
Abstract
Metabolic reprogramming is one of the most critical hallmarks in cancer cells. In the past decades, mounting evidence has demonstrated that, besides the Warburg Effect, lipid metabolism dysregulation is also one of the essential characteristics of cancer cell metabolism. Lipids are water-insoluble molecules with diverse categories of phosphoglycerides, triacylglycerides, sphingolipids, sterols, etc. As the major utilization for energy storage, fatty acids are the primary building blocks for synthesizing triacylglycerides. And phosphoglycerides, sphingolipids, and sterols are the main components constructing biological membranes. More importantly, lipids play essential roles in signal transduction by functioning as second messengers or hormones. Much evidence has shown specific alterations of lipid metabolism in cancer cells. Consequently, the structural configuration of biological membranes, the energy homeostasis under nutrient stress, and the abundance of lipids in the intracellular signal transduction are affected by these alterations. Furthermore, lipid droplets accumulate in cancer cells and function adaptively to different types of harmful stress. This chapter reviews the regulation, functions, and therapeutic benefits of targeting lipid metabolism in cancer cells. Overall, this chapter highlights the significance of exploring more potential therapeutic strategies for malignant diseases by unscrambling lipid metabolism regulation in cancer cells.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
4.1 Introduction
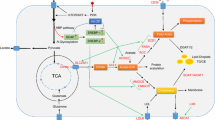
More and more oncology studies have revealed metabolic reprogramming as a hallmark of cancer [1, 2]. It has been shown that cancer cells in rapid proliferation exhibit high demands for macromolecule biosynthesis and energy consumption. The increased demands of glycolysis and glutamine consumption are even more [3,4,5]. For instance, the Warburg Effect decouples glycolysis from pyruvate oxidation in the glucose metabolism of cancer cells. Consequently, despite high oxygen availability in cancer cells, mitochondrial respiration cannot generate maximal ATP through the utilization of carbohydrates. A deeper understanding of these metabolic changes has accelerated new therapeutic approaches to cancer [3]. In recent years, though receiving less attention, lipid metabolic reprogramming in cancer cells has been increasingly recognized (Fig. 4.1). It has been widely accepted that lipid metabolic reprogramming is a critical molecular process in the progression of human malignancies [1, 6, 7]. By promoting the exogenous lipids’ uptake or increasing endogenous lipid synthesis, cancer cells with active proliferation show high lipid avidity [8]. A growing body of studies has proved that, as most of the lipogenic enzymes are activated, cancer cells exhibit a shift in lipid metabolism [9]. For example, it has been reported that cancer cells cultured with a medium containing lipoprotein-deficient serum could significantly inhibit proliferation and increase cell death. However, supplementation of high-density lipoprotein (HDL), low-density lipoprotein (LDL), or very-low-density lipoprotein (VLDL) into the serum could partially restore the growth rate of transformed cells, suggesting the supportive role of lipoproteins in tumor growth [10]. Moreover, it has been reported that prostate cancer cells could significantly elevate the uptake of exogenous cholesterol and lipoproteins, leading to accumulation in lipid droplets (LDs) of cholesteryl ester (CE) and its storage depletion. This metabolic alteration reduces cancer proliferation, impairs its invasion capability, and suppresses tumor growth [11]. The LDs reserve excessive lipids and cholesterol in cancer cells. Therefore, high LDs and stored-CE content are now considered a hallmark of tumor aggressiveness [12]. All these discoveries emphasize the importance of lipid metabolism reprogramming, which plays a critical role in cancer cells.

Summary of the origin of lipids in mammalian cells. Lipid resources in cells are from de novo biosynthesis or the uptake of exogenous lipids. Due to the activation of oncogenic pathways, nutrition stress, or energy requirements for macromolecule biosynthesis in the transformed cells, genes regulating these two biological processes are expressed significantly. The abundance of lipids participates in membrane construction, energy support, signal transduction, etc. in cancer cells
4.2 Overview of Lipid Metabolic Reprogramming in Cancer Cells
Cancer happens in cells with disordered growth and proliferation, requiring nucleic acids, proteins, and lipids as building blocks. As sources of these building blocks, metabolic intermediates are often accumulated in cancer cells due to the disturbed metabolism. In the Warburg Effect, the most understood glucose metabolic disturbance in cancer cells, the carbon from glucose is used to build other molecules instead of complete oxidization to carbon dioxide [13]. During metabolism in normal cells, glucose undergoes glycolysis in the cytoplasm to produce pyruvate when the oxygen is sufficient. Pyruvate is then oxidized to acetyl-CoA after entering the mitochondria. Acetyl-CoA is a component of the Krebs cycle to reduce equivalents for oxidative phosphorylation. In the cytoplasm, excess pyruvate is fermented to lactate when the oxygen is limited. Due to the high efficiency, differentiated cells typically yield 36 ATP molecules with one glucose molecule undergoing the complete oxidative phosphorylation. Meanwhile, 2 ATP molecules are obtained from anaerobic glycolysis. The Warburg Effect fermentates pyruvate even in the presence of oxygen. It is characterized by lactate production, increased glucose uptake and consumption, and a decrease in oxidative phosphorylation. The increased glutamine metabolism marks another commonly observed alteration in cancer cells. By producing α-ketoglutarate to feed into the Krebs cycle, glutamine is a primary energy substrate in mammalian cells. α-ketoglutarate derived from glutamine contributes to citrate production by forwarding flux through the malic enzyme-dependent pyruvate and the Krebs cycle [14]. Through reversing the Krebs cycle reactions catalyzed by isocitrate dehydrogenase and aconitase, glutamine can also be converted to citrate. The citrate can then be used to generate acetyl groups for fatty acids (FAs) synthesis [15,16,17].
Lipid metabolism is altered in rapidly proliferative cells. The products of FAs are the hub in lipid metabolism. In the membrane, storage, or signaling lipids, incorporated intracellular FAs can be found. Otherwise, these FAs can be oxidized to carbon dioxide as an energy source [18,19,20]. In transformed cells from energy production, carbon is diverted to FAs for membranes and signaling molecules’ biosynthesis. In addition to sterols and sphingolipids, many of the cell membrane lipids are phospholipids (PLs), including phosphatidylcholine (PC) and phosphatidylethanolamine (PE). All these lipids are derived partially from acetyl-CoA or contain FAs. Either exogenous sources or de novo FA synthesis constitutes the FA building blocks in cancer cells. Interestingly, normal cells and transformed cells have a distinct preference for the utilization of FAs. Most normal cells prefer to use exogenous sources of FAs, while tumor cells preferentially synthesize FAs de novo, and a shift toward FA synthesis is often seen in these cells [21, 22]. For the transfer to the active status, FAs are covalently modified by CoA via fatty acyl-CoA synthetases. Upon activation, FAs are esterified with glycerol or sterol backbones, thus producing TGs and sterol esters (SEs). Then they are stored in the LDs. Besides de novo FA synthesis pathways, it was also found that some cancer cells scavenge lipids from the environment, indicating that the FA uptake pathways might be a critical molecular event for the malignant behaviors in cancer cells. Due to the difference between in vitro and in vivo conditions for cell culture experiments, the exogenous uptake may be more important in some types of cancer cells. For example, fatty acid-binding protein 4 (FABP4), a lipid chaperone, is involved in providing FAs for tumor growth, chemo-resistance, and even cancer metastasis of ovarian cancer cells from surrounding adipocytes [23,24,25]. In the study of prostate cancer, the utilization of fatty acid synthase (FASN) or ATP citrate lyase (ACLY) inhibitors can only reduce tumor cell viability in cell culture medium deprived with lipoproteins, an exogenous lipid source [26]. And more importantly, a serial of studies in recent years showed that CD36 is related to multiple cancers’ malignant behaviors. This molecule is a widely expressed transmembrane protein with multiple functions, including fatty acid uptake [27,28,29,30]. All the evidence indicates the reprogramming of lipid metabolism in cancer cells is in numerous dimensions (Fig. 4.2). Deeply understanding the undergoing mechanism for it would further extend our knowledge in cancer biology and help us explore more specific strategies to treat these malignant diseases.

Summary of some essential aspects of lipid metabolism in cancer cells. Some critical functional genes regulating lipid uptake, lipid biosynthesis, anabolic and catabolic metabolism of lipids, etc. are shown. All detailed functions and the full name of the genes’ abbreviation will be described in the following context
4.2.1 Signaling Functions of Lipid Products in Cancer Cells
The stimulated biosynthesis of FAs and cholesterol and the mobilization of free FAs from triacylglycerides increase the lipids’ level. As a result, the signaling functions of these lipid products contribute to different aspects of tumorigenesis. As a crucial component of lipid rafts, cholesterol can stimulate receptor-mediated signal transduction pathways [31]. Additionally, farnesyl-pyrophosphate, an intermediate of cholesterol synthesis, is required for protein prenylation. The addition of an isoprenoid chain modifies several vital proteins in the signal transduction. For example, geranyl-geranylation is essential for Rho, Rac, and cdc42 activity, while farnesylation is required to activate Ras and Rheb proteins [32]. Interestingly, suppressing the activity of retinoblastoma tumor suppressor RB causes senescence of cells by increasing the prenylation of N-Ras. This regulation is realized through the E2 transcription factor (TF)-dependent activation of sterol regulatory element-binding proteins (SREBP) [33]. All the evidence emphasizes the crucial role of lipid-mediated modification in cellular signal transduction.
