
Abstract
Despite successive advancement of genome editing technology with zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), the recent breakthrough in the field has been related to clustered regularly interspaced short palindromic repeats/associated proteins (CRISPR/Cas). The high efficiency and convenience of CRIPSR/Cas systems dramatically accelerate pre- and clinical experimentations of dyslipidemia and atherosclerosis. In this chapter, we review the latest state of genome editing in translational research of dyslipidemia and atherosclerosis. We highlight recent progress in therapeutic development for familial dyslipidemia by genome editing. We point to the challenges in maximizing efficacy and minimizing safety issues related to the once-and-done therapy focusing on CRISPR/Cas systems. We give an outlook on the potential gene targets prioritized by large-scale genetic studies of cardiovascular diseases and genome editing in precision medicine of dyslipidemia and atherosclerosis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Dyslipidemia
- Atherosclerosis
- Genome editing
- CRISPR/Cas
- Familial hypercholesterolemia
- Genome-wide association study
- Precision medicine
1 Dyslipidemia and Atherosclerosis
Atherosclerosis represents the major cause of coronary artery disease and thereby mortality worldwide [1]. The complex etiology of atherosclerosis is initiated by dysfunctional endothelial cells lining the arteries that are no longer capable of appropriately regulating vascular tone and permeability for molecules and cells [2]. Progressive infiltration of lipoprotein particles carrying cholesterol into the vessel wall triggers an inflammatory response mediated by cholesterol-loaded macrophages. Proliferation of smooth muscle cells causes vascular remodeling and ultimately leads to narrowing of the vessel and obstruction of blood flow. Dyslipidemia, a common and strong risk factor for atherosclerosis, describes elevated plasma levels of low-density lipoprotein cholesterol (LDL-C), lipoprotein(a) (Lp(a)), and/or triglyceride-rich lipoproteins (TRLs, VLDL, and IDL) [3] and/or decreased levels of high-density lipoprotein cholesterol (HDL-C) [4]. In addition to lifestyle and environmental influences, dyslipidemia is largely determined by genetic factors. Its extreme forms are manifested as familial dyslipidemias caused by gene mutations, including hypercholesterolemia (e.g., LDLR, APOB, PCSK9, LPA, and ANGPTL3) [5,6,7], hypertriglyceridemia (e.g., LPL, APOC3, APOC2, APOA5, ANGTPL4, GPIHBP1, and LMF1), dysbetalipoproteinemia (e.g., APOE), analphalipoproteinemia (e.g., ABCA1), LCAT deficiency [8], and combined hyperlipidemia (e.g., USF1) [9, 10]. Familial hypercholesterolemia (FH), the most common form of the overall rare dyslipidemias, occurs in 1 out of 200,000–250,000 people heterozygously and in 1 out of 160,000–320,000 people homozygously [11, 12]. To fulfill the pressing need of precision medicine, efforts have been increasingly committed to developing targeted therapies for dyslipidemia and atherosclerosis.
2 Current Therapies of Dyslipidemia and Atherosclerosis
2.1 From Traditional Pharmacology to Targeted Therapy
Pharmacological treatment of dyslipidemia and atherosclerosis predominantly focuses on cholesterol lowering [4]. For many years, statin (inhibiting cholesterol synthesis), ezetimibe (suppressing intestine uptake of cholesterol), and bile acid sequestrants have been the major treatments of the conditions [13, 14]. However, a significant proportion of patients do not achieve guideline-recommended cholesterol levels with these medications. Recently approved bempedoic acid further reduces LDL by about ~20% [15]. PCSK9 monoclonal antibodies [16], another new drug type, enable effective LDL reduction in addition to statin therapy but with high costs, hampering the general use. While small molecules targeting PCSK9 are under investigation to bring down the cost, drugs lowering other causal lipids and their inflammatory responses are on the way to treat the residual cardiovascular risk [17]. Revolutionary discoveries of human genetics in the past decade have been a nutritious ground for novel drug developments [18,19,20]. Genetic studies of atherosclerosis, coronary artery disease [1], and myocardial infarction (MI) not only nominated but also validated causal genes, pathways, and risk factors for the conditions. For instance, genetic studies supported Lp(a) and TGs as causal risk factors for atherosclerosis, which led to intensive investigations of related genes, such as LPA, APOC3, ANGPTL3, and ANGPTL4 [3, 21,22,23]. Based on a better understanding of the affected mechanisms, these genes evolved as novel targets for biological drugs, monoclonal antibodies, and nucleic acid-based therapies [24].
2.2 Nucleic Acid-Based Therapy
Nucleic acid-based therapies were initially designed as replacement for dysfunctional genes by delivery of the correct coding sequence [25]. Recently, this concept has been expanded to include gene silencing by antisense oligonucleotides, or short interfering RNAs (siRNA), transcriptional modulation by microRNAs, and long noncoding RNAs (lncRNA), as well as modification of epigenetics and genome editing [25,26,27]. For instance, gene supplementation of LDLR is currently investigated in a phase 1/phase 2a first-in-man trial (NCT02651675) for homozygous FH due to function loss of the gene [28]. AON (antisense oligonucleotide)- and/or siRNA-based therapies targeting several dyslipidemia genes have been intensively tested in large-scale clinical trials for treating atherosclerotic CAD, such as APOA, PCSK9, APOC3, and ANGPTL3 [29,30,31,32,33,34]. LncRNA BM450697 was reported to regulate LDLR via epigenetic-dependent mechanism, and siRNAs targeting the lncRNA enhanced hepatic cholesterol uptake [35]. These novel therapeutic strategies not only expand the druggable genome that previously was largely limited to enzymes, membrane proteins, and circulatory factors but also potentially have advantages of specificity, efficacy, and safety. However, limited half-lives of nucleic acids, requirement of frequent injection, and medication compliance are general limitations. The limitations are not applicable for gene editing-based therapies that could introduce permanent therapeutic changes to specific gene targets. It is conceivable that in the future, a single administration of such drugs mediates durable cure of dyslipidemias and atherosclerosis.
3 Genome Editing
3.1 Evolution of Genome Editing Technology
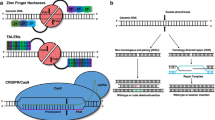
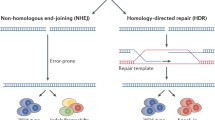
Genome editing generally refers to the specific modification of nucleotide sequences (mainly DNA) by enzymic activities (e.g., nucleases and nickase) [36]. In a broader sense, it also includes RNA editing. Nucleases usually cut a nucleotide sequence and create damage (typically a double-strand break (DSB)), whereas nickases introduce single-strand breaks (SSB) [37]. Both DSB and SSB in turn trigger natural genetic repair mechanisms, such as nonhomologous end joining (NHEJ) and homology-directed repair (HDR) enabled by a homologous-armed template [38]. The cellular repairing machinery is hijacked to install precise nucleotide manipulations.
In the late 1970s, the first generation of gene editing tools was engineered based on hybrid proteins including zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) [39,40,41]. Both types of nucleases rely on a recombinant recognition domain to bind the target DNA sequence. Target-specific ZFN and TALEN engineering involves in tedious designing and screening of the optimal recombinant protein with high binding accuracy and affinity. Of note, TALENs have reached clinical experimentation to generate universal allogeneic CAR T-cells for B-cell lymphoma [42, 43].
Ever since 2012, genome editing has become easier, faster, and more economic, due to the discovery and engineering of RNA-guided gene rewriting technology—the CRISPR/Cas system [44]. The new system holds promise to cure genetic diseases through (1) inactivating detrimental or aberrant gene expression, (2) amending disease-causing or associated mutations, or (3) targeted insertion of therapeutical DNA (Fig. 2a–c). CRISPR/Cas harnesses the marriage of two independent components, the small guide RNA (sgRNA) and a Cas protein. The allocation of two functions of the traditional recombinant nucleases into the nucleotide sequence recognition by a sgRNA and the enzymic cutting by a Cas nuclease dramatically simplified the design and construction of the editing tools. The classic CRISPR/Cas9 system creates DSB and relies on NHEJ for gene knockout and HDR for an error-free DNA retyping. For newer types of CRISPR tools, the nuclease activity of a Cas protein was either inactivated to only bring transcriptional activators or suppressors to the targeted genomic site [46,47,48] or transformed into nickase tandem to other enzymes, such as deaminases in base editors (BEs) [49]. By directly triggering chemical reaction (deamination) on DNA and converting C to T (CBE) or A to G (ABE), BEs allow gene knockout without DSB and individual nucleotide(s) rewriting independent of a template, which hold promise for therapeutic gene editing with minimum off-target effects. In fact, point mutations represent the most common genetic variations associated with human diseases [50]. Recently, more types of Cas proteins, such as Nme2Cas9 and Cas13, have been discovered, extending the coverage of editable genome and enabling RNA manipulation [51, 52].
