
Abstract
Males and females are biologically distinct, and certain pathological conditions affect both sexes differently. Obvious sex difference exists in pulmonary arterial hypertension (PAH). The major sex difference is female sex hormones, especially estrogens, and therefore, the roles of estrogens have been intensively studied in PAH. The incidence of PAH in females is higher than in males, suggesting a female-specific risk factor. However, a general notion that estrogens are cardiovascular protective, and their protective effects demonstrated in animal models, resulted in the emerging concept of “estrogen paradox” in PAH. Later, it was found that female PAH patients live longer, suggesting the survival benefit of estrogens. Questions that need to be answered are (1) Why is PAH more prevalent in females despite the protective effects of estrogens? and (2) Why do female PAH patients show better survival despite the higher incidence of the syndrome? Even with the rigorous research efforts to answer these paradoxical questions, the field has not come to a consensus. This chapter summarizes the current leading theories for the estrogen paradox in PAH.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Estrogen Paradox in PAH
It has long been recognized that the prevalence of PAH is higher in females than in males. Although the ratio of female to male varies depending on subgroups of PAH, epidemiological studies from various countries consistently demonstrate this female predominance in PAH [1–3]. A recent study showed that among male PAH patients, a higher level of estrogens was associated with PAH [4]. Higher female incidence in PAH is not found in pediatric, prepuberty patients [5]. These data collectively suggest that female sex hormones, such as estrogens, have disadvantageous effects and play a role in PAH pathogenesis.
However, it is a general consensus that estrogens are cardiovascular protective. The incidence of atherosclerotic diseases is low in premenopausal females, while it increases after menopause, and postmenopausal use of estrogens is associated with reduced risk of cardiovascular disease [6]. Postmenopausal women also have increased risk of developing PAH [7], which suggests a protective effect of estrogens. Studies with two classical animal models, chronic hypoxia- and monocrotaline-induced pulmonary hypertension, have consistently shown protective effects of estrogens [8–10]. The protective effect of estrogens in hypoxic humans is indirectly supported by the male predominance in the incidence of high altitude-induced pulmonary hypertension [11].
These studies in humans and animals suggest a protective effect of estrogens, which is contrary to the epidemiological data in PAH, which points to an opposite effect of estrogens. These conflicting results led to the concept, “estrogen paradox” [12–14].
Although classical experimental models consistently showed protective effect of estrogens, they presented with limited pathological phenotypes and did not fully recapitulate the human PAH. Therefore, various animal models were developed in recent years in an attempt to obtain a better understanding of the pathogenesis of PAH. The studies with transgenic animal models are unfortunately inconclusive and more confusing, showing protective or detrimental effects of estrogens in PAH. In the Sugen/hypoxia-exposed rat model of PAH, which closely mimics the human hemodynamic profile as well as the pulmonary vascular histopathology [15, 16], the difference between sexes in hemodynamic severity and PAH characteristics is also inconclusive [17, 18]. The data from studies of recent animal models are generally contradictory, and perhaps this reflects the differential effects of estrogens that can be exerted depending on the initiating stimuli for PAH. It also highlights the need for a better animal model that consistently demonstrates higher incidence and better outcome in females, similarly to the human PAH.
The estrogen paradox became more complicated when it was reported that female PAH patients have better survival [19] while male sex is associated with increased risk of death [2, 20]. These findings suggest estrogens have a beneficial effect in PAH, which appears contradictory to the epidemiological finding which indicates that estrogens are a risk factor.
This estrogen paradox in PAH, i.e., females have higher prevalence but longer survival, has been a focus of numerous studies. They have investigated how estrogens exert harmful effects in the pulmonary circulation and how they provide survival benefit in female PAH patients. The exact mechanisms of these sex differences are unclear, and the field has not come to a consensus on whether estrogens are protective or harmful in PAH. This chapter summarizes current understanding of effects of the major female sex steroids, estrogens, on the PAH pathophysiology and the sex difference.
