
Abstract
The phrase “gut microbiome” refers to the huge array of symbiotic bacteria in the gastrointestinal tract of humans as well as their genomes that interact collectively. The latest research suggests that the gut microbes mount multiple crucial biochemical roles for the host as well as those microbiome abnormalities are linked to a wide range of human disease processes. Trillions of microorganisms (altogether referred as the gut microbiota) live in the gastrointestinal system and perform critical roles which are related to host physiology and health. Pathogenesis research has found certain species, bacterial genes, as well as metabolites that have roles in different illnesses and medicinal targets. The gut microbiota has a functional part in macronutrient metabolism, immune system development, and the synthesis of pro or anti-inflammatory signalling molecules and peptides. It has been demonstrated that the gut microbiome has a role in the development of a number of systemic disease states, including obesity and cardiovascular disease, as well as intestinal disorders like inflammatory bowel disease. Active investigation on the roles of the microbes along with the processes underpinning host–microbe interactions will result in a higher understanding function of the microbiota in health moreover illness. Thus, knowing microbiome action is critical for the creation of future customised healthcare methods, and possibly giving novel potential pharmacological targets and research pathways in this rapidly increasing sector in terms of future personalised healthcare strategies. The present chapter focuses on composition, manipulation, and biological factors related to gut microbiota. It also discusses technical challenges related to the gut microbiome and host interactions. Furthermore, future perspectives and utilisations of the gut microbiome are also highlighted.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
The mammalian gastrointestinal system is a suitable habitat to variety of microbes identified as the gut microbiota. The makeup of this microbial population is host-specific, varying during a person’s lifespan in addition to sensitive to both exogenous as well as endogenous alterations (Grond et al. 2018). The recent resurgence of study in the anatomy as well as the role of this “organ” has shed light on its essential role in health along with sickness. Numerous aspects of typical host physiology are impacted by the microbiota, including behaviour, nutritional status, as well as stress response. Furthermore, they could be a primary or secondary reason for many illnesses, affecting both nearby as well as distant organ systems (Moya and Ferrer 2016).
The balance of the gut’s microbial population generally, and the presence and absence of certain strains that might trigger specific responses, is critical in guaranteeing or preventing homeostasis at the gut mucosa (Chow et al. 2010). The methods via which microbes exerts helpful or adverse outcomes are largely unknown, although they include the synthesis of signalling molecules along with the ability of intestinal epithelial and mucosal immune cells to recognise bacterial epitopes (Sekirov et al. 2010). Advancements in gut microflora modelling and examination will expand our understanding of their function in health and illness, enabling for the customisation of present and future therapeutic and preventive methods (Kostic et al. 2014). A population of microorganisms called a microbiota, which includes archaea, viruses, bacteria along with certain unicellular eukaryotes, resides in a particular habitat. Metagenomics is the discipline of molecular study that explores the intricacies of microbiome, whereas a microbiome is the whole of all the genetic components within a given microbiome (Sood et al. 2022).
The curiosity of researchers in microbiota has been increased over the last 15 years. Despite the finding that intestinal bacteria have researched for several years, research into the function of microbes that live within human gut has attained considerable interest outside of typical infectious disorders. Numerous studies have found changes in the gut microbiome throughout not just liver disease, diabetes as well as obesity, but also neurological illnesses and cancer (Cani 2018). The microbiome of the human intestine possibly a reservoir of novel medications. Intestinal bacteria in humans are believed to be potential sources of new treatments and have a profound effect in the metabolism of host (Cani and de Vos 2017).
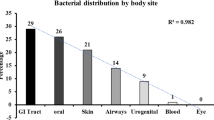
In this context, the human microbiota describes the whole community of microbes that inhabit outside as well as within bodies (Barr 2017). The microbial load in the stomach is substantially smaller, and it rises exponentially along the digestive tract out of the stomach through the duodenum, jejunum, along with ileum, and at last to the colon, which comprises around 109 and 1013 microorganisms (Friedman et al. 2018). These communities play critical roles in digestion, immune system development, human physiology as well as detoxification processes (Tanaka and Nakayama 2017). Furthermore, a few of these bacteria in the gut produce proteins and enzymes important for the digestion of some undigested dietary components, among other essential functions for the host health (Flint et al. 2012).
As a result, we have two genomes in microbiome; one is inherited from parents and another is acquired (Rosenberg and Zilber-Rosenberg 2011). This idea underpins the classification of living being as “superorganisms.” First most crucial distinction among both of these genomes, is that, the inherited genome is still mostly steady throughout life and the microbiome is very active and may be changed with a variety of circumstances, including age, nutrition, hormone cycles, travel, medicines, and sickness (D’Argenio and Salvatore 2015).
2 Biological Factors to Consider While Determining a Healthy Gut Microbiome
The gut of an organism has a particular environment that supports specialised microbes (Kwong and Moran 2016). The gut microbiome, which is frequently regarded as a complex property, is made up of several aspects, and its traits are affected by a mixture of both internal and external forces. Some of them are covered below.
2.1 Infections
The microbiota in the stomach influences viral and bacterial illnesses, these microbial illnesses continue to cause significant morbidity and mortality worldwide (Dudek-Wicher et al. 2018). Several microbes can cause an immune response to be triggered in order to eradicate the microbes. Nonetheless, there is emerging proof that aberrant actions are to blame for catastrophic consequences as well as other inflammatory disorders (Bartold and Van Dyke 2013; Bergström and Lindholm 2000). Patients with severe illness have elevated concentrations of inflammatory cytokines as well as inflammation-related indicators in their blood plasma, for instance, IL 6, 8, and 10, and C reactive protein (CRP) as well as numerous enzymes that represent immunological response and infection-related tissue damage (Kasperska-Zajac et al. 2011). Recent increases in these disorders are almost certainly the consequence of complex and multidimensional external reasons such as changes in climate, greater commodities and people mobility, as well as a quick demographic shift. Parallel to these extrinsic influences, a greater knowledge of the inside components related with immunity against viruses has been acquired. The gut microbiome tract is acknowledged as an imperative component of the host system of defence, operating like a critical regulator of host protection mechanisms along with immune systems (Patel et al. 2022).
2.2 Genetics
The genetic makeup of the host, which impacts host metabolism, as well as health, influences the population of specific bacteria found in microbiome of gut (Boulund et al. 2022; Maglione et al. 2021). Although the variety of gut microbiome varies greatly among humans, members of same family are frequently found to have comparable microbiota than members belong to different family. Familial resemblances are typically linked to common environmental effects, such as food choice, which is a significant shaper of microbiome makeup (Rodríguez-Frías et al. 2021). However, related people have a greater degree of genetic identity, suggesting the likelihood that familial microbiome commonalities are due to shared genetic makeup. Living beings and the associated microorganisms that live in them have developed considerably, resulting in a diverse assemblage of thousands of microbial species coexisting in their digestive tracts in a mutualistic manner (Hernandez and Moeller 2021). Therefore, because gut microbiome is acquired in the environment from birth, it may operate as both an environmental element which interact with the DNA of the host to create phenotypic and a genotypic resolute feature which is influenced via communicates with the host (Maglione et al. 2021; Sedghi et al. 2021). Because the microbes may be manipulated for medicinal purposes, it is an appealing target for modification. In fact, a carefully balanced interaction among microbes as well as human physiologies can have a variety of effects on health and growth, making dysbiosis frequently linked to disease (Rogers et al. 2016; Elias-Oliveira et al. 2020). Due to this, growing data suggests that human genetic variability affects the composition and regulation of their gut flora.
2.3 Drugs
Pharmacological reactions and effectiveness in living being had now been linked to their gut microbiota, as well as the chemicals in these medications could also impact the gut microbes (Weersma et al. 2020). Understanding pharmacological processes and how specific drug side effects occur requires a considerate of the relationship among pharmaceuticals as well as the makeup of gut microbiome. Antibiotics have long been recognised to modify the gut microbiome’s constitution, but research in population-based cohorts has revealed connections between certain drug classes and specific gut microbe’s patterns (Francino 2016; Schwartz et al. 2020; Patangia et al. 2022). Antibiotics are routinely administered treatments that have prevented millions of living beings from illnesses; nonetheless, medications significantly affect the normal gut flora. The impact is immediate and sometimes long-lasting.