For paracrine hormones and growth factors, including leukotrienes, prostaglandins, steroid hormones, and lysophosphatidic acid (LPA), lipids can also be a structural basis. The 20-carbon unit arachidonic acid is the source of leukotrienes and prostaglandins. And this acid is produced from phosphoglycerides by phospholipases A2 and C. Prostaglandin synthesized by the enzyme cyclooxygenase-2 (COX-2) has been implicated in the promotion of tumor growth, neovascularization, and metastatic cancer spread by activation of inflammatory responses [34]. LPA, a water-soluble phospholipid, comprises a phosphate group, a single fatty acid chain, and glycerol. It has been demonstrated that aberrantly high LPA activation can promote the initiation and progression of multiple cancers. Mechanistically, it promotes cancer cell survival, proliferation, and even migration through regulating the G-protein-coupled receptors [35]. In recent years, it has been demonstrated that the alteration of the free FAs level contributes to the progression and tumorigenicity of cancer. The enzyme monoacylglycerol lipase (MAGL), highly expressed in aggressive cancer cell lines and primary tumors, catalyzes monoacylglycerides to free FAs and glycerol. It regulates a FA network promoting the survival, growth, invasion, and migration of tumor cells, enriched with oncogenic signaling lipids. In nonaggressive cancer cells, the overexpression of MAGL can recapitulate this FA network and enhance their pathogenicity phenotypes. A high-fat diet can rescue the impairments of growth in cancer cells lacking MAGL activity, implying the exogenous sources of FAs can contribute to MAGL-dependent malignancies [36]. Together, these findings reveal how a lipolytic enzyme can turn the cancer cells’ lipogenic state into pro-tumor signals. Accumulative evidence has highlighted the importance of lipid products in the intracellular signal transduction of cancer cells.
4.2.2 Alterations in Lipid Metabolism in Cancer Cells
4.2.2.1 Deregulation of Lipid Metabolism in Cancer Cells
The survival and proliferation of many types of cancer require a supply of lipids. The primary pathway exploited by cancer cells for acquiring lipids is FA synthesis, especially the de novo FAs synthesis pathway activation. Boroughs et al. reported that most cultured cancer cells activate the de novo FAs synthesize pathway in the presence of abundant oxygen and extracellular nutrients. However, when there is metabolic stress, cancer cells will scavenge for extracellular lipids as a major adaptive mechanism to maintain viability and growth [1]. Yao et al. observed that rather than de novo synthesis, proliferating fibroblasts, and a serial of tumor cells prefer uptaking lipids directly from the extracellular environment [37]. Besides, many studies have suggested that to promote survival and proliferation, several cancer cells will utilize both lipogenic and lipolytic pathways to acquire lipids [6,7,8,9]. Together, these studies support how cancer cells obtain the lipids depend on the cell type and microenvironment. The obtaining approaches include de novo lipogenesis, uptaking from the environment, and hydrolysis of intracellular TG stored in LDs. They may play a crucial role in cancer initiation and progression. The link between cancer development and elevated level of lipid metabolism has been extensively featured [18]. The ways of lipid metabolism contributing to cancer progression are varied. One documented mechanism implicates the proliferation and growth of transformed cells by alterations in lipid metabolic pathways. These alterations can offer molecules for signaling transduction, substrates for phospholipid synthesis, and metabolic fuels through mitochondria oxidation [38]. Free FAs and cholesterols, the excess intracellular lipids, are esterified to TAG and CEs, respectively, and then incorporated into LDs [39]. In normal cells, the biology of LDs has also been extensively studied. For instance, after the fusion of smaller ones, LDs’ size can vary and increase as their generation proceeds [40]. Moreover, adipose tissue can store abundant LDs in physiological situations [41]. Several studies have proved that cancer cells exhibit a significantly increased number of LDs and LD-related proteins, including adipose differentiation-related protein (ADRP) [42]. Therefore, LDs and ADRP are potential biomarkers of cancer. Also, in harmful situations like drug toxicity, endoplasmic reticulum (ER) stress, and reactive oxygen species (ROS), the increased number of LDs and LD-related proteins can play an adaptive and protective role. Therefore, LDs and LD-related proteins promote cancer cell proliferation and tumor growth.
4.2.2.2 Multiple Steps of Lipid Biosynthesis in Cancer Cells
Citrate is an important mediator to link FA metabolism with other metabolic networks [43]. For example, as an intermediate in the Krebs cycle, citrate is the keypoint of glucose metabolism feeding into the FA metabolism. The citrate’s metabolic fate depends on its subcellular localization. The Krebs cycle is fed by mitochondrial citrate, and the FA synthesis is fed by cytoplasmic citrate. Citrate is transported by the transport protein citrate carrier (CIC) across the mitochondria’s inner membrane for utilization in the cytoplasm. It was found that elevated levels of CIC are correlated with poor prognosis in various cancers. Besides, inhibiting citrate transport by benzene-tricarboxylate analog (BTA) shows antitumor effects in xenograft mice with multiple tumor types [44]. In converting carbons from citrate to bioactive FAs, certain links are necessary, including ACLY, acetyl-CoA carboxylase (ACC), FASN, and acyl-CoA synthetases, also known as fatty acid-CoA ligase (ACS or ACSL). In recent years, a growing body of studies has demonstrated that the high level of some enzymes mentioned above is correlated with poor prognosis. Inhibiting them can have an antitumor effect in the established cancer models, especially in the model of decreased FA availability. When clinically applying these strategies, many enzymes’ inhibitors have minimal impact on non-transformed cells. Here, we would like to describe them one by one in details as the following.
As a central metabolic enzyme, ACLY catalyzes the conversion of citrate to oxaloacetate and coenzyme A (CoA) to acetyl-CoA, both of which are ATP dependent. For cholesterol biosynthesis, FA metabolism, and protein prenylation and acetylation, acetyl-CoA is essential. Due to its relation to the proliferation activity of cancer cells, ACLY has been considered a target for anticancer drugs in many studies. For example, by converting the six-carbon citrate to precursors for FA synthesis, the four-carbon oxaloacetate, and two-carbon acetyl-CoA, ACLY bridges glucose and lipid metabolism. The knockdown of ACLY suppresses the ability of cancer cells to transfer glucose to lipids, which has been observed in murine lymphoid cells and adenocarcinoma cells [45, 46]. By genetic or pharmacological inhibition of ACLY, this metabolic change prevents tumorigenesis and impairs xenograft tumor formation in mice [46, 47]. More importantly, inhibiting acetyl-CoA production may influence other abnormal metabolic pathways in transformed cells, due to its essential role as a substrate for the acetylation of proteins and nucleic acids [48, 49]. ACC carboxylates acetyl-CoA to form malonyl-CoA, one of the most highly regulated enzymes in FA biosynthesis [50]. Citrate and glutamate can allosterically regulate ACC and activate its expression. Meanwhile, this enzyme is negatively and allosterically regulated by long- and short-chain fatty acyl-CoAs, such as palmitoyl-CoA. The AMP-activated protein kinase (AMPK) can inhibit ACC’s activity by phosphorylation. ACC1 and ACC2 are the two isoforms of ACC in the human genome. ACC1 exists in lipogenic tissues in large amounts, while ACC2 is enriched in oxidative tissues. Due to their existence in different tissues, ACC1 and ACC2 have different functions in metabolism. Though malonyl-CoA is a common metabolite derived from both ACC1 and ACC2, the malonyl-CoA catalyzed by AAC1 is a substrate for FA synthesis, whereas the malonyl-CoA catalyzed by ACC2 inhibits CPT1 and prevents FA degradation. The inhibition of ACC1 induces apoptosis in prostate and breast cancer cells, but not in the non-transformed cells [51, 52]. The knockdown of both ACC1 and ACC2 by Soraphen-A in prostate cancer cells reveals similar results [51]. However, in breast cancer cells, where ACC is chemically inhibited by TOFA (5-(tetradecyloxy)-2-furoic acid), a contradictory result is reported [53]. Since TOFA has been observed to block the epidermal growth factor receptor (EGFR)-activated glioblastoma (GBM) cells, but not the EGFR-inactivated cell lines, we may explain the above findings with the role of EGFR signaling [54]. The observation further complicates the situation that, by promoting the NADPH-dependent redox balance, suppression of ACC1 or ACC2 can accelerate lung cancer growth [55]. Researchers still need to elucidate the other aspects of ACC roles in cancer cells. Importantly, it has been demonstrated that ACC activity is regulated in a phosphorylation-dependent manner. It is identified that AMPK phosphorylates ACC1 at Ser79 and ACC2 at Ser212. Thus the conversion of acetyl-CoA to malonyl-CoA can be inhibited [56,57,58]. Since AMPK can be activated by metformin, which is already widely used clinically, more preclinical experiments and clinical trials are needed to further explore the antitumor activity and therapeutic efficacy of metformin [59].
The successive condensation reactions catalyzed by FASN can form FA from malonyl-CoA and acetyl-CoA substrates, and the 16-carbon palmitate is the main product. In many types of cancer cells, the elevated level of FASN is potently correlated with enhanced FA synthesis and a poor prognosis [9]. It is reported that knockdown of FASN decreases TG and phospholipids levels, inhibits proliferation, and stimulates apoptosis of prostate cancer cells, while has no obvious effect on the viability and proliferation of non-transformed fibroblast cells [60]. More studies further confirmed the preferential killing of cancer cells by pharmacological inhibition of FASN [61]. Since most cancer cells depend on FASN-mediated de novo FA synthesis, whereas most non-transformed cells prefer uptake of exogenous FAs, FASN is a particularly appealing therapeutic target. However, data from the mechanistic experiments showed that FASN inhibitor might induce cell death due to the toxic accumulation of malonyl-CoA, rather than a lack of FAs [53].