2 Published Theories for the Estrogen Paradox in PAH
The current knowledge on estrogens and the leading hypotheses on the estrogen paradox based on numerous studies are described below.
2.1 How Estrogens Exert Harmful Effects to the Pulmonary Circulation
2.1.1 Altered Estrogen Receptor (ER) Signaling
Altered estrogen receptor (ER) signaling is thought to contribute to PAH pathology. Estrogens exert their effects mainly via two types of ERs, ERα and ERβ, which mediate various genomic pathways [21]. In the pulmonary circulation, both ERα and ERβ are present and active in humans and rats [22, 23]. Multiple studies demonstrate favorable effects of estrogens, including upregulation of endothelial nitric oxide synthase and prostacyclin synthase in the lungs via those receptors [24, 25]. Estrogen signaling can also be mediated via a G-protein-coupled membrane receptor, GPR30, whose primary function is to activate non-genomic pathways to elicit acute effects of estrogens [12]. The relative proportion of each receptor and additionally their alternative splicing variants affect the overall effect of complex estrogen signaling [14].
In systemic vasculature, single-nucleotide polymorphisms (SNPs) in genes encoding ERα or ERβ are associated with development of myocardial infarction, hypertension, left ventricular hypertrophy, and stroke [12]. Genome-wide RNA expression profiling in the lungs indicated the upregulation of estrogen receptor 1 (ESR1), which encodes ERα, in an idiopathic PAH cohort compared to idiopathic fibrosis and normal cohorts, both in males and females [26]. The ESR1 abnormality is also associated with increased risk of developing pulmonary hypertension in patients with advanced liver disease [27]. The significance of the ERs increases because non-estrogen ligands can also trigger ER activation in the absence of estrogens [14]. This genetic factor may predispose certain populations to increased risk of developing PAH.
Studies in animal models have yielded conflicting results as to which receptor contributes to the PAH pathogenesis. 17β-Estradiol (E2), which is the most important estrogen in premenopausal females, demonstrated a protective effect in the chronic hypoxia model, and this was dependent on both ERs [12, 28]. The protective effect of E2 in the monocrotaline model was mediated by ERβ [10]. On the other hand, in female rats of the same model, downregulation of lung ERα was observed, while no change was found in ERβ [29]. These results indicate that the roles of each receptor may depend on sex and pulmonary hypertensive stimulus. In addition, ER function may be altered as a consequence of mutations in other genes or environment of the vasculature. For example, pulmonary microvascular endothelial cells with BMPR2 mutation showed dysregulation of ERα trafficking [30], which would affect the relative abundance and location of ERα. Hypoxia increased the expression of ERβ, but not ERα, in male rats [31].
Given the genetic alterations observed in human PAH patients, one possibility is that the altered ER signaling contributes to PAH pathogenesis, potentially as an additional “hit” for the onset of PAH, although the cause-and-effect relationship for this clinical observation is still unclear.
2.1.2 Altered Estrogen Metabolites
It has been suggested that an imbalance of estrogen metabolites may explain the estrogen paradox in PAH. Estrogens and their metabolites can elicit various effects that may oppose each other. A distinct feature that separates PAH from other forms of pulmonary hypertension is the extensive pulmonary vascular remodeling. The pathogenesis of this remodeling process is unclear, but antiapoptotic, proproliferative, and angiogenic cells and inflammation are implicated in the disease process. Estrogens and estrogen metabolites are known to play a role in the modulation of these cellular phenotypes and the environment, and, therefore, protective or harmful effects of estrogens and their metabolites are primarily evaluated based on these cellular behaviors.