Specifically, an elevation in bacteria that make fatty acids of short chain that has been linked to shifts in the intestinal microbial makeup including both vivo and in animals after taking the drug Metformin, a type II diabetes medication that is often used (Foretz et al. 2019). A modified gut microbiome composition has been associated with number of medicines, according to a latest report in a general population dataset (Wang et al. 2020). In a similar vein, an in vitro examination of more than thousand commercially available medications revealed that non-antibiotic medicines can indeed hinder the development of gut bacterial strains (Maier et al. 2018). The relationship within intestinal microbiota as well as routinely prescribed non-antibiotic medications is complicated as well as multidirectional: the gut flora can affect a person’s response toward a medication via enzymatically changing the drug’s architecture thus affecting its bioactivity, bioavailability, even toxicity (Le Bastard et al. 2021; Lindell et al. 2022). Indirectly, the gut flora can also affect a person’s response towards immunotherapy for the cancer therapy (Xavier et al. 2020).
3 Delivery Methods at Birth
Right from birth to early life, the gut flora is crucial. Health of neonatal may be impacted by gut microbiome populations because they guard against harmful pathogens (Martin and Sela 2013), assisting in the metabolisation as well as digestion of breast milk but also formula milk (Koropatkin et al. 2012), promoting immune system development (Ge et al. 2021), sustaining intestinal homeostasis (Chen 2014), as well as having an impact on neurodevelopment (Collins et al. 2012). Early in childhood, there is a progressive and dynamic process of microbiome succession. Neonatals tend to have fewer microbial communities than an adolescent because the makeup and complexity of the gut flora settle throughout time (Dicks et al. 2018; Sumich et al. 2022). Microbes in the gut of newborns are mainly made up of the phylum Actinobacteria, Proteobacteria, as well as Bacteroidetes, which comprise the genera Escherichia, Clostridium, Bifidobacterium, Lactobacillus, Prevotella along with Bacteroides (Pushpanathan et al. 2019). Numerous elements, such as delivery method, newborn food (breast milk vs. formula milk), usage of antibiotics, as well as geographical area, may affect the variety of the newborn microbiota (Marques et al. 2010). Sixteen percent of caesarean sections (CS) are performed globally. Microbes from the mother’s vagina as well as gut are initially present in the newborns that are born vaginally (VD). In fact, children delivered by caesarean sections have a higher percentage of genes associated with antibiotic resistance and are invaded by bacteria discovered on the mother’s body (Rutayisire et al. 2016). Birth through caesarean sections may influence the gut microbes of the infant as well as encourage the invasion of harmful microorganisms. In later life, it could increase the chance of immunological and metabolic illnesses such obesity, type II diabetes, and allergies (Takiishi et al. 2017).
4 Nourishment of Infant
Breastfeeding can also have a substantial impact on the bacterial makeup of an infant’s gut. Breast milk is mostly constituted of carbohydrates, particularly HMOs that are human milk oligosaccharides, which encourage Bifidobacterium development in the infant’s stomach (Lawson et al. 2020). Numbers of studies have found that newborn gut flora contains more Bifidobacterium and Lactobacillus during nursing, resulting in a consistent colonisation model when contrasted with the gut microbiome of infants who were not nursed by breastfed (Selvamani et al. 2021; Taylor et al. 2021). As breast feed is not sterile, newborn nutrition is another crucial aspect in creating the bacterial population in the gut (Khor et al. 2020). Breast feeding has been found to be a provider of commensal and possibly probiotics bacterial agents that hinder growth of the newborn gut flora. Human breast milk includes about 700 different types of bacteria. Whereas the bacterial populations in breast milk are frequently diverse as well as differ from person to person, the average microbial load across time is 106 bacterial cells/mL (Wen and Duffy 2017). Thus, nursing newborn ingesting 800 mL breast milk per day might consume up to 8 × 108 bacterial population per day, which is hundred times greater than earlier predictions, as well as an alteration in composition throughout lactation (Hale and Rowe 2016). Even when not exposed to antibiotics, infants have higher amounts of antibiotic resistance genes (ARG) in their gut microbiome than adults (Gibson et al. 2015). Consequently, the shift of antibiotic-resistant bacteria, as well as other assortment mechanisms other than antibiotic therapy, may be driving ARG enrichment in neonates and infants (Ahmad et al. 2021). However, the influence of selection pressure-causing substances other than antibiotics on resistance loading in neonates and infants is little understood. Mobile genetic element (MGEs) transmits ARGs across bacteria, potentially spreading ARGs in the newborn gut microbiota, and ARGs can be passed vertically from the mother or acquired in the hospital environment (Hildebrand et al. 2021). Feed type can have a substantial consequence on microbiota; earlier investigation suggests that food can change the profusion of certain ARGs in the gut of a newborn.
5 Composition of Gut Microbiota
The gut microbiome only contains a small number of phyla. The majority of the gut microbiota is made up of Firmicutes (60–80%), Gram-positive bacteria with more than 200 genera, the most significant of which are Mycoplasma, Ruminococcus, Eubacterium, Lactobacillus, Clostridium, Faecalibacterium along with Roseburia), while Bacteroidetes (20–30%), Gram-negative bacteria with genera Bacteroides, Xylanibacter as well as Prevotella, are also significant (Szablewski 2018). Proteobacteria (1% Gram-negative bacteria with the genera Enterobacteriace, Escherichia along with Desulfovibrio), Actinobacteria (10% Gram-negative bacteria with species Bifidobacterium), as well as Verrucomicrobia are detected in negligible levels (with genus Fusobacteria, Akkermansia, along with Cyanobacteria) (Dinan and Cryan 2015).
In dissimilar parts of the gastrointestinal tract, the luminal contents and microbial composition vary. The main phyla of bacteria that can be found in the gut are Firmicutes, Actinobacteria, Bacteroidetes, Verrucomicrobia along with Proteobacteria (Bruning et al. 2020). The quantity of microbes within body can exceed 1.5 kg or 2% of a typical 75-kg person’s weight, and the bacterial genes number in the stomach is 150 times bigger than those in the genetic material (Farooqui 2021). In animals that are sterile, it has been proved that alterations in the microbiome of gut perform a significant position in the progression of diseases, including diabetes and obesity. There is proof that the gut microbiota’s composition effects on gut permeability, inflammation, along with energy balance, all of which are connected to obesity and other associated illnesses such as T2D (Geurts et al. 2014). Gastric bypass surgery, which causes 70% weight loss and an improvement in glucose metabolism, has been authorised by the American Diabetes Association as an efficient treatment for obesity as well as type 2 diabetes. The original intent of this surgical procedure was to limit meal intake and prevent calorie absorption. The gut flora is significantly altered by gastric bypass, according to studies, which may aid in weight loss (Murphy et al. 2017). Duodenum samples from bypass patients show a substantial difference in the gut flora between those with and those without diabetes, with obese people with T2D having smaller strains of bacteria (Tai et al. 2015). Although the gut microbiota’s makeup may alter as a result of bariatric surgery, weight loss after bypass surgery may not be primarily attributed to the microbiota.
6 Manipulation of Gut Microbiota
Although gut microbes can have a significant impact on its hosts, efforts have been made to comprehend that how diversity in the initial colonisation’s source and timing affects an animal’s function and health throughout ontogeny (Warne et al. 2019). Animals’ immune systems as well as metabolic processes may be affected for the rest of their lives by early colonisation during developmental windows such as birth (Houghteling and Walker 2015). For instance, caesarean delivery in humans and antibiotic-induced microbial community disruption in young mice are linked to higher consequences of metabolic disorders as well as obesity, respectively (Zhou et al. 2019). Although microorganisms often appear to recover from interruptions by antibiotics or other elements, altered host metabolic phenotypes may persist if the interruption occurred early in life, supporting the possibility that important developmental windows may influence both microbial and host interactions. Non-model wildlife offers special opportunity to improve understanding of microbial as well as host interactions in contexts that are appropriate to ecology and evolution, even if the large part of this study regarding mammals and how they relate to agronomic practices as well as people’s health (Le Roux et al. 2016). Additionally, a variety of hosts’ fitness-related features, such as growth and development, behaviour, and vulnerability to infectious diseases, could be profoundly impacted by the gut microbiome diversity throughout ontogeny, which has not yet been thoroughly investigated (Bosch 2013).
