
Abstract
When we closely look at the etiology and pathophysiology of several neoplastic disorders, the weaving role of hormones and the endocrine pathways is very clearly made out. This is particularly true in the sense of breast and prostatic cancers. Hormonal therapies are currently one of the mainstays of therapy for these neoplastic conditions. While selective estrogen receptor modulators (SERMs) and aromatase inhibitors are the hormonal therapeutics used in the management of breast carcinoma, GnRH agonists, GnRH antagonists, combined androgen blockade, and intermittent androgen deprivation therapy are treatment modalities preferred in the management of prostatic malignancies. This chapter elucidates these hormonal therapies in the light of breast and prostate malignancies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Selective estrogen receptor modulators
- Aromatase inhibitors
- Androgens
- Breast neoplasms
- Prostatic neoplasms
1 Historical Timeline
Hormones are involved in the etiology as well as development of many cancers like prostate and breast cancers. Beatson, in 1896, demonstrated that premenopausal women diagnosed with metastatic breast cancer who underwent surgical oophorectomy had tumor regression. In the 1940s, Charles Huggins demonstrated that growth of prostate cancers was dependent on the circulating androgens. Two decades later in 1966, he was awarded the Nobel Prize for medicine for discovering a successful therapy for metastatic prostate cancer in the form of surgical orchidectomy.
2 Hormonal Management of Breast Cancer
2.1 Selective Estrogen Receptor Modulators (SERMs)
-
The main hormone implicated in the growth and development of breast tumor is estrogen. Tamoxifen is a selective estrogen receptor modulator (SERM). It has complex actions. It is a competitive antagonist of estrogen at the estrogen receptor. It suppresses growth of the tumor by virtue of this effect.
-
In addition to competitive antagonistic effect, it also possesses partial agonistic effect. The partial agonistic effect can be beneficial in postmenopausal women as it prevents bone demineralization. At the same time, it might be detrimental as it increases the risk of thromboembolism and uterine cancer (Fig. 65.1).
-
In addition, this partial agonistic effect might have a role in the development of resistance to tamoxifen.
-
Tamoxifen also has a favorable effect on the lipid profile.
-
Tamoxifen being a prodrug is metabolized to endoxifen (active metabolite) by cytochrome P450 enzyme (CYP2D6). Drugs like selective serotonin reuptake inhibitors (SSRIs) inhibit CYP2D6 and interfere with tamoxifen bio-activation rendering the therapy less effective.
-
Tamoxifen can be used both in pre- and postmenopausal women. In early breast cancer patients who are estrogen receptor positive, tamoxifen use post-surgery was associated with 41% reduction in annual recurrence rate and 34% reduction in annual mortality rate.
Fig. 65.1 
Effects of tamoxifen

2.2 Aromatase Inhibitors
Unlike tamoxifen, aromatase inhibitors lower the circulating estrogen levels by inactivating and inhibiting aromatase enzyme. Aromatase is responsible for estrogen synthesis from androgens. In contrast to tamoxifen, aromatase inhibitors do not possess partial agonistic activity (Fig. 65.2).
-
Aromatase inhibitors are divided into three generations:
-
Aminoglutethimide, the first aromatase inhibitor (first generation) was developed as anticonvulsant. It was withdrawn as there were reports of adrenal insufficiency associated with its use. It was found to inhibit multiple cytochrome P450 enzymes which included enzymes linked to adrenal steroidogenesis. It was later redeveloped as an agent for medical adrenalectomy. Side effects like rash and drowsiness limited its use.
-
Formestane and fadrozole were the second generation aromatase inhibitors. They were clinically effective. However, formestane was an injectable to be administered intramuscularly and fadrozole caused aldosterone suppression which limited their use.
-
There are four third generation aromatase inhibitors, namely exemestane, anastrozole, letrozole, and vorozole. Exemestane is a steroidal inactivator (type 1 inhibitor). Anastrozole, letrozole, and vorozole are non-steroidal inhibitors (type 2 inhibitors).
-
-
Aromatase inhibitors should not be used in premenopausal women. In premenopausal women they can be used only for post-ovarian suppression.
-
There are three approaches for their use in patients with early breast cancer. They can be used as initial therapy for 5 years, or after 5 years of tamoxifen use, or they can be switched every 2 to 3 years with tamoxifen therapy. Aromatase inhibitors lower bone mineral density and are associated with increased risk of fractures. Osteoporotic women should be monitored.