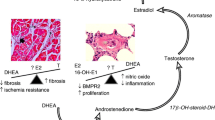
Simplified estrogen metabolism is shown in Fig. 4.1. 17β-Estradiol (E2) is synthesized from precursors by aromatase [14]. E2 is metabolized to 2-hydroxyestradiol (2-OHE2) and 4-hydroxyestradiol (4-OHE2) by the enzyme CYP1A1 and CYP1B1 [12]. These hydroxyestradiols are quickly converted to 2-methoxyestradiol (2-ME2) and 4-methoxyestradiol (4-ME2) by the enzyme, catechol-O-methyltransferase (COMT) [14]. Estrone (E1) is the primary estrogen during menopause and has a weaker estrogenic activity than E2 [32]. E1 is also synthesized from precursors, as well as reversibly converted from E2 by 17β-hydroxysteroid dehydrogenase [12]. E1 subsequently is metabolized to 16α-hydroxyestrone (16α-OHE1) by CYP1B1 [12].

The simplified estrogen metabolism (modified from Lahm et al. (12)). CYP Cytochrome P-450
An increased level of CYP1B1 is found in the lungs of idiopathic and heritable PAH patients as well as in various animal models of pulmonary hypertension, including chronic hypoxia- and Sugen/hypoxia-exposed rats [33]. This leads to increased levels of 16α-OHE1, which has innate effects of antiapoptosis, proproliferation, and pro-inflammation [12]. These properties suggest detrimental effects in pulmonary vascular remodeling by propagating undesirable cellular phenotypes. The unfavorable effects of 16α-OHE1 are demonstrated by studies in CYP1B1-null mice and in chronic hypoxia- and Sugen/hypoxia-exposed rats [33].
In heritable PAH, the higher activity of CYP1B1 results in a higher penetrance of PAH. Among the carriers of BMPR2 mutation, wild-type homozygous genotype of CYP1B1 was associated with lower urinary 2-OHE2/16α-OHE1, suggesting a shift toward proproliferative antiapoptotic metabolites [34]. In addition, high concentrations of E2 and 16α-OHE1 reduce BMPR2 gene expression [30]. Therefore, increased activity of CYP1B1 and a subsequent increase of 16α-OHE1 levels appear to facilitate PAH pathogenesis.
An increased level of CYP1B1 also shifts the pathway toward more synthesis of 2-ME2. 2-ME2 has antiproliferative and proapoptotic properties that suggest a protective role against vascular remodeling [13]. The beneficial effects of 2-ME2 are shown in the monocrotaline, bleomycin, and Sugen/hypoxia animal models [35–37]. However, this theory of estrogen metabolites is challenged due to the weak affinity of 2-ME2 for the receptors [38] and to the study that showed that treatment with E2 attenuated pulmonary hypertension in the presence of a COMT inhibitor, which inhibits the synthesis of 2-ME2 [28].
Among patients with severe liver disease, SNPs in aromatase are associated with development of PAH [27], which suggests a protective role of E2. On the other hand, an increase in aromatase is reported in human PAH, as well as in the chronic hypoxia- and Sugen/hypoxia-exposed models, and the inhibition of aromatase has been shown to have therapeutic effect in both rat models [17].
Taken together, altered estrogen metabolism in the pulmonary circulation can shift the cellular phenotypes from protective to harmful effects of estrogens in PAH pathology. The enzymes that modulate estrogen metabolism are present in vascular cells, which suggest that the local concentrations of the estrogen metabolites likely differ from circulating levels and contribute to cellular modulation [13]. Therefore, the effects of estrogen metabolites should be investigated in a more detailed and specific manner.
2.1.3 Microenvironment
The effect of E2 depends on the microenvironment of the target tissue/organ, affected by environmental and genetic background. This may contribute to the conflicting data among studies and the estrogen paradox.