6.1 Probiotics
Several bacteria and yeasts have long been revered for their health-promoting characteristics (Lordan et al. 2020). The term probiotics was originally described as live microorganisms, when consumed in adequate quantities, impart advantages for the host’s health (Sánchez et al. 2017). The concept distinguishes between live microorganisms employed as processing aids or sources of valuable chemicals and those supplied solely for health advantages (Gurung et al. 2013).
Lactobacilli, streptococci, and bifidobacteria are now the most common probiotics bacteria utilised in human diets, supplements, and/or animal feed (Gibson and Roberfroid 1995). Additionally, E. coli and Saccharomyces boulardii are routinely used (Martins et al. 2009). The European Food Safety Authority (EFSA) acknowledged the advantages of using various probiotics as parts of animal feed for a variety of bacteria, whereas the sole recognised claim for humans at the moment is the advantage on lactose digesting (De Simone 2019). However, scientific evidence is mounting that certain probiotics strains or combinations of strains could be useful in a variety of conditions. Effects might be direct or indirect, depending on whether the probiotics bacteria interact with the commensal microbiota (Timmerman et al. 2007).
6.2 Prebiotics
Prebiotics were initially characterised in 1995 and the recent, improved meaning specifies that a prebiotic is “a preferentially fermented element which leads in particular alteration in the makeup as well as the gut microbiota’s functions, imparting advantages on host health” (Cani and Delzenne 2009). Rather than focusing exclusively on the “bifidogenic impact,” this enlarged definition aims to include changes in additional advantageous gut microbiota members (Gibson et al. 2017). Prebiotics that are now in use are primarily poorly digested carbohydrates with a relatively short chain length that are categorised based on their molecular weight. Prebiotics are classed as mono, oligo, or polysaccharides based on their degree of polymerisation. Carbohydrates, on the other hand, can be classed as digestible or non-digestible depending on their physiological and biochemical features (Yoo et al. 2012). Non-digestible carbohydrates are more commonly used to modify the gut microbiota than digestible carbohydrates because they can have several effects mediated by diverse metabolic pathways, such as glucose and lipid metabolism, inflammatory responses, and even changes in appetite control (Louis et al. 2014). Furthermore, the prebiotic index, which measures the absolute increase in bifidobacteria without mentioning the impact on other microbial inhabitants of the gut, is still often used to describe prebiotic effectiveness (Ouwehand et al. 2005).
Prebiotics work as cultivation medium to specifically raise the figures along with performance of specific microbes present in the colon, enhancing their development or participation. Information from Metagenomic research contrasting the gut flora of healthy and ill persons allows for the identification of bacterial species that are suppressed beneath certain illness states (Grice and Segre 2012). Specific growth tests can then be conducted under increasingly sophisticated settings to uncover substrates that might specifically encourage the functioning or development of specific bacteria, when prebiotics are provided; this will eliminate the dysbiosis related to the condition (Roberts and Darveau 2015).
6.3 Antibiotics
Since the discovery of antibiotics in the 1940s and the start of their mass production, the consequences of these medications on the native gut microorganisms have been extensively studied (Modi et al. 2014). Antibiotics have been used for decades to inhibit the spread of bacterial pathogens and, as a result, to treat bacterial illnesses (Unemo and Shafer 2011). They are used to boost the effectiveness of animal feeding. However, the rapid development of virulence genes in bacteria allows them to withstand these antibiotics. Recent research has proven that antibiotic usage has an influence on gut health (Tuchscherr et al. 2020). These have a lot of negative health implications. Even though antibiotics save millions of lives, they also reduce residential bacteria, which are essential for gut health (Fijan 2014).
Nonetheless, almost the entirety of this research has been on how certain antibiotics influence bacteria that have been grown in a laboratory or on particular kinds of bacteria grown on hosts that have received antibiotic treatment (Valli et al. 2020). Furthermore, the majority of these investigations used rather high doses of these medications compared to their normal proportions in microbial populations that are present in nature, with a particular emphasis on disease causing bacterial species (Engel and Moran 2013). Because of this, the majority of our knowledge of antibiotics consequences is focused pertaining to killing processes as well as particular resistance genotypes as well as phenotypes in the setting of a restricted subgroup of the gut flora separated from the general population.
In the past decade or so, investigation on gut microbiota and antibiotics has shifted to a more ecological as well as systemic viewpoint (Sanders et al. 2019). Current research facilities and healthcare institutions frequently use ecological concepts as well as molecular methods. More investigation regarding the effect of antibiotics on the human gut bacterial population, stressing the implications for lateral transmission of resistance genes is to be focused on.
6.4 Faecal Microbial Transplant (FMT)
The importance of gut flora, also known as microbiota, in preserving human health is becoming better recognised. Beginning with birth, our microbiota develops a life-long close relationship with our nutrition and environment, as well as determining the post-natal anatomical along with purposeful development of the gut (Van Belkum et al. 2020). Furthermore, essential connections between the microbiota and our metabolic processes, as well as the immune machinery that serves as our primary defence against foreign antigens, persist throughout life (Bronzo et al. 2020). The trouble in the gut flora has been associated to a rising diversity of gastrointestinal as well as non-gastrointestinal illness. It has been established that faecal microbiota transplantation (FMT) may repair the dysbiosis that defines various persistent illnesses, resulting in an apparently safe, very affordable, and quickly effective treatment in the great majority of patients treated (Person and Keefer 2021).
Various additional gastrointestinal as well as non-gastrointestinal issues have also been resolved using FMT, however expertise with these diseases is limited. More work is needed with faecal microbial transplant to guarantee its security and appropriate administration route (Sbahi and Di Palma 2016). There is a conceptual shift occurring in regard to bacteria, from being infections to being essential for maintaining health in a dynamic society. Potential research will very certainly reduce the range of organisms that can be administered to patients to treat a variety of disorders. FMT is only the initial stage in this process.
7 Technical Challenges in Studying Gut Microbiome and Host Interactions
We must emphasise that, in addition to bacteria, the stomach includes archeae, viruses, phages, yeast, and fungus (Gurung et al. 2019). These microbes, which are assumed to regulate the host’s activities, more significantly, the activity of the microbes, have been thoroughly studied and could be just as essential as bacteria (Engel and Moran 2013). As a result, our knowledge of host–microorganism interactions is expanded by the addition of the phageome, archaea, mycobiome as well as virome. For example, phages not only exceed bacteria (e.g., phages outnumber bacteria tenfold), however, they are also fresh participants in these intricate connections. Consistent methodologies for analysing faecal phageomes using Metagenomics, on the other hand, have just lately been discovered (Sutton and Hill 2019). As a result, it will take longer time for major basic advancements in this field to be translated into widespread applications for the general public. The following are some of the technological challenges in studying gut microbiota.
7.1 Metagenomics
The term “Metagenome” was coined in 1998, primarily concentrating on the soil bacterial genomes (Yan and Yu 2011). Metagenomic frequently work with complicated microbiome mixtures in comparison to genomics, and as a result, relies primarily on the depth of the sequencing. The fast improvement of sequencing technologies, notably the announcing the use of next-generation sequencing, brought about a decrease in the amount of money and time needed for each sequencing despite increasing the sequencing depth (Bharti and Grimm 2021). As a result, Metagenomic shotgun sequencing has been widely employed in investigations of host–microbes association as well as gut microflora (Karaduta et al. 2021). Metagenomics, as opposed to 16S rRNA sequencing, provides more taxonomic details as well as resolution, particularly at the level of species. Numerous research studies have yet used Metagenomic to analyse microbiota dispersed at the species level (Hillmann et al. 2018). Additionally, Metagenomic can give a straight evaluation of the purposeful potential of the microbiota (Walker et al. 2014). Through Metagenomic, Ye et al. (2018) discovered an altered gut microbe makeup in patients of Behcet’s disease (BD), as well as that processes Patients who suffer from BD have elevated levels of these systems, including the transport mechanism for capsular polysaccharides and the oxidation-reduction process. Nonetheless, Metagenomic has several limits. To begin with, Metagenomic reads the genomes of all bacteria among the sample; therefore, it is impossible to determine whether the microorganisms are alive, dormant, or dead (Quince et al. 2017). Second, during the study, DNA loss, deterioration, as well as contamination may occur, resulting in significant variance. Then, the commonly used new generation sequencing technologies, such Illumina (Solexa) sequencing as well as ABI SOLiD sequencing, cannot prevent space construction in cases where the read length is smaller than repeated sequence (Bao et al. 2011). Furthermore, Metagenomic cannot determine whether or not a gene is expressed, nor does it provide the location and breadth of expression.