Fig. 65.2 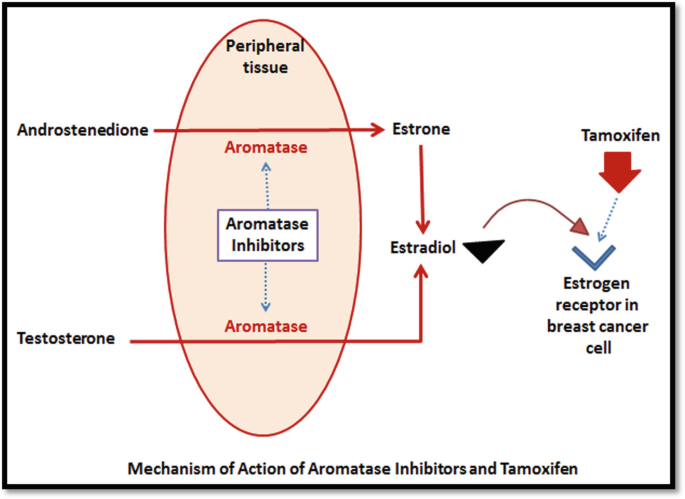
Mechanism of action of aromatase inhibitors and tamoxifen

2.3 Indications for Hormone Therapy in Breast Cancer
-
Neo-adjuvant role—to reduce the tumor size in order to make it operable
-
Adjuvant role—postoperatively to improve survival and reduce recurrence
-
Use in locally advanced tumors—to shrink inoperable large tumors
-
Use in metastasis—palliative management to improve quality of life
3 Hormonal Management of Prostate Cancer
Prostate cancers are the most hormone-sensitive of all the cancers. There is unequivocal evidence to state the dependence of the transformed epithelium of prostate on androgens. Preclinical studies have shown that androgen supplementation is vital for in vivo or in vitro establishment of prostate cancer. Role of testosterone in treatment of prostate cancer is well established so much so that drugs only need to demonstrate reduction in levels of testosterone to get approval by US FDA for treatment of this malignancy.
3.1 Androgen Deprivation Therapy
-
Androgen deprivation therapy (ADT) forms the first line management option for metastatic/advanced prostate cancer. In addition, it is also used commonly for reducing the gland volume in individuals contemplating cryotherapy, high intensity focused ultrasound therapy, and brachytherapy (gland volume > 50 g).
-
ADT can be achieved pharmacologically or surgically. Pharmacological ADT is preferred as they avoid the psychological and physical discomforts which are associated with surgical orchiectomy. Pharmacological therapies include GnRH agonists as well as antagonists. GnRH agonists are associated with shortcomings like testosterone micro-surges and surges (clinical flare).
-
Whether ADT is achieved surgically or pharmacologically, suppression of testosterone results in adverse effects like osteoporosis, cardio-metabolic effects, and hot flushes (Fig. 65.3).
Fig. 65.3 
Adverse effects of androgen deprivation therapy

3.2 Gonadotropin-Releasing Hormone (GnRH) Agonists
-
Native GnRH is produced by hypothalamus and it regulates the serum testosterone levels by modulating the release of luteinizing hormone from the pituitary. They are also known as luteinizing hormone-releasing hormone (LH-RH) agonists (Fig. 65.4).
-
Commercially available GnRH agonists (leuprolide, nafarelin, goserelin, and triptorelin) are produced by modifying the amino acid sequences of native GnRH. They cause an initial flare of testosterone by increased LH stimulation. However, continuous stimulation of GnRH receptors leads to their desensitization resulting in suppression of testosterone.
-
Loss of sensitivity of GnRH receptor over long term can lead to renewed testosterone production which manifests as a breakthrough testosterone escape. GnRH agonists also cause a partial suppression of FSH.
-
Commercial preparations vary in their duration of action (from 1 month to 1 year) and route of administration (subcutaneous injection, subcutaneous implant, or intramuscular injection).
-
GnRH agonists effectively reduce the testosterone levels to <50 ng/dL. Surgical castration reduces the levels of testosterone to around 15 ng/dL. Twelve percent of individuals on GnRH therapy fail to reach the castration levels of <50 ng/dL.
