
Abstract
Bacterial genomes are flexible entities due to the complicated and dynamic makeup of the bacterial chromosomes. The plasticity and evolution of the bacterial genome are facilitated by genome rearrangements, point mutations, and lateral or horizontal gene transfer (HGT). Bacteria benefit from HGT as it facilitates them to adapt to their environment, colonize new niches, and steer evolution in “quantum leaps.” Analyses of thousands of bacterial genome sequences have shown that there exist two major divisions in the bacterial genome: the primary core genome and the accessory gene containing flexible genome. Accessory gene acquisition might be mediated by entities designated as genomic islands (GIs), facilitating the process of HGT. GI represents horizontally acquired genes frequently clustered together in bacterial genomes with different GC content, dinucleotide frequencies, codon use, etc., than the neighboring genes. GIs make the genome flexible enough to adapt novel functions over a short life span and encode a diverse range of accessory genes for improved fitness, pathogenicity, resistance potential, metabolic flexibility, ecological adaptability, symbiosis, etc., in the harboring bacteria. Niche dynamicity is the key to having highly flexible genomes in bacteria. The niche exerts selection pressure to retain only helpful information and optimizes genomes based upon the costs and benefits of different strains. This strain-specific propensity to have high island variability contributes to genome plasticity and subsequent genome evolution that might lead to niche specialization of specific bacterial strains. The most significant evolutionary benefit of GI is fostering the genetic flexibility and ability to transmit multiple genes, enabling more effective adaptation and enhancing pertinence in specific ecological niches. The consecutive acquisition and loss of auxiliary genes within the GI and the consequent transmission of chromosomal DNA from the host appear to be the prerequisites for the evolution of bacterial species.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Genomic Islands
- Bacterial genome evolution
- Pathogenicity Islands
- Horizontal gene transfer
- Genome plasticity
5.1 Genome Plasticity, Horizontal Gene Transfer and Evolution of the Bacterial Genome
Bacteria represent a diverse group of ubiquitous microbial organisms capable of surviving and tolerating a vast range of environments. Bacterial genomes are flexible or plastic, or adaptive entities resulting from the complex and dynamic nature of the bacterial chromosomes (Darmon and Leach 2014). The phenomenon of genome evolution involves different processes through which the content and arrangement of a species’ genetic information modify over time. The plasticity and evolution of the bacterial genome are facilitated by various processes, including genetic mutations or genome reshuffling through deletions, duplications, inversions, translocations, and horizontal gene transfer (HGT) (shown in Fig. 5.1). Of these, HGT is unarguably one of the major creative forces that drive bacterial evolution, molding bacterial species’ gene repertoires and providing and retaining population diversity. HGT takes place across the bacterial genome and causes the generation of incredibly different adaptations (Ochman et al. 2000). The phenomenon of HGT has cropped up significant ecological differences among closely related bacterial strains belonging to a single “species” taxon (Welch et al. 2002). Bacteria get benefitted from HGT due to their adaptation to various environments through colonization in new niches, thus allowing them to steer evolution in “quantum leaps” (Hacker and Carniel 2001). HGT is generally associated with acquiring DNA fragments that can move from one cell to the other or within a genome, or both (Bellanger et al. 2014). HGT in bacteria is brought about by transformation, transduction, and conjugation (Fig. 5.2), transferring genes across potentially distant bacterial lineages (Bazin et al. 2020). HGT is facilitated by mobile genetic components like conjugative plasmids, transposons, insertion elements, bacteriophages, and genomic islands or GIs (Hacker and Carniel 2001; Milkman 2004). Mobile genetic elements can encode different factors relating to drug resistance, bacterial pathogenicity, production of bacteriocins, and specific metabolic functions associated with the breakdown of xenobiotics chemicals, etc.

Graphical representation of the major processes facilitating the plasticity and evolution of the bacterial genome like point mutations, genome rearrangements, and horizontal gene transfer

The processes involved in bringing about horizontal gene transfer in bacteria include transformation (cell-free, one-way DNA uptake), transduction (bacteriophage-mediated one-way genetic material transfer from one bacterium to the other), and conjugation (one-way genetic material transfer via physical association between two living bacterial cells)
The advancement in next-generation shotgun sequencing has expedited the production of large amounts of genome sequence data, revealing insight into genome evolution and speciation. The analyses of thousands of prokaryotic genome sequences have led to the understanding that the bacterial genome is primarily divided into the core genome comprising of the core genes and the flexible genome made up of accessory genes, collectively referred to as “pan-genome,” a term first coined by Tettelin and collaborators in 2005 (Tettelin et al. 2005). Generally, core genes encode essential metabolic activities, while accessory genes encode traits that provide fitness to bacteria to thrive under specific growth or environmental conditions. The magnitude of the flexible prokaryotic genome is astonishing. Only around 50% of the genome of any given strain is core. With hundreds of novel flexible genes contributed by each strain, the compounded flexible pool is enormous (Rodriguez-Valera et al. 2016). Acquisition of these accessory genes might be mediated by entities known as GIs, responsible for facilitating HGT. GI represents the cluster of horizontally acquired genes present in bacterial genomes that vary in dinucleotide frequency, GC content, codon usage pattern, etc., compared to the neighboring genes (Busby et al. 2013). While the core genome reflects the evolutionarily conserved character even under severe selection pressure, microbial genome dynamicity is achieved by recurrent gene acquisition and loss. In a microbial species, the plasticity of the genome results from HGT, which facilitates the acquisition of GIs and accelerates the rate of evolution. GIs make the genome flexible enough to adapt novel functions over a short life span.
Genome plasticity or genome flexibility is the phenomenon where the genome has large tracts of strain-specific polymorphic genes, known as the regions of genomic plasticity (RGPs), in different areas. It is critical for microbial survival in novel ecological niches, pathogenicity, symbiosis, and evolution of the genome (Mathee et al. 2008; Ogier et al. 2010). The key to having highly flexible genomes in bacteria is niche dynamicity. The more availability and proximity of exogenous genetic information result in the acquisition of novel information in the bacterial genome. The niche exerts selection pressure to retain only helpful information and optimizes genomes based upon the costs and benefits of different strains. The RGPs are broadly categorized into hypervariable regions that may arise from the deletions of specific DNA segments in different bacterial strains and mobile genetic elements (Ogier et al. 2010). The mobile genetic elements may get transferred from one place to another within the intracellular genome, or bacteria often use various HGT methods for efficient intercellular transfer. The enzymes transposases and recombinases (site-specific) are responsible for the mobility of mobile genetic elements and are coded by genes positioned on either the commonly shared core genome or the mobile genetic elements themselves.
5.2 GIs: Features, Types, Significance, and Plasticity
A GI is a fragment of exotic DNA that has been inserted into the bacterial genome and has clearly defined boundaries (Novick and Ram 2016). These transferable DNA regions are larger than 10–500 kb base-pair sequences (Osborn and Böltner 2002). Hacker et al. (1997) initially described GIs as gene clusters inside a bacterial genome possessing a specified dinucleotide frequency and GC content. GIs contribute significantly to genome flexibility, evolution, and environmental adaptation (Li and Wang 2021) by armoring bacteria with genes that provide antibiotic resistance, virulence traits and even sometime incorporating them with genes encoding enzymes leading to de novo metabolic pathway(s) formation (da Silva Filho et al. 2018).
5.2.1 Features Attributed to a GI
GIs encode a diverse range of accessory genes for improved fitness, secretion, pathogenicity, resistance potential, metabolic flexibility, ecological adaptability, symbiosis, etc., in the harboring bacteria (Darmon and Leach 2014). GIs have many other distinguishing features which delineate them from the other genomic regions. The specialized components of GIs are the presence of flexible sequences that differ from the core genome, the occurrence of genes for self-mobilization (viz. insertion sequences or ISs, integrases, and transposases), direct repeats (DRs) for flanking, and particular integration sites (Juhas et al. 2009; Schmidt and Hensel 2004). The recombination or transposition events during the integration process of GIs into the chromosome may generate the DRs. A generalized graphical representation of GI is given in Fig. 5.3. The evolution of GIs occurs through genome exchange, gene reduction, or further acquisition of some transposable elements (i.e., mobile genetic elements). The gene content between related strains of a species may vary considerably in each subset of islands. The G + C content (25–75%) of GIs is specific; they differ from the core chromosomal regions of bacteria (Schmidt and Hensel 2004). GIs are mostly inserted at 3′ end of genes coding for tRNA, and the region is often known as a hotspot for GI insertion (Lu and Leong 2016; Williams 2002). However, structurally every GI is almost the same in having a recombination module (integrase/excisionase module), two attachment sites (attR and attL, at the right and left end, respectively), and sometimes a recombination directionality factor (RDF). The tyrosine/serine recombinase enzyme family is involved in the exchange process during GIs integration (Boyd et al. 2009; Desvaux et al. 2020).

A graphical representation of the generalized organization of genomic island integrated within the bacterial chromosome (GI). DR = direct repeats, IS = insertion sequence and int = gene coding for integrase. The functional gene content of GIs are used to classify them into some subtypes, such as (i) pathogenicity island (PAI) that encode genes essential for bacterial pathogenicity/virulence, (ii) resistance island (RI) that encode antimicrobial resistance genes, (iii) symbiosis island (SI), and (iv) metabolic island (MI) that encode for adaptive metabolic abilities
5.2.2 Types of GIs
The flexible gene content of GIs was classified and named based on the types of functions they encipher (Rainey and Oren 2011). These are (i) pathogenicity islands (PAIs) encoding genes relating to bacterial pathogenicity or virulence, (ii) symbiosis islands possessing similar structural attributes to pathogenicity islands though encoding proteins facilitating the mutual relationship of bacteria and multicellular organisms (Rainey and Oren 2011), (iii) antibiotic resistance islands that encode antimicrobial resistance genes, and (iv) metabolic/catabolic genomic islands enabling bacteria for adaptive metabolic abilities in degrading xenobiotic chemicals (Bertelli et al. 2019). Several kinds of mobile transposable elements including integrons, integrative and conjugative elements (ICEs), prophages, etc., are included within this wide dimensions of GI. GIs are mainly distinguished depending on the acquisition mechanism (i.e., transformation, conjugation, or transduction) and accompanying mobile elements (like transposases, ISs, and integrases) facilitating GI mobilization and transmission (Bertelli et al. 2019; Juhas et al. 2009; Langille et al. 2010; Soucy et al. 2015). According to Boyd et al. (2009), the GIs refer to a discrete class of evolutionarily ancient integrative components that are not “degenerate relics of prophages, episomes, integrons or ICEs.”
