sensitivity of refractory or relapsed acute myeloid leukemia cells to anthracyclines via regulation of the HDAC3 -AKT-P21-CDK2 signaling pathway. J Exp Clin Cancer Res. 2020;39(1):278.
Li Y, Wang Y, Zhou Y, Li J, Chen K, Zhang L, et al. Cooperative effect of chidamide and chemotherapeutic drugs induce apoptosis by DNA damage accumulation and repair defects in acute myeloid leukemia stem and progenitor cells. Clin Epigenetics. 2017;9:83.
Chen K, Yang Q, Zha J, Deng M, Zhou Y, Fu G, et al. Preclinical evaluation of a regimen combining chidamide and ABT-199 in acute myeloid leukemia. Cell Death Dis. 2020;11(9):778.
Ye J, Zha J, Shi Y, Li Y, Yuan D, Chen Q, et al. Co-inhibition of HDAC and MLL-menin interaction targets MLL-rearranged acute myeloid leukemia cells via disruption of DNA damage checkpoint and DNA repair. Clin Epigenetics. 2019;11(1):137.
Zhang H, Li L, Li M, Huang X, Xie W, Xiang W, et al. Combination of betulinic acid and chidamide inhibits acute myeloid leukemia by suppression of the HIF1α pathway and generation of reactive oxygen species. Oncotarget. 2017;8(55):94743–58.
Wang L, Luo J, Chen G, Fang M, Wei X, Li Y, et al. Chidamide, decitabine, cytarabine, aclarubicin, and granulocyte colony-stimulating factor (CDCAG) in patients with relapsed/refractory acute myeloid leukemia: a single-arm, phase 1/2 study. Clin Epigenetics. 2020;12(1):132.
Ramsey JM, Kettyle LM, Sharpe DJ, Mulgrew NM, Dickson GJ, Bijl JJ, et al. Entinostat prevents leukemia maintenance in a collaborating oncogene-dependent model of cytogenetically normal acute myeloid leukemia. Stem Cells. 2013;31(7):1434–45.
Nishioka C, Ikezoe T, Yang J, Takeuchi S, Koeffler HP, Yokoyama A. MS-275, a novel histone deacetylase inhibitor with selectivity against HDAC1, induces degradation of FLT3 via inhibition of chaperone function of heat shock protein 90 in AML cells. Leuk Res. 2008;32(9):1382–92.
Nishioka C, Ikezoe T, Yang J, Koeffler HP, Yokoyama A. Inhibition of MEK/ERK signaling synergistically potentiates histone deacetylase inhibitor-induced growth arrest, apoptosis and acetylation of histone H3 on p21waf1 promoter in acute myelogenous leukemia cell. Leukemia. 2008;22(7):1449–52.
Nishioka C, Ikezoe T, Yang J, Koeffler HP, Yokoyama A. Blockade of mTOR signaling potentiates the ability of histone deacetylase inhibitor to induce growth arrest and differentiation of acute myelogenous leukemia cells. Leukemia. 2008;22(12):2159–68.
Nishioka C, Ikezoe T, Yang J, Udaka K, Yokoyama A. Simultaneous inhibition of DNA methyltransferase and histone deacetylase induces p53-independent apoptosis via down-regulation of mcl-1 in acute myelogenous leukemia cells. Leuk Res. 2011;35(7):932–9.
Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood. 2007;109(7):2781–90.
Fandy TE, Herman JG, Kerns P, Jiemjit A, Sugar EA, Choi SH, et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood. 2009;114(13):2764–73.
Prebet T, Sun Z, Figueroa ME, Ketterling R, Melnick A, Greenberg PL, et al. Prolonged administration of azacitidine with or without entinostat for myelodysplastic syndrome and acute myeloid leukemia with myelodysplasia-related changes: results of the US leukemia intergroup trial E1905. J Clin Oncol. 2014;32(12):1242–8.
Lillico R, Lawrence CK, Lakowski TM. Selective DOT1L, LSD1, and HDAC class I inhibitors reduce HOXA9 expression in MLL-AF9 rearranged leukemia cells, but dysregulate the expression of many histone-modifying enzymes. J Proteome Res. 2018;17(8):2657–67.
Garcia-Manero G, Assouline S, Cortes J, Estrov Z, Kantarjian H, Yang H, et al. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood. 2008;112(4):981–9.
Yan B, Chen Q, Shimada K, Tang M, Li H, Gurumurthy A, et al. Histone deacetylase inhibitor targets CD123/CD47-positive cells and reverse chemoresistance phenotype in acute myeloid leukemia. Leukemia. 2019;33(4):931–44.
Shaker S, Bernstein M, Momparler LF, Momparler RL. Preclinical evaluation of antineoplastic activity of inhibitors of DNA methylation (5-aza-2′-deoxycytidine) and histone deacetylation (trichostatin a, depsipeptide) in combination against myeloid leukemic cells. Leuk Res. 2003;27(5):437–44.
Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, et al. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood. 2005;105(3):959–67.
Klimek VM, Fircanis S, Maslak P, Guernah I, Baum M, Wu N, et al. Tolerability, pharmacodynamics, and pharmacokinetics studies of depsipeptide (romidepsin) in patients with acute myelogenous leukemia or advanced myelodysplastic syndromes. Clin Cancer Res. 2008;14(3):826–32.
Craddock C, Tholouli E, Munoz Vicente S, Gbandi E, Houlton AE, Drummond MW, et al. Safety and clinical activity of combined romidepsin and azacitidine therapy in high risk acute myeloid leukemia: preliminary results of the romaza trial. Blood. 2017;130(Suppl 1):2581.
Kosugi H, Towatari M, Hatano S, Kitamura K, Kiyoi H, Kinoshita T, et al. Histone deacetylase inhibitors are the potent inducer/enhancer of differentiation in acute myeloid leukemia: a new approach to anti-leukemia therapy. Leukemia. 1999;13(9):1316–24.
Maeda T, Towatari M, Kosugi H, Saito H. Up-regulation of costimulatory/adhesion molecules by histone deacetylase inhibitors in acute myeloid leukemia cells. Blood. 2000;96(12):3847–56.
Fredly H, Gjertsen BT, Bruserud O. Histone deacetylase inhibition in the treatment of acute myeloid leukemia: the effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents. Clin Epigenetics. 2013;5(1):12.
Trus MR, Yang L, Suarez Saiz F, Bordeleau L, Jurisica I, Minden MD. The histone deacetylase inhibitor valproic acid alters sensitivity towards all trans retinoic acid in acute myeloblastic leukemia cells. Leukemia. 2005;19(7):1161–8.
Liu N, Wang C, Wang L, Gao L, Cheng H, Tang G, et al. Valproic acid enhances the antileukemic effect of cytarabine by triggering cell apoptosis. Int J Mol Med. 2016;37(6):1686–96.
ten Cate B, Samplonius DF, Bijma T, de Leij LF, Helfrich W, Bremer E. The histone deacetylase inhibitor valproic acid potently augments gemtuzumab ozogamicin-induced apoptosis in acute myeloid leukemic cells. Leukemia. 2007;21(2):248–52.
Nie D, Huang K, Yin S, Li Y, Xie S, Ma L, et al. Synergistic/additive interaction of valproic acid with bortezomib on proliferation and apoptosis of acute myeloid leukemia cells. Leuk Lymphoma. 2012;53(12):2487–95.
Wang AH, Wei L, Chen L, Zhao SQ, Wu WL, Shen ZX, et al. Synergistic effect of bortezomib and valproic acid treatment on the proliferation and apoptosis of acute myeloid leukemia and myelodysplastic syndrome cells. Ann Hematol. 2011;90(8):917–31.
Heo SK, Noh EK, Yoon DJ, Jo JC, Park JH, Kim H. Dasatinib accelerates valproic acid-induced acute myeloid leukemia cell death by regulation of differentiation capacity. PLoS One. 2014;9(2):e98859.
McCormack E, Haaland I, Venås G, Forthun RB, Huseby S, Gausdal G, et al. Synergistic induction of p53 mediated apoptosis by valproic acid and nutlin-3 in acute myeloid leukemia. Leukemia. 2012;26(5):910–7.