7.2 Metatranscriptomics
Metatranscriptomics involves the global expression of RNA in the microbiome (Yost et al. 2015; Duran-Pinedo 2021). Metatranscriptomics is likely utilised to analyse gene expression regulation at transcription level, since DNA is transcribed into RNA sequences during transcription, which allows the researchers better understand the role as well as metabolic pathway of the gut microbes (Bashiardes et al. 2016). When compared to Metagenomic, Metatranscriptomics likewise depends on NGS but has higher sensitivity and reproducibility (Shi et al. 2010). In contradictory to the higher variances found in Metagenomic data, Gosalbes et al. (2012) conducted a Metatranscriptomics examination on ten healthy young adults and found that the genetic makeup of functional gut microbiota and prospective functional level was rather similar within people. They proposed a paradigm for investigating the association between the health status and functional microbes, as well as contrasting the makeup of gut microbes under dissimilar physiologic circumstances. Nevertheless, because of mRNA’s brief half-life, enzymatic destruction of mRNA, and difficulties in identifying responses to external stimuli, Metatranscriptomics has certain challenges (Walworth et al. 2021). Additionally, host RNA often pollutes the rRNA sequences of the gastrointestinal microbiota, raising the expense of sequencing and complicating data processing (Morgan and Huttenhower 2014). Finally, whereas Metatranscriptomics analyses the intestinal microbes, at the mRNA level, it does not accurately predict the protein-level, and while mRNA often indicates the presence of protein, this is not always the case.
7.3 Metabolomics
A complete picture of microbial metabolism as well as host–microbiota interactions is provided by Metabolomics. Metabolomics can be used to identify the alive, inactive, or dead states of microbes (Bashiardes et al. 2016). The overall metabolite composition can be evaluated using Metabolomics and to analyse individual metabolite (Withers et al. 2020). Mass spectrometry (MS) as well as nuclear magnetic resonance spectroscopy (NMR) are the two techniques used in Metabolomics the most frequently (Marshall and Powers 2017). Small compounds generated by the gut microbiota are identified and quantified using these approaches. A number of researchers have used 1H NMR to analyse the metabolic composition of faecal water from babies born with hypoxic-ischemic encephalopathy (Watkins et al. 2017). Identification of distinct Metabolomics patterns in intestinal lumen, serum, and hippocampus that correspond with seizure defence, counting decreases in systemic gamma-glutamylated amino acids along with higher hippocampal GABA/glutamate levels have also been done (Ghaffari et al. 2022).
7.4 Metaproteomics
The term “Metaproteomics” was first used to describe the collection of proteins in an environment in 2004 (Schiebenhoefer et al. 2019). Expression of protein variations in gut microbiota could be monitored using Metaproteomics. With hundreds of distinct microbiotas available in a particular sample, the Metaproteomics can be exceedingly complicated (Gonzalez 2020). As a result, the development of Metaproteomics has been more difficult and slower than that of standard proteomics (Wilmes et al. 2015). Weak peptide identifications, limited protein yields, as well as database difficulties are among the key hurdles in Metaproteomics. Metaproteomics has only recently become increasingly pertinent to the investigation of the gut microbes due to advancements in specimen preparation, the advent of elevated Mass spectrometry and the application of novel bioinformatics techniques (Isaac et al. 2019). It can, in particular, indicate alteration in the taxonomy, function, along with metabolic pathway of gut microbes. Recent investigated stated that Metaproteomics was extra useful as compared to Metagenomic in studying the significance of the intestinal microbiota in well-being as well as illness (Lee et al. 2017). Furthermore, not only the gut microbiota’s makeup in addition to metabolic alterations in disease states is studied, other than the dynamic alterations of biochemical pathways linked with illnesses.
8 DNA Sequencing-Based Methodologies
One of the first critical stages in ensuring the integrity and consistency of the obtained material is sample collecting methods (Dakappagari et al. 2017). However, a previous study indicated that there was no variation in community structure following field data collecting; numerous studies demonstrated the significance of the collection procedure, with reliability being boosted by rapid downstream processing (Panek et al. 2018). Faeces is among the most complicated biological resources for the isolation bacterial DNA because it carries residues of human being DNA, dietary DNA, as well as several constraints that impede future PCR amplification as well as New Generation Sequences techniques (Eisenhofer et al. 2019). Extracting DNA from faeces is a critical action in acquiring high-quality DNA along with accurately identifying microbial composition and relative abundance (Wagner Mackenzie et al. 2015). As a result, necessary to analyse the various approaches for enhance procedures and techniques for extraction of bacterial DNA from faeces that offer an adequate volume, clarity, and the DNA’s integrity, resulting in a good quality sample for additional research (Maghini et al. 2021).
The past 10 years have outlined a gradual transition in sequencing techniques from traditional Sanger sequencing to New Generation Sequence and the technologies available using sequencers from Roche, Illumina, Pacific Bioscience, and Thermo Fischer Scientific They have all been utilised with effectiveness for the examination of a variety of biological materials (Morganti et al. 2019).
Therefore, each stage in the experimental pipeline contributes heterogeneity that influences the final result, there is an unmet demand for technique standardisation that would allow for accurate and repeatable investigation of important samples of human biological material for studying intestinal bacteria.
9 Imaging Strategies
The microbial makeup varies from niche to niche even inside the digestive system (Min et al. 2020). Faecal samples cannot disclose such variances. Recent research reveals that to analyse the distribution and colonisation of bacteria in the intestines, a limited range of bacterial species may be fluorescently labelled by genetic and chemical engineering and introduced into uninfected mice through stomach or rectal injection (Zhang et al. 2021). This method, however, necessitates the sacrifice of experimental animals, making it impossible to analyse the regular spatial as well as temporal structure of gut microbe in real time inside the identical mammal prior to as well as after an investigational modification (Grond et al. 2018). Furthermore, as only a small number of gut bacterial species may be genetically modified for fluorescent labelling and the intestinal consists of relatively a significant amount of non-culturable microbes, existing fluorescent labelling approaches are only suited for a subset of gut bacteria (Daliri et al. 2017). Furthermore, it is uncertain how long it takes for colonisation to achieve the usual steady-state position, along with sampled surgically may damage the normal, bacterial species’ three-dimensional dispersion in the gut microbiota (Cryan et al. 2019). Even when fluorescent dyes are used to designate indigenous animals in their native habitat, the fluorescent light absorption is frequently too insufficient to expand beyond tiny-animal investigations into diagnostic and clinical research on humans (Yao et al. 2014).
Thus, new approaches are therefore needed for in vivo imaging of the gut microbes in human beings, with minimum disruption to the natural microbiota.
10 Future Perspectives and Utilisations of Gut Microbiome
Numerous human metabolic processes and our well-being are impacted by the relationship between the human microbiome as well as system of defence (Althani et al. 2016; Lasselin et al. 2016). Furthermore, the connections among people in addition to microbes can be crucial in deciding whether or not a person is healthy or ill (Dorrestein et al. 2014). Several disorders, including cutaneous, inflammatory, metabolic, and neurological problems, are associated with dysbiosis (Scotti et al. 2017). Accurate diagnosis and therapy of many disorders depend on a deeper comprehension of the host–microbe relationship. Because of the consequence of the microbes to host health, novel therapeutic approaches have emerged that concentrate on the recommended alteration of the host microbiome, either through the removal of negative taxa or the restoration of positive taxa and the functional roles they play (Gilbert and Lynch 2019). Large numbers of microbial taxa are difficult to cultivate in the laboratory, if not impossible (Stewart 2012). As a result, it is very challenging to list each individual microbiome member and to comprehend how microbial population work and affect host–pathogen interactions (Dillard et al. 2021). Numerous Metagenomic research studies are now being conducted thanks to recent improvements in sequencing technology and computational tools. These investigations have offered crucial information about the human microbiome as well as numerous other microbial communities in different environments (Langille et al. 2013).