3.2 In Vivo Delivery of Genome Editing Systems
Intracellular delivery of gene editing tools has been the most challenging step in vivo. Adeno-associated virus (AAV)-, adenovirus-, and lentivirus-mediated delivery systems have been tested for CRISPR-based gene therapy [53, 54]. Due to lower immunogenicity, non-integrative and high efficiency, AAVs are widely used in CRISPR-based ex vivo and in vivo biological research and therapeutic development. However, the packaging limit of AAV (~4.7 kb) often hampers its applications. Thus, instead of spCas9 (~4.1 kb), saCas9 (~3.2 kb) is usually employed for AAV-based gene editing, which allows all-in-one CRISPR therapy carrying both saCAS9 and sgRNA sequences on the same vector [55]. Generally, immunogenicity and potential transgene integration are of high concern when viral vectors were chosen for therapeutic development. Therefore, efforts have been exerted in seeking nonviral carriers for CRISPR-mediated gene [53].
Another promising in vivo delivery method involves the encapsulation of CRISPR/Cas into nanocarriers, in the forms of RNA-protein complex (RNP) or coding nucleic acids (DNA plasmids or mRNAs). In particular, delivery by lipid nanoparticles (LNP) achieves efficient targeting of specific tissues and protects the loaded proteins and nucleic acids [56,57,58]. Advanced LNP technologies for gene editing include self-assembled DNA nanoclews [59], cationic LNP and lipoplexes [60,61,62], gold nanoparticles [63,64,65], and zeolitic imidazole frameworks [66]. Most approaches harness electrostatic interactions between guest and host. Despite the promise, delivery of RNPs has been the most challenging due to the strong negative charge of sgRNA, the large size of Cas proteins, and the sensitivity of RNPs to denaturation and degradation during formulation and delivery. To date, the development of stable and organ-specific nanoparticles for delivery of CRISPR toolkits remains elusive.
4 Genome Editing in Dyslipidemia and Atherosclerosis
4.1 Genome Editing: A Driving Force for Dyslipidemia and Atherosclerosis Research
Ever since the applicable invention of CRISPR/Cas9 system in 2012 [67], it has been increasingly used in cardiovascular research (Fig. 1) and fosters delicacy of cellular and animal models for dyslipidemia and atherosclerosis research. Patient-induced pluripotent stem cell (hiPSC) line of carrier of heterozygous p.C310R (c.928 T > C) mutation in LPL, encoding lipoprotein lipase, has been reprogramed to model familial hypertriglyceridemia (FHTG). In parallel, researchers generated a mutation-corrected isogenic iPSC line (AHQUi001-A-1) using CRISPR/Cas9 technology [68]. The isogenic pair could differentiate into relevant cell types, such as adipocyte and endothelial cells, and test therapeutic modifications for the patient. Cell banks, such as WiCell, provide as precious resources of isogenic hiPSCs for dyslipidemia and coronary artery disease. Given that the CRISPR/Cas system relies on open chromatin to screen the matched gene code, the efficiency of the gene editing heavily depends on the proliferation and transcription activity of cells. Hyperproliferative cells, such as stem cell and cancer cell, are relatively easy to target with high efficiency. Therefore, gene editing in hiPSC has been valuable in cardiovascular research. It comes with high efficiency for differentiation of many disease relevant cell types that are challenging to access or target, such as hepatocytes, adipocytes, immune cells, endothelium cells, and vascular smooth muscle cells [69].

PubMed search of “gene editing in cardiovascular diseases” for the last 15 years. Numbers of studies by year as indicated, figure adapted from PubMed statistics
CRISPR/Cas systems have substantially reduced the time and cost to generate animal models of germline gene knockouts or somatic targeting in vivo. The high efficiency of CRIPSR/Cas allows genetic modification of multiple genes at any time points of an animal’s lifespan. The diverse CRISPR tools allow the flexibility in duration of editing, conditional alleles, tissue-specific targeting, and directions of modulation. Yang and Jaenisch et al. have established a protocol to create gene-modified mice by piezo-driven injection of Cas9 mRNA and sgRNA into zygotes. The authors showed that, beginning with target design, the time frame for generation of transgenic mice can be as short as four weeks [70]. Currently, this method and similar others are commonly used for cardiovascular research. For example, Yu and Cowan et al. generated G protein-coupled receptor 146 (GPR146) deficiency mice and showed that the deficiency protected against hypercholesterolemia and atherosclerosis [71]. To establish atherosclerosis mouse models using CRISPR in adult mice, Jarrett et al. performed somatic knockout of Ldlr via AAV8 medicated delivery of all-in-one AAV-CRISPR. The approach robustly disrupted Ldlr and resulted in severe hypercholesterolemia and atherosclerotic lesions in the mouse aorta [72]. Although the cholesterol increase induced by the somatic Ldlr knockdown was not as high as by germline Ldlr knockout, it might better model the chronic condition of atherosclerosis which usually develops at higher age [72]. Similar approaches were adopted to generate atherosclerosis animal models in rabbit, pig, and hamster by knocking out Ldlr, Apoe, or Lcat (lecithin-cholesterol acyltransferase) [73,74,75,76]. The success of the transgene models, on the other hand, suggested the effectiveness of in vivo CRISPR/Cas system in testing novel gene functions in dyslipidemia and atherosclerosis. Indeed, the novel role of CCC(COMMD-CCDC22-CCDC93) complex in hepatic cholesterol metabolism was explored and confirmed by somatic CRISPR/Cas targeting of Commd and Ccdc22 in mice [77, 78].
However, as for point mutation correction, the editing efficiency of CRISPR/Cas remains low. Omer et al. attempted to correct the loss-of-function mutation E208X in Ldlr gene of the mouse liver by AAV-CRISPR/Cas system. The HDR-mediated correction only achieved 6.7% efficiency but resulting in, to some extent, lower serum lipid levels and decreased lesion area [79]. The coming waves of newer types of CRISPR technologies, such as base editor and prime editor, hold potential to improve in this regard.
4.2 Preclinical Investigation of Genome Editing for Dyslipidemia and Atherosclerosis
Gene editing in adult humans, that is, somatic editing, holds the promise to permanently modify one’s risk of dyslipidemia and atherosclerosis. In light of the compliance issue with statins, high costs of PCSK9 monoclonal antibodies, and discomfort of lifetime injection of RNA therapies, such once-and-done strategy is attractive. A poll about the acceptance of the gene editing therapy indicated the support from the majority of the participants [80, 81]. Several gene editing strategies against dyslipidemia and atherosclerosis have been intensively investigated in preclinical settings to inactivate pathogenic gene expression, correct disease-causing mutations, mimic atheroprotective effects of natural genetic variations, or insert beneficial transgenes.
The most intensive testing focused on PCSK9, given the well-studied biology and rare side effect as a therapeutic target. Gene editing-based therapies allow permanent modification of the culprit and therefore are advantageous as a one shot and one cure for dyslipidemia, especially for FH. Disruption of Pcsk9 in mice by CRIPSR/Cas9 has been evaluated by AAV- and nanocarrier-based delivery of spCas9 or saCas9 systems [55, 82,83,84,85,86,87] (Fig. 2d). All led to significant reduction of circulating Pcsk9, plasma total cholesterol (TC), and LDL-C levels. The therapeutic target was further assessed by inactivating the gene using base editing, which result in comparable atheroprotective outcomes [88,89,90,91]. A head-to-head comparison of Pcsk9 gene and base editing in a humanized mouse model showed that the latter introduced no chromosomal translocations, fewer indels, and less new forms of peptides, indicating that it might be a safe strategy for clinical applications [89]. Other gene editing approaches to lower LDL-C level are pursued, such as CRISPR-/Cas9-based targeting of Apob in Ldlr−/− mice [55, 72] and replacement of FH mutation of LDLR E208X in somatic cells of transgenic mice [55, 79], both of which reduced plasma TC level and atherosclerosis development in mice.

Therapeutic genome editing of PCSK9 by CRISPR/Cas and base editing. (a, b) General overview of DNA editing by CRISPR/Cas9, cytosine base editor (CBE), and adenine base editor (ABE). (a) Editing mechanism of CRISPR/Cas9. Cas9 nucleases create double-strand break (DSB) within the guide RNA (gRNA) pairing sequence, usually at 3–4bp ahead of 5′-protospacer adjacent motif [45]. DSB will be fixed through nonhomologous end joining (NHEJ) to create gene knockout or homology directed repair (HDR) to install a genotype or transgene of interest. (b) Mechanism of CBE. Cas9 nickase (Cas9n) nicks the top strand, while the cytidine deaminase domain of CBE convers C to U. Uracil glycosylase inhibitor (UGI, an optional component) protects the U intermediate from excision by uracil DNA glycosylase to boost efficiency of base pair editing assisted by nature DNA repair, which ultimately converts a C•G pair to T•A base pair. (c) Mechanism of ABE. After DNA nicking, adenosine deaminase domain converts A to inosine intermediate, which will be substituted by G in the subsequent DNA repairing process. ABE replaces A•T to G•C pair. gRNA, guide RNA. (d) Overview of strategies investigated to decrease Pcsk9 or PCSK9 in vivo. The editing tool is shown above the arrow line and the delivery approach is described below the line. WT, wild type; LNP, lipid nanoparticle; Adenine BE-Split, ABE separated to two domains (split-ABE-Rma573 and split-ABE-Rma674) for virus package, KRAB Kruppel-associated box (transcriptional repressor)
Given that existing lipid-lowering therapies are centered on optimizing cholesterol levels, drugs to reduce levels of non-LDL lipids including TGs and Lp (a) are of an urgent need, particularly for the dyslipidemia patients suffering from obesity, diabetes, or insulin resistance, whose primary risk of atherosclerosis is often related to elevated TGs and other forms of lipids. The attempts beyond LDL-C lowering by gene editing focus on APOC3 and ANGPTL3 for reducing TG levels and LPA for decreasing Lp(a).