Fuchs O, Provaznikova D, Marinov I, Kuzelova K, Spicka I. Antiproliferative and proapoptotic effects of proteasome inhibitors and their combination with histone deacetylase inhibitors on leukemia cells. Cardiovasc Hematol Disord Drug Targets. 2009;9(1):62–77.
Chen J, Wang G, Wang L, Kang J, Wang J. Curcumin p38-dependently enhances the anticancer activity of valproic acid in human leukemia cells. Eur J Pharm Sci. 2010;41(2):210–8.
Guo SQ, Zhang YZ. Histone deacetylase inhibition: an important mechanism in the treatment of lymphoma. Cancer Biol Med. 2012;9(2):85–9.
Fredly H, Stapnes Bjørnsen C, Gjertsen BT, Bruserud Ø. Combination of the histone deacetylase inhibitor valproic acid with oral hydroxyurea or 6-mercaptopurin can be safe and effective in patients with advanced acute myeloid leukaemia—a report of five cases. Hematology. 2010;15(5):338–43.
Soriano AO, Yang H, Faderl S, Estrov Z, Giles F, Ravandi F, et al. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110(7):2302–8.
Issa JP, Garcia-Manero G, Huang X, Cortes J, Ravandi F, Jabbour E, et al. Results of phase 2 randomized study of low-dose decitabine with or without valproic acid in patients with myelodysplastic syndrome and acute myelogenous leukemia. Cancer. 2015;121(4):556–61.
Blum W, Klisovic RB, Hackanson B, Liu Z, Liu S, Devine H, et al. Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. J Clin Oncol. 2007;25(25):3884–91.
Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, et al. Phase 1/2 study of the combination of 5-aza-2′-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108(10):3271–9.
Corsetti MT, Salvi F, Perticone S, Baraldi A, De Paoli L, Gatto S, et al. Hematologic improvement and response in elderly AML/RAEB patients treated with valproic acid and low-dose Ara-C. Leuk Res. 2011;35(8):991–7.
Lane S, Gill D, McMillan NA, Saunders N, Murphy R, Spurr T, et al. Valproic acid combined with cytosine arabinoside in elderly patients with acute myeloid leukemia has in vitro but limited clinical activity. Leuk Lymphoma. 2012;53(6):1077–83.
Kuendgen A, Schmid M, Schlenk R, Knipp S, Hildebrandt B, Steidl C, et al. The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia. Cancer. 2006;106(1):112–9.
Kuendgen A, Knipp S, Fox F, Strupp C, Hildebrandt B, Steidl C, et al. Results of a phase 2 study of valproic acid alone or in combination with all-trans retinoic acid in 75 patients with myelodysplastic syndrome and relapsed or refractory acute myeloid leukemia. Ann Hematol. 2005;84(Suppl 1):61–6.
Raffoux E, Chaibi P, Dombret H, Degos L. Valproic acid and all-trans retinoic acid for the treatment of elderly patients with acute myeloid leukemia. Haematologica. 2005;90(7):986–8.
Bug G, Ritter M, Wassmann B, Schoch C, Heinzel T, Schwarz K, et al. Clinical trial of valproic acid and all-trans retinoic acid in patients with poor-risk acute myeloid leukemia. Cancer. 2005;104(12):2717–25.
Tassara M, Döhner K, Brossart P, Held G, Götze K, Horst HA, et al. Valproic acid in combination with all-trans retinoic acid and intensive therapy for acute myeloid leukemia in older patients. Blood. 2014;123(26):4027–36.
Gambacorta V, Gnani D, Vago L, Di Micco R. Epigenetic therapies for acute myeloid leukemia and their immune-related effects. Front Cell Dev Biol. 2019;7:207.
Schenk T, Chen WC, Göllner S, Howell L, Jin L, Hebestreit K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18(4):605–11.
Wass M, Göllner S, Besenbeck B, Schlenk RF, Mundmann P, Göthert JR, et al. A proof of concept phase I/II pilot trial of LSD1 inhibition by tranylcypromine combined with ATRA in refractory/relapsed AML patients not eligible for intensive therapy. Leukemia. 2021;35(3):701–11.
Sharma SK, Wu Y, Steinbergs N, Crowley ML, Hanson AS, Casero RA, et al. (Bis)urea and (Bis)thiourea inhibitors of lysine-specific demethylase 1 as epigenetic modulators. J Med Chem. 2010;53(14):5197–212.
Schmitt ML, Hauser A-T, Carlino L, Pippel M, Schulz-Fincke J, Metzger E, et al. Nonpeptidic propargylamines as inhibitors of lysine specific demethylase 1 (LSD1) with cellular activity. J Med Chem. 2013;56(18):7334–42.
Zheng Y-C, Duan Y-C, Ma J-L, Xu R-M, Zi X, Lv W-L, et al. Triazole–dithiocarbamate based selective lysine specific demethylase 1 (LSD1) inactivators inhibit gastric cancer cell growth, invasion, and migration. J Med Chem. 2013;56(21):8543–60.
Ma L-Y, Zheng Y-C, Wang S-Q, Wang B, Wang Z-R, Pang L-P, et al. Design, synthesis, and structure–activity relationship of novel LSD1 inhibitors based on pyrimidine–thiourea hybrids as potent, orally active antitumor agents. J Med Chem. 2015;58(4):1705–16.
Itoh Y, Aihara K, Mellini P, Tojo T, Ota Y, Tsumoto H, et al. Identification of SNAIL1 peptide-based irreversible lysine-specific demethylase 1-selective inactivators. J Med Chem. 2016;59(4):1531–44.
Wu F, Zhou C, Yao Y, Wei L, Feng Z, Deng L, et al. 3-(Piperidin-4-ylmethoxy)pyridine containing compounds are potent inhibitors of lysine specific demethylase 1. J Med Chem. 2016;59(1):253–63.
Borrello MT, Schinor B, Bartels K, Benelkebir H, Pereira S, Al-Jamal WT, et al. Fluorinated tranylcypromine analogues as inhibitors of lysine-specific demethylase 1 (LSD1, KDM1A). Bioorg Med Chem Lett. 2017;27(10):2099–101.
Sartori L, Mercurio C, Amigoni F, Cappa A, Fagá G, Fattori R, et al. Thieno[3,2-b]pyrrole-5-carboxamides as new reversible inhibitors of histone lysine demethylase KDM1A/LSD1. Part 1: high-throughput screening and preliminary exploration. J Med Chem. 2017;60(5):1673–92.
Sugino N, Kawahara M, Tatsumi G, Kanai A, Matsui H, Yamamoto R, et al. A novel LSD1 inhibitor NCD38 ameliorates MDS-related leukemia with complex karyotype by attenuating leukemia programs via activating super-enhancers. Leukemia. 2017;31(11):2303–14.
Yang C, Wang W, Liang J-X, Li G, Vellaisamy K, Wong C-Y, et al. A rhodium(III)-based inhibitor of lysine-specific histone demethylase 1 as an epigenetic modulator in prostate cancer cells. J Med Chem. 2017;60(6):2597–603.
Liu HM, Suo FZ, Li XB, You YH, Lv CT, Zheng CX, et al. Discovery and synthesis of novel indole derivatives-containing 3-methylenedihydrofuran-2(3H)-one as irreversible LSD1 inhibitors. Eur J Med Chem. 2019;175:357–72.
Liang L, Wang H, Du Y, Luo B, Meng N, Cen M, et al. New tranylcypromine derivatives containing sulfonamide motif as potent LSD1 inhibitors to target acute myeloid leukemia: design, synthesis and biological evaluation. Bioorg Chem. 2020;99:103808.
Maes T, Mascaró C, Tirapu I, Estiarte A, Ciceri F, Lunardi S, et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell. 2018;33(3):495–511.e12.
Salamero O, Montesinos P, Willekens C, Pérez-Simón JA, Pigneux A, Récher C, et al. First-in-human phase I study of Iadademstat (ORY-1001): a first-in-class lysine-specific histone demethylase 1A inhibitor, in relapsed or refractory acute myeloid leukemia. J Clin Oncol. 2020;38(36):4260–73.