New GI insertion is followed by subsequent changes in the cellular or colonial morphology, function, or even the lifestyle of the accepting organism. Some GIs are PAIs of the strain Salmonella SPI1 and Listeria monocytogenes, symbiosis islands (SIs) of Bradyrhizobium, Mesorhizobium loti strain R7A, acid fitness islands (AFIs) of Escherichia coli, defense island (DIs) of Microcystis aeruginosa, Shewanella sp. strain ANA-3, resistance islands (RIs) of Haemophilus influenzae, Acinetobacter baumannii, saprophytic islands of some Escherichia coli strains, a phenol degrading ecological island of Pseudomonas putida, xanthan gum production island (a metabolic island) of Xanthomonas (Arashida et al. 2022; Chan et al. 2015; Hadjilouka et al. 2018; Lerminiaux et al. 2020; Lima et al. 2008; Makarova et al. 2011; Mates et al. 2007; Van Elsas et al. 2011; Xiang et al. 2021). Different ecological niches exhibit diverse selection pressure under which bacteria with similar GIs play different functions. A bacterium may contain a variety of GIs within its genome to perform various functions.
5.2.3 Significance of GIs
The evolutionary benefit of GI is that many genes (like the whole operon dealing with novel traits) can be transferred horizontally to the recipient’s genome, instigating significant modifications in the recipient’s characteristics. According to the theory of “selfish operon,” genes concerned with a particular function are grouped to promote their HGT (Lawrence and Roth 1996). GI can improve adaptability and competitiveness within the niche, thus providing selective benefits under certain growth conditions. The most significant evolutionary benefit of GI is fostering the genetic capacity and flexibility to transmit multiple genes, enabling more effective adaptation and enhancing fitness in particular ecological niches (Dobrindt et al. 2004).
5.2.4 Plasticity in GIs
Bacteria harbor IS elements, plasmids, prophages, transposons, GIs, ICEs as MGEs. The ISs are the simplest MGEs (< 2.5 kb in size), which transmit no genes except those that encode machinery essential for their insertion at various DNA sites (Siguier et al. 2014). Plasmids have self-replication ability, and their intercellular transfer involves conjugation in prokaryotic cells (Smillie et al. 2010). Prophages integrate with bacterial chromosomes, and their mobility depends on transduction (Brüssow et al. 2004). Transposons may transfer from one intra-genomic region to another intra-genomic region and do not undergo any HGT. GIs are larger (10–500 kb) MGEs with genes for self-mobility and other essential genes for strain-specific functions. The staphylococcal pathogenicity islands (SaPIs), GTAs, and ICEs are the three most common types of GIs. The first two are presumably descended from prophage forebears and have retained crucial prophage architectural traits. The third group likely originated from conjugative plasmids, which acquired additional characteristics and transformed into mosaics. While GTAs and ICEs independently influence HGT, the SaPIs depend on certain bacteriophages. The ICEs principally transmit their own DNA, whereas the GTAs solely convey the unlinked host DNA, but the SaPIs are a blend of both ICE and GTA. It is assumed that immobile GIs are variations of mobile ones (Novick and Ram 2016).
Not all GIs have genes for autonomous transfer. For example, SaPIs need helper phages for mobility (Lindsay et al. 1998). GIs share discrete segments of DNA between similar strains. However, their formation and ability to acquire accessory genes in the syntenic block lead to bacterial adaptation, genome diversification, and evolution. Different molecular events like recombination, deletion, duplication, inversion, etc., make GIs flexible.
The SaPIs of Gram-positive bacteria have phage integrase and excisionase homologs. The functional relatedness of SaPIs with phage makes them highly mobile with a unique lifestyle. The helper phage-mediated transfer of SaPIbov5 involves a prophage cos site in it. Due to phage interference SaPIs increase the transfer of chromosomal adaptive genes for host virulence. Genomic data from Staphylococcus implies SaPIs have typical phage-related genome organization but are sharply different from their progenitor prophage. Phage-related elements of streptococci and lactococci have orthology patterns similar to the SaPIs. Genome-based analysis also suggests that the widespread and diversified nature of SaPIs all over the bacterial genomic world successfully builds evolutionary strategy (Chen et al. 2015; Dokland 2019; Novick et al. 2010; Novick and Ram 2016).
The ICEs catalyze self-excision through site-specific recombination regardless of conjugation and integration. ICEs have diverse modular structures, i.e., gene clusters with different functions like conjugation, integration or excision, and adaptation. Conjugation modules have two types: a single-stranded DNA-based MOB/MPF module where MOB is the relaxase protein family, and MPF is a mating pair formation protein family. The second is a double-stranded DNA-based module that encodes SpoIIIE/FtsK protein family DNA translocator (Tra proteins) (Álvarez-Rodríguez et al. 2020; Besprozvannaya et al. 2013; Guglielmini et al. 2011). A double-stranded conjugation module containing ICE, regarded as actinomycete ICE (AICE), is found in actinomycetes (Johnson and Grossman 2015; Te Poele et al. 2008). The MOB acts on the 5′ end of ICEs and initiates DNA transfer through the MPF into the recipient bacterial cell. On the other hand, Tra proteins form channels and act on the cis-acting locus of circular AICE to transfer. The integration/excision modules encode enzymes for recombination events, generally, tyrosine/serine recombinase (or DDE transposase) those identify repetitive flanking sequences of ICEs. ICEs containing such integration/excision modules are usually found at the 3′ end of genes coding for tRNA. In case of different housekeeping genes, they can be found either at the 3′ or 5′ end. ICEs Ecoc54N from Escherichia coli, Tn5397 from Clostridium difficile, Tn1806 from Streptococcus pneumoniae, Tn6012 and ICE6013 from Staphylococcus aureus, and TnGBS elements from Streptococcus agalactiae have integration/excision modules with either tyrosine/serine recombinase or DDE transposase (Antonenka et al. 2006; Bellanger et al. 2014; Camilli et al. 2011; Guérillot et al. 2013; Mingoia et al. 2016; Sansevere et al. 2017; Ternan et al. 2012; Wang et al. 2006).
The fundamental mechanisms behind GIs plasticity and evolution are acquiring, exchanging, or deleting different modules. Comparisons of sequences between ICEs provide information about the occurrence of enormous exchanges within different modules. It is suggested that the host specificity of ICEs evolved through the site-specific recombination between conjugation and integration modules of various ICEs (Burrus et al. 2002). Module exchanges also occur between other GIs. Sequence comparisons also reveal many GIs developed by deletion mutations in mobility modules. In some GIs, for example, ICE_2603_tRNALys from S. agalactiae, genes (like orfD) associated with intercellular or intracellular mobility are deleted by incorporating the copy of insertion elements (IS1193). The combination of a GI within another GI, followed by subsequent restructuring, resulted in the acquisition of novel modules. Insertion of non-mobilizable ISs, induce deletion or inversion of neighbor sequences besides inactivation. In the high-pathogenicity island (HPI) of Y. pestis KIM, ISs are connected with conjugation module deletion (Bellanger et al. 2014; Chen et al. 2010; Puymège et al. 2013).
5.2.4.1 Driving Forces for GIs Plasticity
The GIs vary between individual strains and help adapt to new ecological niches, host-cell interaction, and virulence. This strain-specific propensity to have high island variability contributes to genome plasticity and subsequent genome evolution that might lead to niche specialization of specific bacterial strains. The variable strain-specific genomic DNA segments may evolve through rearrangement events like recombination, deletion, insertion, duplication, amplification, inversion, or tandem accretion.
5.2.4.1.1 Recombination
This is a crucial mechanism to keep genome flexibility and fitness. Homologous and non-homologous recombination are genetic information exchange procedures between DNA sequences with higher or lower identity, respectively (Didelot et al. 2012). In homologous recombination, the recombination rate reduces if the sequences are poorly identical. Non-homologous recombination occurs during either DNA synthesis or strand breakage and causes the addition of new genetic material through HGT and site-directed recombination. The insertions of new DNA segments often lead to deletions and subsequent hairpin formation due to strand slippage. The new DNA segments introduce genome diversity and synteny loss in bacteria. In homologous recombination, the involvement of Rec enzymes results in the single-strand or double-strand DNA gap repair; therefore, the recombination rates increase. The Rec enzymes, on the other hand, negatively regulate non-homologous recombination, lowering the recombination rate. Site-directed recombination involves tyrosine/serine recombinase or DDE transposase to recognize the flanking repeats causing the exchange of DNA segments between integration/excision modules of different GIs. Both types of recombination play indispensable roles in bacterial genome evolution. The plasticity of GIs may come through the recombination process between two separate GIs, as shown in Fig. 5.4. A transposon Tn6022 mediated recombination is indicated as the source of GI formation in many Acinetobacter baumannii strains (Patel 2016; Peters et al. 2014). From the ecological perspective, the overlapping niches of strains bring more opportunities to exchange genetic information between GI modules than lineages living in distant niches.