A healthy microbiome is crucial for host organisms, because it facilitates the efficient execution of crucial physiological functions (Foster et al. 2017). Indeed, host organisms and their microbiota have coevolved, with some commensals becoming pathobionts and others becoming symbionts (Ruff et al. 2020). In the human gut, some commensals release signals that encourage appropriate immune system development. Within distinct hosts, as well as various physical surroundings, microbial communities develop a distinct structure (Zheng et al. 2020). Due to this, research on the host–microbiome is primarily concerned with identifying and characterising the bacteria that live there, their distinctive host phenotypes, as well as the biochemical mechanisms with which these microorganisms affect their hosts (Henry et al. 2021).
Investigations into host–microbe interactions can disclose the relationship’s essential properties, such as identification, categorisation, profile prediction, and interaction processes (Starr et al. 2018). Although these microbes’ structure, function, dynamics, and interactions are critical in human metabolism, their identification, measurement, as well as characterisation can be difficult (Rowland et al. 2018). The majority of microbial communities are incredibly varied, and the vast the predominance of each species has yet to be grown. Second, their contact with one another, as well as their proclivity to construct sophisticated networks, makes it impossible to forecast their behaviour (Fierer 2017). Establishing molecular links between gut microbiota and its function provides a new dimension to insight the biology of complex microbial consortia. To research host–microbe interactions, culture-dependent techniques have been the mainstay of traditional approaches to microbial ecology (Gilbert et al. 2016). Even though these are culture-specific methodologies produced intriguing data sets, they also produced a distorted image of microbiota. However, for identifying microorganisms in both qualitative and quantitative ways, a variety of culture-independent approaches, mostly the methods based on PCR, have recently emerged (Hameed et al. 2018). These approaches have completely altered people’s perceptions of the living being microbes and opened the ground for the development of Metagenomic (Lepage et al. 2013). Metagenomic research is expanding our understanding of interactions between host and pathogenic by identifying genes that may permit microorganisms to impact their hosts in unforeseen ways. Pathogen surveillance, biotechnology, host–microbe interactions, functional dysbiosis, and evolutionary biology can all benefit from Metagenomic research of host–microbe interactions (Cruz et al. 2022). Recent Metagenomic investigations of host microbiome has provided important insights into host–microbe interactions.
11 Conclusion
For all forms of life on Earth, microorganisms (host and parasite) are necessary. They both are defined by their surroundings. However, our knowledge of host–pathogen systems is still quite limited. Technologies for bioinformatics as well as sequencing have evolved significantly over the past two decades, creating it possible to investigate microbial populations living inside of various hosts. There is widespread agreement that the diversification of microbes found in harsh environments has mostly gone untouched. Novel approaches are necessary to acquire new information about this “latent” microflora. NGS technology has enabled the quick and low-cost generation of sequencing data, as well as the development of sequencing systems that may be employed in both big genome-sequencing centres and individual laboratories. Updated versions of their specific DNA sequencing technologies have been revealed by Illumina, PacBio, and Applied Biosystems. These enhancements will increase read length as well as high-throughput capability yet dramatically reducing the amount of sequencing each base. Such innovations will considerably benefit research scientists moreover offer them intriguing novel prospects. To address concerns regarding the ecology as well as the complexity of the microbial flora, multiple techniques for biological investigations must be integrated. The enormous amounts of genomic information that are going to be created will provide novel obstacles intended for data analysis, storage, and transport. Sequencing of genome facilities as well as laboratories will grow more reliant on information technology and bioinformatics. To analyse vast volumes of data as well as extract the data for relevant knowledge about bacterial populations, bioinformatics knowledge will become more important. Metagenomic will become increasingly important in health, biological as well as environmental research. A detailed representation and knowledge of the functional microbiome are required for future rational therapies that use the gut microbiome to change host phenotype. Experiments with representative models will be crucial for assessing the influence of various microbial activities on human physiology and explaining their mechanism of action. The authors believe that this chapter provides a thorough summary of gut microbiome of humans, as well as presents sequencing technologies and their prospects, along with their high value and limitations.
References
Ahmad I, Malak HA, Abulreesh HH (2021) Environmental antimicrobial resistance and its drivers: a potential threat to public health. J Glob Antimicrob Resist 27:101–111
Althani AA, Marei HE, Hamdi WS, Nasrallah GK, El Zowalaty ME, Al Khodor S, Al-Asmakh M, Abdel-Aziz H, Cenciarelli C (2016) Human microbiome and its association with health and diseases. J Cell Physiol 231(8):1688–1694
Bao S, Jiang R, Kwan W, Wang B, Ma X, Song YQ (2011) Evaluation of next-generation sequencing software in mapping and assembly. J Hum Genet 56(6):406–414
Barr JJ (2017) A bacteriophage journey through the human body. Immunol Rev 279(1):106–122
Bartold PM, Van Dyke TE (2013) Periodontitis: a host-mediated disruption of microbial homeostasis. Unlearning learned concepts. Periodontol 2000 62(1):203–217
Bashiardes S, Zilberman-Schapira G, Elinav E (2016) Use of metatranscriptomics in microbiome research. Bioinform Biol Insights 10:BBI-S34610
Bergström J, Lindholm B (2000) What are the causes and consequences of the chronic inflammatory state in chronic dialysis patients? In: Seminars in dialysis, vol 13. Blackwell Science Inc., Boston, pp 163–164
Bharti R, Grimm DG (2021) Current challenges and best-practice protocols for microbiome analysis. Brief Bioinform 22(1):178–193
Bosch TC (2013) Cnidarian-microbe interactions and the origin of innate immunity in metazoans. Annu Rev Microbiol 67(1):499–518
Boulund U, Bastos DM, Ferwerda B, van den Born BJ, Pinto-Sietsma SJ, Galenkamp H, Levin E, Groen AK, Zwinderman AH, Nieuwdorp M (2022) Gut microbiome associations with host genotype vary across ethnicities and potentially influence cardiometabolic traits. Cell Host Microbe 30(10):1464–1480
Bronzo V, Lopreiato V, Riva F, Amadori M, Curone G, Addis MF, Cremonesi P, Moroni P, Trevisi E, Castiglioni B (2020) The role of innate immune response and microbiome in resilience of dairy cattle to disease: the mastitis model. Animals 10(8):1397
Bruning J, Chapp A, Kaurala GA, Wang R, Techtmann S, Chen QH (2020) Gut microbiota and short chain fatty acids: influence on the autonomic nervous system. Neurosci Bull 36(1):91–95
Cani PD (2018) Human gut microbiome: hopes, threats and promises. Gut 67(9):1716–1725
Cani PD, de Vos WM (2017) Next-generation beneficial microbes: the case of Akkermansia muciniphila. Front Microbiol 8:1765