As naturally occurring loss-of-function (LoF) mutations in ApoC3 and ANGPTL3 have found to be atheroprotective [92, 93], CRISPR-/Cas9-mediated inactivation of the two genes was tested to treat hyperlipidemia and atherosclerosis. APOC3, a secretory glycoprotein primarily produced by the liver, inhibits LPL- and hepatic lipase-mediated hydrolysis process of triglycerides in circulation and therefore increases TRL levels. In a human-like animal model (hamster), inactivation of ApoC3 by CRISPR-Cas9 significantly decreased triglyceride level with no statistical differences in total cholesterol and HDL-C levels, phenocopying APOC3-deficient humans [94]. ApoC3 knockout hamsters also had less atherosclerotic lesions in both thoracic and abdominal arteries, suggesting clinical relevance of APOC3 targeting for the treatment of hypertriglyceridemia and atherosclerosis [95]. In the case of ANGPTL3, an inhibitor of LPL and endothelial lipase, base editing was employed to introduce LoF mutations at Gln-135 site of Angptl3 in the liver of Ldlr−/− mice. This resulted in a median editing rate of 35% in the liver as well as substantially reduced triglycerides (56%) and cholesterol (51%) [96], suggesting a method to treat combined hyperlipidemia and atherosclerosis [96, 97].
LPA, expressed in the liver, encodes for apo(a) that could covalently bound to APOB100, an essential component for both LDL and Lp(a) [98]. Genetic variation of LPA was estimated to explain 91% of the variation in Lp(a) levels [22, 99]. Serum Lp(a) level could not be modulated by dietary and lifestyle factors, further necessitating the therapeutic intervention [100]. Lp(a) was also shown as a major carrier of oxidized phospholipids and to induce plaque progression [101,102,103]. An earlier pioneer study of RNA editing was explored to transform apoB100 mRNA into its truncated form apoB48 by a recombinant adenovirus encoding cytidine deaminase complex (apoBEC-1) to reduce both atherogenic lipoproteins in humanized apoB/apo(a) transgenic mice. This resulted in hepatic editing of human APOB mRNA and reduced plasma levels of human APOB100 and Lp(a). Similar result was observed when the apoB mRNA was edited accordingly in rabbit. These studies demonstrate mRNA editing by apoBEC-1 as a novel approach for lowering plasma concentrations of the atherogenic lipoproteins LDL and Lp(a) [104]. Furthermore, ongoing preclinical studies are investigating the use of base editing to reduce Lp(a) level by inactivating LPA gene.
These proof-of-concept studies demonstrated the feasibility of in vivo gene editing in reducing phenotypes of dyslipidemia and atherosclerosis and triggered industrial interests in developing these further in clinical experimentations. Currently, base editing of LPA, PCSK9, and ANGPTL3 are under pharmaceutic development, and ABE-PCSK9 has entered the preclinical toxicology studies. So far, all the tested gene targets address familial dyslipidemia. CRISPR-based therapies could provide personalized treatment for the diseases, which currently cannot be cured. However, whether it could be cost effective to treat nonfamilial forms of dyslipidemia and atherosclerosis should be further investigated. Concerns about its advantage over traditional medications and long-term on- and off-target effects need to be addressed before clinical use. Pilot applications might be firstly available for individuals at high risk for myocardial infarction.
4.3 Further Target Discovery for Dyslipidemia and Atherosclerosis
4.3.1 Gene and Variant Targets Inspired by Human Knockout
Phenotypically healthy humans carrying knockouts of a gene provide evidence that pharmacological knockout of this gene may be safe. For example, LoF variants of PCSK9 were associated with strikingly low plasma levels of LDL-C, reduced CAD risk [105], and but no apparent adverse health consequences, thus providing reassurance that therapeutic neutralization of PCSK9 may be safe [106, 107]. Likewise, human knockouts of ANGPTL3 and APOC3 led to the development of pre- and clinical drugs for lowering serum levels of cholesterol and triglycerides, and the corresponding alleles related to hypolipidemia are under investigation for treatments using base editing. Increasing discoveries of such “experiments of nature” will be empowered by exome or whole genome sequencing in large-scale biobank cohorts [3]. More gene and allele targets relevant to dyslipidemia and atherosclerosis will emerge [3, 108,109,110,111].
4.3.2 Candidate Genes and Variants from Large-Scale Genetic Studies
Genome-wide association studies (GWASs) have discovered over 300 CAD loci and more than 900 loci of blood lipid traits including LDL-C, HDL-C, non-HDL-C, total cholesterol and triglycerides, unveiling novel variants, and genes and pathways underlying dyslipidemia and atherosclerosis with unprecedented speed and mechanistic complexity [20, 112, 113]. GWASs also rediscovered rare variants for dyslipidemia and atherosclerosis, suggesting that beyond these, drug targets are tagged by novel GWAS variants and gene candidates at the many loci associated with lipids and atherosclerosis, especially those loci overlapping for the two traits (Fig. 3). When we explored gene loci shared for CAD and lipids including LDL, TGs, TC, and HDL using the latest statistics of GWAS catalog, EMBL-EBI (2021), we identified 83 loci and classified the mapped genes into related pathophysiological pathways (Fig. 3). Surprisingly, other than the largest portion (~30%) of the genes directly involved lipid metabolism, many genes play roles in known pathways linked to CAD, such as inflammation, angiogenesis, and vascular remodeling. Genes for insulin resistance and glucose metabolism were also identified in our analysis, suggesting that novel genes and pathways for the disease are secondary to dysglycemic regulation. The convenience of CRISPR-based technologies will allow investigation of the novel genetic findings in a high-throughput manner.

Genes mapped to 83 shared loci of lipids and CAD GWAS loci and potentially related pathophysiological pathways of atherosclerosis. CAD, coronary artery disease
Furthermore, by testing causality harnessing genetic information, Mendelian randomization (MR) could identify specific genes as potential therapeutic target and assure efficacy and, importantly, safety before the initiation of drug development [114, 115]. Another genetic approach alerting adverse effect is termed phenome-wide association study (PheWAS), which tests associations of a genetic variant or a gene with hundreds of clinical phenotypes linked to all the organ systems [116, 117]. Using integrative data of individual’s genome and electronic health record from large biobank cohort, PheWAS could assess for desirable and adverse clinical outcomes linked to variant and gene of interest. MR and PheWAS provide reassurance for novel gene target selection in pre- and clinical investigations.
4.3.3 Driver Genes and Variants of Systems Genetic Studies
Although compelling efforts have been made to prioritize disease-associated genes utilizing approaches from molecular biology to GWAS, the genetic landscape of atherosclerosis and CAD is not fully elucidated. In the past decade, systems biology based on omic technologies accelerates the understanding of mechanisms underlying complex traits [118, 119]. Systems biology networks, genetic variations, and gene expression with other higher biological layers identify driver variants and genes for complex diseases. Targeting of the key drivers to modulate disease-associated gene or protein networks might enable correction of multiple pathogenic pathways in parallel. Genome editing technologies will play a crucial role in testing related hypothesis and therapeutic potentials [119].
5 Concluding Remarks and Future Perspectives
The possibility of manipulating DNA and RNA has advanced cardiovascular medicine, including understanding gene functions and genetic diseases, as well as the development of novel drug targets. Although the field is still in its infancy, the potentials are exemplified by clinical trials to treat sickle cell disease, to improve effectiveness of chimeric antigen receptor T-cell (CAR-T), or to reverse eye diseases [120]. A clinical trial of base editing targeting PCSK9 to treat heterozygous familial hypercholesterolemia (HeFH) started in July 2022 (clinicaltrials.gov_NCT05398029). Beyond these examples, many rare genetic disorders, in principle, will be treatable with CRISPR-based therapies.
Despite the exciting progress, many challenges should be tackled before its broader applications. First, tissue-specific delivery of genome editors has been a long-standing issue. Although AAV systems could allow relatively specific targeting in the liver, brain, muscle, and eye with low immunogenicity [121, 122], they should be further optimized, and many more tissues need to be considered. A new field of research exploiting nanoparticle-based delivery could provide alternative solutions. Second, current genome editing tools strictly rely on specific recognition sequences as well as specific binding sites on the target, such as the protospacer adjacent motif sequences for Cas proteins [45]. The absence of the assisting recognition sequence limits the targeting capability. Therefore, many research teams focus on discovering or engineering editing tools independent of such sequences. Third, substantial variability of editing efficacy depending on genetic loci and cell types was observed, which are partially caused by differences in chromatin accessibility and DNA repairing mechanisms throughout phases of cell cycle. Forth, off-target mutagenesis, although being rare, were detected within sequences of high similarity. While well-designed gRNAs are critical to minimize off-target events, advanced methods have been established to assess unwanted editing in a genome-wide fashion, such as BLISS, GUIDE-Seq, and DISCOVER-seq [123,124,125]. Finally, a long way has to be gone to fulfill regulatory guidelines and define cost reimbursement for these once-in-a-lifetime therapies. Of note, ongoing therapeutic testing of CRISPR aims to treat patients by modifying their somatic genome. The scientific and social challenges related to human germline editing are discussed elsewhere [126].