Smitheman KN, Severson TM, Rajapurkar SR, McCabe MT, Karpinich N, Foley J, et al. Lysine specific demethylase 1 inactivation enhances differentiation and promotes cytotoxic response when combined with all-trans retinoic acid in acute myeloid leukemia across subtypes. Haematologica. 2019;104(6):1156–67.
Reyes-Garau D, Ribeiro ML, Roué G. Pharmacological targeting of BET bromodomain proteins in acute myeloid leukemia and malignant lymphomas: from molecular characterization to clinical applications. Cancers (Basel). 2019;11(10):1483.
Herrmann H, Blatt K, Shi J, Gleixner KV, Cerny-Reiterer S, Müllauer L, et al. Small-molecule inhibition of BRD4 as a new potent approach to eliminate leukemic stem- and progenitor cells in acute myeloid leukemia AML. Oncotarget. 2012;3(12):1588–99.
Kang C, Kim CY, Kim HS, Park SP, Chung HM. The bromodomain inhibitor JQ1 enhances the responses to all-trans retinoic acid in HL-60 and MV4-11 leukemia cells. Int J Stem Cells. 2018;11(1):131–40.
Pericole FV, Lazarini M, de Paiva LB, Duarte ADSS, Vieira Ferro KP, Niemann FS, et al. BRD4 inhibition enhances azacitidine efficacy in acute myeloid leukemia and myelodysplastic syndromes. Front Oncol. 2019;9:16.
Fiskus W, Sharma S, Qi J, Shah B, Devaraj SG, Leveque C, et al. BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (TKI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD. Mol Cancer Ther. 2014;13(10):2315–27.
Gerlach D, Tontsch-Grunt U, Baum A, Popow J, Scharn D, Hofmann MH, et al. The novel BET bromodomain inhibitor BI 894999 represses super-enhancer-associated transcription and synergizes with CDK9 inhibition in AML. Oncogene. 2018;37(20):2687–701.
Coudé MM, Braun T, Berrou J, Dupont M, Bertrand S, Masse A, et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget. 2015;6(19):17698–712.
Gillberg L, Ørskov AD, Nasif A, Ohtani H, Madaj Z, Hansen JW, et al. Oral vitamin C supplementation to patients with myeloid cancer on azacitidine treatment: normalization of plasma vitamin C induces epigenetic changes. Clin Epigenetics. 2019;11(1):143.
Mastrangelo D, Massai L, Fioritoni G, Coco FL, Noguera N, Testa U. High doses of vitamin c and leukemia: in vitro update. In: Myeloid leukemia. London: InTech; 2018.
Zhao H, Huayuan Z, Yu Z, Li J, Qian S. The synergy of vitamin C with decitabine activates TET2 in leukemic cells and significantly improves overall survival in elderly patients with acute myeloid leukemia. Blood. 2017;130(Suppl 1):1339.
Cao F, Townsend EC, Karatas H, Xu J, Li L, Lee S, et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell. 2014;53(2):247–61.
Borkin D, He S, Miao H, Kempinska K, Pollock J, Chase J, et al. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell. 2015;27(4):589–602.
He S, Senter TJ, Pollock J, Han C, Upadhyay SK, Purohit T, et al. High-affinity small-molecule inhibitors of the menin-mixed lineage leukemia (MLL) interaction closely mimic a natural protein-protein interaction. J Med Chem. 2014;57(4):1543–56.
Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. 2012;8(3):277–84.
Dzama MM, Steiner M, Rausch J, Sasca D, Schönfeld J, Kunz K, et al. Synergistic targeting of FLT3 mutations in AML via combined menin-MLL and FLT3 inhibition. Blood. 2020;136(21):2442–56.
Klossowski S, Miao H, Kempinska K, Wu T, Purohit T, Kim E, et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J Clin Invest. 2020;130(2):981–97.
Rau RE, Rodriguez BA, Luo M, Jeong M, Rosen A, Rogers JH, et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood. 2016;128(7):971–81.
Kühn MW, Hadler MJ, Daigle SR, Koche RP, Krivtsov AV, Olhava EJ, et al. MLL partial tandem duplication leukemia cells are sensitive to small molecule DOT1L inhibition. Haematologica. 2015;100(5):e190–3.
Stein EM, Garcia-Manero G, Rizzieri DA, Tibes R, Berdeja JG, Savona MR, et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood. 2018;131(24):2661–9.
Liu W, Deng L, Song Y, Redell M. DOT1L inhibition sensitizes MLL-rearranged AML to chemotherapy. PLoS One. 2014;9(5):e98270.
Ueda K, Yoshimi A, Kagoya Y, Nishikawa S, Marquez VE, Nakagawa M, et al. Inhibition of histone methyltransferase EZH2 depletes leukemia stem cell of mixed lineage leukemia fusion leukemia through upregulation of p16. Cancer Sci. 2014;105(5):512–9.
Zhou J, Bi C, Cheong LL, Mahara S, Liu SC, Tay KG, et al. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood. 2011;118(10):2830–9.
Jiang X, Lim CZ, Li Z, Lee PL, Yatim SM, Guan P, et al. Functional characterization of D9, a novel Deazaneplanocin a (DZNep) analog, in targeting acute myeloid leukemia (AML). PLoS One. 2015;10(4):e0122983.
Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood. 2015;125(2):346–57.
Cheung N, Fung TK, Zeisig BB, Holmes K, Rane JK, Mowen KA, et al. Targeting aberrant epigenetic networks mediated by PRMT1 and KDM4C in acute myeloid leukemia. Cancer Cell. 2016;29(1):32–48.
Kaushik S, Liu F, Veazey KJ, Gao G, Das P, Neves LF, et al. Genetic deletion or small-molecule inhibition of the arginine methyltransferase PRMT5 exhibit anti-tumoral activity in mouse models of MLL-rearranged AML. Leukemia. 2018;32(2):499–509.
Lin AB, McNeely SC, Beckmann RP. Achieving precision death with cell-cycle inhibitors that target DNA replication and repair. Clin Cancer Res. 2017;23(13):3232–40.
Esposito MT, So CW. DNA damage accumulation and repair defects in acute myeloid leukemia: implications for pathogenesis, disease progression, and chemotherapy resistance. Chromosoma. 2014;123(6):545–61.
Fritz C, Portwood SM, Przespolewski A, Wang ES. PARP goes the weasel! Emerging role of PARP inhibitors in acute leukemias. Blood Rev. 2021;45:100696.
Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–99.
Faraoni I, Compagnone M, Lavorgna S, Angelini DF, Cencioni MT, Piras E, et al. BRCA1, PARP1 and γH2AX in acute myeloid leukemia: role as biomarkers of response to the PARP inhibitor olaparib. Biochim Biophys Acta. 2015;1852(3):462–72.
Yamauchi T, Uzui K, Nishi R, Shigemi H, Ueda T. Gemtuzumab ozogamicin and olaparib exert synergistic cytotoxicity in CD33-positive HL-60 myeloid leukemia cells. Anticancer Res. 2014;34(10):5487–94.
Wang L, Cai W, Zhang W, Chen X, Dong W, Tang D, et al. Inhibition of poly(ADP-ribose) polymerase 1 protects against acute myeloid leukemia by suppressing the myeloproliferative leukemia virus oncogene. Oncotarget. 2015;6(29):27490–504.
Portwood S, Puchalski RA, Walker RM, Wang ES. Combining IMGN779, a novel anti-CD33 antibody-drug conjugate (ADC), with the PARP inhibitor, olaparib, results in enhanced anti-tumor activity in preclinical acute myeloid leukemia (AML) models. Blood. 2016;128(22):1645.
Robert C, Nagaria PK, Pawar N, Adewuyi A, Gojo I, Meyers DJ, et al. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk Res. 2016;45:14–23.
Muvarak NE, Chowdhury K, Xia L, Robert C, Choi EY, Cai Y, et al. Enhancing the cytotoxic effects of PARP inhibitors with DNA demethylating agents—a potential therapy for cancer. Cancer Cell. 2016;30(4):637–50.