Cani PD, Delzenne NM (2009) The role of the gut microbiota in energy metabolism and metabolic disease. Curr Pharm Des 15(13):1546–1558
Chen GY (2014) Role of Nlrp6 and Nlrp12 in the maintenance of intestinal homeostasis. Eur J Immunol 44(2):321–327
Chow J, Lee SM, Shen Y, Khosravi A, Mazmanian SK (2010) Host–bacterial symbiosis in health and disease. Adv Immunol 107:243–274
Collins SM, Surette M, Bercik P (2012) The interplay between the intestinal microbiota and the brain. Nat Rev Microbiol 10(11):735–742
Cruz N, Abernathy GA, Dichosa AE, Kumar A (2022) The age of next-generation therapeutic-microbe discovery: exploiting microbe-microbe and host-microbe interactions for disease prevention. Infect Immun 90:e0058921
Cryan JF, O’Riordan KJ, Cowan CS, Sandhu KV, Bastiaanssen TF, Boehme M, Codagnone MG, Cussotto S, Fulling C, Golubeva AV, Guzzetta KE (2019) The microbiota-gut-brain axis. Physiol Rev 99:1877
D’Argenio V, Salvatore F (2015) The role of the gut microbiome in the healthy adult status. Clinicachim Acta 451:97–102
Dakappagari N, Zhang H, Stephen L, Amaravadi L, Khan MU (2017) Recommendations for clinical biomarker specimen preservation and stability assessments. Bioanalysis 9(8):643–653
Daliri EBM, Wei S, Oh DH, Lee BH (2017) The human microbiome and metabolomics: current concepts and applications. Crit Rev Food Sci Nutr 57(16):3565–3576
De Simone C (2019) The unregulated probiotic market. Clin Gastroenterol Hepatol 17(5):809–817
Dicks LMT, Geldenhuys J, Mikkelsen LS, Brandsborg E, Marcotte H (2018) Our gut microbiota: a long walk to homeostasis. Benefic Microbes 9(1):3–20
Dillard LR, Payne DD, Papin JA (2021) Mechanistic models of microbial community metabolism. Mol Omics 17(3):365–375
Dinan TG, Cryan JF (2015) The impact of gut microbiota on brain and behaviour: implications for psychiatry. Curr Opin Clin Nutr Metab Care 18(6):552–558
Dorrestein PC, Mazmanian SK, Knight R (2014) Finding the missing links among metabolites, microbes, and the host. Immunity 40(6):824–832
Dudek-Wicher RK, Junka A, Bartoszewicz M (2018) The influence of antibiotics and dietary components on gut microbiota. Prz Gastroenterol 13(2):85–92
Duran-Pinedo AE (2021) Metatranscriptomics analyses of the oral microbiome. Periodontol 2000 85(1):28–45
Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, Weyrich LS (2019) Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol 27(2):105–117
Elias-Oliveira J, Leite JA, Pereira ÍS, Guimarães JB, Manso GMDC, Silva JS, Tostes RC, Carlos D (2020) NLR and intestinal dysbiosis-associated inflammatory illness: drivers or dampers? Front Immunol 11:1810
Engel P, Moran NA (2013) The gut microbiota of insects–diversity in structure and function. FEMS Microbiol Rev 37(5):699–735
Farooqui AA (2021) The contribution of microbiota, cerebral blood flow, and sleep deprivation in the pathogenesis of Alzheimer’s disease. In: Gut microbiota in neurologic and visceral diseases. Academic Press, New York, pp 143–158
Fierer N (2017) Embracing the unknown: disentangling the complexities of the soil microbiome. Nat Rev Microbiol 15(10):579–590
Fijan S (2014) Microorganisms with claimed probiotic properties: an overview of recent literature. Int J Environ Res Public Health 11(5):4745–4767
Flint HJ, Scott KP, Duncan SH, Louis P, Forano E (2012) Microbial degradation of complex carbohydrates in the gut. Gut Microbes 3(4):289–306
Foretz M, Guigas B, Viollet B (2019) Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat Rev Endocrinol 15(10):569–589
Foster JA, Rinaman L, Cryan JF (2017) Stress & the gut-brain axis: regulation by the microbiome. Neurobiol Stress 7:124–136
Francino MP (2016) Antibiotics and the human gut microbiome: dysbioses and accumulation of resistances. Front Microbiol 6:1543
Friedman ES, Bittinger K, Esipova TV, Hou L, Chau L, Jiang J, Mesaros C, Lund PJ, Liang X, FitzGerald GA, Goulian M (2018) Microbes vs. chemistry in the origin of the anaerobic gut lumen. Proc Natl Acad Sci 115(16):4170–4175
Ge Y, Wang X, Guo Y, Yan J, Abuduwaili A, Aximujiang K, Yan J, Wu M (2021) Gut microbiota influence tumor development and alter interactions with the human immune system. J Exp Clin Cancer Res 40(1):1–9
Geurts L, Neyrinck AM, Delzenne NM, Knauf C, Cani PD (2014) Gut microbiota controls adipose tissue expansion, gut barrier and glucose metabolism: novel insights into molecular targets and interventions using prebiotics. Benefic Microbes 5(1):3–17
Ghaffari S, Abbasi A, Somi MH, Moaddab SY, Nikniaz L, Kafil HS, Ebrahimzadeh Leylabadlo H (2022) Akkermansia muciniphila: from its critical role in human health to strategies for promoting its abundance in human gut microbiome. Crit Rev Food Sci Nutr:1–21
Gibson GR, Roberfroid MB (1995) Dietary modulation of the human colonic microbiota: introducing the concept of prebiotics. J Nutr 125(6):1401–1412
Gibson MK, Crofts TS, Dantas G (2015) Antibiotics and the developing infant gut microbiota and resistome. Curr Opin Microbiol 27:51–56
Gibson GR, Hutkins RW, Sanders ME, Prescott SL, Reimer RA, Salminen SJ, Scott K, Stanton C, Swanson KS, Cani PD, Verbeke K (2017) The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat Rev Gastroenterol Hepatol 14:491–502
Gilbert JA, Lynch SV (2019) Community ecology as a framework for human microbiome research. Nat Med 25(6):884–889
Gilbert JA, Quinn RA, Debelius J, Xu ZZ, Morton J, Garg N, Jansson JK, Dorrestein PC, Knight R (2016) Microbiome-wide association studies link dynamic microbial consortia to disease. Nature 535(7610):94–103
Gonzalez CG (2020) Revealing host-gut microbiome interactions: a metaproteomic perspective. Stanford University, Stanford
Gosalbes MJ, Abellan JJ, Durban A, Pérez-Cobas AE, Latorre A, Moya A (2012) Metagenomics of human microbiome: beyond 16S rDNA. Clin Microbiol Infect 18:47–49
Grice EA, Segre JA (2012) The human microbiome: our second genome. Annu Rev Genomics Hum Genet 13:151–170
Grond K, Sandercock BK, Jumpponen A, Zeglin LH (2018) The avian gut microbiota: community, physiology and function in wild birds. J Avian Biol 49(11):e01788
Gurung N, Ray S, Bose S, Rai V (2013) A broader view: microbial enzymes and their relevance in industries, medicine, and beyond. Biomed Res Int 2013:329121
Gurung K, Wertheim B, Falcao Salles J (2019) The microbiome of pest insects: it is not just bacteria. Entomol Exp Appl 167(3):156–170
Hale TW, Rowe HE (2016) Medications and mothers’ milk 2017. Springer Publishing Company, Berlin
Hameed S, Xie L, Ying Y (2018) Conventional and emerging detection techniques for pathogenic bacteria in food science: a review. Trends Food Sci Technol 81:61–73
Henry LP, Bruijning M, Forsberg SK, Ayroles JF (2021) The microbiome extends host evolutionary potential. Nat Commun 12(1):1–13
Hernandez CJ, Moeller AH (2021) The microbiome: a heritable contributor to bone morphology? In: Seminars in cell & developmental biology, vol 123. Academic Press, New York, p 82
Hildebrand F, Gossmann TI, Frioux C, Özkurt E, Myers PN, Ferretti P, Kuhn M, Bahram M, Nielsen HB, Bork P (2021) Dispersal strategies shape persistence and evolution of human gut bacteria. Cell Host Microbe 29(7):1167–1176
Hillmann B, Al-Ghalith GA, Shields-Cutler RR, Zhu Q, Gohl DM, Beckman KB, Knight R, Knights D (2018) Evaluating the information content of shallow shotgun metagenomics. mSystems 3(6):e00069-18
Houghteling PD, Walker WA (2015) Why is initial bacterial colonization of the intestine important to infants’ and children’s health? J Pediatr Gastroenterol Nutr 60(3):294–307
Isaac NI, Philippe D, Nicholas A, Raoult D, Eric C (2019) Metaproteomics of the human gut microbiota: challenges and contributions to other OMICS. Clin Mass Spectrom 14:18–30
Karaduta O, Dvanajscak Z, Zybailov B (2021) Metaproteomics—an advantageous option in studies of host-microbiota interaction. Microorganisms 9(5):980