One critical variable that contributes to the environment is the timing of E2 participation in relation to progression status of the disorder. A study on atherosclerosis showed a protective effect of E2 in mild atherosclerotic patients and a harmful effect in advanced atherosclerotic patients [39]. As demonstrated in epidemiological studies in PAH, the patient age has significant effects on the hemodynamic profiles and survival [19, 40]. Environmental changes in the pulmonary circulation, such as oxygen tension, are also important. In pulmonary arterial endothelial cells, E2 decreased VEGF expression upon exposure to hypoxia, while it had no effect on normoxic cells [28]. Genetic background is another key factor to the microenvironment. The genetic alterations that affect estrogen signaling and metabolism, such as SNPs in ESR1, aromatase, and CYP1B1, directly influence the estrogen signaling and metabolism. As seen in the higher penetrance of pulmonary hypertension in BMPR2 mutation carriers, the effects of estrogens are also affected by other genetic alterations. The effects of estrogens depend on the target vascular layers (media vs. intima) and the condition of endothelium (intact/quiescent vs. dysfunctional), as well as the concentrations of estrogens [13]. Therefore, the effects of estrogens are dictated by various factors that compose the microenvironment.
2.2 How Estrogens Exert Survival Benefit in Female PAH Patients
2.2.1 Cardiac Protective Effect of Estrogens
The estrogen paradox may be partly explained by the organ-specific effect of estrogens. The hypothesis is that estrogens may be harmful to the pulmonary arteries, but protective in the heart. Since a major determinant of survival in PAH is right heart function [20], the better cardiac function provided by E2 effects benefits female PAH patients. Right ventricular function, defined by right ventricular ejection fraction, was better in female PAH patients and was improved with PAH-specific therapies in female patients, while males did not benefit [41, 42]. Exogenous hormone therapy is associated with better right ventricular systolic function [43]. In the Sugen/hypoxia rat model, female rats developed significantly less cardiac fibrosis [44], and exogenous E2 treatment improved right heart function [18]. On the other hand, female PAH patients tend to have more severe vascular remodeling and inflammation in pulmonary arteries [45]. In the Sugen/hypoxia model, female rats developed more intimal, angio-proliferative lesions compared to males [44]. These results may indicate the potential roles of estrogens in exacerbating the vascular remodeling while protecting the heart function in PAH.
3 The Perspective
The overall effect of estrogens is determined by numerous factors such as age, genetic background, oxygen tension, cell types, cell condition, concentration of estrogens, expression levels of ERs, estrogen metabolites, co-regulators of estrogens, and organs [46]. Therefore, whether estrogens are protective or detrimental as an end result depends on the balance of all the components in the target tissue/organ.
One of the key features of estrogens is that they apparently exhibit opposite effects depending on the timing/progression of the disorder (i.e., whether the disease was already established or not). Estrogens are protective if present at the time of a disease onset, but ineffective when administered in later stages of the pathology. This is supported by studies of atherosclerosis and Alzheimer’s disease, as well as of balloon injury-induced carotid artery stenosis [46]. It is, therefore, important to fully elucidate exactly what factors determine how estrogens behave, protective or detrimental, in the hypertensive pulmonary circulation. Development of animal models of PAH that recapitulate human pathology and epidemiology may be required to settle this issue.
Additionally, a recent large clinical study has surprisingly revealed that young female PAH patients (<45 years) have better hemodynamic profiles as compared to men [40]. Although more detailed subcategorized analyses of these epidemiological data are needed, such as longitudinal follow-up studies focusing on sex and hormonal status, the implication of this observation could be that estrogens are beneficial, even in the damaged pulmonary circulation in PAH. Then, the question is why PAH is more frequent but less severe in females, or if there is any specific stimuli/stimulus that favors females to trigger less severe PAH. This could be a very critical issue in this field that needs to be carefully addressed in the future.
4 Conclusion
The “estrogen paradox” is not limited to the PAH field. It is now clear that the effects of estrogens in the cardiovascular system are not only beneficial but also detrimental based on the conflicting results of hormone replacement therapy [39, 47]. Estrogens modulate various aspects of the pulmonary circulation, such as vascular tone, cellular proliferation, apoptosis, angiogenesis, as well as inflammatory status, and their effects can be either good or bad depending on various factors. Unfortunately, the current understanding of the roles of estrogens in PAH is incomplete, and many more studies are needed to define their exact roles. Altered estrogen metabolism, altered estrogen signaling, and the microenvironment are likely contributing to the conflicting and paradoxical effects of estrogens. It is thus critical to perform thorough, rigorous studies of the diverse and complex estrogen pharmacology.