GI plasticity via recombination. The plasticity of GIs may come through the recombination process between two separate GIs. Here, the red cross mark in the left diagram depicts the recombination between the integration/excision modules of GI1 and GI2. This event leads to a new GI formation, as shown on the right side of the figure
5.2.4.1.2 Deletion
Inactivation or deletion of part of the genome in bacteria facilitates genome evolution. While acquiring novel genes enhances the bacterial colonization potential, gene loss enables niche specialization. The “Streamlining” and “Black Queen” hypotheses assume that the loss of superfluous genes confers the survival ability or the fitness cost in bacteria lowering the metabolic burden (Giovannoni et al. 2005; Morris et al. 2012). The “Streamlining” theory is dependent on the fact that the challenging environmental circumstances favor robust selection to minimize cell complexity. On the other hand, the “Black Queen” hypothesis specifies that an organism should stop performing its costly function under certain conditions. Both hypotheses support reductive genomic evolution. These trends are visualized when host-specialized, highly virulent bacterial pathogens evolve from a vast range of hosts through massive gene loss. In endosymbionts, genome shrinkage is essential, which occurs possibly by deletion. Many new flagellar genes have been identified in the flagellar operon (fli operon) of Salmonella enterica var. typhimurium LT2. Deletions in the 2.07Mbp region in fli operon resulted in increased fitness (Frye et al. 2006; Koskiniemi et al. 2012). The plasticity and evolution of GIs also depend on the deletion of different modules. GIs are often sensitive to ISs inclusion leading to the inactivation or loss of superfluous genes, thus lowering the energy/mass expenditure.
5.2.4.1.3 Insertion
Insertion is the process of new gene acquisition that induces adaptive alterations in genome architectures and confers bacteria the ability to survive in new ecological niches. The transposition ability of ISs helps them to integrate anywhere in the genome. Their lodging is accidental in the genome, and several mechanisms control their disrupting activity resulting in genome innovation. The introduction of ISs in the genome creates an opportunity to evolve and accommodate better adaptive ability to the stressors. In the genome of nosocomial isolates of Staphylococcus epidermidis, insertion element IS256 facilitates the tolerance to drugs making them multi-drug resistant. The insertion occurs in the GIs, consequently acquiring novel gene clusters (Dengler Haunreiter et al. 2019; Espadinha et al. 2019; Otto 2009). Some GIs may integrate within other GIs, followed by subsequent reorganization. Several mechanisms are responsible for the evolution of such mobilizable GIs. The non-mobilizable GIs that do not encode self-conjugative machinery can insert within ICEs. In the genome of Mesorhizobium loti USDA110, about 64 ISs are found within the 680 kb ICEMlSymR7A (Kaneko et al. 2002; Sullivan et al. 2013). Varieties of transposable elements have also been recognized in many ICEs or related GIs. Some ICEs also bear prophages. For example, Tn6164 from C. difficile bears a complete prophage (Hargreaves et al. 2016). A graphical representation of deletion and insertion in GI is shown in Fig. 5.5.

Schematic presentation of deletion and insertion in GI. Insertion of region 2 from GIA into the GIB and region A from GIB into the GIA leads to the loss and gain of regions islands (Melnyk et al. 2019)
5.2.4.1.4 Duplication and Inversion
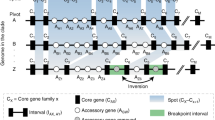
Duplications are dynamic forces that aid in surviving in an unfriendly environment. Multiple amplification of the genomic regions results in tandem repetitive sequences. Inversion is another process when a DNA fragment is excised and reconnected in the opposite direction elsewhere in the genome. In general, the inversion sequences are bordered by inverted repetitions and are occasionally found inside a coding region. This reversal modifies the gene expression profile and alters the phenotypic characteristics of bacterial species. The evolution of distinct bacterial lineages confers adaptability by coupled gene duplication-amplification in response to drugs. It has been found that multistep adaptive development is preceded by gene amplification. As a result, mutation occurs in the additional copies while stabilizing the other copies of essential genes, enhancing fitness. Both duplication and inversion frequently target GIs, leading to plasticity. A schematic presentation of deletion and insertion in GI is depicted in Fig. 5.6. E. coli ST58 contains two GIs, PAI-1 and PAI-2, both islands sharing a great deal of genetic information. A duplication event at two distinct tRNA–Phe–GAA sites led to the development of these two progenitor islands from a single island. This duplication event was followed by inversion, and PAI-1 and PAI-2 acquired separate sets of genes throughout time (Wyrsch et al. 2020). IS-mediated duplication has been found in the symbiosis island of WN105 mutant of Bradyrhizobium diazoefficiens USDA110 (Arashida et al. 2022).

Duplication and inversion in GI. Left diagram shows normal GI, whereas right shows GI with duplicated and inverted region 1 (Wyrsch et al. 2020)
5.2.4.1.5 Tandem Accretion
GIs also originate from site-specific recombination and subsequent tandem accretion-deletion of CIMEs and ICEs (Fig. 5.7). The site-specific accretion resulting from gene gain and loss is a key tool for GI flexibility and evolution. The composite structure of ICESt1 and related GIs from Streptococcus thermophilus demonstrated these components developed via site-specific recombinations and deletions. At the 3′ end of the fda locus in seven distinct strains of S. thermophilus, four forms of ICESt1-related ICEs with comparable conjugation and recombination modules were identified. These elements are flanked by different site-specific attachment sites (att) that are strongly connected to attachment sites of two distinct cis-mobilizable elements (CIMEs) CIME19258 and CIME302. This results in site-specific recombination, which results in the excision of ICEs and the subsequent incorporation of CIMEs at the 3′ end of fda. Moreover, genome analysis showed identically shortened sequences at the att regions of these ICEs and CIMEs (Bellanger et al. 2014; Pavlovic et al. 2004).

Tandem accretion of GIs. The diagram shows the integration of ICE adjacent to the att1 of the resident CIME by site-specific recombination, causing composite GI formation (Bellanger et al. 2014)
5.3 GIs and Bacterial Evolution
The idea of prokaryotic species is complicated, and it is commonly assumed that such species are formed because of continuous processes combining gene loss and gain facilitated by HGT (Lawrence 2001). The acquisition and loss of auxiliary genes within the GI and the probable transmission of chromosomal DNA from the host may be a precondition for bacterial species evolution. Since GIs may integrate themselves into the host chromosomes by excision, conjugated-mediated self-transfer into a new host, and reintegration, they can transmit a piece of the host genome into the recipient bacteria. This process of receiving donor (foreign) DNA may open the way for bacterial evolution when donor DNA is integrated into the genome of the host via transformation. The unique self-transfer type IV secretion system (T4SS) of Neisseria gonorrhoeae (encoded by a horizontally acquired huge gonococcal genetic island or GGI), enables both secretion and spread of the host chromosomal DNA. Later, when taken up by the transformation process, the secreted chromosomal DNA (via GGI-encoded type T4SS) can undergo recombination along with the host’s chromosome, adding to antigenic variation and drug resistance. Thus, GIs appear to influence the evolution of the host bacteria. This change occurs in several Gram-negative or Gram-positive, environmental or pathogenic bacteria like N. gonorrhoeae, Acinetobacter sp. ADP1, Haemophilus influenza, Pseudomonas stutzeri, Bacillus subtilis, Streptococcus pneumoniae, and Ralstonia solanacearum. In addition to transformation, the transmission of GIs across bacterial species can also be mediated through conjugation and bacteriophages. The SaPIs are the most well-known example of this phenomenon. There have been several reports of such bacteriophage-mediated transfers of GIs, that includes the HPI and the GIs of Yersinia pseudotuberculosis and Prochlorococcus sp. (marine cyanobacteria), respectively (Juhas et al. 2009).
5.3.1 GIs in Bacterial Genome Evolution and Shapiro’s Geographical Metaphors
In his article “How clonal are bacteria over time?,” Shapiro (2016) has convincingly employed different geographical metaphors to explain how GIs are potentially associated with genome evolution. According to Shapiro, horizontal transfer (recombination) rates fluctuate significantly across the genome so that an entire population, except for a few loci, can be clonal. These loci are referred to as GIs. The term “peninsula” provides a simile that might better depict the connection of the islands to the microbial genomes. An island remains evolutionarily independent compared to the mainland genome, but their fates may become associated. For instance, a bacterium may obtain any gene from the enormous microbial gene pool. The acquired gene enables the bacteria to colonize into a new ecological niche, initiating a clonal expansion where the acquired gene’s fate and its new host genome are inextricably intertwined, at least during the clonal expansion period. Certain microbial genomes may include an inordinate number of islands to the point where there is no mainland but a large archipelago. Archipelagos are not always stable throughout time, and they can occasionally combine into continents. When the ecological conditions are favorable, any genome from the panmictic gene pool can break free from the “gravitational pull” of recombination and embark on clonal growth (Shapiro 2016).
With the help of bioinformatics, it became apparent that novel genes with unknown functions are present within GIs. These novel genes lack orthologs in other bacterial species and might furnish the host bacterium with various adaptive functions. Different accessory functions like additional metabolic pathways, resistance toward antibiotics and harmful drugs, pathogenesis, symbiosis, or traits involved in increasing microbial fitness, are encoded by GIs.
Although plasticity (Dobrindt et al. 2004) within GI alters bacterial lifestyle or behavior, it is not unfamiliar that bacterial “quantum leaps” in context with evolution is mediated by GI (Juhas et al. 2009), as it has the intrinsic property to transfer a large set of genes and integrate as a whole into the recipient genome, thereby promoting bacterial diversity and speciation (Guo et al. 2012). Some of the GI-mediated features that can offer a significant selective benefit to the host bacteria and thus leap a step toward evolution and speciation are discussed briefly below.