ACS activates FAs to generate FA-CoA, thus entering the bioactive pools. The bioactive FAs also participate in protein palmitoylation, a crucial posttranslational modification in several cancers [62]. ACSL1, ACSL3, ACSL4, ACSL5, and ACSL6 are the five isoforms of ACS genes in mammals. Among them, ACSL4 is upregulated in colon adenocarcinoma, and ACSL5 level is increased in GBM [63, 64]. Overexpression of ACSL4 promotes tumor cell survival by preventing apoptosis, likely through depletion of unesterified arachidonic acid, which yields a pro-apoptotic signal [63]. Chemical inhibition of ACS by Triacsin C (inhibitor targeting ACSL1, ACSL3, and ACSL4, but not ACSL5 or ACSL6) preferentially induces apoptotic cell death in lung, colon, and brain cancer cells [65,66,67]. Several thiazolidinediones (TZDs) can inhibit the activity of ACSL4 (not ACSL1 or ACSL5) by direct binding in vitro. TZDs, widely used in diabetes treatment, can stimulate the peroxisome proliferator-activated receptors (PPARs), especially PPARɣ. Strikingly, TZD utilization, in a PPARɣ-independent manner, is related to decreased incidence of several cancers [68]. Since different isoforms of ACS have different tissue specificities, responses to nutritional state, and preferred substrates, this is a noteworthy point when considering treatment through ACS inactivation [69]. Stearoyl-CoA desaturase (SCD) mainly catalyzes stearoyl-CoA to oleoyl-CoA by introducing the double bonds into short-chain FAs in the C9 position [70]. By changing the physical properties of FAs, this introduction has profound effects on lipid function. SCD1 and SCD5 are the two isoforms of SCD in human beings. The frequently increased SCD1 expression and activity and its importance for cancer biology are increasingly recognized [71]. By induction of unsaturated FAs, inhibiting SCD1 can promote the death of cancer cells [72]. Pharmacological inhibition of SCD1 limits tumor growth in preclinical cancer models without affecting the overall body weight [73, 74]. Interestingly, FAs are also substrates for sphingolipid synthesis. Sphingolipids such as ceramides and sphingosine-1-phosphate can actively suppress or promote tumor growth [75]. Furthermore, the accumulation of ceramides is involved in the therapeutic effects of various cancers [76,77,78].
4.2.3 The Contribution of Lipid Metabolism for the Malignant Behaviors in Cancer Cells
In recent years, accumulating evidence has demonstrated lipid metabolism influence in several aspects of cancer cells. Owing to the diversity of malignant behaviors in cancer cells, some of the potential roles and mechanisms of altered lipid metabolism on tumor growth, energy adaption, redox homeostasis, etc. are described below.
4.2.3.1 Altered Lipid Metabolism for Cancer Cell Proliferation and Tumor Growth
As the building blocks for biological membranes, lipids are primarily required for the highly proliferative cancer cells. In cultured mammalian cells, following Akt activation or interleukin-3 administration, lipid synthesis is also necessary for cell growth [79]. Researchers have also found the essential role of SREBP in maintaining the cell and organ size of Drosophila melanogaster, indicating the conserved significance of lipogenesis for growth [80]. The lipogenic gene SREBP is activated during mitosis, thus affecting the cell cycle progression [81]. SCD, the target gene of SREBP, has been reported to be overexpressed in ontogenically transformed cells and in several human cancers [82,83,84]. SCD is essential for cell transformation in vitro and is associated with the genetic predisposition and growth of cancer in the mouse model [85]. The knockdown of SCD or interruption of the Scd gene inhibits lipid synthesis and enhances β-oxidation by activating AMPK in mice [83, 86, 87]. And the cell cycle progression can be blocked, and cell death can be induced by a chemical inhibitor of SCD1 in lung cancer cells [88].
4.2.3.2 Altered Lipid Metabolism for Energy Homeostasis in Cancer Cells
Compelling evidence has shown the necessity of de novo lipid synthesis in cancer cell proliferation. However, why the uptake of exogenous lipids fails to meet the enhanced lipid demand needs to be elucidated. Therefore, researchers reasonably hypothesize that lipid synthesis may be involved in the tumourigenic process. Cancer cells produce and secrete a high level of lactate due to their large glucose consumption for energetic and biosynthetic use [89]. Thus mechanisms equilibrating the intracellular pH are required, which leads to acidification of the tumor microenvironment [90]. In some cancer cells and conditions, lipid synthesis functions as a carbon sink to sequester excess pyruvate and avoid lactate production but maintains the glycolytic rate at a high level. Furthermore, this metabolic process may also participate in redox balance. When oxygen is not available, hypoxia-tolerant organisms use NADP+, a metabolite of lipid synthesis, as an electron acceptor. And the hypoxic cancer cells may adopt a similar strategy, where lipid synthesis-derived NADP+ can elevate the cytoplasmic NAD+ level to maintain glycolysis [91]. The cytosolic NADP+ is used by isocitrate dehydrogenase-1 to produce α-ketoglutarate since a mitochondria-cytosolic NADPH shuttle may exist [92]. After being transported to the mitochondria, this metabolite is converted back to isocitrate with NADP+’s concomitant production. This recently described inverse reaction is catalyzed by isocitrate dehydrogenase-2 [16, 17, 93]. When there is not enough oxygen to maintain flux through the electron transport chain, the mitochondrial NADH/NAD+ ratio goes up. In this way, the mitochondrial nicotinamide nucleotide transhydrogenase can take up the excess NADH to transfer a proton to NAPD+ and generate NAD+. Through the malate-aspartate or the glycerol phosphate shuttles, this product is available to maintain glycolysis [94]. Thus, lipid synthesis is involved in redox balance between the mitochondria and cytoplasm and plays a role in maximizing glycolysis.
4.2.3.3 Altered Lipid Metabolism for Resistance to Oxidative Stress in Cancer Cells
Oxidative stress is harmful to the survival and proliferation of mammalian cells. The resistance to oxidative stress is one of the critical characteristics of transformed cells. Much evidence has demonstrated the resistance to oxidative stress can be promoted by de novo lipid biosynthesis in cancer cells [95,96,97]. Due to the absence of desaturase, mammalian cells are incapable of synthesizing polyunsaturated FAs. Therefore, compared with those obtained through diet, a high de novo lipid synthesis rate can elevate the relative amount of monounsaturated and saturated FAs. Polyunsaturated acyl chains are more susceptible to peroxidation. Studies show that lipid synthesis inhibition makes cancer cells sensitive to death induced by oxidative stress or chemotherapy drugs, indicating a novel therapeutic target for cancer. However, this intriguing observation still needs further investigation.
4.2.3.4 Altered Lipid Metabolism for Resistance to Energy Stress in Cancer Cells
Most cancer cells have a high glucose utilization rate to meet the increased demands of energy and biosynthesis. In contrast, some cancer cells display a mounting dependence on lipid oxidation as their primary energy source [1]. For instance, prostate cancer cells exhibit a low glucose consumption rate in general. But the uptake of FAs, such as palmitate, is increased, and some β-oxidation enzymes are overexpressed in these cells [98]. The specialized metabolism of prostate epithelial cells, which secrete high levels of citrate into the prostatic fluid, may explain the above observation. And prostate cancer cells will reactivate the TCA cycle to oxidize the secreted citrate during transformation. Moreover, β-oxidation has been demonstrated to play a role in the proliferation and survival of leukemia cells. Under energy stress, the activation of β-oxidation plays a crucial role in supporting cancer cell viability [99]. The hematopoietic cells can be sensitized to withdraw glucose or growth factors through the constitutive activation of the PI3K/Akt pathway [100]. However, under these circumstances, activating β-oxidation alone is enough to maintain cell viability [101]. In GBM cells, β-oxidation has also been proved to participate in ATP production and oxidative stress resistance by providing substrates for glutathione and NADPH production, thus allowing cells to remove ROS [102].