Nevertheless, gene editing therapies have to be evaluated carefully case-by-case in extensive pre- and clinical experimentations. Given the recent progress and efforts around the globe to tackle the related issues, genome editing will certainly expand into a new class of therapy to treat many diseases, including dyslipidemia and atherosclerosis.
References
Liao HK, Hatanaka F, Araoka T, Reddy P, Wu MZ, Sui Y, Yamauchi T, Sakurai M, O'Keefe DD, Nunez-Delicado E, Guillen P, Campistol JM, Wu CJ, Lu LF, Esteban CR, Izpisua Belmonte JC (2017) In vivo target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell 171(7):1495–1507
Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, Tokgozoglu L, Lewis EF (2019) Atherosclerosis. Nat Rev Dis Primers 5(1):56
Stitziel NO, Stirrups KE, Masca NG, Erdmann J, Ferrario PG, Konig IR, Weeke PE, Webb TR, Auer PL, Schick UM, Lu Y, Zhang H, Dube MP, Goel A, Farrall M, Peloso GM, Won HH, Do R, van Iperen E, Kanoni S, Kruppa J, Mahajan A, Scott RA, Willenberg C, Braund PS, van Capelleveen JC, Doney AS, Donnelly LA, Asselta R, Merlini PA, Duga S, Marziliano N, Denny JC, Shaffer CM, El-Mokhtari NE, Franke A, Gottesman O, Heilmann S, Hengstenberg C, Hoffman P, Holmen OL, Hveem K, Jansson JH, Jockel KH, Kessler T, Kriebel J, Laugwitz KL, Marouli E, Martinelli N, McCarthy MI, Van Zuydam NR, Meisinger C, Esko T, Mihailov E, Escher SA, Alver M, Moebus S, Morris AD, Muller-Nurasyid M, Nikpay M, Olivieri O, Lemieux Perreault LP, AlQarawi A, Robertson NR, Akinsanya KO, Reilly DF, Vogt TF, Yin W, Asselbergs FW, Kooperberg C, Jackson RD, Stahl E, Strauch K, Varga TV, Waldenberger M, Zeng L, Kraja AT, Liu C, Ehret GB, Newton-Cheh C, Chasman DI, Chowdhury R, Ferrario M, Ford I, Jukema JW, Kee F, Kuulasmaa K, Nordestgaard BG, Perola M, Saleheen D, Sattar N, Surendran P, Tregouet D, Young R, Howson JM, Butterworth AS, Danesh J, Ardissino D, Bottinger EP, Erbel R, Franks PW, Girelli D, Hall AS, Hovingh GK, Kastrati A, Lieb W, Meitinger T, Kraus WE, Shah SH, McPherson R, Orho-Melander M, Melander O, Metspalu A, Palmer CN, Peters A, Rader D, Reilly MP, Loos RJ, Reiner AP, Roden DM, Tardif JC, Thompson JR, Wareham NJ, Watkins H, Willer CJ, Kathiresan S, Deloukas P, Samani NJ, Schunkert H (2016) Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N Engl J Med 374(12):1134–1144
Authors/Task Force M, Guidelines ESCCfP, Societies ESCNC (2019) 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Atherosclerosis 290:140–205
Braenne I, Kleinecke M, Reiz B, Graf E, Strom T, Wieland T, Fischer M, Kessler T, Hengstenberg C, Meitinger T, Erdmann J, Schunkert H (2016) Systematic analysis of variants related to familial hypercholesterolemia in families with premature myocardial infarction. Eur J Hum Genet 24(2):191–197
Schunkert H, Bourier F (2015) Deciphering unexplained familial dyslipidemias: do we have the tools? Circulation 8(2):250–252
Schmidt N, Dressel A, Grammer TB, Gouni-Berthold I, Julius U, Kassner U, Klose G, Konig C, Koenig W, Otte B, Parhofer KG, Reinhard W, Schatz U, Schunkert H, Steinhagen-Thiessen E, Vogt A, Laufs U, Marz W (2018) Lipid-modifying therapy and low-density lipoprotein cholesterol goal attainment in patients with familial hypercholesterolemia in Germany: the CaReHigh Registry. Atherosclerosis 277:314–322
Di Minno A, Lupoli R, Calcaterra I, Poggio P, Forte F, Spadarella G, Ambrosino P, Iannuzzo G, Di Minno MND (2020) Efficacy and safety of bempedoic acid in patients with hypercholesterolemia: systematic review and meta-analysis of randomized controlled trials. J Am Heart Assoc 9(15):e016262
Ripatti P, Ramo JT, Soderlund S, Surakka I, Matikainen N, Pirinen M, Pajukanta P, Sarin AP, Laurila PP, Ehnholm C, Salomaa V, Wilson RK, Palotie A, Freimer NB, Taskinen MR, Ripatti S (2016) The contribution of GWAS loci in familial dyslipidemias. PLoS Genet 12(5):e1006078
Klarin D, Damrauer SM, Cho K, Sun YV, Teslovich TM, Honerlaw J, Gagnon DR, DuVall SL, Li J, Peloso GM, Chaffin M, Small AM, Huang J, Tang H, Lynch JA, Ho YL, Liu DJ, Emdin CA, Li AH, Huffman JE, Lee JS, Natarajan P, Chowdhury R, Saleheen D, Vujkovic M, Baras A, Pyarajan S, Di Angelantonio E, Neale BM, Naheed A, Khera AV, Danesh J, Chang KM, Abecasis G, Willer C, Dewey FE, Carey DJ, Concato J, Gaziano JM, O’Donnell CJ, Tsao PS, Kathiresan S, Rader DJ, Wilson PWF, Assimes TL (2018) Genetics of blood lipids among ~300,000 multi-ethnic participants of the million veteran program. Nat Genet 50(11):1514–1523
Akioyamen LE, Genest J, Shan SD, Reel RL, Albaum JM, Chu A, Tu JV (2017) Estimating the prevalence of heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. BMJ Open 7(9):e016461
Cuchel M, Bruckert E, Ginsberg HN, Raal FJ, Santos RD, Hegele RA, Kuivenhoven JA, Nordestgaard BG, Descamps OS, Steinhagen-Thiessen E, Tybjaerg-Hansen A, Watts GF, Averna M, Boileau C, Boren J, Catapano AL, Defesche JC, Hovingh GK, Humphries SE, Kovanen PT, Masana L, Pajukanta P, Parhofer KG, Ray KK, Stalenhoef AF, Stroes E, Taskinen MR, Wiegman A, Wiklund O, Chapman MJ (2014) Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J 35(32):2146–2157
Lent-Schochet D, Jialal I (2021) Antilipemic agent bile acid sequestrants. In: StatPearls. StatPearls Publishing, Treasure Island
Schunkert H, Samani NJ (2015) Statin treatment: can genetics sharpen the focus? Lancet 385(9984):2227–2229
Ray KK, Bays HE, Catapano AL, Lalwani ND, Bloedon LT, Sterling LR, Robinson PL, Ballantyne CM, Trial CH (2019) Safety and efficacy of bempedoic acid to reduce LDL cholesterol. N Engl J Med 380(11):1022–1032
Sabatine MS (2019) PCSK9 inhibitors: clinical evidence and implementation. Nat Rev Cardiol 16(3):155–165
Libby P (2021) The changing landscape of atherosclerosis. Nature 592(7855):524–533
Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE, Barrett JH, Konig IR, Stevens SE, Szymczak S, Tregouet DA, Iles MM, Pahlke F, Pollard H, Lieb W, Cambien F, Fischer M, Ouwehand W, Blankenberg S, Balmforth AJ, Baessler A, Ball SG, Strom TM, Braenne I, Gieger C, Deloukas P, Tobin MD, Ziegler A, Thompson JR, Schunkert H (2007) Genomewide association analysis of coronary artery disease. N Engl J Med 357(5):443–453
Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, Stewart AF, Barbalic M, Gieger C, Absher D, Aherrahrou Z, Allayee H, Altshuler D, Anand SS, Andersen K, Anderson JL, Ardissino D, Ball SG, Balmforth AJ, Barnes TA, Becker DM, Becker LC, Berger K, Bis JC, Boekholdt SM, Boerwinkle E, Braund PS, Brown MJ, Burnett MS, Buysschaert I, Cardiogenics CJF, Chen L, Cichon S, Codd V, Davies RW, Dedoussis G, Dehghan A, Demissie S, Devaney JM, Diemert P, Do R, Doering A, Eifert S, Mokhtari NE, Ellis SG, Elosua R, Engert JC, Epstein SE, de Faire U, Fischer M, Folsom AR, Freyer J, Gigante B, Girelli D, Gretarsdottir S, Gudnason V, Gulcher JR, Halperin E, Hammond N, Hazen SL, Hofman A, Horne BD, Illig T, Iribarren C, Jones GT, Jukema JW, Kaiser MA, Kaplan LM, Kastelein JJ, Khaw KT, Knowles JW, Kolovou G, Kong A, Laaksonen R, Lambrechts D, Leander K, Lettre G, Li M, Lieb W, Loley C, Lotery AJ, Mannucci PM, Maouche S, Martinelli N, PP MK, Meisinger C, Meitinger T, Melander O, Merlini PA, Mooser V, Morgan T, Muhleisen TW, Muhlestein JB, Munzel T, Musunuru K, Nahrstaedt J, Nelson CP, Nothen MM, Olivieri O, Patel RS, Patterson CC, Peters A, Peyvandi F, Qu L, Quyyumi AA, Rader DJ, Rallidis LS, Rice C, Rosendaal FR, Rubin D, Salomaa V, Sampietro ML, Sandhu MS, Schadt E, Schafer A, Schillert A, Schreiber S, Schrezenmeir J, Schwartz SM, Siscovick DS, Sivananthan M, Sivapalaratnam S, Smith A, Smith TB, Snoep JD, Soranzo N, Spertus JA, Stark K, Stirrups K, Stoll M, Tang WH, Tennstedt S, Thorgeirsson G, Thorleifsson G, Tomaszewski M, Uitterlinden AG, van Rij AM, Voight BF, Wareham NJ, Wells GA, Wichmann HE, Wild PS, Willenborg C, Witteman JC, Wright BJ, Ye S, Zeller T, Ziegler A, Cambien F, Goodall AH, Cupples LA, Quertermous T, Marz W, Hengstenberg C, Blankenberg S, Ouwehand WH, Hall AS, Deloukas P, Thompson JR, Stefansson K, Roberts R, Thorsteinsdottir U, Erdmann J, Samani NJ (2011) Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet 43(4):333–338