Gaymes TJ, Shall S, MacPherson LJ, Twine NA, Lea NC, Farzaneh F, et al. Inhibitors of poly ADP-ribose polymerase (PARP) induce apoptosis of myeloid leukemic cells: potential for therapy of myeloid leukemia and myelodysplastic syndromes. Haematologica. 2009;94(5):638–46.
Garcia TB, Snedeker JC, Baturin D, Gardner L, Fosmire SP, Zhou C, et al. A small-molecule inhibitor of WEE1, AZD1775, synergizes with olaparib by impairing homologous recombination and enhancing DNA damage and apoptosis in acute leukemia. Mol Cancer Ther. 2017;16(10):2058–68.
Gojo I, Beumer JH, Pratz KW, McDevitt MA, Baer MR, Blackford AL, et al. A phase 1 study of the PARP inhibitor veliparib in combination with temozolomide in acute myeloid leukemia. Clin Cancer Res. 2017;23(3):697–706.
Pratz KW, Rudek MA, Gojo I, Litzow MR, McDevitt MA, Ji J, et al. A phase I study of topotecan, carboplatin and the PARP inhibitor veliparib in acute leukemias, aggressive myeloproliferative neoplasms, and chronic myelomonocytic leukemia. Clin Cancer Res. 2017;23(4):899–907.
Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med. 2017;9(375):eaal2463.
Fordham SE, Blair HJ, Elstob CJ, Plummer R, Drew Y, Curtin NJ, et al. Inhibition of ATR acutely sensitizes acute myeloid leukemia cells to nucleoside analogs that target ribonucleotide reductase. Blood Adv. 2018;2(10):1157–69.
Morgado-Palacin I, Day A, Murga M, Lafarga V, Anton ME, Tubbs A, et al. Targeting the kinase activities of ATR and ATM exhibits antitumoral activity in mouse models of MLL-rearranged AML. Sci Signal. 2016;9(445):ra91.
Ma J, Li X, Su Y, Zhao J, Luedtke DA, Epshteyn V, et al. Mechanisms responsible for the synergistic antileukemic interactions between ATR inhibition and cytarabine in acute myeloid leukemia cells. Sci Rep. 2017;7(1):41950.
Grosjean-Raillard J, Tailler M, Adès L, Perfettini JL, Fabre C, Braun T, et al. ATM mediates constitutive NF-kappaB activation in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene. 2009;28(8):1099–109.
David L, Fernandez-Vidal A, Bertoli S, Grgurevic S, Lepage B, Deshaies D, et al. CHK1 as a therapeutic target to bypass chemoresistance in AML. Sci Signal. 2016;9(445):ra90.
Vincelette ND, Ding H, Huehls AM, Flatten KS, Kelly RL, Kohorst MA, et al. Effect of CHK1 inhibition on CPX-351 cytotoxicity in vitro and ex vivo. Sci Rep. 2019;9(1):3617.
Zhao J, Niu X, Li X, Edwards H, Wang G, Wang Y, et al. Inhibition of CHK1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Oncotarget. 2016;7(23):34785–99.
David L, Manenti S, Récher C, Hoffmann JS, Didier C. Targeting ATR/CHK1 pathway in acute myeloid leukemia to overcome chemoresistance. Mol Cell Oncol. 2017;4(5):e1289293.
Dai Y, Chen S, Kmieciak M, Zhou L, Lin H, Pei XY, et al. The novel Chk1 inhibitor MK-8776 sensitizes human leukemia cells to HDAC inhibitors by targeting the intra-S checkpoint and DNA replication and repair. Mol Cancer Ther. 2013;12(6):878–89.
Webster JA, Tibes R, Morris L, Blackford AL, Litzow M, Patnaik M, et al. Randomized phase II trial of cytosine arabinoside with and without the CHK1 inhibitor MK-8776 in relapsed and refractory acute myeloid leukemia. Leuk Res. 2017;61:108–16.
Porter CC, Kim J, Fosmire S, Gearheart CM, van Linden A, Baturin D, et al. Integrated genomic analyses identify WEE1 as a critical mediator of cell fate and a novel therapeutic target in acute myeloid leukemia. Leukemia. 2012;26(6):1266–76.
Tibes R, McDonagh KT, Lekakis L, Bogenberger JM, Kim S, Frazer N, et al. Phase I study of the novel Cdc2/CDK1 and AKT inhibitor terameprocol in patients with advanced leukemias. Investig New Drugs. 2015;33(2):389–96.
Yang C, Boyson CA, Di Liberto M, Huang X, Hannah J, Dorn DC, et al. CDK4/6 inhibitor PD 0332991 sensitizes acute myeloid leukemia to cytarabine-mediated cytotoxicity. Cancer Res. 2015;75(9):1838–45.
Uras IZ, Walter GJ, Scheicher R, Bellutti F, Prchal-Murphy M, Tigan AS, et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood. 2016;127(23):2890–902.
Uras IZ, Maurer B, Nebenfuehr S, Zojer M, Valent P, Sexl V. Therapeutic vulnerabilities in FLT3-mutant AML unmasked by palbociclib. Int J Mol Sci. 2018;19(12):3987.
Li C, Liu L, Liang L, Xia Z, Li Z, Wang X, et al. AMG 925 is a dual FLT3/CDK4 inhibitor with the potential to overcome FLT3 inhibitor resistance in acute myeloid leukemia. Mol Cancer Ther. 2015;14(2):375–83.
Fröhling S, Agrawal M, Jahn N, Fransecky LR, Baldus CD, Wäsch R, et al. CDK4/6 inhibitor palbociclib for treatment of KMT2A-rearranged acute myeloid leukemia: interim analysis of the AMLSG 23-14 trial. Blood. 2016;128(22):1608.
Kadia TM, Konopleva MY, Garcia-Manero G, Benton CB, Wierda WG, Bose P, et al. Phase I study of palbociclib alone and in combination in patients with relapsed and refractory (R/R) leukemias. Blood. 2018;132(Suppl 1):4057.
Sorf A, Sucha S, Morell A, Novotna E, Staud F, Zavrelova A, et al. Targeting pharmacokinetic drug resistance in acute myeloid leukemia cells with CDK4/6 inhibitors. Cancers (Basel). 2020;12(6):1596.
Borthakur GM, Donnellan WB, Solomon SR, Abboud C, Nazha A, Mazan M, et al. SEL120—a first-in-class CDK8/19 inhibitor as a novel option for the treatment of acute myeloid leukemia and high-risk myelodysplastic syndrome—data from preclinical studies and introduction to a phase Ib clinical trial. Blood. 2019;134(Suppl_1):2651.
Pelish HE, Liau BB, Nitulescu II, Tangpeerachaikul A, Poss ZC, Da Silva DH, et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature. 2015;526(7572):273–6.
Chantkran W, Zheleva D, Frame S, Hsieh Y-C, Copland M. Combination of CYC065, a second generation CDK2/9 inhibitor, with venetoclax or standard chemotherapies—a novel therapeutic approach for acute myeloid leukaemia (AML). Blood. 2019;134(Suppl_1):3938.
Cidado J, Boiko S, Proia T, Ferguson D, Criscione SW, San Martin M, et al. AZD4573 is a highly selective CDK9 inhibitor that suppresses MCL-1 and induces apoptosis in hematologic cancer cells. Clin Cancer Res. 2020;26(4):922–34.
Luedtke DA, Su Y, Ma J, Li X, Buck SA, Edwards H, et al. Inhibition of CDK9 by voruciclib synergistically enhances cell death induced by the Bcl-2 selective inhibitor venetoclax in preclinical models of acute myeloid leukemia. Signal Transduct Target Ther. 2020;5(1):17.
Li KL, Bray SC, Iarossi D, Adams J, Zhong L, Noll B, et al. Investigation of a novel cyclin-dependent-kinase (CDK) inhibitor Cdki-73 as an effective treatment option for MLL-AML. Blood. 2015;126(23):1365.
Phillips DC, Jin S, Gregory GP, Zhang Q, Xue J, Zhao X, et al. A novel CDK9 inhibitor increases the efficacy of venetoclax (ABT-199) in multiple models of hematologic malignancies. Leukemia. 2020;34(6):1646–57.