Fig. 65.4 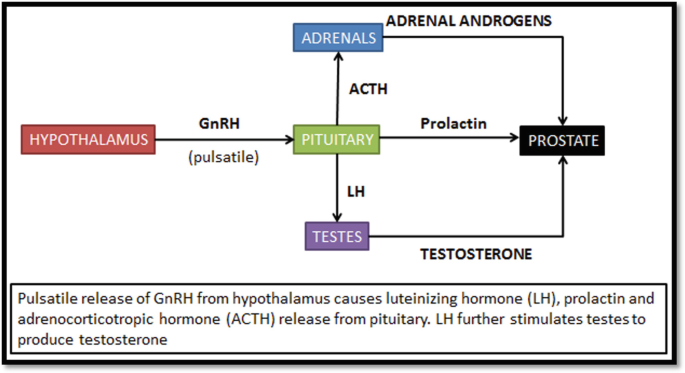
Effects of gonadotropin-releasing hormone (GnRH)

3.3 GnRH Antagonists
-
GnRH antagonists competitively bind to the GnRH receptors in the pituitary, producing reduced amounts of LH and FSH.
-
Unlike GnRH agonists, GnRH antagonists reduce testosterone levels without the initial flare.
-
Degarelix, a new agent for ADT in advanced prostate cancer, is administered as a monthly subcutaneous injection. Use of this drug is modest due to severity and frequency of local reactions at the injection site. As it causes immediate fall in testosterone levels, it has been used for initiation of ADT and later switched to GnRH agonists which are better tolerated.
-
Adverse events due to suppression of testosterone levels are similar GnRH agonists. However, risks of cardiovascular events are lower.
3.4 Combined Androgen Blockade
Combined androgen blockade (CAB) simultaneously eliminates all the androgen sources. It includes use of GnRH agonists/antagonists to block testosterone production from testes along with antiandrogens (flutamide, bicalutamide) to block the androgen production from adrenal glands.
3.5 Intermittent Androgen Deprivation Therapy
-
The concept of intermittent androgen deprivation therapy (iADT) is now being increasingly used in the management of prostate cancer. By causing intermittent androgen deprivation, patients experience lesser treatment related adverse effects and it is cheaper.
-
In individuals where the disease burden is lower, intermittent ADT is as effective as continuous ADT.
-
Ideal candidates for intermittent ADT are ones with poor tolerability to continuous ADT, non-metastatic or locally advanced tumor, cancer with limited metastasis, longer prostate specific antigen doubling time.
4 Resistance to Hormonal Therapy
Almost all prostate and metastatic breast cancer patients who respond to hormonal agents develop resistance during the course of therapy. Mechanisms behind the development of this resistance are unclear. Proposed mechanisms are activation of pathways without hormone exposure, increased receptor sensitivity, and upregulation of downstream pathways. Intermittent ADT is one regimen that delays development of hormone resistance.
Bibliography
Abraham J, Straffurth J (2011) Hormonal therapy for cancer. Medicine 39:723–727
Abrahamsson P (2017) Intermittent androgen deprivation therapy in patients with prostate cancer: connecting the dots. Asian J Urol 4:208–222
Brawer MK (2006) Hormonal therapy for prostate cancer. Rev Urol 8(Suppl 2):S35–S47
Crawford ED, Heidenreich A, Lawrentschuk N, Tombal B, Pompeo ACL, Mendoza-Valdes A (2019) Androgen-targeted therapy in men with prostate cancer: evolving practice and future considerations. Prostate Cancer Prostatic Dis 22:24–38
Goktas S, Ziada A, Crawford ED (1999) Combined androgen blockade for advanced prostatic carcinoma. Prostate Cancer Prostatic Dis 2:172–179
Kunath F, Borgmann H, Blumle A, Keck B, Wullich B, Schmucker C (2015) Gonadotropin-releasing hormone antagonists versus standard androgen suppression therapy for advanced prostate cancer a systematic review with meta-analysis. BMJ Open 5:e008217
Labrie F (2010) Hormonal therapy of prostate cancer. Prog Brain Res 182:321–341
Lepore H, Shore MD (2012) LHRH agonists for the treatment of prostate cancer: 2012. Rev Urol 14:1–12
Riggs BL, Hartmann LC (2003) Selective estrogen-receptor modulators — mechanisms of action and application to clinical practice. N Engl J Med 348:618–629
Smith IE, Dowsett M (2003) Aromatase inhibitors in breast cancer. N Engl J Med 348:2431–2442
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Kunder, S.K., Arivazhahan, A. (2021). Hormonal Agents in the Pharmacotherapy of Cancer. In: Paul, A., Anandabaskar, N., Mathaiyan, J., Raj, G.M. (eds) Introduction to Basics of Pharmacology and Toxicology. Springer, Singapore. https://doi.org/10.1007/978-981-33-6009-9_65
Download citation
DOI: https://doi.org/10.1007/978-981-33-6009-9_65
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-33-6008-2
Online ISBN: 978-981-33-6009-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)