5.3.2 GI and Heavy Metal Tolerance
GIs may provide an advantage for living in harsh toxic environments leading to the evolution of resistant strains. One of the most critical toxic environments includes acid mine drainage (AMD) which contains high concentrations of toxic heavy metals (including arsenic). A comparative genomic study on different isolates of Thiomonas bacteria obtained from AMD of Carnoulès, France, revealed the presence of more than 20 GIs. The findings also indicated that arsenic-associated GIs had evolved differently in two closely related Thiomonas strains, resulting in varied survival capabilities in As-rich environments (Freel et al. 2015).
GIs contributing to heavy metal tolerance in Mucilaginibacter spp. have been observed in isolates obtained from gold/copper mines. Clusters of genes that may be connected with mobile genetic elements were discovered by analyzing the location of heavy metal resistance determinants. These loci contained genes for tyrosine recombinases (integrases) and subunits of T4SS, letting integration/excision and conjugative transfer of many GIs, respectively. The supposed presence of many CTnDOT-related GIs in the genomes of Mucilaginibacter may have a crucial task in genome evolution and subsequent adaptation (Vásquez-Ponce et al. 2018).
In Cupriavidus metallidurans, GI variation has contributed to the variable plasticity of the genome. C. metallidurans represents a versatile multi-metal resistant bacteria. Comparative genomic hybridization of sixteen C. metallidurans strains revealed that the broad arsenal of heavy metal resistance factors was well preserved across all strains of C. metallidurans. Contrarily, the transposable elements found in strain CH34 were not observed in the other strains but displayed an indirect pattern associated with a specific geographical location or biotope. One set of strains had nearly all transposable components, whereas the second group had a substantially lower proportion. This was also manifested in their capacity to break down toluene and thrive on carbon dioxide and hydrogen gas in an autotrophic manner. Both of these are connected to distinct GIs of the Tn4371 family (Van Houdt et al. 2012).
5.3.3 GIs in Secondary Metabolism, Pathway Evolution and Xenobiotic Degradation
GIs have been found in a distinct environmental (marine) bacteria Salinispora, belonging to the phylum Actinobacteria. Genomic comparison of S. tropica with S. arenicola displayed the distribution of three-quarters of species-specific genes within 21 GIs associated with the production of secondary metabolites, also establishing a connection between secondary metabolism and functional adaptation. All species-specific biosynthetic pathways are found in GIs, most of which are found in S. arenicola, contributing to its worldwide distribution in different habitats. Gene duplication and acquisition dominate genome evolution, which provides rapid chances for generating novel bioactive compounds in the case of secondary metabolism. The horizontal sharing of secondary metabolic pathways performs a major functional role in acquiring natural product biosynthetic gene clusters, which also serve as the driving force for maintaining bacterial diversity (Penn et al. 2009). GIs have been identified as hotspots for biosynthetic gene cluster acquisition in Salinispora (Letzel et al. 2017). Using something apparently like a plug-and-play paradigm of evolution, clusters of acquired biosynthetic genes are targeted to certain GI and can replace each other (Letzel et al. 2017).
GIs seem to have a significant role in developing novel pathways through “patchwork assembly” (a novel combination of previously existing pathways) (Dobrindt et al. 2004; Guzman and Harris 2015; Mingoia et al. 2016). Studies reveal that plasmids, transposons, and GIs include catabolic genes that encode functionalities with the capacity to digest xenobiotic substances. For instance, Ralstonia oxalatica Tn437, a GI, might break down chlorobiphenyl. First discovered in Pseudomonas knackmussii B13, the clc element could utilize chloroaromatic chemicals as a carbon source. In the field of biodegradation, ICEclc is the best-known ICE. It bears selective genes for the ortho-cleavage of chlorocatechols and aminophenol metabolism, and these are clc and amn genes respectively). This component is capable of metabolizing 3-chlorocatechol, 3-chlorobenzoate, 4-chlorocatechol, as well as aminophenol. Due to its self-transfer capabilities, it can insert itself into the genomes of different Proteobacteria based on environmental circumstances (Klockgether et al. 2006). Based on amino acid homology searches, similar clc-like elements from bacterial genomes have already been isolated around the world, including in Xylella fastidiosa 9a5c (a plant pathogen), Pseudomonas aeruginosa C (a clinical isolate), P. aeruginosa S17GM (an environmental isolate) and Xanthomonas campestris (Lacour et al. 2006). Pseudomonas-like clc components have also been identified in a Ralstonia sp. JS705 isolate (reported from contaminated groundwater). This clc element codes for chlorobenzene to chlorocatechol metabolizing enzymes with 85–100% nucleotide similarity in the conserved area (Klockgether et al. 2006). These discoveries demonstrate that the clc element can spread to new environments and obtain new functionalities within its current location (van der Meer and Sentchilo 2003). GIs conferring the capacity to degrade xenobiotics and toxic compounds biologically has been detected in many bacterial populations. Some examples of GI with biodegradation functions are listed in Table 5.1.
5.3.4 GIs and Siderophore Expressing Bacteria
Bacteria are well known for expressing iron uptake systems, known as siderophores. Siderophores represent low molecular weight secondary metabolites synthesized and then released into their environment, where they chelate ferric iron to combat iron deficiency (Neilands 1995; Thode et al. 2018). This is an adaptation to survive in an iron-restricted environment and is associated with virulence. Genes encoding for siderophores are distributed in several pathogenic and non-pathogenic bacterial species harboring GIs. Examples include HPI in Yersinia sp.; SHI-2, SRL, and SHI-3 in various species of the genus Shigella; and PPI-1 in S. pneumoniae (Dobrindt et al. 2004). Such GIs can serve as fitness islands in environmental bacteria or PAI in pathogenic bacteria.
5.3.5 GIs and Bacterial Secretion Systems
The evolution of pathogenicity in bacteria through acquiring GIs (virulence-carrying genes) is a well-established phenomenon. During evolution, bacteria may have gained new genes by HGT, or their current genes may have acquired mutation. One fine example of the successful host-pathogen interaction is represented by several classes of protein secretory systems encoded by GIs (Martínez 2013). The type III secretion systems (T3SS) or “contact-dependent” secretion systems are complex multiprotein machinery (Scherer and Miller 2001). T3SS is generally expressed by pathogenic bacteria infecting plants and animals, including genera like Yersinia, Shigella, Salmonella, Pseudomonas, enteropathogenic E. coli (EPEC), Erwinia and Rhizobium. In some exceptional cases, two T3SSs are expressed within a single pathogen, each necessary at a different infection stage. In S. enterica, out of the two T3SS (harbored by SPI-1 and 2), one is essential for the initial interaction and penetration into the eukaryotic target cell (intestinal epithelium cells). At the same time, the other is essential for systemic infection (Juhas et al. 2009).
GIs of many bacterial pathogens encode T4SSs, translocating bacterial effector proteins through the bacterial membrane and plasma membrane into eukaryotic host cells. T4SSs, in turn, mediate HGT, which contributes to plasticity of the genome, development of infectious diseases, and the spread of drug resistance and other attributes related to virulence. The architecture of the genetic determinants of T4SS is diverse and comprises numerous genes grouped as a single functional unit (Juhas et al. 2007). The T4SS has been extensively studied in Agrobacterium tumefaciens. Unlike the T3SS system, this complex system is unique as it delivers nucleoprotein complexes and effector proteins into plant cells, contributing to pathogenicity directly (Dobrindt et al. 2004).
5.3.6 GIs and Antimicrobial Resistance
One of the most significant routes for acquiring drug resistance is GIs. The advent of methicillin-resistant Staphylococcus aureus (MRSA) has largely been attributed to the so-called staphylococcal cassette chromosome methicillin-resistant (SCCmec) islands present in the genome of S. aureus. This island can also integrate with other MGEs and might confer resistance against additional antibiotics, thus representing a hotspot with variable size (20 kb to ≥60 kb) (Dobrindt et al. 2004). MRSA is resistant to various antibiotics like methicillin, penicillins, kanamycin, tobramycin, bleomycin, tetracycline, macrolide, lincosamide, streptogramin, vancomycin and also to heavy metals (Juhas et al. 2009). Origin of the SSCmec island in S. aureus is yet to be established; however, comparative bioinformatic studies have proposed that it could have originated from other staphylococcal species via HGT, such as S. sciuri, S. fleuretti, S. epidermidis, or S. haemolyticus. Reports have shown the existence of SSCmec in S. epidermidis well before its discovery from S. aureus. Thus, SCCmec in S. epidermidis might act like a pool of resistance genes contributing to the evolution of multi-drug-resistant S. aureus. In another case, mecA was naturally found to be present in the chromosome of S. fleuretti and therefore was supposed to be the original source of the mecA in the SCCmec. Many dynamic SCCmec islands have been discovered from S. haemolyticus genome, thus indicating S. haemolyticus to be a potent carrier for methicillin-resistant genes (Juhas 2019).