Nishi R, Shigemi H, Negoro E, Okura M, Hosono N, Yamauchi T. Venetoclax and alvocidib are both cytotoxic to acute myeloid leukemia cells resistant to cytarabine and clofarabine. BMC Cancer. 2020;20(1):984.
Karp JE, Ross DD, Yang W, Tidwell ML, Wei Y, Greer J, et al. Timed sequential therapy of acute leukemia with flavopiridol: in vitro model for a phase I clinical trial. Clin Cancer Res. 2003;9(1):307–15.
Bogenberger J, Whatcott C, Hansen N, Delman D, Shi CX, Kim W, et al. Combined venetoclax and alvocidib in acute myeloid leukemia. Oncotarget. 2017;8(63):107206–22.
Karp JE, Passaniti A, Gojo I, Kaufmann S, Bible K, Garimella TS, et al. Phase I and pharmacokinetic study of flavopiridol followed by 1-beta-D-arabinofuranosylcytosine and mitoxantrone in relapsed and refractory adult acute leukemias. Clin Cancer Res. 2005;11(23):8403–12.
Zeidner JF, Karp JE. Clinical activity of alvocidib (flavopiridol) in acute myeloid leukemia. Leuk Res. 2015;39(12):1312–8.
Zeidner JF, Foster MC, Blackford AL, Litzow MR, Morris LE, Strickland SA, et al. Randomized multicenter phase II study of flavopiridol (alvocidib), cytarabine, and mitoxantrone (FLAM) versus cytarabine/daunorubicin (7+3) in newly diagnosed acute myeloid leukemia. Haematologica. 2015;100(9):1172–9.
Zeidner JF, Foster MC, Blackford AL, Litzow MR, Morris LE, Strickland SA, et al. Final results of a randomized multicenter phase II study of alvocidib, cytarabine, and mitoxantrone versus cytarabine and daunorubicin (7 + 3) in newly diagnosed high-risk acute myeloid leukemia (AML). Leuk Res. 2018;72:92–5.
Baker A, Gregory GP, Verbrugge I, Kats L, Hilton JJ, Vidacs E, et al. The CDK9 inhibitor dinaciclib exerts potent apoptotic and antitumor effects in preclinical models of MLL-rearranged acute myeloid leukemia. Cancer Res. 2016;76(5):1158–69.
Gojo I, Sadowska M, Walker A, Feldman EJ, Iyer SP, Baer MR, et al. Clinical and laboratory studies of the novel cyclin-dependent kinase inhibitor dinaciclib (SCH 727965) in acute leukemias. Cancer Chemother Pharmacol. 2013;72(4):897–908.
Willems E, Dedobbeleer M, Digregorio M, Lombard A, Lumapat PN, Rogister B. The functional diversity of aurora kinases: a comprehensive review. Cell Div. 2018;13(1):7.
Brunner AM, Blonquist TM, DeAngelo DJ, McMasters M, Winer ES, Hobbs GS, et al. Phase II clinical trial of alisertib, an Aurora a kinase inhibitor, in combination with induction chemotherapy in high-risk, untreated patients with acute myeloid leukemia. Blood. 2018;132(Suppl 1):766.
Löwenberg B, Muus P, Ossenkoppele G, Rousselot P, Cahn JY, Ifrah N, et al. Phase 1/2 study to assess the safety, efficacy, and pharmacokinetics of barasertib (AZD1152) in patients with advanced acute myeloid leukemia. Blood. 2011;118(23):6030–6.
Kantarjian HM, Sekeres MA, Ribrag V, Rousselot P, Garcia-Manero G, Jabbour EJ, et al. Phase I study assessing the safety and tolerability of barasertib (AZD1152) with low-dose cytosine arabinoside in elderly patients with AML. Clin Lymphoma Myeloma Leuk. 2013;13(5):559–67.
Ghelli Luserna di Rora’ A, Iacobucci I, Martinelli G. The cell cycle checkpoint inhibitors in the treatment of leukemias. J Hematol Oncol. 2017;10(1):77.
Brandwein JM. Targeting polo-like kinase 1 in acute myeloid leukemia. Ther Adv Hematol. 2015;6(2):80–7.
Gjertsen BT, Schöffski P. Discovery and development of the polo-like kinase inhibitor volasertib in cancer therapy. Leukemia. 2015;29(1):11–9.
Gumireddy K, Reddy MV, Cosenza SC, Boominathan R, Baker SJ, Papathi N, et al. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell. 2005;7(3):275–86.
Navada SC, Fruchtman SM, Odchimar-Reissig R, Demakos EP, Petrone ME, Zbyszewski PS, et al. A phase 1/2 study of rigosertib in patients with myelodysplastic syndromes (MDS) and MDS progressed to acute myeloid leukemia. Leuk Res. 2018;64:10–6.
Navada SC, Garcia-Manero G, OdchimarReissig R, Pemmaraju N, Alvarado Y, Ohanian MN, et al. Rigosertib in combination with azacitidine in patients with myelodysplastic syndromes or acute myeloid leukemia: results of a phase 1 study. Leuk Res. 2020;94:106369.
Rudolph D, Steegmaier M, Hoffmann M, Grauert M, Baum A, Quant J, et al. BI 6727, a polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res. 2009;15(9):3094–102.
Bug G, Schlenk RF, Müller-Tidow C, Lübbert M, Krämer A, Fleischer F, et al. Phase I/II study of BI 6727 (volasertib), an intravenous polo-like kinase-1 (Plk1) inhibitor, in patients with acute myeloid leukemia (AML): results of the dose finding for BI 6727 in combination with low-dose cytarabine. Blood. 2010;116(21):3316.
Bug G, Müller-Tidow C, Schlenk RF, Krämer A, Lübbert M, Krug U, et al. Phase I/II study of volasertib (BI 6727), an intravenous polo-like kinase (Plk) inhibitor, in patients with acute myeloid leukemia (AML): updated results of the dose finding phase I part for volasertib in combination with low-dose cytarabine (LD-Ara-C) and as monotherapy in relapsed/refractory AML. Blood. 2011;118(21):1549.
Döhner H, Lübbert M, Fiedler W, Fouillard L, Haaland A, Brandwein JM, et al. Randomized, phase 2 trial of low-dose cytarabine with or without volasertib in AML patients not suitable for induction therapy. Blood. 2014;124(9):1426–33.
Cortes J, Podoltsev N, Kantarjian H, Borthakur G, Zeidan AM, Stahl M, et al. Phase 1 dose escalation trial of volasertib in combination with decitabine in patients with acute myeloid leukemia. Int J Hematol. 2021;113(1):92–9.
Ridgefield C. Results of phase III study of volasertib for the treatment of acute myeloid leukemia presented at European Hematology Association Annual Meeting Boehringer Ingelheim. 2016. https://www.boehringer-ingelheim.us/press-release/results-phase-iii-study-volasertib-treatment-acute-myeloid-leukemia-presented-european.
Zeidan AM, Ridinger M, Lin TL, Becker PS, Schiller GJ, Patel PA, et al. A phase Ib study of onvansertib, a novel oral PLK1 inhibitor, in combination therapy for patients with relapsed or refractory acute myeloid leukemia. Clin Cancer Res. 2020;26(23):6132–40.
Lee K-H, Schlenk RF, Bug G, Müller-Tidow C, Waesch RM, Nachbaur D, et al. Polo-like kinase-1 (Plk-1) inhibitor BI 2536 induces mitotic arrest and apoptosis in vivo: first demonstration of target inhibition in the bone marrow of AML patients. Blood. 2008;112(11):2641.
Müller-Tidow C, Bug G, Schlenk R, Lübbert M, Krämer A, Krauter J, et al. Phase I/II study of BI 2536, an intravenous polo-like Kinase-1 (Plk-1) inhibitor, in elderly patients with relapsed or refractory acute myeloid leukemia (AML): first results of a multi-center trial. Blood. 2008;112(11):2973.