References
Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. 2010;137(2):376–87. Epub 2009/10/20
Olsson KM, Delcroix M, Ghofrani HA, Tiede H, Huscher D, Speich R, et al. Anticoagulation and survival in pulmonary arterial hypertension: results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation. 2014;129(1):57–65. Epub 2013/10/02
Alves Jr JL, Gavilanes F, Jardim C, Fernandes CJ, Morinaga LT, Dias B, et al. Pulmonary arterial hypertension in the southern hemisphere: results from a registry of incident Brazilian cases. Chest. 2015;147(2):495–501. Epub 2014/10/16
Ventetuolo CE, Baird GL, Barr RG, DA B, JS F, NS H, et al. Higher estradiol and lower dehydroepiandrosterone-sulfate levels are associated with pulmonary arterial hypertension in men. Am J Respir Crit Care Med. 2015; Epub 2015/12/15
van Loon RL, Roofthooft MT, Hillege HL, ten Harkel AD, van Osch-Gevers M, Delhaas T, et al. Pediatric pulmonary hypertension in the Netherlands: epidemiology and characterization during the period 1991 to 2005. Circulation. 2011;124(16):1755–64. Epub 2011/09/29
Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. 1999;340(23):1801–11. Epub 1999/06/11
Scorza R, Caronni M, Bazzi S, Nador F, Beretta L, Antonioli R, et al. Post-menopause is the main risk factor for developing isolated pulmonary hypertension in systemic sclerosis. Ann N Y Acad Sci. 2002;966:238–46. Epub 2002/07/13
Rabinovitch M, Gamble WJ, Miettinen OS, Reid L. Age and sex influence on pulmonary hypertension of chronic hypoxia and on recovery. Am J Physiol. 1981;240(1):H62–72. Epub 1981/01/01
Farhat MY, Chen MF, Bhatti T, Iqbal A, Cathapermal S, Ramwell PW. Protection by oestradiol against the development of cardiovascular changes associated with monocrotaline pulmonary hypertension in rats. Br J Pharmacol. 1993;110(2):719–23. Epub 1993/10/01
Umar S, Iorga A, Matori H, Nadadur RD, Li J, Maltese F, et al. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am J Respir Crit Care Med. 2011;184(6):715–23. Epub 2011/06/28
Aldashev AA, Sarybaev AS, Sydykov AS, Kalmyrzaev BB, Kim EV, Mamanova LB, et al. Characterization of high-altitude pulmonary hypertension in the Kyrgyz: association with angiotensin-converting enzyme genotype. Am J Respir Crit Care Med. 2002;166(10):1396–402. Epub 2002/10/31
Lahm T, Tuder RM, Petrache I. Progress in solving the sex hormone paradox in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2014;307(1):L7–26. Epub 2014/05/13
Tofovic SP. Estrogens and development of pulmonary hypertension: interaction of estradiol metabolism and pulmonary vascular disease. J Cardiovasc Pharmacol. 2010;56(6):696–708. Epub 2010/10/01
Austin ED, Lahm T, West J, Tofovic SP, Johansen AK, Maclean MR, et al. Gender, sex hormones and pulmonary hypertension. Pulm Circ. 2013;3(2):294–314. Epub 2013/09/10
Toba M, Alzoubi A, O’Neill KD, Gairhe S, Matsumoto Y, Oshima K, et al. Temporal hemodynamic and histological progression in Sugen5416/hypoxia/normoxia-exposed pulmonary arterial hypertensive rats. Am J Physiol Heart Circ Physiol. 2014;306(2):H243–50. Epub 2013/11/19
Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, et al. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121(25):2747–54. Epub 2010/06/16
Mair KM, Wright AF, Duggan N, Rowlands DJ, Hussey MJ, Roberts S, et al. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. Am J Respir Crit Care Med. 2014;190(4):456–67. Epub 2014/06/24
Frump AL, Goss KN, Vayl A, Albrecht M, Fisher A, Tursunova R, et al. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am J Physiol Lung Cell Mol Physiol. 2015;308(9):L873–90. Epub 2015/02/26