The genus Enterococcus has become a chief cause of nosocomial infections, and the prime player in the swift expansion of such enterococcal infection comprises those of drug-resistant strains. Besides genomic modification and HGT, GIs also play a crucial role in the acquisition of drug resistance. In studies where the whole genome sequences of some E. faecium and E. faecalis (carrying many resistance genes) were screened to analyze the correlation between antibiotic resistance genes (ARGs) and GI transmission, two observations became distinct, firstly the prevalent nature of GIs in Enterococcus, and secondly, antibiotic-resistant genomic islands (ARGIs) contributing significantly to the dissemination of some ARGs. The above study has clearly shown the existence of 119 GIs in 37 strains, with an average value of 3.2 in each strain (universal presence of GI in Enterococcus). GIs in these strains was found to harbor variant ARGs, including aminoglycosides, chloramphenicol, glycopeptides/peptides, lincosamides, streptomycin and multi-resistant efflux pumps. The ARGs identified in the enterococcal ARGIs are, mdtG (encodes an efflux pump providing resistance against with fosfomycin), tetM (tetracycline resistance), dfrG (diaminopyrimidine antibiotic resistance), lnuG (lincosamide resistance), fexA, (an efflux pump providing chloramphenicol resistance). Besides encoding for drug-resistant, some of the GIs have been credited with mobility-related elements, like genes for conjugation, transposase or excisionase. Credible relationships among enterococcal strains and GIs were found to indicate frequent genetic exchanges within and between Enterococcus strains. Regular genetic exchanges among all the strains (comprising E. faecium and E. faecalis) mediated by GIs were not unusual (Li and Wang 2021). The high plasticity of Enterococcus genome has been allocated to the conjoint action of HGT, and either gain or loss of genetic information. According to Darwin, for the evolution of an organism, environmental selection pressure must have served as the driving force. This hypothesis fits well in the case of the MDR Enterococcus, isolated from complex ecological niches, like hospitals, medical clinics, farmlands, contaminated water, stools, humans, and pigs, and where harsh environmental factors always exist (such as antimicrobial compounds, organic and inorganic biocides, and heavy metals). The GIs harboring numerous novel genes due to their self-mobility or non-mobility might have integrated into the bacterial genome. Consequently, the recipient organism develops a new metabolic potential to enhance fitness or adaptability (Li and Wang 2021).
Many Salmonella enterica serovars responsible for gastrointestinal sickness are resistant to antibiotics due to GIs bearing a class 1 integron that contains the resistance genes. Studies have suggested that Salmonella genomic island 1 (SGI-1) retains a complex multi-drug resistance segment, imparting resistance against many antibiotics, including tetracycline, ampicillin, chloramphenicol/florfenicol, sulfamethoxazole and streptomycin/spectinomycin. The SGI1-associated MDR region comprises a complex integron harboring the aadA2, floR, blaPSE, tetR, and tetG genes (Vo et al. 2010). Many Salmonella serovars and Proteus mirablis possess SGI1 or similar islands harboring diverse resistance gene sets. SGI1 is a mobilizable integrative element transferable experimentally into E. coli (Hall 2010).
Cholera, caused by Vibrio cholerae, is a dreadful disease. Reports of multidrug-resistant V. cholerae strains have been frequent over the past few decades. The spread of determinants related to resistance is primarily due to mobile genetic elements such as the SXT / R391 integrated conjugate element, IncC plasmid, and GI. Transmission of the IncC plasmid is activated by the master activator AcaCD (Rivard et al. 2020). The regulatory network of AcaCD extends to the chromosomally integrated GIs. A discrete and novel mobile genomic island (MGI) MGIVchHai6 integrated into the chromosome of a multidrug-resistant V. cholerae HC-36A1 isolate (Carraro et al. 2016) contains an integron In104-like multi-drug resistance element and a mercury resistance transposon, analogous to SGI1. Acquisition of MGIVchHai6 plays a vital role in resistance against β-lactams, chloramphenicol, trimethoprim, tetracycline, sulfamethoxazole, and streptomycin/spectinomycin (Carraro et al. 2016).
Pseudomonas aeruginosa is a severe threat to burn patients and the immune-compromised. Different high-risk clonal strains, like ST111, ST175, and ST235 carry genes that give resistance to β-lactam antibiotics (Roy Chowdhury et al. 2016). GIs play a determining role in the spread of resistance to a wide variety of effective antibiotics, such as metallo-β-lactams and extended-spectrum β-lactams. Strains of P. aeruginosa (ST) 235 carry Tn6162 and Tn6163 in GI1 and GI2, respectively. The class 1 integron coupled with Tn6163 in GI2 carries a blaGES-5–aacA4–gcuE15–aphA15 cassette range that confers resistance to aminoglycosides, including carbapenems. Studies suggest that the evolution of GI2 could have occurred from a novel ICE. GI2 is winged by a repeat motif region (direct) of 12 nucleotide bases and codes for integration, conjugative transfer and ICE-specific proteins (Roy Chowdhury et al. 2016).
Another Gram-negative opportunistic pathogen, Acinetobacter baumannii, is a nosocomial pathogen that causes serious health hazards to immunocompromised patients. Nowadays, A. baumannii has been garnering considerable attention widely due to its rapid capacity to build up multi-drug resistance. The sequences of many A. baumannii genomes have divulged a vast collection of ARGs, several of which are connected with transposable elements and ISs, and it might be found in GIs, known as AbaR (Leal et al. 2020; Liu et al. 2014). Different AbaR islands have been found that vary in size, and are dynamically reshaped primarily because of recombinases, transposases, and integrases (Leal et al. 2020). Few resistance genes are present within plasmid, which can be intra- and interspecies exchanged, even by prophages (Leal et al. 2020). Studies have detected the presence of a novel GI (GIBJ4) in the drug-sensitive strain BJ4 possessing metal resistance genes inserted into the position where AbaR-like RIs commonly reside in other strains of A. baumannii (Liu et al. 2014). In A. baumannii several antibiotic resistance determinants are also present outside the RIs, such as integrons, chromosomal intrinsic antibiotic resistance genes, and the blaOXA-23-containing transposon Tn2009 (Liu et al. 2014).
5.3.7 In Planta GI Mediated Bacterial Evolution
GIs can transmit across bacteria in vitro but not during the infection process in the host. Lovell et al. (2009) have demonstrated that horizontal transmission of a GI (PPHGI-1) occurs in planta between strains of the plant pathogen Pseudomonas syringae pv. Phaseolicola (Pph). This study reveals that the transfer of PPHGI-1 across Pph strains by transformation involves four unique steps: (i) excision of the GI from the bacterial chromosome, (ii) release of the circular episome from the bacterium, (iii) relocation into competent bacterial cells, and (iv) integration at a particular att site. Transformation, the simplest method of DNA exchange, may thus accomplish the evolution of bacterial pathogens via HGT (Lovell et al. 2009).
5.3.8 GIs in Evolution of Pathogenic Bacteria
Many bacteria incorporate PAIs, a subset of GI, in their chromosomes. PAIs are specialized islands comprising arrays of genes whose expression leads to pathogenicity (virulence) and disease. These PAIs provide fitness to the PAI-positive bacteria directly or indirectly by increasing their chances of survival in vivo and/or transmission to new hosts and contributing to genome evolution. This PAI-mediated fitness can be well observed during the onset of clinical symptoms, which is directly linked to the pathogenicity or lesions triggered by the pathogenic or virulent bacteria (Hacker and Carniel 2001). PAIs are the most well-known GI which has been most exhaustively studied. The various determinants of pathogenic bacteria responsible for the pathogenesis or disease are embedded within these PAIs and also on various mobile elements like extra-chromosomal plasmids, phages, insertion elements, and transposons (Schmidt and Hensel 2004). PAIs are ubiquitous in both Gram-positive and Gram-negative pathogens. Some examples of Gram-negative bacteria harboring PAIs include Salmonella spp., Neisseria spp., Shigella spp., Yersinia spp., Helicobacter pylori, E. coli, Pseudomonas spp., Vibrio cholerae, Porphyromonas gingivalis, and Francisella spp. Examples of Gram-positive pathogenic bacteria having PAIs include Staphylococcus aureus, Streptococcus spp., Listeria spp., Clostridium spp., and Enterococcus spp. The presence of a certain PAI, on the whole, is specific to a pathogenic bacterium or a specific strain of bacteria. A particular bacteria can also have more than one PAI in its genome (Gal-Mor and Finlay 2006). Bacterial species or strains equipped with PAI have an inherent advantage over their non-PAI-bearing counterparts when it comes to pathogenicity. Most virulence determinants for the typical Salmonella enterica are present both in the chromosome and within PAI, termed as Salmonella pathogenicity islands (SPIs). Both SPI-1 and SPI-2 are essential in determining virulence. SPI-1 encoded T3SS proteins (Shea et al. 1996) build up complex machinery for the translocation of various effector proteins from the extracellular S. enterica into the host (eukaryotes) cells (Schmidt and Hensel 2004). SPI-1 also codes for various regulators, some acting as transcriptional activators and others as inhibitors or repressors of SPI-1 genes. The most crucial are HilA, HilE, and LeuO; these gene products and many more work in a complex way to tightly regulate SPI-1 genes (Lou et al. 2019). HPI, a discrete PAI, is naturally found in all the virulent serotypes of Yersinia sp., namely Y. pestis, Y. enterocolitica and Y. pseudotuberculosis but is completely lacking in serotypes with low virulence. Due to the high instability of HPI, it has also been established in various other members of enterobacteria (Carniel et al. 1996). Clostridium difficile, an aerobic bacterium, produces various toxins leading to diarrhea and pseudo-membranous colitis. However, toxin production is restricted only to the virulent (toxigenic) variant but is lacking in the non-virulent type. Comparing both variants revealed that the genes coding for the toxins are integrated into a PAI termed PaLoc (pathogenicity locus) (Braun et al. 1996). The first case of vancomycin resistance in bacterial pathogen was described from a clinical isolate of E. faecalis. Genes related to virulence determinants are incorporated in a 154 kb PAI. It has been found that commensal E. faecalis got transformed into virulent ones by acquiring GI-encoding virulence factors from the virulent E. faecalis (Juhas et al. 2009; Shankar et al. 2002). Members of the genus Pseudomonas have been discovered from multiple habitats, and some species are even considered opportunistic pathogens. Genomic analysis has shown the presence of different types of PAI in strains of P. aeruginosa, termed PAGI (viz., PAGI-1, 2, 3), thereby conferring various adaptive traits (Battle et al. 2009). PAGI-1 (from strain PAO1 and patients with urinary infection) was found to contain genes coding for numerous dehydrogenases (with unknown potentials) and proteins able to sense redox-cycling agents, thus, indicating a protective role of this island against reactive oxygen species (ROS) damage. The latter two, PAGI-2 and PAGI-3, were discovered from the type C strain of P. aeruginosa (isolated from cystic fibrosis patients) and strain SG17M (aquatic strain), respectively. The island PAGI-2 contains numerous genes encoding transporters, regulators, and proteins needed for biosynthetic pathways. The crucial one seems to be the proteins involved in the biogenesis of cytochrome C, thus providing a selective advantage to the bacteria to thrive in an environment with oxidative stress and deprived of iron. Cytochrome C-mediated iron uptake and inactivation of free radicals appeared to be the player behind the scenes. Furthermore, PAGI-3 was found to be a metabolic island without any virulence factors (Schmidt and Hensel 2004). Six more novel islands were identified through a subtractive hybridization approach from P. aeruginosa clinical isolates (Battle et al. 2009).