Müller-Tidow C, Bug G, Lübbert M, Krämer A, Krauter J, Valent P, et al. A randomized, open-label, phase I/II trial to investigate the maximum tolerated dose of the polo-like kinase inhibitor BI 2536 in elderly patients with refractory/relapsed acute myeloid leukaemia. Br J Haematol. 2013;163(2):214–22.
Hikichi Y, Honda K, Hikami K, Miyashita H, Kaieda I, Murai S, et al. TAK-960, a novel, orally available, selective inhibitor of polo-like kinase 1, shows broad-spectrum preclinical antitumor activity in multiple dosing regimens. Mol Cancer Ther. 2012;11(3):700–9.
Casolaro A, Golay J, Albanese C, Ceruti R, Patton V, Cribioli S, et al. The polo-like kinase 1 (PLK1) inhibitor NMS-P937 is effective in a new model of disseminated primary CD56+ acute monoblastic leukaemia. PLoS One. 2013;8(3):e58424.
Brenner AK, Reikvam H, Rye KP, Hagen KM, Lavecchia A, Bruserud Ø. CDC25 inhibition in acute myeloid leukemia-a study of patient heterogeneity and the effects of different inhibitors. Molecules. 2017;22(3):446.
Chae H-D, Dutta R, Tiu B, Hoff FW, Accordi B, Serafin V, et al. RSK inhibitor BI-D1870 inhibits acute myeloid leukemia cell proliferation by targeting mitotic exit. Oncotarget. 2020;11(25):2387–403.
Dutta R, Castellanos M, Tiu B, Chae H-D, Davis KL, Sakamoto KM. RSK inhibition suppresses AML proliferation through activation of DNA damage pathways and S phase arrest. Blood. 2016;128(22):2894.
Rashidi A, Uy GL. Targeting the microenvironment in acute myeloid leukemia. Curr Hematol Malig Rep. 2015;10(2):126–31.
Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119(17):3917–24.
Uy GL, Avigan D, Cortes JE, Becker PS, Chen RW, Liesveld JL, et al. Safety and tolerability of plerixafor in combination with cytarabine and daunorubicin in patients with newly diagnosed acute myeloid leukemia- preliminary results from a phase I study. Blood. 2011;118(21):82.
Roboz GJ, Scandura JM, Ritchie E, Dault Y, Lam L, Xie W, et al. Combining decitabine with plerixafor yields a high response rate in newly diagnosed older patients with AML. Blood. 2013;122(21):621.
Andreeff M, Borthakur G, Zeng Z, Kelly MA, Wang R-Y, McQueen TJ, et al. Mobilization and elimination of FLT3-ITD+ acute myelogenous leukemia (AML) stem/progenitor cells by plerixafor, G-CSF, and sorafenib: phase I trial results in relapsed/refractory AML patients. J Clin Oncol. 2014;32(15_suppl):7033.
Barbier V, Erbani J, Fiveash C, Davies JM, Tay J, Tallack MR, et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat Commun. 2020;11(1):2042.
DeAngelo DJ, Liesveld JL, Jonas BA, O’Dwyer ME, Bixby DL, Magnani JL, et al. A phase I/II study of GMI-1271, a novel E-selectin antagonist, in combination with induction chemotherapy in relapsed/refractory and elderly previously untreated acute myeloid leukemia; results to date. Blood. 2016;128(22):4049.
DeAngelo DJ, Jonas BA, Liesveld JL, Bixby DL, Advani AS, Marlton P, et al. GMI-1271 improves efficacy and safety of chemotherapy in R/R and newly diagnosed older patients with AML: results of a Phase 1/2 study. Blood. 2017;130(Suppl 1):894.
DeAngelo DJ, Jonas BA, Becker PS, O’Dwyer M, Advani AS, Marlton P, et al. GMI-1271, a novel E-selectin antagonist, combined with induction chemotherapy in elderly patients with untreated AML. J Clin Oncol. 2017;35(15_suppl):2560.
Stanchina M, Soong D, Zheng-Lin B, Watts JM, Taylor J. Advances in acute myeloid leukemia: recently approved therapies and drugs in development. Cancers (Basel). 2020;12(11):3225.
Castaigne S, Pautas C, Terré C, Raffoux E, Bordessoule D, Bastie J-N, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012;379(9825):1508–16.
Lambert J, Pautas C, Terré C, Raffoux E, Turlure P, Caillot D, et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica. 2019;104(1):113–9.
Amadori S, Suciu S, Selleslag D, Aversa F, Gaidano G, Musso M, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34(9):972–9.
Schlenk RF, Paschka P, Krzykalla J, Weber D, Kapp-Schwoerer S, Gaidzik VI, et al. Gemtuzumab ozogamicin in NPM1-mutated acute myeloid leukemia: early results from the prospective randomized AMLSG 09-09 Phase III study. J Clin Oncol. 2020;38(6):623–32.
Kung Sutherland MS, Walter RB, Jeffrey SC, Burke PJ, Yu C, Kostner H, et al. SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood. 2013;122(8):1455–63.
Bixby DL, Stein AS, Fathi AT, Kovacsovics TJ, Levy MY, Erba HP, et al. Vadastuximab talirine monotherapy in older patients with treatment naive CD33-positive acute myeloid leukemia (AML). Blood. 2016;128(22):590.
Stein EM, Walter RB, Erba HP, Fathi AT, Advani AS, Lancet JE, et al. A phase 1 trial of vadastuximab talirine as monotherapy in patients with CD33-positive acute myeloid leukemia. Blood. 2018;131(4):387–96.
Fathi AT, Erba HP, Lancet JE, Stein EM, Ravandi F, Faderl S, et al. Vadastuximab talirine plus hypomethylating agents: a well-tolerated regimen with high remission rate in frontline older patients with acute myeloid leukemia (AML). Blood. 2016;128(22):591.
Fathi AT, Erba HP, Lancet JE, Stein EM, Ravandi F, Faderl S, et al. A phase 1 trial of vadastuximab talirine combined with hypomethylating agents in patients with CD33-positive AML. Blood. 2018;132(11):1125–33.
Hofland P. Phase III CASCADE clinical trial of vadastuximab talirine in frontline acute myeloid leukemia discontinued ADC review. Journal of Antibody-Drug Conjugates. 2017. https://www.adcreview.com/news/phase-iii-cascade-clinical-trial-vadastuximab-talirine-frontline-acute-myeloid-leukemia-discontinued/.
Whiteman KR, Noordhuis P, Walker R, Watkins K, Kovtun Y, Harvey L, et al. The antibody-drug conjugate (ADC) IMGN779 is highly active in vitro and in vivo against acute myeloid leukemia (AML) with FLT3-ITD mutations. Blood. 2014;124(21):2321.
Kovtun Y, Noordhuis P, Whiteman KR, Watkins K, Jones GE, Harvey L, et al. IMGN779, a novel CD33-targeting antibody-drug conjugate with DNA-alkylating activity, exhibits potent antitumor activity in models of AML. Mol Cancer Ther. 2018;17(6):1271–9.
Cortes JE, DeAngelo DJ, Erba HP, Traer E, Papadantonakis N, Arana-Yi C, et al. Maturing clinical profile of IMGN779, a next-generation CD33-targeting antibody-drug conjugate, in patients with relapsed or refractory acute myeloid leukemia. Blood. 2018;132(Suppl 1):26.
Daver NG, Erba HP, Papadantonakis N, DeAngelo DJ, Wang ES, Konopleva MY, et al. A phase I, first-in-human study evaluating the safety and preliminary Antileukemia activity of IMGN632, a novel CD123-targeting antibody-drug conjugate, in patients with relapsed/refractory acute myeloid leukemia and other CD123-positive hematologic malignancies. Blood. 2018;132(Suppl 1):27.
Kuruvilla VM, Zhang Q, Daver N, Watkins K, Sloss CM, Zweidler-McKay PA, et al. Combining IMGN632, a novel CD123-targeting antibody drug conjugate with azacitidine and venetoclax facilitates apoptosis in vitro and prolongs survival in vivo in AML models. Blood. 2020;136(Suppl 1):32–3.
Williams BA, Law A, Hunyadkurti J, Desilets S, Leyton JV, Keating A. Antibody therapies for acute myeloid leukemia: unconjugated, toxin-conjugated, radio-conjugated and multivalent formats. J Clin Med. 2019;8(8):1261.