4.2.4 Upstream Regulatory Mechanisms of Lipid Synthesis in Cancer Cells
Most of the FA and cholesterol synthesis enzymes are regulated by SREBPs, TFs of the helix-loop-helix leucine zipper family [103]. SREBP1a, SREBP1c, and SREBP2 are the three SREBP isoforms identified in mammalian cells. Among them, SREBP1a is the isoform with the greatest abundance in most cultured cells and mainly controls FA, phospholipid, and TG synthesis. Both SREBP1a and SREBP1c are alternatively spliced, and their levels vary across different tissues. Meanwhile, SREBP2 regulates the expression of genes associated with cholesterol synthesis [104]. The SREBPs’ activity is closely related to the intracellular sterol concentration [105]. The SREBP/SCAP complex binds to COPII-coated vesicles when the sterol level is low and would be translocated to the Golgi apparatus. In this organelle, the transcriptionally active fragment of the 65-kDa N-terminal is released by a two-step proteolytic cleavage [106]. This mature protein enters the nucleus and binds to the promoter region of SREBP target genes with the sterol regulatory elements [107]. When the sterol level is saturated, the combination between SREBP/SCAP complex and COPII is blocked due to insulin-induced genes binding. Thus, the complex stays in the endoplasmic reticulum and cannot enter the nucleus. This classic sterol-dependent regulation termed “regulated intramembrane proteolysis,” is mainly applicable to SREBP2 target genes. It is highly conserved from flies to mammals. However, Drosophila’s SREBP processing is regulated by phosphatidylcholine and phosphatidylethanolamine rather than sterols [108]. Interestingly, even in the presence of cholesterol, the depletion of phosphatidylcholine in mammalian cells leads to SREBP1 accumulation in the nuclear, but not SREBP2. And SREBP1 accumulates through a SCAP-independent mechanism, indicating phospholipid level as the principal regulator of SREBP1 [109]. Besides proteolysis regulation, SREBPs’ activity is regulated by their interaction with transcriptional coactivators, such as p300 [110]. To transcriptionally activate specific target genes, SREBP can also recruit and bind to cofactor or mediator complexes [111]. Furthermore, with a cdc4 phospho-degron motif, SREBPs can be phosphorylated by glycogen synthase kinase 3, leading to the polyubiquitination and degradation of mature proteins [112, 113]. The phosphoinositide 3-kinase/Akt/PKB signaling pathway is often stimulated in human cancer cells. By phosphorylating the ATP-citrate lyase, Akt can elevate the expression of several FA and cholesterol synthesis-related genes [114]. As critical downstream effector of Akt, the mammalian target of rapamycin complex I (mTORC1) is a multiprotein kinase implicating in certain metabolic processes [115]. Intriguingly, the accumulation of mature SREBP1 in the nucleus needs an activated mTORC1, a downstream target of Akt phosphorylation. The metabolically regulatory role of mTORC1 has also been proved in cells lacking the tuberous sclerosis complex 1 or 2 genes, which are two negative regulators of mTORC1 [80]. mTORC1 also modulates the level of SREBP1 and is essential for stimulating hepatic lipogenesis [116]. In mammalian cells and the developing wing of D. melanogaster, SREBP is also required for the cell size control dependent on Akt activation. This finding explains the coordinated manner of the Akt/mTORC axis in regulating protein and lipid synthesis during cell growth [80]. SREBP is also found downstream of certain tumor-suppressive pathways. Downstream of the tumor suppressor liver kinase B1 (LKB1), AMPK can directly phosphorylate SREBP and inhibit its proteolytic function [117]. Through SREBP1 and SREBP2 induction, the retinoblastoma protein loss increases the expression of genes implicated in the isoprenylation of N-Ras [35]. Moreover, mutant p53 binds to SREBP at the promoter region of genes on the mevalonate pathway, promoting its expression. Through disturbing the tissue architecture and promoting breast cancer formation, this hyperactivation reveals the crucial role of SREBP-dependent lipogenesis during transformation [118]. Both SREBP1 and SREBP2 are upregulated in many cancers. Though independent of mTORC1, SREBP1 is activated by aberrant EGFR signaling in human GBM. Meanwhile, SREBP1-dependent induction of low-density lipoprotein receptors is critical for the survival of some cancer [119, 120]. These findings have verified the primary role of activated SREBP in oncogenic signaling pathways. The microenvironment around solid tumors is often hypoxic due to the increased tumor volume. Low oxygen level induces hypoxia-inducible factors (HIFs), two heterodimeric TFs formed by an a-subunit (HIF1-α or HIF2-α), and a β-subunit. In the presence of oxygen, the oxygen-sensitive prolylhydroxylases target and mark HIF-1α and HIF-2α. These two TFs are then degraded by the tumor suppressor, Von Hippel–Lindau (VHL)-dependent ubiquitination [121]. In renal cell carcinomas, VHL is frequently mutated, thus creating a pseudo-hypoxic state that stabilizes the level of HIF1α and HIF2α, even under normoxia [122]. HIFs can also be induced by tumorigenic pathways [123]. Interestingly, HIFs activity can also be attributed to metabolic activity. TCA cycle enzymes of fumarate, hydratase, and succinate dehydrogenase with inactivating mutations can lead to the collection of succinate, which blocks prolyl-hydroxylases and promotes the assembly of HIF1α [124]. By promoting the expression of vascular endothelial growth factor (VEGF), HIF activation can induce angiogenesis. And through a metabolic shift to anaerobic energy production, HIFs can also adapt to the hypoxic microenvironment [125, 126]. HIF also increases the expression of glucose transporter 1 (GLUT1) and other glycolytic enzymes [127, 128]. HIF also upregulates the level of pyruvate dehydrogenase kinase 1 (PDK1), a kinase that phosphorylates and suppresses pyruvate dehydrogenase, thus blocking the entry of pyruvate into the TCA cycle and the glucose-derived lipid synthesis [129]. However, it has been observed that in breast cancer cells, HIF1 increases the level of FASN, which is upregulated in the hypoxic tumor environment [130]. Since hypoxia attenuates the carbon flow from glucose to FAs, the FA synthesis under this condition requires other carbon sources. Indeed, the bidirectional enzyme Acetyl-CoA synthetase 2, which catalyzes acetyl-CoA synthesis from cytoplasmic acetate, is upregulated under this hypoxic condition to facilitate the survival of cancer cells [131]. More importantly, two back-to-back papers published in Nature have reported that when mitochondria are unfunctional, the primary carbon source for lipid synthesis falls in glutamine. Researchers found that by reductive carboxylation of glutamine-derived α-ketoglutarate in cancer cells, isocitrate dehydrogenase-1 can produce cytoplasmic citrate, which is an active metabolic phenotype in conditions of hypoxia and defective mitochondria [16, 17]. In different tissues, hypoxia has been demonstrated to have an inhibitory effect on β-oxidation. In the heart cells, ischemia inhibits β-oxidation by blocking the oxidation of NADH and FADH2. When the macrophages are exposed to hypoxic conditions, the storage of triacylglycerides is enhanced [132, 133]. By introducing hypoxia-inducible protein 2 implicated in the deposition of neutral lipids into LDs, HIF1 can also promote lipid accumulation [134]. Through the induction of PPARɣ, HIF1α can also enhance the uptake of free FAs and TG production in the liver and adipose tissue, respectively [134]. When researchers specifically knockout VHL in mouse liver, they observe steatosis with increased LD formation and downregulated β-oxidation genes. They have reported that HIF2α is responsible for these metabolic changes as well [135]. HIF2a has also been found to downregulate SREBP1c and its target genes in the liver. Interestingly, it has been reported that the accumulation of lipids is frequently occurring in renal cancer cells, where VHL is mutated and the HIF1α level is stabilized [136]. However, the exact role of LDs in promoting tumor cell proliferation and progression is not fully elucidated. Researchers have proposed that under intermittent hypoxia, the increased storage of triacylglycerides could be beneficial as a ready-to-use fuel source after reoxygenation.
4.2.5 Molecular Events of Fatty Acid Degradation in Cancer Cells
FA levels might be reduced in cancer cells due to their increased degradation, broken down by β-oxidation in the mitochondria. By carnitine palmitoyltransferase 1 (CPT1), FA-CoAs are converted to FA carnitines at the outer membrane of mitochondria after transported from the cytoplasm. In the mitochondria, acetyl-CoAs are produced following the repeat cleavage of FAs. The acetyl-CoAs can enter the TCA cycle and reduce equivalents for oxidative phosphorylation. Limitations on FA abundance by enhancing its oxidation could be theoretically beneficial, but experimental data have revealed mixed ideas. Carnitine palmitoyl transferase-1 (CPT1) participates in the rate-limiting and first step of FA transportation into mitochondria for oxidation to carbon dioxide, which is inhibited by malonyl-CoA. As the direct product derived from ACC, malonyl-CoA depletion can enhance the β-oxidation of FAs and inhibit the activity of ACC2. Therefore, the increased degradation of FAs may partially contribute to the slowed proliferation of cancer cells by inhibiting ACC. Whether the elevated FA oxidation will block the growth of cancer cells remains unclear. Depending on the energy needs and ACC isoforms, cancer types might differ in their clinical responses to the enhanced FA oxidation, diminishing the FA availability. Moreover, the CPT1 and FA oxidation inhibitors, etomoxir and ranolazine, may both kill cancer cells [102, 137]. Another significance of the increased FA oxidation rate lies in the elevated cellular ATP level, which provides energy for further cell proliferation. Indeed under energy stress, CPT1C, the brain isoform of CPT1, is vital for the existence of cancer cells [138]. PPARα is a central transcriptional regulator of FA oxidation. Extended PPARα activation causes liver cancer in mice and rats by an unclear mechanism that implicates cell cycle disturbance and ROS production [139]. However, PPARα agonists administered in humans have not caused similar cancers, and on the contrary, PPARα activation suppresses tumor growth in the established cancer models [140].
4.2.6 Diverting Fatty Acids to Storage in Cancer Cells
Once produced, FAs can be utilized for membrane lipid synthesis, be degraded, or be stored. Intriguingly, elevated storage of FAs belonging to neutral lipids, such as TGs or SEs, can decrease availability the FAs to be used as membrane building or signaling blocks, thus blocking cell proliferation. Most cells store TGs in the cytosolic LDs, the prominent lipid storage organelle [141]. The function of LDs in cancer cells remains still unclear. In many cancer cells, researchers have observed an increased number of LD and proposed them to be pathogenic. However, LD accumulation per se may not be the reason for cancer development but might reflect a cellular response to energy stress [42]. The location of LD accumulation, whether it occurs in cancer or surrounding cells, needs to be elucidated in future studies.
The major pathway of TG synthesis is known as the glycerol phosphate or Kennedy pathway. It condenses FAs with enzymes of acylglycerolphosphate acyltransferase (AGPAT), glycerol-3-phosphate acyltransferase (GPAT), diacylglycerol acyltransferase (DGAT), and phosphatidic acid phosphohydrolase (PAP). Except for the most distal enzyme (DGAT), all the other products will feed into the PL synthesis. Therefore, the PL production may be limited by blocking AGPAT, GPAT, and PAP, while activating DGAT might enhance the FA storage. Additionally, only with concomitant suppression of FA release can the potential benefits of improving FA storage be realized.
AGPAT esterifies lysophosphatidic acid (LPA) and an FA-CoA to form phosphatidic acid (PA). There are 11 members of AGPATs documented [142]. An elevated expression of AGPAT2 predicts a poor prognosis of ovarian cancer, and AGPAT2 inhibitors exhibit antitumor activity in the xenograft mouse model. Additionally, AGPAT9 and AGPAT11 have been reported to be upregulated in various cancers [143].
PAP, also known as Lipin, produces diacylglycerol (DG) by removing a PA’s phosphate group. As one of the least studied enzymes in terms of cancer, it is still unclear how this step of lipid synthesis and storage influences cancer progression. However, PAP is implicated in regulating SREBP activity, a TF family modulating the expression of several enzymes participating in FA and cholesterol biosynthesis [144]. PAP is phosphorylated and then inactivated by mTORC1, leading to SREBP activation [145]. This discovery suggests that PAP may therefore have a significant effect on maintaining cellular lipid homeostasis.