In conclusion, it may be stated that GIs are still an enigma. The evolution of prokaryotes, particularly that of eubacteria, is primarily impacted by forces like HGT, and the GIs contribute vastly to this direction. With the availability of a deluge of whole genome sequence information due to next-generation sequence techniques, the puzzle of bacterial evolution and speciation has started to unwrap, albeit slowly. With more than 400,000 prokaryotic whole genome sequences in global databases at present, biologists have an enormous amount of data to analyze and decipher newer paradigms in bacterial evolution.
References
Álvarez-Rodríguez I, Arana L, Ugarte-Uribe B, Gómez-Rubio E, Martín-Santamaría S, Garbisu C, Alkorta I (2020) Type IV coupling proteins as potential targets to control the dissemination of antibiotic resistance. Front Mol Biosci 7:201
Antonenka U, Nölting C, Heesemann J, Rakin A (2006) Independent acquisition of site-specific recombination factors by asn tRNA gene-targeting genomic islands. Int J Med Microbiol 296:341–352
Arashida H, Odake H, Sugawara M, Noda R, Kakizaki K, Ohkubo S, Mitsui H, Sato S, Minamisawa K (2022) Evolution of rhizobial symbiosis islands through insertion sequence-mediated deletion and duplication. ISME J 16:112–121
Battle SE, Rello J, Hauser AR (2009) Genomic islands of Pseudomonas aeruginosa. FEMS Microbiol Lett 290:70–78. https://doi.org/10.1111/j.1574-6968.2008.01406.x
Bazin A, Gautreau G, Médigue C, Vallenet D, Calteau A (2020) panRGP: a pangenome-based method to predict genomic islands and explore their diversity. Bioinformatics 36:i651–i658. https://doi.org/10.1093/bioinformatics/btaa792
Bellanger X, Payot S, Leblond-Bourget N, Guédon G (2014) Conjugative and mobilizable genomic islands in bacteria: evolution and diversity. FEMS Microbiol Rev 38:720–760. https://doi.org/10.1111/1574-6976.12058
Bertelli C, Tilley KE, Brinkman FSL (2019) Microbial genomic island discovery, visualization and analysis. Brief Bioinform 20:1685–1698. https://doi.org/10.1093/bib/bby042
Besprozvannaya M, Pivorunas VL, Feldman Z, Burton BM (2013) SpoIIIE protein achieves directional DNA translocation through allosteric regulation of ATPase activity by an accessory domain. J Biol Chem 288:28962–28974
Boyd EF, Almagro-Moreno S, Parent MA (2009) Genomic islands are dynamic, ancient integrative elements in bacterial evolution. Trends Microbiol 17:47–53. https://doi.org/10.1016/j.tim.2008.11.003
Braun V, Hundsberger T, Leukel P, Sauerborn M, Von Eichel-Streiber C (1996) Definition of the single integration site of the pathogenicity locus in Clostridium difficile. Gene 181:29–38. https://doi.org/10.1016/S0378-1119(96)00398-8
BrüSsow H, Canchaya C, Hardt W-D (2004) Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev 68:560–602
Burrus V, Pavlovic G, Decaris B, Guédon G (2002) Conjugative transposons: the tip of the iceberg. Mol Microbiol 46:601–610
Busby B, Kristensen DM, Koonin EV (2013) Contribution of phage-derived genomic islands to the virulence of facultative bacterial pathogens. Environ Microbiol 15:307–312. https://doi.org/10.1111/j.1462-2920.2012.02886.x
Camilli R, Bonnal RJ, Del Grosso M, Iacono M, Corti G, Rizzi E, Marchetti M, Mulas L, Iannelli F, Superti F (2011) Complete genome sequence of a serotype 11A, ST62 Streptococcus pneumoniae invasive isolate. BMC Microbiol 11:1–13
Carniel E, Guilvout I, Prentice M (1996) Characterization of a large chromosomal “high-pathogenicity island” in biotype 1B Yersinia enterocolitica. J Bacteriol 178:6743–6751. https://doi.org/10.1128/jb.178.23.6743-6751.1996
Carraro N, Rivard N, Ceccarelli D, Colwell RR, Burrus V (2016) IncA/C conjugative plasmids mobilize a new family of multidrug resistance islands in clinical vibrio cholerae non-O1/non-O139 isolates from Haiti. MBio 7:e00509–e00516
Chan AP, Sutton G, Depew J, Krishnakumar R, Choi Y, Huang X-Z, Beck E, Harkins DM, Kim M, Lesho EP (2015) A novel method of consensus pan-chromosome assembly and large-scale comparative analysis reveal the highly flexible pan-genome of Acinetobacter baumannii. Genome Biol 16:1–28
Chen J, Ram G, Penadés JR, Brown S, Novick RP (2015) Pathogenicity island-directed transfer of unlinked chromosomal virulence genes. Mol Cell 57:138–149
Chen PE, Cook C, Stewart AC, Nagarajan N, Sommer DD, Pop M, Thomason B, Thomason MPK, Lentz S, Nolan N (2010) Genomic characterization of the Yersinia genus. Genome Biol 11:1–18
Da Silva Filho AC, Raittz RT, Guizelini D, De Pierri CR, Augusto DW, Dos Santos-Weiss ICR, Marchaukoski JN (2018) Comparative analysis of Genomic Island prediction tools. Front Genet 9:619. https://doi.org/10.3389/fgene.2018.00619
Darmon E, Leach DRF (2014) Bacterial genome instability. Microbiol Mol Biol Rev 78:1–39. https://doi.org/10.1128/MMBR.00035-13
Dengler Haunreiter V, Boumasmoud M, Häffner N, Wipfli D, Leimer N, Rachmühl C, Kühnert D, Achermann Y, Zbinden R, Benussi S (2019) In-host evolution of Staphylococcus epidermidis in a pacemaker-associated endocarditis resulting in increased antibiotic tolerance. Nat Commun 10:1–14
Desvaux M, Dalmasso G, Beyrouthy R, Barnich N, Delmas J, Bonnet R (2020) Pathogenicity factors of genomic islands in intestinal and extraintestinal Escherichia coli. Front Microbiol 11:2065
Didelot X, Méric G, Falush D, Darling AE (2012) Impact of homologous and non-homologous recombination in the genomic evolution of Escherichia coli. BMC Genomics 13:1–15
Dobrindt U, Hochhut B, Hentschel U, Hacker J (2004) Genomic islands in pathogenic and environmental microorganisms. Nat Rev Microbiol 2:414–424
Dokland T (2019) Molecular piracy: redirection of bacteriophage capsid assembly by mobile genetic elements. Viruses 11:1003
Espadinha D, Sobral RG, Mendes CI, Méric G, Sheppard SK, Carriço JA, De Lencastre H, Miragaia M (2019) Distinct phenotypic and genomic signatures underlie contrasting pathogenic potential of Staphylococcus epidermidis clonal lineages. Front Microbiol 10:1971
Freel KC, Krueger MC, Farasin J, Brochier-Armanet C, Barbe V, Andrès J, Cholley P-E, Dillies M-A, Jagla B, Koechler S, Leva Y, Magdelenat G, Plewniak F, Proux C, Coppée J-Y, Bertin PN, Heipieper HJ, Arsène-Ploetze F (2015) Adaptation in toxic environments: arsenic Genomic Islands in the bacterial genus Thiomonas. PLoS One 10:e0139011. https://doi.org/10.1371/journal.pone.0139011
Frye J, Karlinsey JE, Felise HR, Marzolf B, Dowidar N, Mcclelland M, Hughes KT (2006) Identification of new flagellar genes of Salmonella enterica serovar Typhimurium. J Bacteriol 188:2233–2243
Gaillard M, Vallaeys T, Vorhölter FJ, Minoia M, Werlen C, Sentchilo V, Pühler A, Van Der Meer JR (2006) The clc element of Pseudomonas sp. strain B13, a genomic island with various catabolic properties. J Bacteriol 188:1999–2013. https://doi.org/10.1128/JB.188.5.1999-2013.2006
Gal-Mor O, Finlay BB (2006) Pathogenicity islands: a molecular toolbox for bacterial virulence. Cell Microbiol 8:1707–1719. https://doi.org/10.1111/j.1462-5822.2006.00794.x
Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vergin KL, Baptista D, Bibbs L, Eads J, Richardson TH, Noordewier M (2005) Genome streamlining in a cosmopolitan oceanic bacterium. Science 309:1242–1245
Guérillot R, Da Cunha V, Sauvage E, Bouchier C, Glaser P (2013) Modular evolution of Tn GBS s, a new family of integrative and conjugative elements associating insertion sequence transposition, plasmid replication, and conjugation for their spreading. J Bacteriol 195:1979–1990
Guglielmini J, Quintais L, Garcillán-Barcia MP, De La Cruz F, Rocha EP (2011) The repertoire of ICE in prokaryotes underscores the unity, diversity, and ubiquity of conjugation. PLoS Genet 7:e1002222
Guo FB, Wei W, Wang XL, Lin H, Ding H, Huang J, Rao N (2012) Co-evolution of genomic islands and their bacterial hosts revealed through phylogenetic analyses of 17 groups of homologous genomic islands. Genet Mol Res 11:3735–3743. https://doi.org/10.4238/2012.October.15.5
Guzman MG, Harris E (2015) Dengue. Lancet 385:453–465. https://doi.org/10.1016/s0140-6736(14)60572-9