Matthews DC, Appelbaum FR, Eary JF, Fisher DR, Durack LD, Hui TE, et al. Phase I study of (131)I-anti-CD45 antibody plus cyclophosphamide and total body irradiation for advanced acute leukemia and myelodysplastic syndrome. Blood. 1999;94(4):1237–47.
Pagel JM, Appelbaum FR, Eary JF, Rajendran J, Fisher DR, Gooley T, et al. 131I-anti-CD45 antibody plus busulfan and cyclophosphamide before allogeneic hematopoietic cell transplantation for treatment of acute myeloid leukemia in first remission. Blood. 2006;107(5):2184–91.
Pagel JM, Gooley TA, Rajendran J, Fisher DR, Wilson WA, Sandmaier BM, et al. Allogeneic hematopoietic cell transplantation after conditioning with 131I-anti-CD45 antibody plus fludarabine and low-dose total body irradiation for elderly patients with advanced acute myeloid leukemia or high-risk myelodysplastic syndrome. Blood. 2009;114(27):5444–53.
Mawad R, Gooley TA, Rajendran JG, Fisher DR, Gopal AK, Shields AT, et al. Radiolabeled anti-CD45 antibody with reduced-intensity conditioning and allogeneic transplantation for younger patients with advanced acute myeloid leukemia or myelodysplastic syndrome. Biol Blood Marrow Transplant. 2014;20(9):1363–8.
Orozco JJ, Kenoyer A, Balkin ER, Gooley TA, Hamlin DK, Wilbur DS, et al. Anti-CD45 radioimmunotherapy without TBI before transplantation facilitates persistent haploidentical donor engraftment. Blood. 2016;127(3):352–9.
Orozco JJ, Zeller J, Pagel JM. Radiolabeled antibodies directed at CD45 for conditioning prior to allogeneic transplantation in acute myeloid leukemia and myelodysplastic syndrome. Ther Adv Hematol. 2012;3(1):5–16.
Jurcic JG, Ravandi F, Pagel JM, Park JH, Smith BD, Douer D, et al. Phase I trial of α-particle therapy with actinium-225 (225Ac)-lintuzumab (anti-CD33) and low-dose cytarabine (LDAC) in older patients with untreated acute myeloid leukemia (AML). J Clin Oncol. 2015;33(15_suppl):7050.
Ravandi F, Assi R, Daver N, Benton CB, Kadia T, Thompson PA, et al. Idarubicin, cytarabine, and nivolumab in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: a single-arm, phase 2 study. Lancet Haematol. 2019;6(9):e480–8.
Guy DG, Uy GL. Bispecific antibodies for the treatment of acute myeloid leukemia. Curr Hematol Malig Rep. 2018;13(6):417–25.
Taghiloo S, Asgarian-Omran H. Immune evasion mechanisms in acute myeloid leukemia: a focus on immune checkpoint pathways. Crit Rev Oncol Hematol. 2021;157:103164.
Daver N, Garcia-Manero G, Basu S, Boddu PC, Alfayez M, Cortes JE, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-label, phase II study. Cancer Discov. 2019;9(3):370–83.
Daver NG, Garcia-Manero G, Konopleva MY, Alfayez M, Pemmaraju N, Kadia TM, et al. Azacitidine (AZA) with nivolumab (Nivo), and AZA with Nivo + ipilimumab (Ipi) in relapsed/refractory acute myeloid leukemia: a non-randomized, prospective, phase 2 study. Blood. 2019;134(Suppl_1):830.
Liao D, Wang M, Liao Y, Li J, Niu T. A review of efficacy and safety of checkpoint inhibitor for the treatment of acute myeloid leukemia. Front Pharmacol. 2019;10:609.
Gojo I, Stuart RK, Webster J, Blackford A, Varela JC, Morrow J, et al. Multi-center phase 2 study of pembroluzimab (Pembro) and azacitidine (AZA) in patients with relapsed/refractory acute myeloid leukemia (AML) and in newly diagnosed (≥65 years) AML patients. Blood. 2019;134(Suppl_1):832.
Lindblad KE, Thompson J, Gui G, Valdez J, Worthy T, Tekleab H, et al. Pembrolizumab and Decitabine for refractory or relapsed acute myeloid leukemia. Blood. 2018;132(Suppl 1):1437.
Zeidner JF, Vincent BG, Esparza S, Ivanova A, Moore DT, Foster MC, et al. Final clinical results of a phase II study of high dose cytarabine followed by pembrolizumab in relapsed/refractory AML. Blood. 2019;134(Suppl_1):831.
Zheng H, Mineishi S, Claxton DF, Zhu J, Zhao C, Jia B, et al. Effect of avelumab to immune response in AML: a phase I study of avelumab in combination with decitabine as first line treatment of unfit patients. Blood. 2019;134(Suppl_1):3939.
Zeidan AM, Cavenagh J, Voso MT, Taussig D, Tormo M, Boss I, et al. Efficacy and safety of azacitidine (AZA) in combination with the anti-PD-L1 durvalumab (durva) for the front-line treatment of older patients (pts) with acute myeloid leukemia (AML) who are unfit for intensive chemotherapy (IC) and Pts with higher-risk myelodysplastic syndromes (HR-MDS): results from a large, international, randomized phase 2 study. Blood. 2019;134(Suppl_1):829.
Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, Savell A, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med. 2016;375(2):143–53.
Borate U, Esteve J, Porkka K, Knapper S, Vey N, Scholl S, et al. Phase Ib study of the anti-TIM-3 antibody MBG453 in combination with decitabine in patients with high-risk myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). Blood. 2019;134(Suppl_1):570.
Chao MP, Takimoto CH, Feng DD, McKenna K, Gip P, Liu J, et al. Therapeutic targeting of the macrophage immune checkpoint CD47 in myeloid malignancies. Front Oncol. 2019;9:1380.
Sallman DA, Asch AS, Al Malki MM, Lee DJ, Donnellan WB, Marcucci G, et al. The first-in-class anti-CD47 antibody magrolimab (5F9) in combination with azacitidine is effective in MDS and AML patients: ongoing phase 1b results. Blood. 2019;134(Suppl_1):569.
Deng J, Zhao S, Zhang X, Jia K, Wang H, Zhou C, He Y. OX40 (CD134) and OX40 ligand, important immune checkpoints in cancer. OncoTargets and therapy. 2019;12:7347.
Fan G, Wang Z, Hao M, Li J. Bispecific antibodies and their applications. J Hematol Oncol. 2015;8:130.
Krupka C, Kufer P, Kischel R, Zugmaier G, Bögeholz J, Köhnke T, et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood. 2014;123(3):356–65.
Laszlo GS, Gudgeon CJ, Harrington KH, Dell’Aringa J, Newhall KJ, Means GD, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood. 2014;123(4):554–61.
Subklewe M, Stein A, Walter RB, Bhatia R, Wei AH, Ritchie D, et al. Preliminary results from a phase 1 first-in-human study of AMG 673, a novel half-life extended (HLE) anti-CD33/CD3 BiTE® (bispecific T-cell engager) in patients with relapsed/refractory (R/R) acute myeloid leukemia (AML). Blood. 2019;134(Suppl_1):833.
Reusch U, Harrington KH, Gudgeon CJ, Fucek I, Ellwanger K, Weichel M, et al. Characterization of CD33/CD3 tetravalent bispecific tandem diabodies (TandAbs) for the treatment of acute myeloid leukemia. Clin Cancer Res. 2016;22(23):5829–38.
Westervelt P, Cortes JE, Altman JK, Long M, Oehler VG, Gojo I, et al. Phase 1 first-in-human trial of AMV564, a bivalent bispecific (2:2) CD33/CD3 T-cell engager, in patients with relapsed/refractory acute myeloid leukemia (AML). Blood. 2019;134(Suppl_1):834.
Uy GL, Godwin J, Rettig MP, Vey N, Foster M, Arellano ML, et al. Preliminary results of a phase 1 study of flotetuzumab, a CD123 x CD3 bispecific Dart® protein, in patients with relapsed/refractory acute myeloid leukemia and myelodysplastic syndrome. Blood. 2017;130(Suppl 1):637.