Shapiro S, Traiger GL, Turner M, McGoon MD, Wason P, Barst RJ. Sex differences in the diagnosis, treatment, and outcome of patients with pulmonary arterial hypertension enrolled in the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Chest. 2012;141(2):363–73. Epub 2011/07/16
Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010;122(2):164–72. Epub 2010/06/30
Umar S, Rabinovitch M, Eghbali M. Estrogen paradox in pulmonary hypertension: current controversies and future perspectives. Am J Respir Crit Care Med. 2012;186(2):125–31. Epub 2012/05/09
Hamidi SA, Dickman KG, Berisha H, Said SI. 17beta-estradiol protects the lung against acute injury: possible mediation by vasoactive intestinal polypeptide. Endocrinology. 2011;152(12):4729–37. Epub 2011/10/20
Mollerup S, Jorgensen K, Berge G, Haugen A. Expression of estrogen receptors alpha and beta in human lung tissue and cell lines. Lung Cancer. 2002;37(2):153–9. Epub 2002/07/26
Lahm T, Crisostomo PR, Markel TA, Wang M, Wang Y, Tan J, et al. Selective estrogen receptor-alpha and estrogen receptor-beta agonists rapidly decrease pulmonary artery vasoconstriction by a nitric oxide-dependent mechanism. Am J Physiol Regul Integr Comp Physiol. 2008;295(5):R1486–93. Epub 2008/10/04
Sherman TS, Chambliss KL, Gibson LL, Pace MC, Mendelsohn ME, Pfister SL, et al. Estrogen acutely activates prostacyclin synthesis in ovine fetal pulmonary artery endothelium. Am J Respir Cell Mol Biol. 2002;26(5):610–6. Epub 2002/04/24
Rajkumar R, Konishi K, Richards TJ, Ishizawar DC, Wiechert AC, Kaminski N, et al. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2010;298(4):H1235–48. Epub 2010/01/19
Roberts KE, Fallon MB, Krowka MJ, Brown RS, Trotter JF, Peter I, et al. Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Am J Respir Crit Care Med. 2009;179(9):835–42. Epub 2009/02/17
Lahm T, Albrecht M, Fisher AJ, Selej M, Patel NG, Brown JA, et al. 17beta-Estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med. 2012;185(9):965–80. Epub 2012/03/03
Yuan P, Wu WH, Gao L, Zheng ZQ, Liu D, Mei HY, et al. Oestradiol ameliorates monocrotaline pulmonary hypertension via NO, prostacyclin and endothelin-1 pathways. Eur Respir J. 2013;41(5):1116–25. Epub 2012/09/01
Fessel JP, Chen X, Frump A, Gladson S, Blackwell T, Kang C, et al. Interaction between bone morphogenetic protein receptor type 2 and estrogenic compounds in pulmonary arterial hypertension. Pulm Circ. 2013;3(3):564–77. Epub 2014/03/13
Mona S, Jordan W, Angelia DL, Marjorie A, Kelly S, Irina P, et al. Hypoxia increases expression of estrogen receptor (ER)-Beta in vivo and in vitro. A107 pulmonary hypertension associated with hypoxia and paranchymal lung disease. Am Thorac Soc. 2013:A2257
Kuijper EA, Ket JC, Caanen MR, Lambalk CB. Reproductive hormone concentrations in pregnancy and neonates: a systematic review. Reprod Biomed Online. 2013;27(1):33–63. Epub 2013/05/15
White K, Johansen AK, Nilsen M, Ciuclan L, Wallace E, Paton L, et al. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation. 2012;126(9):1087–98. Epub 2012/08/04
Austin ED, Cogan JD, West JD, Hedges LK, Hamid R, Dawson EP, et al. Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir J. 2009;34(5):1093–9. Epub 2009/04/10
Tofovic SP, Rafikova O. Preventive and therapeutic effects of 2-Methoxyestradiol, but not estradiol, in severe occlusive pulmonary arterial hypertension in female rats. A51 experimental models of pulmonary hypertension. Am Thorac Soc. 2009:A1802.