Hacker J, Blum-Oehler G, Mühldorfer I, Tschäpe H (1997) Pathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol Microbiol 23:1089–1097
Hacker J, Carniel E (2001) Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep 2:376–381. https://doi.org/10.1093/embo-reports/kve097
Hadjilouka A, Paramithiotis S, Drosinos EH (2018) Genetic analysis of the Listeria Pathogenicity Island 1 of Listeria monocytogenes 1/2a and 4b isolates. Curr Microbiol 75:857–865
Hall RM (2010) Salmonella genomic islands and antibiotic resistance in Salmonella enterica. Future Microbiol 5:1525–1538. https://doi.org/10.2217/fmb.10.122
Hargreaves KR, Thanki AM, Jose BR, Oggioni MR, Clokie MR (2016) Use of single molecule sequencing for comparative genomics of an environmental and a clinical isolate of Clostridium difficile ribotype 078. BMC Genomics 17:1–12
Hickey W, Chen S, Zhao J (2012) The phn island: a new Genomic Island encoding catabolism of Polynuclear aromatic hydrocarbons. Front Microbiol 3:125. https://doi.org/10.3389/fmicb.2012.00125
Jin HM, Jeong H, Moon EJ, Math RK, Lee K, Kim HJ, Jeon CO, Oh TK, Kim JF (2011) Complete genome sequence of the polycyclic aromatic hydrocarbon-degrading bacterium Alteromonas sp. strain SN2. J Bacteriol 193:4292–4293. https://doi.org/10.1128/jb.05252-11
Johnson CM, Grossman AD (2015) Integrative and conjugative elements (ICEs): what they do and how they work. Annu Rev Genet 49:577–601. https://doi.org/10.1146/annurev-genet-112414-055018
Juhas M (2019) Genomic Islands and the evolution of multidrug-resistant bacteria. In: Villa TG, Viñas M (eds) Horizontal gene transfer breaking borders between living kingdoms. Springer, Cham, pp 143–153. https://doi.org/10.1007/978-3-030-21862-1_4
Juhas M, Crook DW, Dimopoulou ID, Lunter G, Harding RM, Ferguson DJP, Hood DW (2007) Novel type IV secretion system involved in propagation of genomic islands. J Bacteriol 189:761–771. https://doi.org/10.1128/JB.01327-06
Juhas M, Van Der Meer JR, Gaillard M, Harding RM, Hood DW, Crook DW (2009) Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev 33:376–393. https://doi.org/10.1111/j.1574-6976.2008.00136.x
Kaneko T, Nakamura Y, Sato S, Minamisawa K, Uchiumi T, Sasamoto S, Watanabe A, Idesawa K, Iriguchi M, Kawashima K (2002) Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110. DNA Res 9:189–197
Klockgether J, Reva O, Tümmler B (2006) Spread of genomic islands between clinical and environmental strains. Paper presented at the Prokaryotic Diversity: Mechanisms and Significance: Published for the Society for General Microbiology, held at The University of Warwick April 2006, 01/01
Koskiniemi S, Sun S, Berg OG, Andersson DI (2012) Selection-driven gene loss in bacteria. PLoS Genet 8:e1002787
Lacour S, Gaillard M, Meer J (2006) Genomic islands and evolution of catabolic pathways. In: Logan NA, Lappin-Scott HM, Oyston PCF (eds) Prokaryotic diversity: mechanisms and significance. Cambridge University Press, Cambridge, pp 255–273. https://doi.org/10.1017/CBO9780511754913.016
Langille MG, Hsiao WW, Brinkman FS (2010) Detecting genomic islands using bioinformatics approaches. Nat Rev Microbiol 8:373–382
Lawrence JG (2001) Catalyzing bacterial speciation: correlating lateral transfer with genetic headroom. Syst Biol 50:479–496. https://doi.org/10.1080/10635150120286
Lawrence JG, Roth JR (1996) Selfish operons: horizontal transfer may drive the evolution of gene clusters. Genetics 143:1843–1860
Leal NC, Campos TL, Rezende AM, Docena C, Mendes-Marques CL, De Sá Cavalcanti FL, Wallau GL, Rocha IV, Cavalcanti CL, Veras DL (2020) Comparative genomics of Acinetobacter baumannii clinical strains from Brazil reveals polyclonal dissemination and selective exchange of mobile genetic elements associated with resistance genes. Front Microbiol 11:1176
Lerminiaux NA, Mackenzie KD, Cameron AD (2020) Salmonella pathogenicity island 1 (SPI-1): the evolution and stabilization of a core genomic type three secretion system. Microorganisms 8:576
Letzel A-C, Li J, Amos GCA, Millán-Aguiñaga N, Ginigini J, Abdelmohsen UR, Gaudêncio SP, Ziemert N, Moore BS, Jensen PR (2017) Genomic insights into specialized metabolism in the marine actinomycete Salinispora. Environ Microbiol 19:3660–3673. https://doi.org/10.1111/1462-2920.13867
Li W, Wang A (2021) Genomic islands mediate environmental adaptation and the spread of antibiotic resistance in multiresistant Enterococci - evidence from genomic sequences. BMC Microbiol 21:55. https://doi.org/10.1186/s12866-021-02114-4
Lima WC, Paquola AC, Varani AM, Van Sluys M-A, Menck CF (2008) Laterally transferred genomic islands in Xanthomonadales related to pathogenicity and primary metabolism. FEMS Microbiol Lett 281:87–97
Lindsay JA, Ruzin A, Ross HF, Kurepina N, Novick RP (1998) The gene for toxic shock toxin is carried by a family of mobile pathogenicity islands in Staphylococcus aureus. Mol Microbiol 29:527–543
Liu F, Zhu Y, Yi Y, Lu N, Zhu B, Hu Y (2014) Comparative genomic analysis of Acinetobacter baumannii clinical isolates reveals extensive genomic variation and diverse antibiotic resistance determinants. BMC Genomics 15:1–14
Lou L, Zhang P, Piao R, Wang Y (2019) Salmonella pathogenicity island 1 (SPI-1) and its complex regulatory network. Front Cell Infect Microbiol 9:270. https://doi.org/10.3389/fcimb.2019.00270
Lovell HC, Mansfield JW, Godfrey SA, Jackson RW, Hancock JT, Arnold DL (2009) Bacterial evolution by genomic island transfer occurs via DNA transformation in planta. Curr Biol 19:1586–1590. https://doi.org/10.1016/j.cub.2009.08.018
Lu B, Leong HW (2016) Computational methods for predicting genomic islands in microbial genomes. Comput Struct Biotechnol J 14:200–206
Makarova KS, Wolf YI, Snir S, Koonin EV (2011) Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J Bacteriol 193:6039–6056
Martínez JL (2013) Bacterial pathogens: from natural ecosystems to human hosts. Environ Microbiol 15:325–333. https://doi.org/10.1111/j.1462-2920.2012.02837.x
Mates AK, Sayed AK, Foster JW (2007) Products of the Escherichia coli acid fitness island attenuate metabolite stress at extremely low pH and mediate a cell density-dependent acid resistance. J Bacteriol 189:2759–2768
Mathee K, Narasimhan G, Valdes C, Qiu X, Matewish JM, Koehrsen M, Rokas A, Yandava CN, Engels R, Zeng E (2008) Dynamics of Pseudomonas aeruginosa genome evolution. Proc Natl Acad Sci U S A 105:3100–3105
Melnyk RA, Engelbrektson A, Clark IC, Carlson HK, Byrne-Bailey K, Coates JD (2011) Identification of a perchlorate reduction genomic island with novel regulatory and metabolic genes. Appl Environ Microbiol 77:7401–7404. https://doi.org/10.1128/AEM.05758-11
Melnyk RA, Hossain SS, Haney CH (2019) Convergent gain and loss of genomic islands drive lifestyle changes in plant-associated Pseudomonas. ISME J 13:1575–1588. https://doi.org/10.1038/s41396-019-0372-5
Milkman R (2004) Horizontal transfer, genomic diversity, and genomic differentiation. In: Microbial evolution. Wiley, Hoboken, pp 295–318
Mingoia M, Morici E, Marini E, Brenciani A, Giovanetti E, Varaldo PE (2016) Macrolide resistance gene erm (TR) and erm (TR)-carrying genetic elements in Streptococcus agalactiae: characterization of ICE Sag TR7, a new composite element containing IME Sp 2907. J Antimicrob Chemother 71:593–600
Morris JJ, Lenski RE, Zinser ER (2012) The Black Queen Hypothesis: evolution of dependencies through adaptive gene loss. MBio 3:e00036–e00012
Neilands JB (1995) Siderophores: structure and function of microbial iron transport compounds. J Biol Chem 270:26723–26726. https://doi.org/10.1074/jbc.270.45.26723
Nishi A, Tominaga K, Furukawa K (2000) A 90-kilobase conjugative chromosomal element coding for biphenyl and salicylate catabolism in Pseudomonas putida KF715. J Bacteriol 182:1949–1955
Novick RP, Christie GE, Penadés JR (2010) The phage-related chromosomal islands of Gram-positive bacteria. Nat Rev Microbiol 8:541–551
Novick RP, Ram G (2016) The floating (pathogenicity) island: a genomic dessert. Trends Genet 32:114–126. https://doi.org/10.1016/j.tig.2015.11.005