DGAT encodes a multipass transmembrane protein that functions as a critical metabolic enzyme. It catalyzes the conversion of DG and FA-CoA to TG. DGAT1 and DGAT2 are the two isoforms of DGATs Mammals have. DGAT catalyzes the only dedicated step in TG conversion, resulting in decreased available lipids via increased lipid storage. In transformed human fibroblasts, increased TG, reduced phospholipids level, and inhibited cell growth and invasiveness can be observed due to DGAT1 overexpression [146]. Inhibiting DGAT1 may also facilitate the accumulation of its substrate DG in cancer cells, which is of great significance in the signal transduction [147]. These findings would give cautious concerns in treating cancers with DGAT1 inhibitors, which are under clinical trials for metabolic diseases.
4.2.7 Lipid Uptake in Cancer cells
Transformed cells require more lipids and would uptake more lipids than normal cells. One way of increasing the uptake is through the upregulation of receptors for plasma lipids on the cell surface. Among these receptors, the cluster of differentiation 36 (CD36) can facilitate lipid uptake from the extracellular environment [148]. In a recent study, Pascual et al. revealed that with lipid receptor CD36, cancer cells display a poor prognosis. It has also demonstrated that inhibition of CD36 impairs the metastasis of cancer cells. Along with other findings, the FA receptor CD36 has been identified as a metastasis-initiating marker and driver in a lipid metabolism-dependent manner [29].
Another approach to enhance lipid uptake in cancer cells is through the increased level of fatty acid-binding proteins (FABPs) [149]. In regulating lipid uptake and tumor development, different isoforms of FABPs expressed in various tissues may play distinctive roles. FABP3 and FABP7 have been shown to promote the uptake of extracellular FAs under hypoxia, thus forming an increased level of LDs [150]. Additionally, through activating the intracellular receptor PPARβ/δ, overexpression of FABP5 plays a critical role in promoting cell proliferation and tumor growth in prostate cancer [151]. These findings extend our knowledge of lipid metabolism, thus providing potential targets for novel antitumor therapeutics.
4.2.8 Release of Lipids from Storage in Cancer Cells
Specific lipases can release FAs from storage for utilization. By inhibiting lipolysis, the available pool of FA for cancer cell proliferation becomes smaller. Lipolysis can also produce FAs to serve as precursors of essential signaling lipids [152]. In adipocytes, sequential reactions through adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoacylglycerol lipase (MAGL) can fully hydrolyze one TG molecule to release three FAs in the LD. Moreover, each of these lipases also has specific functions in other tissues.
Currently, most studies addressing the lipases–cancer relation have focused on MAGL. Upon leaving the glycerol backbone, MG is hydrolyzed by MAGL to the final FA. Researchers have observed enhanced MAGL expression and activity in primary tumors and aggressive human cancer cells [36]. Chemically inhibiting MAGL by JZL184 can lower the free FA level and prevent melanoma and ovarian cancer cells’ tumorigenicity both in vitro and in vivo. In contrast, the upregulation of MAGL showed the opposite phenotype. Interestingly, when the MAGL inhibitor suppresses the mouse model’s tumor growth, a high-fat diet can reverse the phenotype. This observation raises whether a specific diet can influence the efficacy of targeting lipid metabolism as a cancer treatment [36]. Furthermore, MAGL also functions in regulating signaling lipids. For example, invasive tumors have increased levels of LPA and PGE2, which can be lowered by MAGL inhibitors.
4.2.9 Formation of Lipid Droplets in Cancer Cells
LDs are major energy storage organelles of excess cellular lipids in esterified form. Cancer cells contain remarkably more LDs than normal cells. The upregulation of certain LD-decorating proteins (i.e., hypoxia-inducible protein 2 (HIG2), Perilipin, ADRP, and Tip47) has been proved to facilitate the formation and accumulation of LDs in different types of cancer cell [153]. These proteins may participate in scavenging ROS, maintaining ER homeostasis, and drug resistance in cancer cells. LD density stimulation enhances the proliferation of colon cancer cells (CRC), whereas perilipin 2 (PLIN2) knockdown suppresses tumor growth [154]. Another research has reported that the HIF-2α/PLIN2/lipid storage axis is essential for ER homeostasis and resistance against cytotoxic ER stress [155]. It has also revealed that loss of HIF-2a/PLIN2-dependent lipid storage enhances sensitivity to ER stress. Moreover, suppressing lipid synthesis or silencing PLIN2 can noticeably attenuate CRC proliferation [156]. All of the studies above suggest LD-associated proteins as potential targets for cancer treatment. However, more studies are needed to explain further the LD-associated stress responses mechanically.
4.2.10 Lipid Scavenging and Fatty Acid Oxidation in Cancer Cells
Lipids, required for cell survival and growth, comprise a large fraction of mammalian cells’ dry weight. With oxygen and abundant extracellular nutrients, most cancer cells choose the de novo synthesis to produce FAs. However, to maintain viability and proliferation, cancer cells have to scavenge extracellular lipids to adapt to metabolic stress. Scavenging, rather than synthesizing lipids, spare cells from the need to supply carbon. The oncogenic Ras stimulates the uptake and consumption of lysophospholipids, providing an intracellular lipid pool for tumor growth, which also occurs under hypoxia [157, 158]. As a result, cancer cells driven by KRas are resistant to the silencing of SCD1. This enzyme generally desaturates the de novo FAs synthesis before their incorporation into complex lipids [159]. Extracellular lipids, especially desaturated FAs, are also critical in cells with inactivated mTORC1 under hypoxia. In this context, protein synthesis is enhanced and lipid desaturation is decreased, resulting in activation of the unfolded protein response and cell death, but can be rescued by unsaturated FAs [160]. These findings have been repeated in renal carcinoma, GBM, and bladder cancer, suggesting solid tumors’ dependence on the extracellular environment to uptake FAs. However, a subset of diffuse large B-cell lymphomas prefer FA oxidation as a fuel source even under nutritious conditions, which express high levels of related enzymes [161]. Autophagy and related processes enable other cells to utilize FAs for fuel. In a genetic lung cancer mouse model, impaired autophagy leads to lipid accumulation, dysfunctional mitochondria, defective FA oxidation, and enhanced starvation sensitivity. Moreover, FA oxidation and other mitochondrial oxidative pathways are likely to enable cancer cells to survive through the regression period [162]. In a pancreatic cancer model driven by KRas, tumor regression induced by kinase inhibitors or knockdown of KRas produce a dormant population of cancer cells, which largely rely on mitochondrial respiration for survival. Inhibition of either autophagy or FA oxidation decreases the tumor-initiating potential of this population, thus mimicking a condition after an initial round of treatment and suggesting the importance of these catabolic pathways in enabling cancer cells to form tumors [163]. Another study has shown the coordinated mechanisms of stromal cells providing FAs to tumor cells as the fuel source, especially in ovarian cancer [28]. These tumor cells generally metastasize to the omentum, the large fold of fatty tissue in the abdomen. The co-culture of ovarian cancer cells with adipocytes has revealed that FAs transferred from adipocytes can activate AMPK and FA oxidation in the cancer cells, thus promoting cell proliferation. These findings raise many questions regarding the possibility of metabolite transfer between cells and the role of tumor microenvironment in promoting cell metabolism.
4.3 Conclusion and Perspective
Compelling evidence has revealed the critical role of lipid metabolism reprogramming in supporting transformed cells’ malignant behaviors. They are obtained from lipidomics studies, established cancer models, and clinical trials. Disrupting lipid metabolism can induce the regression of tumors and inhibit their metastatic spread. The approaches include limiting lipids’ origins, blocking lipid utilization, breaking down LDs, or deactivating certain enzymes involved in lipid metabolism. Based on these findings, the lipid metabolic pathways have been targeted to develop several cancer treatment drugs. However, many of the mechanisms involved have not been elucidated clearly. Certain inhibitors could suppress the proliferation and growth of cancer cells, but along with cytotoxicity in the normal cells. Thus, an all-around and in-depth understanding of lipid metabolism in cancer cells is necessary for future studies. The potential challenges are as the following: (1) identify different lipid metabolic processes and the hub genes involved in the initiation and progression of cancer, (2) avoid toxicity to normal cells while developing targeted drugs, and (3) clarify the relationship and crosstalk of lipid, glucose, protein, and energy metabolism. The oncogenic signalings and lipid metabolism are interwound with each other, and the lipids function in a broad spectrum at both cellular and organismal levels. These facts highlight the importance of targeting lipid metabolism in offering novel cancer treatment strategies in the future.
References
Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17(4):351–9.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619–34.
Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35(8):427–33.
Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15(21):6479–83.
Cao Y. Adipocyte and lipid metabolism in cancer drug resistance. J Clin Invest. 2019;129(8):3006–17.
Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, Cao Y. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer. 2017;16(1):76.
Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis. 2016;5(1):e189.
Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7(10):763–77.
Huang J, Li L, Lian J, Schauer S, Vesely PW, Kratky D, Hoefler G, Lehner R. Tumor-Induced hyperlipidemia contributes to tumor growth. Cell Rep. 2016;15(2):336–48.
Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, Cheng L, Masterson TA, Liu X, Ratliff TL, Cheng JX. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014;19(3):393–406.
de Gonzalo-Calvo D, López-Vilaró L, Nasarre L, Perez-Olabarria M, Vázquez T, Escuin D, Badimon L, Barnadas A, Lerma E, Llorente-Cortés V. Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: a molecular and clinicopathological study. BMC Cancer. 2015;15:460.
Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14.
DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104(49):19345–50.
Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105(48):18782–7.
Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481(7381):385–8.
Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481(7381):380–4.
Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care. 2006;9(4):358–65.
DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. 2012;148(6):1132–44.
Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. 2012;279(15):2610–23.
Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953;13(1):27–9.
Ookhtens M, Kannan R, Lyon I, Baker N. Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor. Am J Phys. 1984;247(1 Pt 2):R146–53.
Gharpure KM, Pradeep S, Sans M, Rupaimoole R, Ivan C, Wu SY, Bayraktar E, Nagaraja AS, Mangala LS, Zhang X, Haemmerle M, Hu W, Rodriguez-Aguayo C, McGuire M, Mak CSL, Chen X, Tran MA, Villar-Prados A, Pena GA, Kondetimmanahalli R, Nini R, Koppula P, Ram P, Liu J, Lopez-Berestein G, Baggerly K, Eberlin LS, Sood AK. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat Commun. 2018;9(1):2923.
Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, Yamada SD, Peter ME, Gwin K, Lengyel E. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17(11):1498–503.
Mukherjee A, Chiang CY, Daifotis HA, Nieman KM, Fahrmann JF, Lastra RR, Romero IL, Fiehn O, Lengyel E. Adipocyte-induced FABP4 expression in ovarian cancer cells promotes metastasis and mediates carboplatin resistance. Cancer Res. 2020;80(8):1748–61.
Ros S, Santos CR, Moco S, Baenke F, Kelly G, Howell M, Zamboni N, Schulze A. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov. 2012;2(4):328–43.
DeFilippis RA, Chang H, Dumont N, Rabban JT, Chen YY, Fontenay GV, Berman HK, Gauthier ML, Zhao J, Hu D, et al. CD36 repression activates a multicellular stromal program shared by high mammographic density and tumor tissues. Cancer Discov. 2012;2:826–39.
Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, Nieman KM, Pascual G, Benitah SA, Montag A, Yamada SD, Abumrad NA, Lengyel E. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene. 2018;37(17):2285–301.
Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A, Hueto JA, Bescós C, Di Croce L, Benitah SA. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017;541(7635):41–5.
Watt MJ, Clark AK, Selth LA, Haynes VR, Lister N, Rebello R, Porter LH, Niranjan B, Whitby ST, Lo J, Huang C, Schittenhelm RB, Anderson KE, Furic L, Wijayaratne PR, Matzaris M, Montgomery MK, Papargiris M, Norden S, Febbraio M, Risbridger GP, Frydenberg M, Nomura DK, Taylor RA. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci Transl Med. 2019;11(478):eaau5758.
Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1(1):31–9.
Sebti SM. Protein farnesylation: implications for normal physiology, malignant transformation, and cancer therapy. Cancer Cell. 2005;7(4):297–300.
Shamma A, Takegami Y, Miki T, Kitajima S, Noda M, Obara T, Okamoto T, Takahashi C. Rb Regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer Cell. 2009;15(4):255–69.
Gupta GP, Nguyen DX, Chiang AC, Bos PD, Kim JY, Nadal C, Gomis RR, Manova-Todorova K, Massagué J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007;446(7137):765–70.
Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3(8):582–91.
Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140(1):49–61.
Yao CH, Fowle-Grider R, Mahieu NG, Liu GY, Chen YJ, Wang R, Singh M, Potter GS, Gross RW, Schaefer J, Johnson SL, Patti GJ. Exogenous fatty acids are the preferred source of membrane lipids in proliferating fibroblasts. Cell Chem Biol. 2016;23(4):483–93.
Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res. 2013;52(4):585–9.
Koizume S, Miyagi Y. Lipid droplets: a key cellular organelle associated with cancer cell survival under normoxia and hypoxia. Int J Mol Sci. 2016;17(9):1430.
Wilfling F, Haas JT, Walther TC, Farese RV Jr. Lipid droplet biogenesis. Curr Opin Cell Biol. 2014;29:39–45.
Ahmadian M, Duncan RE, Jaworski K, Sarkadi-Nagy E, Sul HS. Triacylglycerol metabolism in adipose tissue. Future Lipidol. 2007;2(2):229–37.
Bozza PT, Viola JP. Lipid droplets in inflammation and cancer. Prostaglandins Leukot Essent Fatty Acids. 2010;82(4–6):243–50.
Mycielska ME, Dettmer K, Rümmele P, Schmidt K, Prehn C, Milenkovic VM, Jagla W, Madej GM, Lantow M, Schladt M, Cecil A, Koehl GE, Eggenhofer E, Wachsmuth CJ, Ganapathy V, Schlitt HJ, Kunzelmann K, Ziegler C, Wetzel CH, Gaumann A, Lang SA, Adamski J, Oefner PJ, Geissler EK. Extracellular citrate affects critical elements of cancer cell metabolism and supports cancer development in vivo. Cancer Res. 2018;78(10):2513–23.
Catalina-Rodriguez O, Kolukula VK, Tomita Y, Preet A, Palmieri F, Wellstein A, Byers S, Giaccia AJ, Glasgow E, Albanese C, Avantaggiati ML. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget. 2012;3(10):1220–35.
Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005;24(41):6314–22.
Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8(4):311–21.
Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, Ushijima M, Mashima T, Seimiya H, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008;68(20):8547–54.
Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP- citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324(5930):1076–80.
Zaidi N, Swinnen JV, Smans K. ATP-citrate lyase: a key player in cancer metabolism. Cancer Res. 2012;72(15):3709–14.
Wakil SJ, Abu-Elheiga LA. Fatty acid metabolism: target for metabolic syndrome. J Lipid Res. 2009;50 Suppl(Suppl):S138–43.
Brusselmans K, De Schrijver E, Verhoeven G, Swinnen JV. RNA interference- mediated silencing of the acetyl-CoA-carboxylase-alpha gene induces growth inhibition and apoptosis of prostate cancer cells. Cancer Res. 2005;65(15):6719–25.
Chajès V, Cambot M, Moreau K, Lenoir GM, Joulin V. Acetyl-CoA carboxylase alpha is essential to breast cancer cell survival. Cancer Res. 2006;66(10):5287–94.
Pizer ES, Thupari J, Han WF, Pinn ML, Chrest FJ, Frehywot GL, Townsend CA, Kuhajda FP. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res. 2000;60(2):213–8.
Guo D, Hildebrandt IJ, Prins RM, Soto H, Mazzotta MM, Dang J, Czernin J, Shyy JY, Watson AD, Phelps M, Radu CG, Cloughesy TF, Mischel PS. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc Natl Acad Sci U S A. 2009;106(31):12932–7.
Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485(7400):661–5.
Carlson CA, Kim KH. Regulation of hepatic acetyl coenzyme A carboxylase by phosphorylation and dephosphorylation. J Biol Chem. 1973;248(1):378–80.
Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987;223(2):217–22.
Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, O’Neill HM, Ford RJ, Palanivel R, O’Brien M, Hardie DG, Macaulay SL, Schertzer JD, Dyck JR, van Denderen BJ, Kemp BE, Steinberg GR. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med. 2013;19(12):1649–54.
Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2012;2(9):778–90.
De Schrijver E, Brusselmans K, Heyns W, Verhoeven G, Swinnen JV. RNA interference-mediated silencing of the fatty acid synthase gene attenuates growth and induces morphological changes and apoptosis of LNCaP prostate cancer cells. Cancer Res. 2003;63(13):3799–804.
Lupu R, Menendez JA. Pharmacological inhibitors of Fatty Acid Synthase (FASN)—catalyzed endogenous fatty acid biogenesis: a new family of anti-cancer agents? Curr Pharm Biotechnol. 2006;7(6):483–93.
Resh MD. Targeting protein lipidation in disease. Trends Mol Med. 2012;18(4):206–14.
Cao Y, Pearman AT, Zimmerman GA, McIntyre TM, Prescott SM. Intracellular unesterified arachidonic acid signals apoptosis. Proc Natl Acad Sci U S A. 2000;97(21):11280–5.
Yamashita Y, Kumabe T, Cho YY, Watanabe M, Kawagishi J, Yoshimoto T, Fujino T, Kang MJ, Yamamoto TT. Fatty acid induced glioma cell growth is mediated by the acyl-CoA synthetase 5 gene located on chromosome 10q25.1-q25.2, a region frequently deleted in malignant gliomas. Oncogene. 2000;19(51):5919–25.
Van Horn CG, Caviglia JM, Li LO, Wang S, Granger DA, Coleman RA. Characterization of recombinant long-chain rat acyl-CoA synthetase isoforms 3 and 6: identification of a novel variant of isoform 6. Biochemistry. 2005;44(5):1635–42. https://doi.org/10.1021/bi047721l.
Kim JH, Lewin TM, Coleman RA. Expression and characterization of recombinant rat Acyl-CoA synthetases 1, 4, and 5. Selective inhibition by triacsin C and thiazolidinediones. J Biol Chem. 2001;276(27):24667–73.
Mashima T, Oh-hara T, Sato S, Mochizuki M, Sugimoto Y, Yamazaki K, Hamada J, Tada M, Moriuchi T, Ishikawa Y, Kato Y, Tomoda H, Yamori T, Tsuruo T. p53-defective tumors with a functional apoptosome-mediated pathway: a new therapeutic target. J Natl Cancer Inst. 2005;97(10):765–77.
Weng JR, Chen CY, Pinzone JJ, Ringel MD, Chen CS. Beyond peroxisome proliferator-activated receptor gamma signaling: the multi-facets of the anti-tumor effect of thiazolidinediones. Endocr Relat Cancer. 2006;13(2):401–13.
Mashek DG, Li LO, Coleman RA. Rat long-chain acyl-CoA synthetase mRNA, protein, and activity vary in tissue distribution and in response to diet. J Lipid Res. 2006;47(9):2004–10.
Paton CM, Ntambi JM. Biochemical and physiological function of stearoyl-CoA desaturase. Am J Physiol Endocrinol Metab. 2009;297(1):E28–37.