Tofovic SP, Salah EM, Mady HH, Jackson EK, Melhem MF. Estradiol metabolites attenuate monocrotaline-induced pulmonary hypertension in rats. J Cardiovasc Pharmacol. 2005;46(4):430–7. Epub 2005/09/15
Tofovic SP, Zhang X, Jackson EK, Zhu H, Petrusevska G. 2-methoxyestradiol attenuates bleomycin-induced pulmonary hypertension and fibrosis in estrogen-deficient rats. Vascul Pharmacol. 2009;51(2–3):190–7. Epub 2009/06/23
Tofovic SP, Zhang X, Jackson EK, Dacic S, Petrusevska G. 2-Methoxyestradiol mediates the protective effects of estradiol in monocrotaline-induced pulmonary hypertension. Vascul Pharmacol. 2006;45(6):358–67. Epub 2006/07/29
Mendelsohn ME, Karas RH. Molecular and cellular basis of cardiovascular gender differences. Science. 2005;308(5728):1583–7. Epub 2005/06/11
Ventetuolo CE, Praestgaard A, Palevsky HI, Klinger JR, Halpern SD, Kawut SM. Sex and haemodynamics in pulmonary arterial hypertension. Eur Respir J. 2014;43(2):523–30. Epub 2013/08/21
Jacobs W, van de Veerdonk MC, Trip P, de Man F, Heymans MW, Marcus JT, et al. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest. 2014;145(6):1230–6. Epub 2013/12/07
Kawut SM, Al-Naamani N, Agerstrand C, Rosenzweig EB, Rowan C, Barst RJ, et al. Determinants of right ventricular ejection fraction in pulmonary arterial hypertension. Chest. 2009;135(3):752–9. Epub 2008/10/14
Ventetuolo CE, Ouyang P, Bluemke DA, Tandri H, Barr RG, Bagiella E, et al. Sex hormones are associated with right ventricular structure and function: The MESA-right ventricle study. Am J Respir Crit Care Med. 2011;183(5):659–67. Epub 2010/10/05
Rafikova O, Rafikov R, Meadows ML, Kangath A, Jonigk D, Black SM. The sexual dimorphism associated with pulmonary hypertension corresponds to a fibrotic phenotype. Pulm Circ. 2015;5(1):184–97. Epub 2015/05/21
Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186(3):261–72. Epub 2012/06/09
Straub RH. The complex role of estrogens in inflammation. Endocr Rev. 2007;28(5):521–74. Epub 2007/07/21
Phillips GB. Is atherosclerotic cardiovascular disease an endocrinological disorder? The estrogen-androgen paradox. J Clin Endocrinol Metab. 2005;90(5):2708–11. Epub 2005/02/03
Acknowledgment
We thank Dr. Ivan F. McMurtry for the critical reading and suggestions for this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media Singapore
About this chapter
Cite this chapter
Oshima, K., Oka, M. (2017). Sex Hormones. In: Fukumoto, Y. (eds) Diagnosis and Treatment of Pulmonary Hypertension. Springer, Singapore. https://doi.org/10.1007/978-981-287-840-3_4
Download citation
DOI: https://doi.org/10.1007/978-981-287-840-3_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-287-839-7
Online ISBN: 978-981-287-840-3
eBook Packages: MedicineMedicine (R0)