Xencor. Xencor announces partial clinical hold on phase 1 study of XmAb14045. 2019. https://investors.xencor.com/news-releases/news-release-details/xencor-announces-partial-clinical-hold-phase-1-study-xmab14045.
Wire B. Xencor announces partial clinical hold lifted on phase 1 study of XmAb®14045. 2019. https://www.businesswire.com/news/home/20190430005312/en/Xencor-Announces-Partial-Clinical-Hold-Lifted-on-Phase-1-Study-of-XmAb®14045.
Ravandi F, Bashey A, Foran JM, Stock W, Mawad R, Blum W, et al. Complete responses in relapsed/refractory acute myeloid leukemia (AML) patients on a weekly dosing schedule of XmAb14045, a CD123 x CD3 T cell-engaging bispecific antibody: initial results of a phase 1 study. Blood. 2018;132(Suppl 1):763.
Przespolewski AC, Griffiths EA. BITES and CARS and checkpoints, oh my! Updates regarding immunotherapy for myeloid malignancies from the 2018 annual ASH meeting. Blood Rev. 2020;43:100654.
Sallman DA, Brayer J, Sagatys EM, Lonez C, Breman E, Agaugué S, et al. NKG2D-based chimeric antigen receptor therapy induced remission in a relapsed/refractory acute myeloid leukemia patient. Haematologica. 2018;103(9):e424–e6.
Sallman DA, Kerre T, Poire X, Havelange V, Lewalle P, Davila ML, et al. Remissions in relapse/refractory acute myeloid leukemia patients following treatment with NKG2D CAR-T therapy without a prior preconditioning chemotherapy. Blood. 2018;132(Suppl 1):902.
Liu F, Cao Y, Pinz K, Ma Y, Wada M, Chen K, et al. First-in-human CLL1-CD33 compound CAR T cell therapy induces complete remission in patients with refractory acute myeloid leukemia: update on Phase 1 clinical trial. Blood. 2018;132(Suppl 1):901.
Fang Liu HZ, Lihua Sun, Yecheng Li, Shan Zhang, Guangcui He, Hai Yi, Masayuki Wada, Kevin G Pinz, Kevin H Chen, Yu Ma, Yisong Xiong, Yi Su, Yupo Ma. First-in-human CLL1-CD33 compound car (CCAR) T cell therapy in relapsed and refractory acute myeloid leukemia. 2020. https://library.ehaweb.org/eha/2020/eha25th/294969/fang.liu.first-in-human.cll1-cd33.compound.car.28ccar29.t.cell.therapy.in.html?f=listing%3D0%2Abrowseby%3D8%2Asortby%3D1%2Asearch%3Ds149.
Myburgh R, Kiefer JD, Russkamp NF, Magnani CF, Nuñez N, Simonis A, et al. Anti-human CD117 CAR T-cells efficiently eliminate healthy and malignant CD117-expressing hematopoietic cells. Leukemia. 2020;34(10):2688–703.
Sommer C, Cheng H-Y, Yeung YA, Nguyen D, Sutton J, Melton Z, et al. Preclinical evaluation of ALLO-819, an allogeneic CAR T cell therapy targeting FLT3 for the treatment of acute myeloid leukemia. Blood. 2019;134(Suppl_1):3921.
Dos Santos C, Xiaochuan S, Chenghui Z, Habineza Ndikuyeze G, Glover J, Secreto T, et al. Anti-leukemic activity of daratumumab in acute myeloid leukemia cells and patient-derived Xenografts. Blood. 2014;124(21):2312.
Mistry JJ, Hellmich C, Moore JA, Marlein CR, Pillinger G, Collins A, et al. Daratumumab inhibits AML metabolic capacity and tumor growth through inhibition of CD38 mediated mitochondrial transfer from bone marrow stromal cells to blasts in the leukemic microenvironment. Blood. 2019;134(Suppl_1):1385.
Jelinek T, Zabaleta A, Perez C, Ajona D, Alignani D, Rodriguez I, et al. Pre-clinical efficacy of the anti-CD38 monoclonal antibody (mAb) Isatuximab in acute myeloid leukemia (AML). Blood. 2017;130(Suppl 1):2655.
Busfield SJ, Biondo M, Wong M, Ramshaw HS, Lee EM, Ghosh S, et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia. 2014;28(11):2213–21.
Kubasch AS, Schulze F, Giagounidis A, Götze KS, Krönke J, Sockel K, et al. Single agent talacotuzumab demonstrates limited efficacy but considerable toxicity in elderly high-risk MDS or AML patients failing hypomethylating agents. Leukemia. 2020;34(4):1182–6.
Montesinos P, Roboz GJ, Bulabois CE, Subklewe M, Platzbecker U, Ofran Y, et al. Safety and efficacy of talacotuzumab plus decitabine or decitabine alone in patients with acute myeloid leukemia not eligible for chemotherapy: results from a multicenter, randomized, phase 2/3 study. Leukemia. 2021;35(1):62–74.
Bjordahl R, Gaidarova S, Woan K, Cichocki F, Bonello G, Robinson M, et al. FT538: preclinical development of an off-the-shelf adoptive NK cell immunotherapy with targeted disruption of CD38 to prevent anti-CD38 antibody-mediated fratricide and enhance ADCC in multiple myeloma when combined with daratumumab. Blood. 2019;134(Suppl_1):133.
Janakiram M, Vij R, Siegel DS, Shih T, Weymer S, Valamehr B, et al. A phase I study of FT538, a first-of-kind, off-the-shelf, multiplexed engineered, iPSC-derived NK cell therapy as monotherapy in relapsed/refractory acute myelogenous leukemia and in combination with daratumumab or elotuzumab in relapsed/refractory multiple myeloma. Blood. 2020;136(Suppl 1):3.
Maslak PG, Dao T, Bernal Y, Chanel SM, Zhang R, Frattini M, et al. Phase 2 trial of a multivalent WT1 peptide vaccine (galinpepimut-S) in acute myeloid leukemia. Blood Adv. 2018;2(3):224–34.
SELLAS. SELLAS announces positive follow-up phase 1/2 clinical data for galinpepimut-S (GPS) in acute myeloid leukemia (AML). 2020. https://www.globenewswire.com/news-release/2020/02/26/1990972/0/en/SELLAS-Announces-Positive-Follow-Up-Phase-1-2-Clinical-Data-for-Galinpepimut-S-GPS-in-Acute-Myeloid-Leukemia-AML.html.
Kobayashi Y, Sakura T, Miyawaki S, Toga K, Sogo S, Heike Y. A new peptide vaccine OCV-501: in vitro pharmacology and phase 1 study in patients with acute myeloid leukemia. Cancer Immunol Immunother. 2017;66(7):851–63.
van de Loosdrecht AA, van Wetering S, Santegoets S, Singh SK, Eeltink CM, den Hartog Y, et al. A novel allogeneic off-the-shelf dendritic cell vaccine for post-remission treatment of elderly patients with acute myeloid leukemia. Cancer Immunol Immunother. 2018;67(10):1505–18.
Dhodapkar MV, Sznol M, Zhao B, Wang D, Carvajal RD, Keohan ML, et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci Transl Med. 2014;6(232):232ra51.
Saxena M, Sabado RL, La Mar M, Mohri H, Salazar AM, Dong H, et al. Poly-ICLC, a TLR3 agonist, induces transient innate immune responses in patients with treated HIV-infection: a randomized double-blinded placebo controlled trial. Front Immunol. 2019;10:725.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Gill, H., Yip, A. (2023). In the Pipeline: Emerging Therapy for Acute Myeloid Leukaemia. In: Gill, H., Kwong, YL. (eds) Pathogenesis and Treatment of Leukemia. Springer, Singapore. https://doi.org/10.1007/978-981-99-3810-0_16
Download citation
DOI: https://doi.org/10.1007/978-981-99-3810-0_16
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-3809-4
Online ISBN: 978-981-99-3810-0
eBook Packages: MedicineMedicine (R0)