Igal RA. Stearoyl-CoA desaturase-1: a novel key player in the mechanisms of cell proliferation, programmed cell death and transformation to cancer. Carcinogenesis. 2010;31(9):1509–15.
Ariyama H, Kono N, Matsuda S, Inoue T, Arai H. Decrease in membrane phospholipid unsaturation induces unfolded protein response. J Biol Chem. 2010;285(29):22027–35.
Fritz V, Benfodda Z, Rodier G, Henriquet C, Iborra F, Avancès C, Allory Y, de la Taille A, Culine S, Blancou H, Cristol JP, Michel F, Sardet C, Fajas L. Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Mol Cancer Ther. 2010;9(6):1740–54.
Roongta UV, Pabalan JG, Wang X, Ryseck RP, Fargnoli J, Henley BJ, Yang WP, Zhu J, Madireddi MT, Lawrence RM, Wong TW, Rupnow BA. Cancer cell dependence on unsaturated fatty acids implicates stearoyl-CoA desaturase as a target for cancer therapy. Mol Cancer Res. 2011;9(11):1551–61.
Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer. 2004;4(8):604–16.
Li G, Liu D, Kimchi ET, Kaifi JT, Qi X, Manjunath Y, Liu X, Deering T, Avella DM, Fox T, Rockey DC, Schell TD, Kester M, Staveley-O’Carroll KF. Nanoliposome C6-ceramide increases the anti-tumor immune response and slows growth of liver tumors in mice. Gastroenterology. 2018;154(4):1024–1036.e9.
Ordoñez R, Fernández A, Prieto-Domínguez N, Martínez L, García-Ruiz C, Fernández-Checa JC, Mauriz JL, González-Gallego J. Ceramide metabolism regulates autophagy and apoptotic cell death induced by melatonin in liver cancer cells. J Pineal Res. 2015;59(2):178–89.
Tagaram HR, Divittore NA, Barth BM, Kaiser JM, Avella D, Kimchi ET, Jiang Y, Isom HC, Kester M, Staveley-O’Carroll KF. Nanoliposomal ceramide prevents in vivo growth of hepatocellular carcinoma. Gut. 2011;60(5):695–701.
Griffiths B, Lewis CA, Bensaad K, Ros S, Zhang Q, Ferber EC, Konisti S, Peck B, Miess H, East P, Wakelam M, Harris AL, Schulze A. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 2013;1(1):3.
Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8(3):224–36.
Bengoechea-Alonso MT, Ericsson J. Cdk1/cyclin B-mediated phosphorylation stabilizes SREBP1 during mitosis. Cell Cycle. 2006;5(15):1708–18.
Scaglia N, Caviglia JM, Igal RA. High stearoyl-CoA desaturase protein and activity levels in simian virus 40 transformed-human lung fibroblasts. Biochim Biophys Acta. 2005;1687(1–3):141–51.
Scaglia N, Igal RA. Stearoyl-CoA desaturase is involved in the control of proliferation, anchorage-independent growth, and survival in human transformed cells. J Biol Chem. 2005;280(27):25339–49.
Li J, Ding SF, Habib NA, Fermor BF, Wood CB, Gilmour RS. Partial characterization of a cDNA for human stearoyl-CoA desaturase and changes in its mRNA expression in some normal and malignant tissues. Int J Cancer. 1994;57(3):348–52.
Falvella FS, Pascale RM, Gariboldi M, Manenti G, De Miglio MR, Simile MM, Dragani TA, Feo F. Stearoyl-CoA desaturase 1 (Scd1) gene overexpression is associated with genetic predisposition to hepatocarcinogenesis in mice and rats. Carcinogenesis. 2002;23(11):1933–6.
Scaglia N, Chisholm JW, Igal RA. Inhibition of stearoylCoA desaturase-1 inactivates acetyl-CoA carboxylase and impairs proliferation in cancer cells: role of AMPK. PLoS One. 2009;4(8):e6812.
Dobrzyn P, Dobrzyn A, Miyazaki M, Cohen P, Asilmaz E, Hardie DG, Friedman JM, Ntambi JM. Stearoyl-CoA desaturase 1 deficiency increases fatty acid oxidation by activating AMP-activated protein kinase in liver. Proc Natl Acad Sci U S A. 2004;101(17):6409–14.
Hess D, Chisholm JW, Igal RA. Inhibition of stearoylCoA desaturase activity blocks cell cycle progression and induces programmed cell death in lung cancer cells. PLoS One. 2010;5(6):e11394.
Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13(6):472–82.
Parks SK, Chiche J, Pouyssegur J. pH control mechanisms of tumor survival and growth. J Cell Physiol. 2011;226(2):299–308.
Hochachka PW, Rupert JL, Goldenberg L, Gleave M, Kozlowski P. Going malignant: the hypoxia-cancer connection in the prostate. Bioessays. 2002;24(8):749–57.
Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21(3):297–308.
Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108(49):19611–6.
Rydström J. Mitochondrial NADPH, transhydrogenase and disease. Biochim Biophys Acta. 2006;1757(5–6):721–6.
Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, Van Veldhoven PP, Waltregny D, Daniëls VW, Machiels J, Vanderhoydonc F, Smans K, Waelkens E, Verhoeven G, Swinnen JV. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010;70(20):8117–26.
Rodrigues C, Milkovic L, Bujak IT, Tomljanovic M, Soveral G, Cipak GA. Lipid profile and aquaporin expression under oxidative stress in breast cancer cells of different malignancies. Oxidative Med Cell Longev. 2019;2019:2061830.
Riahi H, Brekelmans C, Foriel S, Merkling SH, Lyons TA, Itskov PM, Kleefstra T, Ribeiro C, van Rij RP, Kramer JM, Schenck A. The histone methyltransferase G9a regulates tolerance to oxidative stress-induced energy consumption. PLoS Biol. 2019;17(3):e2006146.
Schlaepfer IR, Rider L, Rodrigues LU, Gijón MA, Pac CT, Romero L, Cimic A, Sirintrapun SJ, Glodé LM, Eckel RH, Cramer SD. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol Cancer Ther. 2014;13(10):2361–71.
Kim S, Yang X, Yin A, Zha J, Beharry Z, Bai A, Bielawska A, Bartlett MG, Yin H, Cai H. Dietary palmitate cooperates with Src kinase to promote prostate tumor progression. Prostate. 2019;79(8):896–908.
Deberardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem. 2006;281(49):37372–80.
Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene. 2005;24(26):4165–73.
Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim Biophys Acta. 2011;1807(6):726–34.
Eberlé D, Hegarty B, Bossard P, Ferré P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie. 2004;86(11):839–48.
Shimomura I, Shimano H, Horton JD, Goldstein JL, Brown MS. Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest. 1997;99(5):838–45.
Bengoechea-Alonso MT, Ericsson J. SREBP in signal transduction: cholesterol metabolism and beyond. Curr Opin Cell Biol. 2007;19(2):215–22.
Espenshade PJ, Li WP, Yabe D. Sterols block binding of COPII proteins to SCAP, thereby controlling SCAP sorting in ER. Proc Natl Acad Sci U S A. 2002;99(18):11694–9.
Rawson RB. The SREBP pathway—insights from Insigs and insects. Nat Rev Mol Cell Biol. 2003;4(8):631–40.
Dobrosotskaya IY, Seegmiller AC, Brown MS, Goldstein JL, Rawson RB. Regulation of SREBP processing and membrane lipid production by phospholipids in Drosophila. Science. 2002;296(5569):879–83.
Walker AK, Jacobs RL, Watts JL, Rottiers V, Jiang K, Finnegan DM, Shioda T, Hansen M, Yang F, Niebergall LJ, Vance DE, Tzoneva M, Hart AC, Näär AM. A conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell. 2011;147(4):840–52.
Oliner JD, Andresen JM, Hansen SK, Zhou S, Tjian R. SREBP transcriptional activity is mediated through an interaction with the CREB-binding protein. Genes Dev. 1996;10(22):2903–11. https://doi.org/10.1101/gad.10.22.2903.
Yang F, Vought BW, Satterlee JS, Walker AK, Jim Sun ZY, Watts JL, DeBeaumont R, Saito RM, Hyberts SG, Yang S, Macol C, Iyer L, Tjian R, van den Heuvel S, Hart AC, Wagner G, Näär AM. An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature. 2006;442(7103):700–4.
Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, Ericsson J. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab. 2005;1(6):379–91.
Bengoechea-Alonso MT, Ericsson J. A phosphorylation cascade controls the degradation of active SREBP1. J Biol Chem. 2009;284(9):5885–95.
Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem. 2002;277(37):33895–900.
Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, Schulze A. PKB / Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 2005;24:6465–81.
Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A. 2010;107(8):3441–6.
Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, Gao B, Wierzbicki M, Verbeuren TJ, Shaw RJ, Cohen RA, Zang M. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13(4):376–88.
Freed-Pastor William A, Mizuno H, Zhao X, Langerød A, Moon S-H, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244–58.
Guo D, Prins RM, Dang J, Kuga D, Iwanami A, Soto H, Lin KY, Huang TT, Akhavan D, Hock MB, Zhu S, Kofman AA, Bensinger SJ, Yong WH, Vinters HV, Horvath S, Watson AD, Kuhn JG, Robins HI, Mehta MP, Wen PY, DeAngelis LM, Prados MD, Mellinghoff IK, Cloughesy TF, Mischel PS. EGFR signaling through an Akt- SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal. 2009;2(101):ra82.
Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, Kuga D, Amzajerdi AN, Soto H, Zhu S, Babic I, Tanaka K, Dang J, Iwanami A, Gini B, Dejesus J, Lisiero DD, Huang TT, Prins RM, Wen PY, Robins HI, Prados MD, Deangelis LM, Mellinghoff IK, Mehta MP, James CD, Chakravarti A, Cloughesy TF, Tontonoz P, Mischel PS. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011;1(5):442–56.
Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393–402.
Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol. 2010;7(5):277–85.
Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21(12):3995–4004.
Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85.
Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia- inducible factor 1. Mol Cell Biol. 1996;16(9):4604–13.
Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000;14(1):34–44.
Ebert BL, Firth JD, Ratcliffe PJ. Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct Cis-acting sequences. J Biol Chem. 1995;270(49):29083–9.
Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8(9):705–13.
Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–85.
Furuta E, Pai SK, Zhan R, Bandyopadhyay S, Watabe M, Mo YY, Hirota S, Hosobe S, Tsukada T, Miura K, Kamada S, Saito K, Iiizumi M, Liu W, Ericsson J, Watabe K. Fatty acid synthase gene is upregulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008;68(4):1003–11.
Yoshii Y, Furukawa T, Yoshii H, Mori T, Kiyono Y, Waki A, Kobayashi M, Tsujikawa T, Kudo T, Okazawa H, Yonekura Y, Fujibayashi Y. Cytosolic acetyl-CoA synthetase affected tumor cell survival under hypoxia: the possible function in tumor acetyl-CoA/acetate metabolism. Cancer Sci. 2009;100(5):821–7.
Whitmer JT, Idell-Wenger JA, Rovetto MJ, Neely JR. Control of fatty acid metabolism in ischemic and hypoxic hearts. J Biol Chem. 1978;253(12):4305–9.
Boström P, Magnusson B, Svensson PA, Wiklund O, Borén J, Carlsson LM, Ståhlman M, Olofsson SO, Hultén LM. Hypoxia converts human macrophages into triglyceride-loaded foam cells. Arterioscler Thromb Vasc Biol. 2006;26(8):1871–6.
Gimm T, Wiese M, Teschemacher B, Deggerich A, Schödel J, Knaup KX, Hackenbeck T, Hellerbrand C, Amann K, Wiesener MS, Höning S, Eckardt KU, Warnecke C. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. 2010;24(11):4443–58.
Rankin EB, Rha J, Selak MA, Unger TL, Keith B, Liu Q, Haase VH. Hypoxia- inducible factor 2 regulates hepatic lipid metabolism. Mol Cell Biol. 2009;29(16):4527–38.
Du W, Zhang L, Brett-Morris A, Aguila B, Kerner J, Hoppel CL, Puchowicz M, Serra D, Herrero L, Rini BI, Campbell S, Welford SM. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat Commun. 2017;8(1):1769.
Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, Kaluarachchi K, Bornmann W, Duvvuri S, Taegtmeyer H, Andreeff M. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120(1):142–56.
Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J, Huang P, Sawyer SK, Fuerth B, Faubert B, Kalliomäki T, Elia A, Luo X, Nadeem V, Bungard D, Yalavarthi S, Growney JD, Wakeham A, Moolani Y, Silvester J, Ten AY, Bakker W, Tsuchihara K, Berger SL, Hill RP, Jones RG, Tsao M, Robinson MO, Thompson CB, Pan G, Mak TW. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011;25(10):1041–51.
Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. 2004;4(1):61–70.
Yokoyama Y, Mizunuma H. Peroxisome proliferator-activated receptor and epithelial ovarian cancer. Eur J Gynaecol Oncol. 2010;31(6):612–5.
Farese RV Jr, Walther TC. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell. 2009;139(5):855–60.
Takeuchi K, Reue K. Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am J Physiol Endocrinol Metab. 2009;296(6):E1195–209.
Agarwal AK, Garg A. Enzymatic activity of the human 1-acylglycerol-3-phosphate-O-acyltransferase isoform 11: upregulated in breast and cervical cancers. J Lipid Res. 2010;51(8):2143–52.
Ishimoto K, Nakamura H, Tachibana K, Yamasaki D, Ota A, Hirano K, Tanaka T, Hamakubo T, Sakai J, Kodama T, Doi T. Sterol-mediated regulation of human lipin 1 gene expression in hepatoblastoma cells. J Biol Chem. 2009;284(33):22195–205.
Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, Sabatini DM. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146(3):408–20.
Bagnato C, Igal RA. Overexpression of diacylglycerol acyltransferase-1 reduces phospholipid synthesis, proliferation, and invasiveness in simian virus 40-transformed human lung fibroblasts. J Biol Chem. 2003;278(52):52203–11.
Cheng X, Geng F, Pan M, Wu X, Zhong Y, Wang C, Tian Z, Cheng C, Zhang R, Puduvalli V, Horbinski C, Mo X, Han X, Chakravarti A, Guo D. Targeting DGAT1 ameliorates glioblastoma by increasing fat catabolism and oxidative stress. Cell Metab. 2020;32(2):229–242.e8.
Wang J, Li Y. CD36 tango in cancer: signaling pathways and functions. Theranostics. 2019;9(17):4893–908.
McKillop IH, Girardi CA, Thompson KJ. Role of fatty acid binding proteins (FABPs) in cancer development and progression. Cell Signal. 2019;62:109336.
Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li JL, Zhang Q, Wakelam MJO, Karpe F, Schulze A, Harris AL. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014;9(1):349–65.
Forootan FS, Forootan SS, Gou X, Yang J, Liu B, Chen D, Al Fayi MS, Al-Jameel W, Rudland PS, Hussain SA, Ke Y. Fatty acid activated PPARγ promotes tumorigenicity of prostate cancer cells by up regulating VEGF via PPAR responsive elements of the promoter. Oncotarget. 2016;7(8):9322–39.
Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2008;9(2):162–76.
Togashi A, Katagiri T, Ashida S, Fujioka T, Maruyama O, Wakumoto Y, Sakamoto Y, Fujime M, Kawachi Y, Shuin T, Nakamura Y. Hypoxia-inducible protein 2 (HIG2), a novel diagnostic marker for renal cell carcinoma and potential target for molecular therapy. Cancer Res. 2005;65(11):4817–26.
Qi W, Fitchev PS, Cornwell ML, Greenberg J, Cabe M, Weber CR, Roy HK, Crawford SE, Savkovic SD. FOXO3 growth inhibition of colonic cells is dependent on intraepithelial lipid droplet density. J Biol Chem. 2013;288(23):16274–81.
Qiu B, Ackerman D, Sanchez DJ, Li B, Ochocki JD, Grazioli A, Bobrovnikova-Marjon E, Diehl JA, Keith B, Simon MC. HIF2α-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov. 2015;5(6):652–67.
Penrose H, Heller S, Cable C, Makboul R, Chadalawada G, Chen Y, Crawford SE, Savkovic SD. Epidermal growth factor receptor mediated proliferation depends on increased lipid droplet density regulated via a negative regulatory loop with FOXO3/Sirtuin6. Biochem Biophys Res Commun. 2016;469(3):370–6.
Falasca M, Corda D. Elevated levels and mitogenic activity of lysophosphatidylinositol in k-ras-transformed epithelial cells. Eur J Biochem. 1994;221(1):383–9.
Ha JH, Radhakrishnan R, Jayaraman M, Yan M, Ward JD, Fung KM, Moxley K, Sood AK, Isidoro C, Mukherjee P, Song YS, Dhanasekaran DN. LPA induces metabolic reprogramming in ovarian cancer via a pseudohypoxic response. Cancer Res. 2018;78(8):1923–34.
Morgan-Lappe SE, Tucker LA, Huang X, Zhang Q, Sarthy AV, Zakula D, Vernetti L, Schurdak M, Wang J, Fesik SW. Identification of Ras-related nuclear protein, targeting protein for xenopus kinesin-like protein 2, and stearoyl-CoA desaturase 1 as promising cancer targets from an RNAi-based screen. Cancer Res. 2007;67(9):4390–8.
Young RM, Ackerman D, Quinn ZL, Mancuso A, Gruber M, Liu L, Giannoukos DN, Bobrovnikova-Marjon E, Diehl JA, Keith B, Simon MC. Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev. 2013;27(10):1115–31.
Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, Polak K, Tondera D, Gounarides J, Yin H, Zhou F, Green MR, Chen L, Monti S, Marto JA, Shipp MA, Danial NN. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell. 2012;22(4):547–60.
Bhatt V, Khayati K, Hu ZS, Lee A, Kamran W, Su X, Guo JY. Autophagy modulates lipid metabolism to maintain metabolic flexibility for LKB1-deficient Kras-driven lung tumorigenesis. Genes Dev. 2019;33(3–4):150–65.
Patra KC, Kato Y, Mizukami Y, Widholz S, Boukhali M, Revenco I, Grossman EA, Ji F, Sadreyev RI, Liss AS, Screaton RA, Sakamoto K, Ryan DP, Mino-Kenudson M, Castillo CF, Nomura DK, Haas W, Bardeesy N. Mutant GNAS drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat Cell Biol. 2018;20(7):811–22.
Krishnan J, Suter M, Windak R, Krebs T, Felley A, Montessuit C, Tokarska-Schlattner M, Aasum E, Bogdanova A, Perriard E, Perriard JC, Larsen T, Pedrazzini T, Krek W. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009;9(6):512–24.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81773262 and 81902897) and the Independent Program of the State Key Laboratory of Cancer Biology, China (CBSKL2019ZZ21).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Zheng, M., Wang, W., Liu, J., Zhang, X., Zhang, R. (2021). Lipid Metabolism in Cancer Cells. In: Li, Y. (eds) Lipid Metabolism in Tumor Immunity. Advances in Experimental Medicine and Biology, vol 1316. Springer, Singapore. https://doi.org/10.1007/978-981-33-6785-2_4
Download citation
DOI: https://doi.org/10.1007/978-981-33-6785-2_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-33-6784-5
Online ISBN: 978-981-33-6785-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)