Erdmann J, Kessler T, Munoz Venegas L, Schunkert H (2018) A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res 114(9):1241–1257
Silbernagel G, Scharnagl H, Kleber ME, Hoffmann MM, Delgado G, Stojakovic T, Gary T, Zeng L, Ritsch A, Zewinger S, Speer T, Schunkert H, Landmesser U, Marz W, Grammer TB (2020) Common APOC3 variants are associated with circulating ApoC-III and VLDL cholesterol but not with total apolipoprotein B and coronary artery disease. Atherosclerosis 311:84–90
Zeng L, Moser S, Mirza-Schreiber N, Lamina C, Coassin S, Nelson CP, Annilo T, Franzen O, Kleber ME, Mack S, Andlauer TFM, Jiang B, Stiller B, Li L, Willenborg C, Munz M, Kessler T, Kastrati A, Laugwitz KL, Erdmann J, Moebus S, NoThen MM, Peters A, Strauch K, MuLler-Nurasyid M, Gieger C, Meitinger T, Steinhagen-Thiessen E, MaRz W, Metspalu A, BjoRkegren JLM, Samani NJ, Kronenberg F, Muller-Myhsok B, Schunkert H (2021) Cis-epistasis at the LPA locus and risk of cardiovascular diseases. Cardiovasc Res 118(4):1088–1102
Stitziel NO, Khera AV, Wang X, Bierhals AJ, Vourakis AC, Sperry AE, Natarajan P, Klarin D, Emdin CA, Zekavat SM, Nomura A, Erdmann J, Schunkert H, Samani NJ, Kraus WE, Shah SH, Yu B, Boerwinkle E, Rader DJ, Gupta N, Frossard PM, Rasheed A, Danesh J, Lander ES, Gabriel S, Saleheen D, Musunuru K, Kathiresan S (2017) ANGPTL3 deficiency and protection against coronary artery disease. J Am Coll Cardiol 69(16):2054–2063
Valanti EK, Dalakoura-Karagkouni K, Siasos G, Kardassis D, Eliopoulos AG, Sanoudou D (2021) Advances in biological therapies for dyslipidemias and atherosclerosis. Metabolism 116:154461
Wirth T, Parker N, Yla-Herttuala S (2013) History of gene therapy. Gene 525(2):162–169
Al Mahmeed W, Bakir S, Beshyah SA, Morcos B, Wajih S, Horack M, Lautsch D, Ambegaonkar B, Brudi P, Baxter CA, Vyas A, Gitt AK (2019) Prevalence of lipid abnormalities and cholesterol target value attainment in patients with stable and acute coronary heart disease in the United Arab Emirates. Heart Views 20(2):37–46
Landmesser U, Poller W, Tsimikas S, Most P, Paneni F, Luscher TF (2020) From traditional pharmacological towards nucleic acid-based therapies for cardiovascular diseases. Eur Heart J 41(40):3884–3899
Rodriguez-Calvo R, Masana L (2019) Review of the scientific evolution of gene therapy for the treatment of homozygous familial hypercholesterolaemia: past, present and future perspectives. J Med Genet 56(11):711–717
Tsimikas S, Viney NJ, Hughes SG, Singleton W, Graham MJ, Baker BF, Burkey JL, Yang Q, Marcovina SM, Geary RS, Crooke RM, Witztum JL (2015) Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. Lancet 386(10002):1472–1483
Viney NJ, van Capelleveen JC, Geary RS, Xia S, Tami JA, Yu RZ, Marcovina SM, Hughes SG, Graham MJ, Crooke RM, Crooke ST, Witztum JL, Stroes ES, Tsimikas S (2016) Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 388(10057):2239–2253
Ray KK, Landmesser U, Leiter LA, Kallend D, Dufour R, Karakas M, Hall T, Troquay RP, Turner T, Visseren FL, Wijngaard P, Wright RS, Kastelein JJ (2017) Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N Engl J Med 376(15):1430–1440
Ray KK, Wright RS, Kallend D, Koenig W, Leiter LA, Raal FJ, Bisch JA, Richardson T, Jaros M, Wijngaard PLJ, Kastelein JJP (2020) Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol. N Engl J Med 382(16):1507–1519
Witztum JL, Gaudet D, Freedman SD, Alexander VJ, Digenio A, Williams KR, Yang Q, Hughes SG, Geary RS, Arca M, Stroes ESG, Bergeron J, Soran H, Civeira F, Hemphill L, Tsimikas S, Blom DJ, O'Dea L, Bruckert E (2019) Volanesorsen and triglyceride levels in familial chylomicronemia syndrome. N Engl J Med 381(6):531–542
Alexander VJ, Xia S, Hurh E, Hughes SG, O'Dea L, Geary RS, Witztum JL, Tsimikas S (2019) N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur Heart J 40(33):2785–2796
Ray RM, Hansen AH, Slott S, Taskova M, Astakhova K, Morris KV (2019) Control of LDL uptake in human cells by targeting the LDLR regulatory long non-coding RNA BM450697. Mol Ther 17:264–276
Fu BXH, Smith JD, Fuchs RT, Mabuchi M, Curcuru J, Robb GB, Fire AZ (2019) Target-dependent nickase activities of the CRISPR-Cas nucleases Cpf1 and Cas9. Nat Microbiol 4(5):888–897
Kessler T, Graf T, Hilgendorf I, Rizas K, Martens E, von Zur MC, Kraemer P, Meyer-Saraei R, Neumann FJ, Bode C, Laugwitz KL, Massberg S, Schunkert H, Weil J, Kastrati A, Sager HB (2020) Hospital admissions with acute coronary syndromes during the COVID-19 pandemic in German cardiac care units. Cardiovasc Res 116(11):1800–1801
Carusillo A, Mussolino C (2020) DNA damage: from threat to treatment. Cell 9:7
Hinnen A, Hicks JB, Fink GR (1978) Transformation of yeast. Proc Natl Acad Sci U S A 75(4):1929–1933
Kim YG, Cha J, Chandrasegaran S (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A 93(3):1156–1160
Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF (2010) Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186(2):757–761
Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, Potrel P, Bas C, Lemaire L, Galetto R, Lebuhotel C, Eyquem J, Cheung GW, Duclert A, Gouble A, Arnould S, Peggs K, Pule M, Scharenberg AM, Smith J (2015) Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res 75(18):3853–3864
Waseem Qasim PJ, Samarasinghe S, Ghorashian S, Zhan H, Stafford S, Butler K, Ahsan G, Gilmour K, Adams S, Pinner D, Chiesa R, Chatters S, Swift S, Goulden N, Peggs K, Thrasher AJ, Veys P, Pule M (2015) First clinical application of Talen engineered universal CAR19 T Cells in B-ALL. Blood 126:23
Doudna JA, Charpentier E (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346(6213):1258096
Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg-Gresham JL, Chang HY, Demirkan A, Den Hertog HM, Donnelly LA, Ehret GB, Esko T, Feitosa MF, Ferreira T, Fischer K, Fontanillas P, Fraser RM, Freitag DF, Gurdasani D, Heikkila K, Hypponen E, Isaacs A, Jackson AU, Johansson A, Johnson T, Kaakinen M, Kettunen J, Kleber ME, Li X, Luan J, Lyytikainen LP, Magnusson PK, Mangino M, Mihailov E, Montasser ME, Muller-Nurasyid M, Nolte IM, O’Connell JR, Palmer CD, Perola M, Petersen AK, Sanna S, Saxena R, Shah S, Shungin D, Sidore C, Song C, Strawbridge RJ, Surakka I, Tanaka T, Teslovich TM, Thorleifsson G, Van den Herik EG, Voight BF, Volcik KA, Waite LL, Wong A, Wu Y, Zhang W, Absher D, Asiki G, Barroso I, Been LF, Bolton JL, Bonnycastle LL, Brambilla P, Burnett MS, Cesana G, Dimitriou M, Doney AS, Doring A, Elliott P, Epstein SE, Eyjolfsson GI, Gigante B, Goodarzi MO, Grallert H, Gravito ML, Groves CJ, Hallmans G, Hartikainen AL, Hayward C, Hernandez D, Hicks AA, Holm H, Hung YJ, Illig T, Jones MR, Kaleebu P, Kastelein JJ, Khaw KT, Kim E, Klopp N, Komulainen P, Kumari M, Langenberg C, Lehtimaki T, Lin SY, Lindstrom J, Loos RJ, Mach F, McArdle WL, Meisinger C, Mitchell BD, Muller G, Nagaraja R, Narisu N, Nieminen TV, Nsubuga RN, Olafsson I, Ong KK, Palotie A, Papamarkou T, Pomilla C, Pouta A, Rader DJ, Reilly MP, Ridker PM, Rivadeneira F, Rudan I, Ruokonen A, Samani N, Scharnagl H, Seeley J, Silander K, Stancakova A, Stirrups K, Swift AJ, Tiret L, Uitterlinden AG, van Pelt LJ, Vedantam S, Wainwright N, Wijmenga C, Wild SH, Willemsen G, Wilsgaard T, Wilson JF, Young EH, Zhao JH, Adair LS, Arveiler D, Assimes TL, Bandinelli S, Bennett F, Bochud M, Boehm BO, Boomsma DI, Borecki IB, Bornstein SR, Bovet P, Burnier M, Campbell H, Chakravarti A, Chambers JC, Chen YD, Collins FS, Cooper RS, Danesh J, Dedoussis G, de Faire U, Feranil AB, Ferrieres J, Ferrucci L, Freimer NB, Gieger C, Groop LC, Gudnason V, Gyllensten U, Hamsten A, Harris TB, Hingorani A, Hirschhorn JN, Hofman A, Hovingh GK, Hsiung CA, Humphries SE, Hunt SC, Hveem K, Iribarren C, Jarvelin MR, Jula A, Kahonen M, Kaprio J, Kesaniemi A, Kivimaki M, Kooner JS, Koudstaal PJ, Krauss RM, Kuh D, Kuusisto J, Kyvik KO, Laakso M, Lakka TA, Lind L, Lindgren CM, Martin NG, Marz W, McCarthy MI, Mckenzie CA, Meneton P, Metspalu A, Moilanen L, Morris AD, Munroe PB, Njolstad I, Pedersen NL, Power C, Pramstaller PP, Price JF, Psaty BM, Quertermous T, Rauramaa R, Saleheen D, Salomaa V, Sanghera DK, Saramies J, Schwarz PE, Sheu WH, Shuldiner AR, Siegbahn A, Spector TD, Stefansson K, Strachan DP, Tayo BO, Tremoli E, Tuomilehto J, Uusitupa M, van Duijn CM, Vollenweider P, Wallentin L, Wareham NJ, Whitfield JB, Wolffenbuttel BH, Altshuler D, Ordovas JM, Boerwinkle E, Palmer CN, Thorsteinsdottir U, Chasman DI, Rotter JI, Franks PW, Ripatti S, Cupples LA, Sandhu MS, Rich SS, Boehnke M, Deloukas P, Mohlke KL, Ingelsson E, Abecasis GR, Daly MJ, Neale BM, Kathiresan S (2013) Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet 45(11):1345–1352
Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, Lin S, Kiani S, Guzman CD, Wiegand DJ, Ter-Ovanesyan D, Braff JL, Davidsohn N, Housden BE, Perrimon N, Weiss R, Aach J, Collins JJ, Church GM (2015) Highly efficient Cas9-mediated transcriptional programming. Nat Methods 12(4):326–328
Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F (2015) Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517(7536):583–588
Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154(2):442–451
Rees HA, Liu DR (2018) Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet 19(12):770–788
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, Maglott DR (2016) ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44(1):862–868
Edraki A, Mir A, Ibraheim R, Gainetdinov I, Yoon Y, Song CQ, Cao Y, Gallant J, Xue W, Rivera-Perez JA, Sontheimer EJ (2019) A compact, high-accuracy Cas9 with a dinucleotide PAM for in vivo genome editing. Mol Cell 73(4):714–726
Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, Zhang F (2017) RNA editing with CRISPR-Cas13. Science 358(6366):1019–1027
Liu C, Zhang L, Liu H, Cheng K (2017) Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J Control Release 266:17–26
Luther DC, Lee YW, Nagaraj H, Scaletti F, Rotello VM (2018) Delivery approaches for CRISPR/Cas9 therapeutics in vivo: advances and challenges. Expert Opin Drug Deliv 15(9):905–913
Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F (2015) In vivo genome editing using Staphylococcus aureus Cas9. Nature 520(7546):186–191
Wei T, Cheng Q, Min YL, Olson EN, Siegwart DJ (2020) Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat Commun 11(1):3232
Finn JD, Smith AR, Patel MC, Shaw L, Youniss MR, van Heteren J, Dirstine T, Ciullo C, Lescarbeau R, Seitzer J, Shah RR, Shah A, Ling D, Growe J, Pink M, Rohde E, Wood KM, Salomon WE, Harrington WF, Dombrowski C, Strapps WR, Chang Y, Morrissey DV (2018) A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep 22(9):2227–2235
Rosenblum D, Gutkin A, Kedmi R, Ramishetti S, Veiga N, Jacobi AM, Schubert MS, Friedmann-Morvinski D, Cohen ZR, Behlke MA, Lieberman J, Peer D (2020) CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci Adv 6:47
Sun W, Ji W, Hall JM, Hu Q, Wang C, Beisel CL, Gu Z (2015) Self-assembled DNA nanoclews for the efficient delivery of CRISPR-Cas9 for genome editing. Angew Chem Int Ed 54(41):12029–12033
Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen ZY, Liu DR (2015) Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol 33(1):73–80
Wang M, Zuris JA, Meng F, Rees H, Sun S, Deng P, Han Y, Gao X, Pouli D, Wu Q, Georgakoudi I, Liu DR, Xu Q (2016) Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc Natl Acad Sci U S A 113(11):2868–2873
Gao X, Tao Y, Lamas V, Huang M, Yeh WH, Pan B, Hu YJ, Hu JH, Thompson DB, Shu Y, Li Y, Wang H, Yang S, Xu Q, Polley DB, Liberman MC, Kong WJ, Holt JR, Chen ZY, Liu DR (2018) Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553(7687):217–221
Lee K, Conboy M, Park HM, Jiang F, Kim HJ, Dewitt MA, Mackley VA, Chang K, Rao A, Skinner C, Shobha T, Mehdipour M, Liu H, Huang WC, Lan F, Bray NL, Li S, Corn JE, Kataoka K, Doudna JA, Conboy I, Murthy N (2017) Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat Biomed Eng 1:889–901
Mout R, Ray M, Yesilbag Tonga G, Lee YW, Tay T, Sasaki K, Rotello VM (2017) Direct cytosolic delivery of CRISPR/Cas9-ribonucleoprotein for efficient gene editing. ACS Nano 11(3):2452–2458
Wang P, Zhang L, Xie Y, Wang N, Tang R, Zheng W, Jiang X (2017) Genome editing for cancer therapy: delivery of Cas9 protein/sgRNA plasmid via a gold nanocluster/lipid core-shell nanocarrier. Adv Sci 4(11):1700175
Alsaiari SK, Patil S, Alyami M, Alamoudi KO, Aleisa FA, Merzaban JS, Li M, Khashab NM (2018) Endosomal escape and delivery of CRISPR/Cas9 genome editing machinery enabled by nanoscale zeolitic imidazolate framework. J Am Chem Soc 140(1):143–146
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816–821
Sun X, Zhou X, Dong B, Wang C, Xiao X, Wang Y (2021) Generation of a gene-corrected isogenic iPSC line (AHQUi001-A-1) from a patient with familial hypertriglyceridemia (FHTG) carrying a heterozygous p.C310R (c.928 T > C) mutation in LPL gene using CRISPR/Cas9. Stem Cell Res 52:102230
Musunuru K, Sheikh F, Gupta RM, Houser SR, Maher KO, Milan DJ, Terzic A, Wu JC (2018) Induced pluripotent stem cells for cardiovascular disease modeling and precision medicine: a scientific statement from the American Heart Association. Circulation 11(1):e000043
Yang H, Wang H, Jaenisch R (2014) Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat Protoc 9(8):1956–1968
Yu H, Rimbert A, Palmer AE, Toyohara T, Xia Y, Xia F, LMR F, Chen Z, Chen T, Loaiza N, Horwitz NB, Kacergis MC, Zhao L, Consortium B, Soukas AA, Kuivenhoven JA, Kathiresan S, Cowan CA (2019) GPR146 deficiency protects against hypercholesterolemia and atherosclerosis. Cell 179(6):1276–1288
Jarrett KE, Lee C, De Giorgi M, Hurley A, Gillard BK, Doerfler AM, Li A, Pownall HJ, Bao G, Lagor WR (2018) Somatic editing of Ldlr with adeno-associated viral-CRISPR is an efficient tool for atherosclerosis research. Arterioscler Thromb Vasc Biol 38(9):1997–2006