Kasperska-Zajac A, Sztylc J, Machura E, Jop G (2011) Plasma IL-6 concentration correlates with clinical disease activity and serum C-reactive protein concentration in chronic urticaria patients. Clin Exp Allergy 41(10):1386–1391
Khor GL, Tan SS, Stoutjesdijk E, Ng KWT, Khouw I, Bragt M, Schaafsma A, Dijck-Brouwer DJ, Muskiet FA (2020) Temporal changes in breast milk fatty acids contents: a case study of Malay breastfeeding women. Nutrients 13(1):101
Koropatkin NM, Cameron EA, Martens EC (2012) How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol 10(5):323–335
Kostic AD, Xavier RJ, Gevers D (2014) The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology 146(6):1489–1499
Kwong WK, Moran NA (2016) Gut microbial communities of social bees. Nat Rev Microbiol 14(6):374–384
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31(9):814–821
Lasselin J, Alvarez-Salas E, Grigoleit JS (2016) Well-being and immune response: a multi-system perspective. Curr Opin Pharmacol 29:34–41
Lawson MA, O’Neill IJ, Kujawska M, Gowrinadh Javvadi S, Wijeyesekera A, Flegg Z, Chalklen L, Hall LJ (2020) Breast milk-derived human milk oligosaccharides promote Bifidobacterium interactions within a single ecosystem. ISME J 14(2):635–648
Le Bastard Q, Berthelot L, Soulillou JP, Montassier E (2021) Impact of non-antibiotic drugs on the human intestinal microbiome. Expert Rev Mol Diagn 21(9):911–924
Le Roux F, Wegner KM, Polz MF (2016) Oysters and vibrios as a model for disease dynamics in wild animals. Trends Microbiol 24(7):568–580
Lee PY, Chin SF, Neoh HM, Jamal R (2017) Metaproteomic analysis of human gut microbiota: where are we heading? J Biomed Sci 24(1):1–8
Lepage P, Leclerc MC, Joossens M, Mondot S, Blottière HM, Raes J, Ehrlich D, Doré J (2013) A metagenomic insight into our gut’s microbiome. Gut 62(1):146–158
Lindell AE, Zimmermann-Kogadeeva M, Patil KR (2022) Multimodal interactions of drugs, natural compounds and pollutants with the gut microbiota. Nat Rev Microbiol 20:1–13
Lordan C, Thapa D, Ross RP, Cotter PD (2020) Potential for enriching next-generation health-promoting gut bacteria through prebiotics and other dietary components. Gut Microbes 11(1):1–20
Louis P, Hold GL, Flint HJ (2014) The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 12(10):661–672
Maghini DG, Moss EL, Vance SE, Bhatt AS (2021) Improved high-molecular-weight DNA extraction, nanopore sequencing and metagenomic assembly from the human gut microbiome. Nat Protoc 16(1):458–471
Maglione A, Zuccalà M, Tosi M, Clerico M, Rolla S (2021) Host genetics and gut microbiome: perspectives for multiple sclerosis. Genes 12(8):1181
Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, Brochado AR, Fernandez KC, Dose H, Mori H, Patil KR (2018) Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 555(7698):623–628
Marques TM, Wall R, Ross RP, Fitzgerald GF, Ryan CA, Stanton C (2010) Programming infant gut microbiota: influence of dietary and environmental factors. Curr Opin Biotechnol 21(2):149–156
Marshall DD, Powers R (2017) Beyond the paradigm: combining mass spectrometry and nuclear magnetic resonance for metabolomics. Prog Nucl Magn Reson Spectrosc 100:1–16
Martin MA, Sela DA (2013) Infant gut microbiota: developmental influences and health outcomes. In: Building babies. Springer, New York, pp 233–256
Martins FS, Silva AA, Vieira AT, Barbosa FH, Arantes RM, Teixeira MM, Nicoli JR (2009) Comparative study of Bifidobacterium animalis, Escherichia coli, Lactobacillus casei and Saccharomyces boulardii probiotic properties. Arch Microbiol 191(8):623–630
Min S, Kim S, Cho SW (2020) Gastrointestinal tract modeling using organoids engineered with cellular and microbiota niches. Exp Mol Med 52(2):227–237
Modi SR, Collins JJ, Relman DA (2014) Antibiotics and the gut microbiota. J Clin Invest 124(10):4212–4218
Morgan XC, Huttenhower C (2014) Meta’omic analytic techniques for studying the intestinal microbiome. Gastroenterology 146(6):1437–1448
Morganti S, Tarantino P, Ferraro E, D’Amico P, Duso BA, Curigliano G (2019) Next generation sequencing (NGS): a revolutionary technology in pharmacogenomics and personalized medicine in cancer. In: Translational research and onco-omics applications in the era of cancer personal genomics. Springer, Berlin, pp 9–30
Moya A, Ferrer M (2016) Functional redundancy-induced stability of gut microbiota subjected to disturbance. Trends Microbiol 24(5):402–413
Murphy R, Tsai P, Jüllig M, Liu A, Plank L, Booth M (2017) Differential changes in gut microbiota after gastric bypass and sleeve gastrectomy bariatric surgery vary according to diabetes remission. Obes Surg 27(4):917–925
Ouwehand AC, Derrien M, de Vos W, Tiihonen K, Rautonen N (2005) Prebiotics and other microbial substrates for gut functionality. Curr Opin Biotechnol 16(2):212–217
Panek M, ČipčićPaljetak H, Barešić A, Perić M, Matijašić M, Lojkić I, Vranešić Bender D, Krznarić Ž, Verbanac D (2018) Methodology challenges in studying human gut microbiota—effects of collection, storage, DNA extraction and next generation sequencing technologies. Sci Rep 8(1):1–13
Patangia DV, Anthony Ryan C, Dempsey E, Paul Ross R, Stanton C (2022) Impact of antibiotics on the human microbiome and consequences for host health. Microbiol Open 11(1):e1260
Patel M, McAllister M, Nagaraju R, Al Badran SSF, Edwards J, McBain AJ, Barriuso J, Aziz O (2022) The intestinal microbiota in colorectal cancer metastasis–passive observer or key player? Crit Rev Oncol Hematol 180:103856
Person H, Keefer L (2021) Psychological comorbidity in gastrointestinal diseases: update on the brain-gut-microbiome axis. Prog Neuro-Psychopharmacol Biol Psychiatry 107:110209
Pushpanathan P, Mathew GS, Selvarajan S, Seshadri KG, Srikanth P (2019) Gut microbiota and its mysteries. Indian J Med Microbiol 37(2):268–277
Quince C, Walker AW, Simpson JT, Loman NJ, Segata N (2017) Shotgun metagenomics, from sampling to analysis. Nat Biotechnol 35(9):833–844
Roberts FA, Darveau RP (2015) Microbial protection and virulence in periodontal tissue as a function of polymicrobial communities: symbiosis and dysbiosis. Periodontol 2000 69(1):18–27
Rodríguez-Frías F, Quer J, Tabernero D, Cortese MF, Garcia-Garcia S, Rando-Segura A, Pumarola T (2021) Microorganisms as shapers of human civilization, from pandemics to even our genomes: villains or friends? A historical approach. Microorganisms 9(12):2518
Rogers GB, Keating DJ, Young RL, Wong ML, Licinio J, Wesselingh S (2016) From gut dysbiosis to altered brain function and mental illness: mechanisms and pathways. Mol Psychiatry 21(6):738–748
Rosenberg E, Zilber-Rosenberg I (2011) Symbiosis and development: the hologenome concept. Birth Defects Res C Embryo Today 93(1):56–66
Rowland I, Gibson G, Heinken A, Scott K, Swann J, Thiele I, Tuohy K (2018) Gut microbiota functions: metabolism of nutrients and other food components. Eur J Nutr 57(1):1–24
Ruff WE, Greiling TM, Kriegel MA (2020) Host–microbiota interactions in immune-mediated diseases. Nat Rev Microbiol 18(9):521–538
Rutayisire E, Huang K, Liu Y, Tao F (2016) The mode of delivery affects the diversity and colonization pattern of the gut microbiota during the first year of infants’ life: a systematic review. BMC Gastroenterol 16(1):1–12
Sánchez B, Delgado S, Blanco-Míguez A, Lourenço A, Gueimonde M, Margolles A (2017) Probiotics, gut microbiota, and their influence on host health and disease. Mol Nutr Food Res 61(1):1600240
Sanders ME, Merenstein DJ, Reid G, Gibson GR, Rastall RA (2019) Probiotics and prebiotics in intestinal health and disease: from biology to the clinic. Nat Rev Gastroenterol Hepatol 16(10):605–616
Sbahi H, Di Palma JA (2016) Faecal microbiota transplantation: applications and limitations in treating gastrointestinal disorders. BMJ Open Gastroenterol 3(1):e000087