Ochman H, Lawrence JG, Groisman EA (2000) Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304. https://doi.org/10.1038/35012500
Ogier J-C, Calteau A, Forst S, Goodrich-Blair H, Roche D, Rouy Z, Suen G, Zumbihl R, Givaudan A, Tailliez P (2010) Units of plasticity in bacterial genomes: new insight from the comparative genomics of two bacteria interacting with invertebrates, Photorhabdus and Xenorhabdus. BMC Genomics 11:1–21
Osborn AM, Böltner D (2002) When phage, plasmids, and transposons collide: genomic islands, and conjugative- and mobilizable-transposons as a mosaic continuum. Plasmid 48(3):202–212. https://doi.org/10.1016/s0147-619x(02)00117-8
Otto M (2009) Staphylococcus epidermidis—the ‘accidental’ pathogen. Nat Rev Microbiol 7:555–567
Patel S (2016) Drivers of bacterial genomes plasticity and roles they play in pathogen virulence, persistence and drug resistance. Infect Genet Evol 45:151–164
Pavlovic G, Burrus V, Gintz B, Decaris B, Guedon G (2004) Evolution of genomic islands by deletion and tandem accretion by site-specific recombination: ICESt1-related elements from Streptococcus thermophilus. Microbiology 150:759–774
Penn K, Jenkins C, Nett M, Udwary DW, Gontang EA, Mcglinchey RP, Foster B, Lapidus A, Podell S, Allen EE, Moore BS, Jensen PR (2009) Genomic islands link secondary metabolism to functional adaptation in marine Actinobacteria. ISME J 3:1193–1203. https://doi.org/10.1038/ismej.2009.58
Peters JE, Fricker AD, Kapili BJ, Petassi MT (2014) Heteromeric transposase elements: generators of genomic islands across diverse bacteria. Mol Microbiol 93:1084–1092
Puymège A, Bertin S, Chuzeville S, Guédon G, Payot S (2013) Conjugative transfer and cis-mobilization of a genomic island by an integrative and conjugative element of Streptococcus agalactiae. J Bacteriol 195:1142–1151
Rainey FA, Oren A (2011) 1 - Taxonomy of prokaryotes – introduction. In: Rainey F, Oren A (eds) Methods in microbiology, vol 38. Academic Press, Cambridge, pp 1–5. https://doi.org/10.1016/B978-0-12-387730-7.00001-2
Rivard N, Colwell RR, Burrus V (2020) Antibiotic resistance in vibrio cholerae: mechanistic insights from IncC plasmid-mediated dissemination of a novel family of genomic islands inserted at trmE. mSphere 5:e00748–e00720
Rodriguez-Valera F, Martin-Cuadrado AB, López-Pérez M (2016) Flexible genomic islands as drivers of genome evolution. Curr Opin Microbiol 31:154–160. https://doi.org/10.1016/j.mib.2016.03.014
Roy Chowdhury P, Scott M, Worden P, Huntington P, Hudson B, Karagiannis T, Charles IG, Djordjevic SP (2016) Genomic islands 1 and 2 play key roles in the evolution of extensively drug-resistant ST235 isolates of Pseudomonas aeruginosa. Open Biol 6:150175
Sansevere EA, Luo X, Park JY, Yoon S, Seo KS, Robinson DA (2017) Transposase-mediated excision, conjugative transfer, and diversity of ICE 6013 elements in Staphylococcus aureus. J Bacteriol 199:e00629–e00616
Scherer CA, Miller SI (2001) Chapter 7 - Molecular pathogenesis of salmonellae. In: Groisman EA (ed) Principles of bacterial pathogenesis. Academic Press, San Diego, pp 265–333. https://doi.org/10.1016/B978-012304220-0/50008-X
Schmidt H, Hensel M (2004) Pathogenicity islands in bacterial pathogenesis. Clin Microbiol Rev 17:14–56
Shankar N, Baghdayan AS, Gilmore MS (2002) Modulation of virulence within a pathogenicity island in vancomycin-resistant Enterococcus faecalis. Nature 417:746–750. https://doi.org/10.1038/nature00802
Shapiro BJ (2016) How clonal are bacteria over time? Curr Opin Microbiol 31:116–123. https://doi.org/10.1016/j.mib.2016.03.013
Shea JE, Hensel M, Gleeson C, Holden DW (1996) Identification of a virulence locus encoding a second type III secretion system in Salmonella typhimurium. Proc Natl Acad Sci U S A 93:2593–2597. https://doi.org/10.1073/pnas.93.6.2593
Siguier P, Gourbeyre E, Chandler M (2014) Bacterial insertion sequences: their genomic impact and diversity. FEMS Microbiol Rev 38:865–891
Smillie C, Garcillán-Barcia MP, Francia MV, Rocha EP, De La Cruz F (2010) Mobility of plasmids. Microbiol Mol Biol Rev 74:434–452
Soucy SM, Huang J, Gogarten JP (2015) Horizontal gene transfer: building the web of life. Nat Rev Genet 16:472–482
Sullivan JT, Brown SD, Ronson CW (2013) The NifA-RpoN regulon of Mesorhizobium loti strain R7A and its symbiotic activation by a novel LacI/GalR-family regulator. PLoS One 8:e53762
Te Poele EM, Bolhuis H, Dijkhuizen L (2008) Actinomycete integrative and conjugative elements. Antonie Van Leeuwenhoek 94:127–143
Ternan NG, Jain S, Srivastava M, Mcmullan G (2012) Comparative transcriptional analysis of clinically relevant heat stress response in Clostridium difficile strain 630. PLoS One 7:e42410
Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS (2005) Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A 102:13950–13955
Thode SK, Rojek E, Kozlowski M, Ahmad R, Haugen P (2018) Distribution of siderophore gene systems on a Vibrionaceae phylogeny: database searches, phylogenetic analyses and evolutionary perspectives. PLoS One 13:e0191860. https://doi.org/10.1371/journal.pone.0191860
Toussaint A, Merlin C, Monchy S, Benotmane MA, Leplae R, Mergeay M, Springael D (2003) The biphenyl- and 4-chlorobiphenyl-catabolic transposon Tn4371, a member of a new family of genomic islands related to IncP and Ti plasmids. Appl Environ Microbiol 69:4837–4845. https://doi.org/10.1128/AEM.69.8.4837-4845.2003
Van Der Meer JR, Sentchilo V (2003) Genomic islands and the evolution of catabolic pathways in bacteria. Curr Opin Biotechnol 14:248–254. https://doi.org/10.1016/s0958-1669(03)00058-2
Van Elsas JD, Semenov AV, Costa R, Trevors JT (2011) Survival of Escherichia coli in the environment: fundamental and public health aspects. ISME J 5:173–183
Van Houdt R, Monsieurs P, Mijnendonckx K, Provoost A, Janssen A, Mergeay M, Leys N (2012) Variation in genomic islands contribute to genome plasticity in Cupriavidus metallidurans. BMC Genomics 13:111. https://doi.org/10.1186/1471-2164-13-111
Vásquez-Ponce F, Higuera-Llantén S, Pavlov MS, Marshall SH, Olivares-Pacheco J (2018) Phylogenetic MLSA and phenotypic analysis identification of three probable novel Pseudomonas species isolated on King George Island, South Shetland, Antarctica. Braz J Microbiol 49:695–702. https://doi.org/10.1016/j.bjm.2018.02.005
Vo ATT, Van Duijkeren E, Gaastra W, Fluit AC (2010) Antimicrobial resistance, class 1 integrons, and genomic island 1 in salmonella isolates from Vietnam. PLoS One 5:e9440. https://doi.org/10.1371/journal.pone.0009440
Wang H, Smith MC, Mullany P (2006) The conjugative transposon Tn 5397 has a strong preference for integration into its Clostridium difficile target site. J Bacteriol 188:4871–4878
Welch RA, Burland V, Plunkett G, Redford P, Roesch P, Rasko D, Buckles EL, Liou S-R, Boutin A, Hackett J, Stroud D, Mayhew GF, Rose DJ, Zhou S, Schwartz DC, Perna NT, Mobley HLT, Donnenberg MS, Blattner FR (2002) Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci 99:17020–17024. https://doi.org/10.1073/pnas.252529799
Williams KP (2002) Integration sites for genetic elements in prokaryotic tRNA and tmRNA genes: sublocation preference of integrase subfamilies. Nucleic Acids Res 30:866–875
Wyrsch ER, Chowdhury PR, Jarocki VM, Brandis KJ, Djordjevic SP (2020) Duplication and diversification of a unique chromosomal virulence island hosting the subtilase cytotoxin in Escherichia coli ST58. Microb Genom 6:e000387
Xiang L, Li G, Wen L, Su C, Liu Y, Tang H, Dai J (2021) Biodegradation of aromatic pollutants meets synthetic biology. Synth Syst Biotechnol 6:153–162
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Pal, A., Roy, V., Dutta, P., Adhikary, S., Saha, B.K., Saha, J. (2023). Genomic Islands in Bacterial Genome Evolution and Speciation. In: Mani, I., Singh, V., Alzahrani, K.J., Chu, DT. (eds) Microbial Genomic Islands in Adaptation and Pathogenicity. Springer, Singapore. https://doi.org/10.1007/978-981-19-9342-8_5
Download citation
DOI: https://doi.org/10.1007/978-981-19-9342-8_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-9341-1
Online ISBN: 978-981-19-9342-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)