Niimi M, Yang D, Kitajima S, Ning B, Wang C, Li S, Liu E, Zhang J, Eugene Chen Y, Fan J (2016) ApoE knockout rabbits: a novel model for the study of human hyperlipidemia. Atherosclerosis 245:187–193
Yuan T, Zhong Y, Wang Y, Zhang T, Lu R, Zhou M, Lu Y, Yan K, Chen Y, Hu Z, Liang J, Fan J, Cheng Y (2019) Generation of hyperlipidemic rabbit models using multiple sgRNAs targeted CRISPR/Cas9 gene editing system. Lipids Health Dis 18(1):69
Huang L, Hua Z, Xiao H, Cheng Y, Xu K, Gao Q, Xia Y, Liu Y, Zhang X, Zheng X, Mu Y, Li K (2017) CRISPR/Cas9-mediated ApoE-/- and LDLR-/- double gene knockout in pigs elevates serum LDL-C and TC levels. Oncotarget 8(23):37751–37760
Dong Z, Shi H, Zhao M, Zhang X, Huang W, Wang Y, Zheng L, Xian X, Liu G (2018) Loss of LCAT activity in the golden Syrian hamster elicits pro-atherogenic dyslipidemia and enhanced atherosclerosis. Metabolism 83:245–255
Fedoseienko A, Wijers M, Wolters JC, Dekker D, Smit M, Huijkman N, Kloosterhuis N, Klug H, Schepers A, Willems van Dijk K, Levels JHM, Billadeau DD, Hofker MH, van Deursen J, Westerterp M, Burstein E, Kuivenhoven JA, van de Sluis B (2018) The COMMD family regulates plasma LDL levels and attenuates atherosclerosis through stabilizing the CCC complex in endosomal LDLR trafficking. Circ Res 122(12):1648–1660
Bartuzi P, Billadeau DD, Favier R, Rong S, Dekker D, Fedoseienko A, Fieten H, Wijers M, Levels JH, Huijkman N, Kloosterhuis N, van der Molen H, Brufau G, Groen AK, Elliott AM, Kuivenhoven JA, Plecko B, Grangl G, McGaughran J, Horton JD, Burstein E, Hofker MH, van de Sluis B (2016) CCC- and WASH-mediated endosomal sorting of LDLR is required for normal clearance of circulating LDL. Nat Commun 7:10961
Zhao H, Li Y, He L, Pu W, Yu W, Li Y, Wu YT, Xu C, Wei Y, Ding Q, Song BL, Huang H, Zhou B (2020) In vivo AAV-CRISPR/Cas9-mediated gene editing ameliorates atherosclerosis in familial hypercholesterolemia. Circulation 141(1):67–79
Musunuru K, Lagor WR, Miano JM (2017) What do we really think about human germline genome editing, and what does it mean for medicine? Circulation 10:5
Delhove J, Osenk I, Prichard I, Donnelley M (2020) Public acceptability of gene therapy and gene editing for human use: a systematic review. Hum Gene Ther 31(1-2):20–46
Ibraheim R, Song CQ, Mir A, Amrani N, Xue W, Sontheimer EJ (2018) All-in-one adeno-associated virus delivery and genome editing by Neisseria meningitidis Cas9 in vivo. Genome Biol 19(1):137
Wang X, Raghavan A, Chen T, Qiao L, Zhang Y, Ding Q, Musunuru K (2016) CRISPR-Cas9 targeting of PCSK9 in human hepatocytes in vivo-brief report. Arterioscler Thromb Vasc Biol 36(5):783–786
Yin H, Song CQ, Suresh S, Wu Q, Walsh S, Rhym LH, Mintzer E, Bolukbasi MF, Zhu LJ, Kauffman K, Mou H, Oberholzer A, Ding J, Kwan SY, Bogorad RL, Zatsepin T, Koteliansky V, Wolfe SA, Xue W, Langer R, Anderson DG (2017) Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat Biotechnol 35(12):1179–1187
Ding Q, Strong A, Patel KM, Ng SL, Gosis BS, Regan SN, Cowan CA, Rader DJ, Musunuru K (2014) Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res 115(5):488–492
Zhang L, Wang L, Xie Y, Wang P, Deng S, Qin A, Zhang J, Yu X, Zheng W, Jiang X (2019) Triple-targeting delivery of CRISPR/Cas9 to reduce the risk of cardiovascular diseases. Angew Chem Int Ed 58(36):12404–12408
Li Q, Su J, Liu Y, Jin X, Zhong X, Mo L, Wang Q, Deng H, Yang Y (2021) In vivo PCSK9 gene editing using an all-in-one self-cleavage AAV-CRISPR system. Mol Ther 20:652–659
Chadwick AC, Wang X, Musunuru K (2017) In vivo base editing of PCSK9 (proprotein convertase subtilisin/kexin Type 9) as a therapeutic alternative to genome editing. Arterioscler Thromb Vasc Biol 37(9):1741–1747
Carreras A, Pane LS, Nitsch R, Madeyski-Bengtson K, Porritt M, Akcakaya P, Taheri-Ghahfarokhi A, Ericson E, Bjursell M, Perez-Alcazar M, Seeliger F, Althage M, Knoll R, Hicks R, Mayr LM, Perkins R, Linden D, Boren J, Bohlooly YM, Maresca M (2019) In vivo genome and base editing of a human PCSK9 knock-in hypercholesterolemic mouse model. BMC Biol 17(1):4
Zhang X, Zhao W, Nguyen GN, Zhang C, Zeng C, Yan J, Du S, Hou X, Li W, Jiang J, Deng B, McComb DW, Dorkin R, Shah A, Barrera L, Gregoire F, Singh M, Chen D, Sabatino DE, Dong Y (2020) Functionalized lipid-like nanoparticles for in vivo mRNA delivery and base editing. Sci Adv 6:34
Yuxi Chen SZ, Liu W, Wen J, Sihui H, Cao T, Sun H, Li Y, Huang L, Liu Y, Liang P, Huang J (2020) Development of highly efficient dual-AAV split adenosine base editor for in vivo gene therapy. Small Methods 4:2000309
Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A (2014) Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med 371(1):32–41
Musunuru K, Pirruccello JP, Do R, Peloso GM, Guiducci C, Sougnez C, Garimella KV, Fisher S, Abreu J, Barry AJ, Fennell T, Banks E, Ambrogio L, Cibulskis K, Kernytsky A, Gonzalez E, Rudzicz N, Engert JC, DePristo MA, Daly MJ, Cohen JC, Hobbs HH, Altshuler D, Schonfeld G, Gabriel SB, Yue P, Kathiresan S (2010) Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med 363(23):2220–2227
Saleheen D, Natarajan P, Armean IM, Zhao W, Rasheed A, Khetarpal SA, Won HH, Karczewski KJ, O’Donnell-Luria AH, Samocha KE, Weisburd B, Gupta N, Zaidi M, Samuel M, Imran A, Abbas S, Majeed F, Ishaq M, Akhtar S, Trindade K, Mucksavage M, Qamar N, Zaman KS, Yaqoob Z, Saghir T, Rizvi SNH, Memon A, Hayyat Mallick N, Ishaq M, Rasheed SZ, Memon FU, Mahmood K, Ahmed N, Do R, Krauss RM, MacArthur DG, Gabriel S, Lander ES, Daly MJ, Frossard P, Danesh J, Rader DJ, Kathiresan S (2017) Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature 544(7649):235–239
Guo M, Xu Y, Dong Z, Zhou Z, Cong N, Gao M, Huang W, Wang Y, Liu G, Xian X (2020) Inactivation of ApoC3 by CRISPR/Cas9 protects against atherosclerosis in hamsters. Circ Res 127(11):1456–1458
Chadwick AC, Evitt NH, Lv W, Musunuru K (2018) Reduced blood lipid levels with in vivo CRISPR-Cas9 base editing of ANGPTL3. Circulation 137(9):975–977
Rhee JW, Wu JC (2018) Dyslipidaemia: in vivo genome editing of ANGPTL3: a therapy for atherosclerosis? Nat Rev Cardiol 15(5):259–260
Trieu VN, McConathy WJ (1995) A two-step model for lipoprotein(a) formation. J Biol Chem 270(26):15471–15474
Boerwinkle E, Leffert CC, Lin J, Lackner C, Chiesa G, Hobbs HH (1992) Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J Clin Investig 90(1):52–60
Nordestgaard BG, Langsted A (2016) Lipoprotein (a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res 57(11):1953–1975
Leibundgut G, Scipione C, Yin H, Schneider M, Boffa MB, Green S, Yang X, Dennis E, Witztum JL, Koschinsky ML, Tsimikas S (2013) Determinants of binding of oxidized phospholipids on apolipoprotein (a) and lipoprotein (a). J Lipid Res 54(10):2815–2830
Bergmark C, Dewan A, Orsoni A, Merki E, Miller ER, Shin MJ, Binder CJ, Horkko S, Krauss RM, Chapman MJ, Witztum JL, Tsimikas S (2008) A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J Lipid Res 49(10):2230–2239
van der Steeg WA, Holme I, Boekholdt SM, Larsen ML, Lindahl C, Stroes ES, Tikkanen MJ, Wareham NJ, Faergeman O, Olsson AG, Pedersen TR, Khaw KT, Kastelein JJ (2008) High-density lipoprotein cholesterol, high-density lipoprotein particle size, and apolipoprotein A-I: significance for cardiovascular risk: the IDEAL and EPIC-Norfolk studies. J Am Coll Cardiol 51(6):634–642
Hughes SD, Rouy D, Navaratnam N, Scott J, Rubin EM (1996) Gene transfer of cytidine deaminase apoBEC-1 lowers lipoprotein(a) in transgenic mice and induces apolipoprotein B editing in rabbits. Hum Gene Ther 7(1):39–49
Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH (2006) Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 354(12):1264–1272
Zhao Z, Tuakli-Wosornu Y, Lagace TA, Kinch L, Grishin NV, Horton JD, Cohen JC, Hobbs HH (2006) Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet 79(3):514–523
Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR (2007) The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 193(2):445–448
Peloso GM, Lange LA, Varga TV, Nickerson DA, Smith JD, Griswold ME, Musani S, Polfus LM, Mei H, Gabriel S, Quarells RC, Altshuler D, Boerwinkle E, Daly MJ, Neale B, Correa A, Reiner AP, Wilson JG, Kathiresan S (2016) Association of exome sequences with cardiovascular traits among blacks in the Jackson Heart Study. Circulation 9(4):368–374
Abramowitz Y, Roth A, Keren G, Isakov O, Shomron N, Laitman Y, Weissglas-Volkov D, Arbel Y, Banai S, Finkelstein A, Friedman E (2016) Whole-exome sequencing in individuals with multiple cardiovascular risk factors and normal coronary arteries. Coron Artery Dis 27(4):257–266
Hu X, Chen L, Gong C, Guo J, Chen Y, Wang Q, Guo R, Li W, Hao C (2021) Whole exome sequencing for non-selective pediatric patients with hyperlipidemia. Gene 768:145310
Hixson JE, Jun G, Shimmin LC, Wang Y, Yu G, Mao C, Warren AS, Howard TD, Heide RSV, Van Eyk J, Wang Y, Herrington DM (2017) Whole exome sequencing to identify genetic variants associated with raised atherosclerotic lesions in young persons. Sci Rep 7(1):4091
Liu DJ, Peloso GM, Yu H, Butterworth AS, Wang X, Mahajan A, Saleheen D, Emdin C, Alam D, Alves AC, Amouyel P, Di Angelantonio E, Arveiler D, Assimes TL, Auer PL, Baber U, Ballantyne CM, Bang LE, Benn M, Bis JC, Boehnke M, Boerwinkle E, Bork-Jensen J, Bottinger EP, Brandslund I, Brown M, Busonero F, Caulfield MJ, Chambers JC, Chasman DI, Chen YE, Chen YI, Chowdhury R, Christensen C, Chu AY, Connell JM, Cucca F, Cupples LA, Damrauer SM, Davies G, Deary IJ, Dedoussis G, Denny JC, Dominiczak A, Dube MP, Ebeling T, Eiriksdottir G, Esko T, Farmaki AE, Feitosa MF, Ferrario M, Ferrieres J, Ford I, Fornage M, Franks PW, Frayling TM, Frikke-Schmidt R, Fritsche LG, Frossard P, Fuster V, Ganesh SK, Gao W, Garcia ME, Gieger C, Giulianini F, Goodarzi MO, Grallert H, Grarup N, Groop L, Grove ML, Gudnason V, Hansen T, Harris TB, Hayward C, Hirschhorn JN, Holmen OL, Huffman J, Huo Y, Hveem K, Jabeen S, Jackson AU, Jakobsdottir J, Jarvelin MR, Jensen GB, Jorgensen ME, Jukema JW, Justesen JM, Kamstrup PR, Kanoni S, Karpe F, Kee F, Khera AV, Klarin D, Koistinen HA, Kooner JS, Kooperberg C, Kuulasmaa K, Kuusisto J, Laakso M, Lakka T, Langenberg C, Langsted A, Launer LJ, Lauritzen T, Liewald DC, Lin LA, Linneberg A, Loos RJ, Lu Y, Lu X, Magi R, Malarstig A, Manichaikul A, Manning AK, Mantyselka P, Marouli E, Masca NG, Maschio A, Meigs JB, Melander O, Metspalu A, Morris AP, Morrison AC, Mulas A, Muller-Nurasyid M, Munroe PB, Neville MJ, Nielsen JB, Nielsen SF, Nordestgaard BG, Ordovas JM, Mehran R, O'Donnell CJ, Orho-Melander M, Molony CM, Muntendam P, Padmanabhan S, Palmer CN, Pasko D, Patel AP, Pedersen O, Perola M, Peters A, Pisinger C, Pistis G, Polasek O, Poulter N, Psaty BM, Rader DJ, Rasheed A, Rauramaa R, Reilly DF, Reiner AP, Renstrom F, Rich SS, Ridker PM, Rioux JD, Robertson NR, Roden DM, Rotter JI, Rudan I, Salomaa V, Samani NJ, Sanna S, Sattar N, Schmidt EM, Scott RA, Sever P, Sevilla RS, Shaffer CM, Sim X, Sivapalaratnam S, Small KS, Smith AV, Smith BH, Somayajula S, Southam L, Spector TD, Speliotes EK, Starr JM, Stirrups KE, Stitziel N, Strauch K, Stringham HM, Surendran P, Tada H, Tall AR, Tang H, Tardif JC, Taylor KD, Trompet S, Tsao PS, Tuomilehto J, Tybjaerg-Hansen A, van Zuydam NR, Varbo A, Varga TV, Virtamo J, Waldenberger M, Wang N, Wareham NJ, Warren HR, Weeke PE, Weinstock J, Wessel J, Wilson JG, Wilson PW, Xu M, Yaghootkar H, Young R, Zeggini E, Zhang H, Zheng NS, Zhang W, Zhang Y, Zhou W, Zhou Y, Zoledziewska M, Charge Diabetes Working G, Consortium EP-I, Consortium E-C, Danesh J, McCarthy MI, Cowan CA, Abecasis G, Deloukas P, Musunuru K, Willer CJ, Kathiresan S (2017) Exome-wide association study of plasma lipids in >300,000 individuals. Nat Genet 49(12):1758–1766
Tcheandjieu C, et al (2021) A large-scale multi-ethnic genome-wide association study of coronary artery disease. Research Square
Jansen H, Samani NJ, Schunkert H (2014) Mendelian randomization studies in coronary artery disease. Eur Heart J 35(29):1917–1924
Jansen H, Lieb W, Schunkert H (2016) Mendelian randomization for the identification of causal pathways in atherosclerotic vascular disease. Cardiovasc Drugs Ther 30(1):41–49
Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, Wang D, Masys DR, Roden DM, Crawford DC (2010) PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 26(9):1205–1210
Nikpay M, Mohammadzadeh S (2020) Phenome-wide screening for traits causally associated with the risk of coronary artery disease. J Hum Genet 65(4):371–380
Franzen O, Ermel R, Cohain A, Akers NK, Di Narzo A, Talukdar HA, Foroughi-Asl H, Giambartolomei C, Fullard JF, Sukhavasi K, Koks S, Gan LM, Giannarelli C, Kovacic JC, Betsholtz C, Losic B, Michoel T, Hao K, Roussos P, Skogsberg J, Ruusalepp A, Schadt EE, Bjorkegren JL (2016) Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science 353(6301):827–830
Rau CD, Lusis AJ, Wang Y (2020) Systems genetics for mechanistic discovery in heart diseases. Circ Res 126(12):1795–1815
Porteus MH (2019) A new class of medicines through DNA editing. N Engl J Med 380(10):947–959
Kotterman MA, Schaffer DV (2014) Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet 15(7):445–451
Samulski RJ, Muzyczka N (2014) AAV-mediated gene therapy for research and therapeutic purposes. Ann Rev Virol 1(1):427–451
Yan WX, Mirzazadeh R, Garnerone S, Scott D, Schneider MW, Kallas T, Custodio J, Wernersson E, Li Y, Gao L, Federova Y, Zetsche B, Zhang F, Bienko M, Crosetto N (2017) BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat Commun 8:15058
Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, Aryee MJ, Joung JK (2015) GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33(2):187–197
Wienert B, Wyman SK, Yeh CD, Conklin BR, Corn JE (2020) CRISPR off-target detection with DISCOVER-seq. Nat Protoc 15(5):1775–1799
Doudna JA (2020) The promise and challenge of therapeutic genome editing. Nature 578(7794):229–236
Acknowledgments
We acknowledge the Servier Medical Art for providing basic graph elements for our figures, PubMed for publication statistics, and NHGRI-EBI GWAS Catalog for the latest statistics of lipid traits and CAD.
Competing Financial Interests
The authors declare no competing financial interests.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Chen, Z., Lehertshuber, C., Schunkert, H. (2023). Genome Editing in Dyslipidemia and Atherosclerosis. In: Xiao, J. (eds) Genome Editing in Cardiovascular and Metabolic Diseases. Advances in Experimental Medicine and Biology, vol 1396. Springer, Singapore. https://doi.org/10.1007/978-981-19-5642-3_10
Download citation
DOI: https://doi.org/10.1007/978-981-19-5642-3_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-5641-6
Online ISBN: 978-981-19-5642-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)