Schiebenhoefer H, Van Den Bossche T, Fuchs S, Renard BY, Muth T, Martens L (2019) Challenges and promise at the interface of metaproteomics and genomics: an overview of recent progress in metaproteogenomic data analysis. Expert Rev Proteomics 16(5):375–390
Schwartz DJ, Langdon AE, Dantas G (2020) Understanding the impact of antibiotic perturbation on the human microbiome. Genome Med 12(1):1–12
Scotti E, Boué S, Sasso GL, Zanetti F, Belcastro V, Poussin C, Sierro N, Battey J, Gimalac A, Ivanov NV, Hoeng J (2017) Exploring the microbiome in health and disease: implications for toxicology. Toxicol Res Appl 1:2397847317741884
Sedghi L, DiMassa V, Harrington A, Lynch SV, Kapila YL (2021) The oral microbiome: role of key organisms and complex networks in oral health and disease. Periodontol 2000 87(1):107–131
Sekirov I, Russell SL, Antunes LCM, Finlay BB (2010) Gut microbiota in health and disease. Physiol Rev 90:859
Selvamani S, Mehta D, Deb C, El Enshasy H, Abomoelak B (2021) Role of the gut microbiota in the digestive tract diseases. In: Probiotics, the natural microbiota in living organisms. CRC Press, Boca Raton, pp 30–57
Shi W, Syrenne R, Sun JZ, Yuan JS (2010) Molecular approaches to study the insect gut symbiotic microbiota at the ‘omics’ age. Insect Sci 17(3):199–219
Sood U, Dhingra GG, Anand S, Hira P, Kumar R, Kaur J, Verma M, Singhvi N, Lal S, Rawat CD, Singh VK (2022) Microbial journey: Mount Everest to Mars. Indian J Microbiol 62:1–15
Starr AE, Deeke SA, Li L, Zhang X, Daoud R, Ryan J, Ning Z, Cheng K, Nguyen LV, Abou-Samra E, Lavallée-Adam M (2018) Proteomic and metaproteomic approaches to understand host–microbe interactions. Anal Chem 90(1):86–109
Stewart EJ (2012) Growing unculturable bacteria. J Bacteriol 194(16):4151–4160
Sumich A, Heym N, Lenzoni S, Hunter K (2022) Gut microbiome-brain axis and inflammation in temperament, personality and psychopathology. Curr Opin Behav Sci 44:101101
Sutton TD, Hill C (2019) Gut bacteriophage: current understanding and challenges. Front Endocrinol 10:784
Szablewski L (2018) Human gut microbiota in health and Alzheimer’s disease. J Alzheimers Dis 62(2):549–560
Tai N, Wong FS, Wen L (2015) The role of gut microbiota in the development of type 1, type 2 diabetes mellitus and obesity. Rev Endocr Metab Disord 16(1):55–65
Takiishi T, Fenero CIM, Câmara NOS (2017) Intestinal barrier and gut microbiota: shaping our immune responses throughout life. Tissue Barriers 5(4):e1373208
Tanaka M, Nakayama J (2017) Development of the gut microbiota in infancy and its impact on health in later life. Allergol Int 66(4):515–522
Taylor SL, Simpson JL, Rogers GB (2021) The influence of early-life microbial exposures on long-term respiratory health. Paediatr Respir Rev 40:15–23
Timmerman HM, Niers LE, Ridwan BU, Koning CJ, Mulder L, Akkermans LM, Rombouts FM, Rijkers GT (2007) Design of a multispecies probiotic mixture to prevent infectious complications in critically ill patients. Clin Nutr 26(4):450–459
Tuchscherr L, Löffler B, Proctor RA (2020) Persistence of Staphylococcus aureus: multiple metabolic pathways impact the expression of virulence factors in small-colony variants (SCVs). Front Microbiol 11:1028
Unemo M, Shafer WM (2011) Antibiotic resistance in Neisseria gonorrhoeae: origin, evolution, and lessons learned for the future. Ann N Y Acad Sci 1230(1):E19–E28
Valli RX, Lyng M, Kirkpatrick CL (2020) There is no hiding if you Seq: recent breakthroughs in Pseudomonas aeruginosa research revealed by genomic and transcriptomic next-generation sequencing. J Med Microbiol 69(2):162–175
Van Belkum M, Mendoza Alvarez L, Neu J (2020) Preterm neonatal immunology at the intestinal interface. Cell Mol Life Sci 77(7):1209–1227
Wagner Mackenzie B, Waite DW, Taylor MW (2015) Evaluating variation in human gut microbiota profiles due to DNA extraction method and inter-subject differences. Front Microbiol 6:130
Walker AW, Duncan SH, Louis P, Flint HJ (2014) Phylogeny, culturing, and metagenomics of the human gut microbiota. Trends Microbiol 22(5):267–274
Walworth NG, Saito MA, Lee MD, McIlvin MR, Moran DM, Kellogg RM, Fu FX, Hutchins DA, Webb EA (2021) Why environmental biomarkers work: transcriptome–proteome correlations and modeling of multistressor experiments in the marine Bacterium Trichodesmium. J Proteome Res 21(1):77–89
Wang J, Xiong K, Zhao S, Zhang C, Zhang J, Xu L, Ma A (2020) Long-term effects of multi-drug-resistant tuberculosis treatment on gut microbiota and its health consequences. Front Microbiol 11:53
Warne RW, Kirschman L, Zeglin L (2019) Manipulation of gut microbiota during critical developmental windows affects host physiological performance and disease susceptibility across ontogeny. J Anim Ecol 88(6):845–856
Watkins C, Murphy K, Yen S, Carafa I, Dempsey EM, O’Shea CA, Vercoe EA, Ross RP, Stanton C, Ryan CA (2017) Effects of therapeutic hypothermia on the gut microbiota and metabolome of infants suffering hypoxic-ischemic encephalopathy at birth. Int J Biochem Cell Biol 93:110–118
Weersma RK, Zhernakova A, Fu J (2020) Interaction between drugs and the gut microbiome. Gut 69(8):1510–1519
Wen L, Duffy A (2017) Factors influencing the gut microbiota, inflammation, and type 2 diabetes. J Nutr 147(7):1468S–1475S
Wilmes P, Heintz-Buschart A, Bond PL (2015) A decade of metaproteomics: where we stand and what the future holds. Proteomics 15(20):3409–3417
Withers E, Hill PW, Chadwick DR, Jones DL (2020) Use of untargeted metabolomics for assessing soil quality and microbial function. Soil Biol Biochem 143:107758
Xavier JB, Young VB, Skufca J, Ginty F, Testerman T, Pearson AT, Macklin P, Mitchell A, Shmulevich I, Xie L, Caporaso JG (2020) The cancer microbiome: distinguishing direct and indirect effects requires a systemic view. Trends Cancer 6(3):192–204
Yan Q, Yu Y (2011) Metagenome-based analysis: a promising direction for plankton ecological studies. Sci China Life Sci 54(1):75–81
Yao J, Yang M, Duan Y (2014) Chemistry, biology, and medicine of fluorescent nanomaterials and related systems: new insights into biosensing, bioimaging, genomics, diagnostics, and therapy. Chem Rev 114(12):6130–6178
Ye Z, Zhang N, Wu C, Zhang X, Wang Q, Huang X, Du L, Cao Q, Tang J, Zhou C, Hou S (2018) A metagenomic study of the gut microbiome in Behcet’s disease. Microbiome 6(1):1–13
Yoo HD, Kim D, Paek SH (2012) Plant cell wall polysaccharides as potential resources for the development of novel prebiotics. Biomol Ther 20(4):371
Yost S, Duran-Pinedo AE, Teles R, Krishnan K, Frias-Lopez J (2015) Functional signatures of oral dysbiosis during periodontitis progression revealed by microbial metatranscriptome analysis. Genome Med 7(1):1–19
Zhang Z, Xu D, Fang J, Wang D, Zeng J, Liu X, Hong S, Xue Y, Zhang X, Zhao X (2021) In situ live imaging of gut microbiota. mSphere 6(5):e0054521
Zheng D, Liwinski T, Elinav E (2020) Interaction between microbiota and immunity in health and disease. Cell Res 30(6):492–506
Zhou X, Du L, Shi R, Chen Z, Zhou Y, Li Z (2019) Early-life food nutrition, microbiota maturation and immune development shape life-long health. Crit Rev Food Sci Nutr 59(Suppl 1):S30–S38
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Salaria, N., Neeraj, Furhan, J., Kumar, R. (2023). Gut Microbiome: Perspectives and Challenges in Human Health. In: Sobti, R., Kuhad, R.C., Lal, R., Rishi, P. (eds) Role of Microbes in Sustainable Development. Springer, Singapore. https://doi.org/10.1007/978-981-99-3126-2_3
Download citation
DOI: https://doi.org/10.1007/978-981-99-3126-2_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-3125-5
Online ISBN: 978-981-99-3126-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)